
Abstract
Aims/hypothesis
Under normal physiological conditions, glucagon signalling is important in glucose homeostasis. Hyperglucagonaemia or altered insulin:glucagon ratio plays a role in maintaining hyperglycaemia in subjects with type 2 diabetes. It has been reported that glucagon receptor knockout (Gcgr −/−) mice develop normally and have lower plasma glucose on a normal diet. The goal of the current research was to further investigate the role of glucagon signalling in metabolic control and glucose homeostasis.
Methods
Gcgr −/− mice were challenged with a high-fat diet (HFD) and with streptozotocin, which induces beta cell damage. They were then analysed for whole-body and serum metabolic phenotypes as well as pancreatic islet morphology.
Results
In comparison with wild-type mice, Gcgr −/− mice exhibited decreased body weight and food intake, reduced plasma glucose levels, and improved oral and intraperitoneal glucose tolerance. Elevated glucagon-like peptide-1 levels and reduced gastric emptying were also observed in Gcgr −/− mice, which also had reduced HFD-induced hyperinsulinaemia and hyperleptinaemia, and were resistant to the development of hepatic steatosis. In addition, Gcgr −/− mice were resistant to STZ-induced hyperglycaemia and pancreatic beta cell destruction.
Conclusions/interpretation
This study demonstrates that blocking glucagon signalling by targeted Gcgr gene deletion leads to an improvement in metabolic control in this mouse model.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Glucagon is a 29-amino acid peptide hormone secreted by alpha cells of the pancreatic islets. This hormone is secreted into portal blood supply and acts as a counter-regulatory hormone to insulin. Acting through the glucagon receptor (GCGR), a seven-transmembrane G-protein coupled receptor, glucagon increases hepatic glucose output during fasting, a process critically important in maintaining euglycaemic states under normal physiological conditions. On the other hand, hyperglucagonaemia has been known to be associated with the maintenance of hyperglycaemia and type 2 diabetes under pathophysiological conditions [1].
It has been reported that the absolute level of glucagon or the ratio of glucagon:insulin are often elevated in diabetic animals and human subjects [2–4]. Chronic hyperglucagonaemia is correlated with and is, at least partially, responsible for increased hepatic glucose output and hyperglycaemia in type 2 diabetes [5]. While the level of glucagon decreases immediately after a meal under normal physiological conditions, it is either not suppressed or even elevated in subjects with type 2 diabetes [6–8]. Furthermore, experimental suppression of postprandial hyperglucagonaemia corrects postprandial hyperglycaemia in subjects with type 2 diabetes [9].
Inhibition of glucagon signalling represents one of the potential approaches to the treatment of type 2 diabetes [1, 10]. Gcgr knockout mice (Gcgr −/− mice) have lower plasma glucose without hypoglycaemia [11, 12]. High-affinity glucagon-neutralising antibodies that effectively reduced free glucagon reduced glycaemia in animal models [13–15]. Treatment with glucagon receptor antisense oligonucleotide improves glucose metabolism in ob/ob mice [16, 17]. Finally, both peptidyl and non-peptidyl glucagon receptor antagonists have been shown to lower blood glucose in animal models and to block glucagon-induced elevation of plasma glucose in human subjects [18–26].
While it is generally accepted that inhibition of glucagon signalling reduces plasma glucose by suppressing hepatic glucose production, emerging evidence suggests that inhibition of glucagon signalling may have additional benefits due to increased circulating levels of glucagon-like peptide 1 (GLP-1). Specifically, both Gcgr −/− mice and mice treated with Gcgr antisense oligonucleotides showed dramatically elevated circulating levels of both glucagon and GLP-1 apparently due to a compensatory response [11, 12, 16, 17]. Both GLP-1 and glucagon are encoded by the same preproglucagon gene but processed by different subtilisin-like proprotein convertases. While glucagon increases blood glucose, GLP-1 lowers postprandial glucose by promoting insulin secretion from pancreatic beta cells [1, 27]. The observations in Gcgr −/− mice and mice treated with Gcgr antisense oligonucleotide suggest that inhibition of the glucagon receptor has dual benefits from both glucagon receptor-mediated hepatic and GLP-1 receptor-mediated pancreatic effects [17]. However, it remains uncertain whether the elevated circulating GLP-1 levels associated with blockade of GCGR function do indeed alter beta cell function.
In the current study, we demonstrate that Gcgr −/− mice are resistant to obesity induced by a high-fat diet (HFD). In comparison with wild-type (WT) mice on a HFD, Gcgr −/− mice had decreased body weight, improved oral and intraperitoneal glucose tolerance, and were resistant to streptozotocin (STZ)-induced diabetes. This study suggests that inhibition of GCGR function improves metabolic control by multiple mechanisms in the mouse model.
Materials and methods
Animal maintenance Gcgr −/−
mice were bred and backcrossed for seven generations on a C57/BL6 background as previously described [12]. Male WT and Gcgr −/− mice (5 weeks of age) were housed 10 per cage in a room maintained at constant temperature (25°C). Mice had free access to matched low-fat diet (LFD) or HFD with 10% (Diet: D12450B) or 45% (Diet: 12451) of energy from fat, respectively (Research Diets, New Brunswick, NJ, USA). All animal procedures were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of Merck Research Laboratories, Rahway, NJ, USA. All mice were bred at and obtained from Taconic Farm (Germantown, NY, USA).
Food intake measurement
Mice were housed individually and allowed to acclimatise to this condition for 1 week before food intake measurements were made. Both WT and Gcgr −/− mice were ~9 weeks of age at the onset of the food intake study, and were maintained on a LFD or HFD. Free access to food was provided, with consumption measured each morning (at 10.00 h) for a period of 4 days.
Glucose tolerance test
Mice were fed a HFD or LFD for 3 months and were fasted for 4 h prior to glucose tolerance tests. Gcgr −/− and WT mice were dosed with glucose at 2 g/kg of body weight either orally or intraperitoneally. Glucose levels were measured from tail bleeds with a glucometer (Lifescan, Milpitas, CA, USA) at specified time points after glucose administration. In addition, postprandial glucose measurement was made before the fasting period.
Measurement of metabolic parameters
Body composition and fat content were assessed by a Fat/Lean Mice Whole-Body Magnetic Resonance Analyser (Bruker, Billerica, MA, USA). Plasma insulin, leptin, and corticosterone were measured with ELISA kits (Alpco, Windham, NC, USA). Triacylglycerol and NEFA were measured with ELISA kits (Roche Diagnostic, Indianapolis, Indiana, USA and Wako, Neuss, Germany, respectively). Glucagon and GLP-1(7–36) were also measured with ELISA kits (Linco Research Immunoassay, St. Charles, MO, USA). Liver glycogen levels were determined by a method previously described [28].
Xylose absorption test
After 4 h of fasting, Gcgr −/− and WT mice were orally gavaged with xylose at 1 g/kg. Blood was collected from tail bleeds after xylose administration for determination of xylose concentration. Xylose assay was performed using 5 μl serum samples or standard (xylose 45 mg/ml stock solution serially diluted with serum) placed in 12 × 75 mm borosilicate test-tubes. Next, 500 μl of phloroglucinol (1,3,5-trihydroxybenzene) reagent (5 mg/ml in HCl/acetic acid) was added to each tube and incubated for 4 min at 105°C. Samples were cooled to room temperature before reading via spectrophotometer at 554 nm.
Streptozotocin treatment in mice
Mice fed either a LFD or HFD for 6 months were treated with a single intraperitoneal injection of either vehicle (saline) or STZ (Sigma Chemical, St. Louis, MO, USA) at 100 mg/kg and 125 mg/kg, respectively. The animals were then continued on the same diet for another 20 days during which some of the metabolic parameters were measured. At the end of the experiment, the animals were killed for measurement of additional parameters.
Histological and immunohistochemical analysis
Liver tissue was fixed in fixative (Prefer; Anatech, Battle Creek, MI, USA) and embedded in paraffin according to standard haematoxylin/eosin and Oil Red O staining procedure. Following removal the entire pancreas was fixed for 48 h in 4% paraformaldehyde in 0.01 mol/l phosphate buffer pH 7.3 and embedded in paraffin blocks. Sections (7 μm) were cut and mounted on coated glass slides (Plus-X; Fisher Scientific, Hampton, NH, USA), deparaffinised and rehydrated through xylene, ethanol and PBS. Prior to immuno-labelling the sections were treated sequentially with 3% hydrogen peroxide, citrate based antigen retrieval solution (Biogenex, San Ramon, CA, USA), 0.1% Triton X-100 and TSA blocking dolution (Perkin Elmer, Boston, MA, USA). The sections were then immuno-labelled by sequential 60-min incubations with rabbit anti-glucagon (Biogenex, San Ramon, CA, USA), Cy3-conjugated donkey anti-rabbit IgG (Jackson Labs, West Grove, PA, USA), guinea pig anti-insulin (Dako, Carpinteria, CA, USA) and Cy2-conjugated donkey anti-guinea pig IgG (Jackson Labs). All antibody incubations and washes were facilitated using the Shandon Coverplate system (Thermo Electron, Pittsburgh, PA, USA). After the final wash, coverslips were mounted using PermaFluor aqueous mounting media (Thermo Electron). The resultant fluorescently labelled slides were examined and digital images captured in a Zeiss Axioplan II microscope equipped with a Slidebook Image analysis workstation (Intelligent Imaging Innovations, Denver, CO, USA).
Statistics
Data are expressed as means ± SEM. Statistical analysis was conducted using Student’s t test or ANOVA. Statistical significance was defined as p < 0.05.
Results
Gcgr −/− mice are resistant to diet-induced obesity
Male C57/B6 (WT) mice and Gcgr −/− mice were fed a LFD or HFD for 12 weeks and plasma parameters and other characterisations are show in Table 1. As shown in Fig. 1a, WT mice on LFD were slightly but not significantly heavier than Gcgr −/− mice. In contrast, the WT mice on HFD were about 30% heavier than their Gcgr −/− counterparts (Fig. 1b). Whole-body nuclear magnetic resonance analysis indicated that WT mice on HFD had significantly higher body fat composition than the WT mice on LFD (35% vs 23% total body mass, p < 0.05) (Table 1). Although Gcgr −/− mice on HFD also had significantly higher body fat composition than the Gcgr −/− mice on LFD, the body fat composition of Gcgr −/− mice was significantly lower than that of the WT mice, regardless of diet. The nuclear magnetic resonance analysis also showed that, in comparison with the WT mice, Gcgr −/− mice on both the LFD and HFD exhibited a 10 to 20% higher level of lean mass (Table 1). Taken together, these results clearly demonstrate that Gcgr −/− mice are leaner on LFD and are largely resistant to HFD-induced body weight gain. A food intake study showed that the WT mice consumed a similar amount of energy per day on LFD and HFD (Fig. 1c). In contrast, Gcgr −/− mice consumed slightly but significantly lower amounts of energy on HFD than on LFD. Considering that on HFD the WT mice consumed about 30% more energy than Gcgr −/− mice (Fig. 1b) and that WT mice gained about 30% more weight than the Gcgr −/− mice (Fig. 1c), we conclude that the difference in body weight between the WT and the Gcgr −/− mice on HFD can be explained largely by the differences in their food intake.

Gcgr −/− mice are resistant to HFD-induced obesity and insulin resistance. WT (filled circles) and Gcgr −/− (open circles) mice were fed a LFD (10% energy as fat) and at 2 months either continued on the LFD or switched to a LFD or a HFD (45% energy as fat) for 11 weeks. Body weights of the animals during the 11 week period on LFD (a) and HFD (b) are shown. c Average food consumption per mouse per day during a 4-day period. Mice were being fed with LFD or HFD for 4 weeks at the time of this study. n = 8 per group. *p < 0.05 in t test comparing WT with Gcgr −/− on the same diet; # p < 0.05 comparing same mice of the same genotype on LFD with those on HFD
Terminal evaluation of the epididymal white adipose tissue and interscapular brown adipose tissue showed that the WT mice on HFD had significantly higher white adipose tissue and brown adipose tissue mass than the WT mice on LFD (Fig. 2). In contrast, Gcgr −/− mice on HFD had slightly but not significantly higher white adipose tissue and brown adipose tissue mass than the Gcgr −/− mice on LFD (Fig. 2). Finally, Gcgr −/− mice had significantly lower white adipose tissue and brown adipose tissue mass than the WT mice on both HFD and LFD (Fig. 2). Consistent with the reduced adipose tissue weight, plasma leptin levels were significantly lower in Gcgr −/− mice, both on LFD and, more strikingly, on HFD (Table 1).

Gcgr −/− mice are resistant to HFD-induced hypertrophy of white adipose tissue (WAT) and brown adipose tissue (BAT). WAT (a) and BAT (b) mass in the same WT and Gcgr −/− mice fed either a LFD or HFD for 12 weeks. Fat mass was determined by nuclear magnetic resonance. Values are means ± SD. *p < 0.05 in t test comparing WT and Gcgr −/− mice on the same diet; # p < 0.05 in t test comparing like mice on LFD versus HFD. n = 8 per group
Liver histological preparations and Oil Red O staining demonstrated that the WT mice on HFD readily developed intrahepatocyte lipid deposits shown by the collection of Oil Red O positive spots in these intracellular locations (Fig. 3), suggesting the development of HFD-induced liver steatosis. In contrast, the liver of the Gcgr −/− mice on both LFD and HFD were devoid of steatosis. While Gcgr −/− mice had lower hepatic lipid content than the WT mice, the hepatic glycogen content was not statistically different between the Gcgr −/− mice and the WT mice under ambient conditions (Table 1).

Gcgr −/− mice are resistant to HFD-induced hepatic steatosis. Oil Red O staining of liver sections from WT and Gcgr −/− mice fed either LFD or HFD for 12 weeks. Arrow, lipid drops that stained positive by Oil Red O. The bars in all four panels indicate the scale of the images
Gcgr −/− mice have improved glucose homeostasis
WT mice on HFD had similar plasma glucose levels but about threefold higher levels of insulin than WT mice on LFD (Table 1), consistent with HFD-induced insulin resistance commonly observed in WT mice. In comparison with WT mice, Gcgr −/− mice on LFD had significantly lower levels of plasma glucose as well as insulin, suggesting that Gcgr −/− mice are more insulin-sensitive. Furthermore, in contrast to WT mice, Gcgr −/− mice on HFD did not develop hyperinsulinaemia. The circulating insulin levels in Gcgr −/− mice on HFD were actually lower than those of WT mice on LFD. As expected, there were significant elevations of plasma glucagon and GLP-1 levels in Gcgr −/− mice on both diets. There were no significant changes in plasma triacylglycerol, NEFA, or corticosterone in Gcgr −/− mice compared with WT mice.
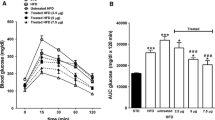
OGTTs were performed on WT and Gcgr −/− mice fed either LFD or HFD for 3 months. Results from LFD-fed mice showed that WT mice had significantly higher glycaemic excursions than their Gcgr −/− counterparts (Fig. 4a). Similar results were obtained from another set of experiments comparing the WT with the Gcgr −/− mice on HFD (Fig. 4c). Moreover, when intraperitoneal glucose tolerance tests were administered, the Gcgr −/− mice also had significantly reduced glucose excursion (Fig. 4b,d). These results suggest that Gcgr −/− mice are more insulin-sensitive than the WT mice and that the Gcgr −/− mice are resistant to diet-induced insulin resistance.

Gcgr −/− mice have improved glucose tolerance. OGTT (a) and intraperitoneal glucose tolerance tests (IPGTT) (b) on WT (circles) and Gcgr −/− (squares) mice fed LFD as described in Materials and methods. c, d OGTT and IPGTT tests, respectively, on WT and Gcgr −/− mice fed HFD for 4 weeks as described. Glucose was given at time 0. *p < 0.05 in t test comparing WT versus Gcgr −/− in the same diet. n = 8 per group
Gcgr −/− mice have reduced gastric emptying
While the lower plasma glucose and improved OGTT discussed above indicate that Gcgr −/− mice are more insulin-sensitive than WT mice, the interpretation of the observations was complicated by the fact that, in comparison with the WT mice, the Gcgr −/− mice have between ~100- and 75-fold higher levels of circulating biologically active GLP-1(7–36) and glucagon, respectively (Table 1). The elevated glucagon is not expected to have any biological consequences due to the absence of the glucagon receptor in these mice. On the other hand, GLP-1 is known, among other functions, to stimulate insulin secretion from the pancreas and slow gastric emptying. Therefore, it is likely that the reduced plasma glucose (Table 1) and improved OGTT (Fig. 4) in the Gcgr −/− mice are at least partially due to slower gastric emptying consequential to elevated circulating GLP-1 levels.
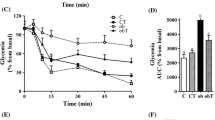
To compare gastric emptying between WT and Gcgr −/− mice, we employed a xylose absorption assay which has been used to determine intestinal glucose absorption under various conditions in humans, primates and rodents [29–31]. The assay was performed on both WT and Gcgr −/− mice. Xylose is used in the assay because it is absorbed like glucose but not metabolised. Therefore, xylose levels in plasma reflect glucose absorption. The results showed that, in comparison with WT mice, the Gcgr −/− mice had significantly slower xylose absorption (Fig. 5), indicating that Gcgr −/− mice indeed have slower gastric emptying.

Gastric emptying/intestinal motility in Gcgr −/− mice. WT (open symbols) and Gcgr −/− (filled symbols) mice fed the LFD were dosed orally with xylose at 1 g per kg body weight. Blood samples were obtained by tail vein bleeding at 20, 40, 60 and 120 min post dosing. Xylose levels in the blood were assayed as described in Materials and methods. Values are mean ± SD. n = 8 per group. *p < 0.05 in t test comparing WT with Gcgr −/− on the same diet
Gcgr −/− mice are resistant to streptozotocin-induced hyperglycaemia and beta cell injury
Streptozotocin is a selective pancreatic beta cell toxin widely used to induce experimental diabetes in animals. The insulinopenic effect of STZ resembles the impaired insulin secretion associated with insufficient beta cell function in type 2 diabetes. In this study, mice were fed either a LFD or a HFD, followed by an intraperitoneal injection of a moderate dose of STZ to induce hyperglycaemia. Mice fed the LFD were exposed to a 125 mg/kg dose of STZ, while the HFD-fed groups received a 100 mg/kg dose of STZ. The mice on the HFD were given a lower dose of STZ to compensate for the anticipated synergistic effects of HFD and STZ in negatively affecting pancreatic beta cell function.
The results showed that, during the entire course of the experiment, saline-treated WT mice had slightly but significantly higher plasma glucose than the Gcgr−/− mice, regardless of the diet (Fig. 6), consistent with the observation shown in Table 1. STZ treatment led to progressive hyperglycaemia in the WT mice on both diets. On the other hand, the Gcgr −/− mice remained euglycaemic throughout the experiment regardless of STZ treatment and HFD (Fig. 6). Subsequent analysis showed that STZ treatment sharply reduced circulating insulin levels in the WT mice, particularly those on HFD (Fig. 7a). On the other hand, STZ treatment had no detectable effects on the circulating insulin levels in the Gcgr −/− mice (Fig. 7b). Immunohistochemical analysis was performed on pancreatic tissues collected from WT and Gcgr −/− mice. When fed on LFD, the islets in the Gcgr −/− mice were significantly larger in size and richer in glucagon-positive alpha cells than islets from the WT mice (Fig. 7c). This is consistent with previously reported alpha cell hyperplasia in the pancreas of the Gcgr −/− mice [12]. HFD feeding expectedly resulted in hyperinsulinaemia in the WT mice (Fig. 7a) and beta cell hypertrophy (Fig. 7c in the current study). Quite strikingly, STZ treatment significantly altered the architecture of the islets in the WT mice but not in the Gcgr −/− mice (Fig. 7c). Specifically, STZ treatment dramatically reduced the size of the islets as well as the number of insulin-positive beta cells in WT mice, but not in the Gcgr −/− mice. Taken together, these results clearly demonstrate that the islets of the Gcgr −/− mice are protected from STZ-mediated toxicity and beta cell damage in vivo.

Gcgr −/− mice are resistant to STZ-induced hyperglycaemia on both LFD and HFD. WT and Gcgr −/− mice that had been fed either LFD (a) or a HFD (b) for 6 months were given a single injection of either saline (Sal) or STZ on day 0 at 125 and 100 mg/kg for mice on LFD and HFD, respectively. Filled circles, WT Sal; open circles, WT STZ; filled squares, Gcgr −/− Sal; open squares, Gcgr −/− STZ. Graphs show blood glucose levels on the subsequent 20 days post injection. *p < 0.05 in t test comparing WT mice injected with saline with STZ. # p < 0.05 in t test comparing WT with Gcgr −/− mice (n = 8) on the same diet

Gcgr −/− mice are resistant to STZ-induced pancreatic beta cell destruction. Plasma insulin levels (a WT, b Gcgr −/−) and representative images (c) of an immunohistochemical analysis of islet sections from WT and Gcgr −/− mice on either LFD or HFD and treated with saline (Sal) or STZ. Results were obtained 20 days post saline or STZ injection. Bars (a) labelled with a common letter are not significantly different from each other (at p > 0.05 in t test)
Discussion
Given the important role of glucagon in hepatic glucose production under normal as well pathophysiological conditions, inhibition of the glucagon signalling pathway should reduce plasma glucose in vivo. Indeed, lower plasma glucose has been shown in mice treated with glucagon antagonistic peptides, glucagon neutralising antibodies [13–15], glucagon receptor antisense oligonucleotides [16, 17], glucagon receptor non-peptidyl antagonists [18–25], and glucagon receptor knockout mice on normal chow [11, 12]. While the glucose-lowering effects derived from blockade of glucagon signalling are well documented, it remains less clear whether such inhibition has other beneficial or adverse consequences. This study investigated how glucagon receptor deficiency affects in vivo physiological characteristics, including body weight, beta cell function, and hepatic lipid and glycogen contents.
It was reported previously that Gcgr −/− mice have normal body weight when fed normal chow [12]. In the current study, we showed that Gcgr −/− mice are resistant to HFD-induced obesity. Since the degree of reduction in body weight gain (~30%) is largely parallel to that in food intake (Fig. 1c), it appears that reduced food intake is largely responsible for resistance to HFD-induced obesity in the Gcgr −/− mice. Consistent with reduced food intake, we also found that Gcgr −/− mice have reduced gastric emptying as revealed in a xylose absorption test. The mechanisms of reduced food intake and gastric emptying are not entirely clear. However, it could be at least in part due to the elevated circulating GLP-1 levels (Fig. 5), since GLP-1 has been shown to affect these parameters [32–34]. In addition to the current findings that Gcgr −/− have reduced body weight, reduced hepatic steatosis, and normal hepatic glycogen content, we previously showed that epididymal fat from Gcgr −/− mice exhibits enhanced lipolytic glycerol release in response to treatment with epinephrine ex vivo [12]. These results suggest that glucagon inhibition may have beneficial effects on body weight control in addition to lowering glucose levels.
Since glucagon reduces lipogenesis by multiple mechanisms, it was thought that reduction of glucagon signalling, i.e. via the use of glucagon receptor antagonists, may lead to the unfavourable accumulation of lipids in the liver [1]. In fact, glucagon has been proposed as a therapy for hepatic steatosis [35, 36]. However, the results from the current study show that Gcgr −/− mice have reduced hepatic lipid contents compared with WT mice on HFD (Fig. 3). Similarly, since glucagon plays a critical role in glycogen metabolism in liver, it was thought that inhibition of glucagon signalling may lead to glycogen accumulation in liver. The current study showed that Gcgr −/− mice have hepatic glycogen contents that are statistically indistinguishable from those in the WT mice under ambient fed conditions (Table 1). These results provide the evidence to alleviate certain theoretical concerns over some of the key potential adverse effects associated with inhibition of glucagon signalling.
Given that elevated glucagon:insulin ratios are known to be associated with insulin resistance in vivo [1], inhibition of glucagon signalling should improve insulin sensitivity in vivo. Indeed, it was reported previously in two independent studies that Gcgr −/− mice have improved insulin sensitivity on normal chow [11, 12]. These results are confirmed by the current study (Fig. 4a,b), which has further demonstrated that Gcgr −/− mice manifest improved insulin sensitivity when challenged with the high-fat feeding that induces insulin resistance (Fig. 4c,d).
Finally, results from the current study have also demonstrated that Gcgr −/− mice are resistant to STZ-induced beta cell destruction and hyperglycaemia (Figs. 6 and 7), suggesting that inhibition of glucagon signalling promotes beta cell survival and function. However, the potential implications of such pancreatic protective effects against STZ, a chemical toxin, on the development and treatment of type 2 diabetes require further investigation. The mechanism for such resistance to STZ remains unclear. It is possible that the resistance of Gcgr −/− mice to STZ may at least partially be mediated via increased circulating GLP-1. This would be consistent with a previous report that mice with dipeptidyl peptidase 4 gene deletion manifest increased circulating GLP-1 levels and are resistant to STZ-induced beta cell destruction [37]. It is also consistent with the observation that GLP-1 treatment prevents STZ-induced apoptosis of a beta-(INS-1)-cell line in vitro in a dose-dependent fashion [38]. Future studies will define the precise mechanism underlying the beta cell protection from STZ that was seen in Gcgr −/− mice.
In summary, the results from the current study indicate that GCGR deficiency in mice results in prevention of diet-induced obesity as well as protection against STZ-mediated beta cell destruction. These results suggest that antagonising glucagon function may provide beneficial metabolic effects beyond glucose control.
Abbreviations
- GCGR:
-
glucagon receptor
- Gcgr −/− mice:
-
Gcgr knockout mice
- GLP-1:
-
glucagon-like peptide-1
- HFD:
-
high-fat diet
- LFD:
-
low-fat diet
- STZ:
-
streptozotocin
- WT:
-
wild-type
References
Jiang G, Zhang BB (2003) Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab 284:E671–E678
Burcelin R, Katz EB, Charron MJ (1996) Molecular and cellular aspects of the glucagon receptor: role in diabetes and metabolism. Diabetes Metab 22:373–396
Toft I, Gerich JE, Jenssen T (2002) Autoregulation of endogenous glucose production during hyperglucagonemia. Metabolism 51:1128–1134
Unger RH (1985) Glucagon physiology and pathophysiology in the light of new advances. Diabetologia 28:574–578
Consoli A (1992) Role of liver in pathophysiology of NIDDM. Diabetes Care 15:430–441
Basu A, Alzaid A, Dinneen S, Caumo A, Cobelli C, Rizza RA (1996) Effects of a change in the pattern of insulin delivery on carbohydrate tolerance in diabetic and nondiabetic humans in the presence of differing degrees of insulin resistance. J Clin Invest 97:2351–2361
Butler PC, Rizza RA (1991) Contribution to postprandial hyperglycaemia and effect on initial splanchnic glucose clearance of hepatic glucose cycling in glucose-intolerant or NIDDM patients. Diabetes 40:73–81
Larsson H, Ahren B (2000) Islet dysfunction in insulin resistance involves impaired insulin secretion and increased glucagon secretion in postmenopausal women with impaired glucose tolerance. Diabetes Care 23:650–657
Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA (2000) Lack of suppression of glucagon contributes to postprandial hyperglycaemia in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab 85:4053–4059
Zhang BB, Moller DE (2000) New approaches in the treatment of type 2 diabetes. Curr Opin Chem Biol 4:461–467
Parker JC, Andrews KM, Allen MR, Stock JL, McNeish JD (2002) Glycemic control in mice with targeted disruption of the glucagon receptor gene. Biochem Biophys Res Commun 290:839–843
Gelling RW, Du XQ, Dichmann DS et al (2003) Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA 100:1438–1443
Brand CL, Jorgensen PN, Knigge U et al (1995) Role of glucagon in maintenance of euglycaemia in fed and fasted rats. Am J Physiol 269:E469–E477
Brand CL, Jorgensen PN, Svendsen I, Holst JJ (1996) Evidence for a major role for glucagon in regulation of plasma glucose in conscious, nondiabetic, and alloxan-induced diabetic rabbits. Diabetes 45:1076–1083
Brand CL, Rolin B, Jorgensen PN, Svendsen I, Kristensen JS, Holst JJ (1994) Immunoneutralization of endogenous glucagon with monoclonal glucagon antibody normalizes hyperglycaemia in moderately streptozotocin-diabetic rats. Diabetologia 37:985–993
Liang Y, Osborne MC, Monia BP et al (2004) Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 53:410–417
Sloop KW, Cao JX, Siesky AM et al (2004) Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest 113:1571–1581
Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi D (1982) Hyperglycaemia of diabetic rats decreased by a glucagon receptor antagonist. Science 215:1115–1116
Unson CG, Gurzenda EM, Merrifield RB (1989) Biological activities of des-His1[Glu9]glucagon amide, a glucagon antagonist. Peptides 10:1171–1177
Van Tine BA, Azizeh BY, Trivedi D et al (1996) Low level cyclic adenosine 3′,5′-monophosphate accumulation analysis of [des-His1, des-Phe6, Glu9] glucagon-NH2 identifies glucagon antagonists from weak partial agonists/antagonists. Endocrinology 137:3316–3322
Ling A, Hong Y, Gonzalez J et al (2001) Identification of alkylidene hydrazides as glucagon receptor antagonists. J Med Chem 44:3141–3149
Ling A, Plewe M, Gonzalez J et al (2002) Human glucagon receptor antagonists based on alkylidene hydrazides. Bioorg Med Chem Lett 12:663–666
Petersen KF, Sullivan JT (2001) Effects of a novel glucagon receptor antagonist (Bay 27-9955) on glucagon-stimulated glucose production in humans. Diabetologia 44:2018–2024
Qureshi SA, Rios Candelore M, Xie D et al (2004) A novel glucagon receptor antagonist inhibits glucagon-mediated biological effects. Diabetes 53:3267–3273
Dallas-Yang Q, Shen X, Strowski M et al (2004) Hepatic glucagon receptor binding and glucose-lowering in vivo by peptidyl and non-peptidyl glucagon receptor antagonists. Eur J Pharmacol 501:225–234
Shen DM, Zhang F, Brady EJ et al (2005) Discovery of novel, potent, and orally active spiro-urea human glucagon receptor antagonists. Bioorg Med Chem Lett 15:4564–4569
Baggio LL, Drucker DJ (2002) Harnessing the therapeutic potential of glucagon-like peptide-1: a critical review. Treat Endocrinol 1:117–125
Walaas O, Walaas E (1950) Effect of epinephrine on rat diaphragm. J Biol Chem 187:769–776
Fujita Y, Kojima H, Hidaka H, Fujimiya M, Kashiwagi A, Kikkawa R (1998) Increased intestinal glucose absorption and postprandial hyperglycaemia at the early step of glucose intolerance in Otsuka Long-Evans Tokushima Fatty rats. Diabetologia 41:1459–1466
D’Alessio DA, Vogel R, Prigeon R et al (1996) Elimination of the action of glucagon-like peptide 1 causes an impairment of glucose tolerance after nutrient ingestion by healthy baboons. J Clin Invest 97:133–138
Ehrenpreis ED, Salvino M, Craig RM (2001) Improving the serum D-xylose test for the identification of patients with small intestinal malabsorption. J Clin Gastroenterol 33:36–40
Drucker DJ (2001) Minireview: the glucagon-like peptides. Endocrinology. 142:521–527
Nakade Y, Tsukamoto K, Pappas TN, Takahashi T (2006) Central glucagon like peptide-1 delays solid gastric emptying via central CRF and peripheral sympathetic pathway in rats. Brain Res 1111:117–121
Nauck MA, Holst JJ, Willms B, Schmiegel W (1997) Glucagon-like peptide 1 (GLP-1) as a new therapeutic approach for type 2-diabetes. Exp Clin Endocrinol Diabetes 105:187–195
Hippen AR (2000) Glucagon as a potential therapy for ketosis and fatty liver. Vet Clin North Am Food Anim Pract 16:267–282
Hippen AR, She P, Young JW et al (1999) Alleviation of fatty liver in dairy cows with 14-day intravenous infusions of glucagon. J Dairy Sci 82:1139–1152
Conarello SL, Li Z, Ronan J et al (2003) Mice lacking dipeptidyl peptidase IV are protected against obesity and insulin resistance. Proc Natl Acad Sci USA 100:6825–6830
Pospisilik JA, Martin J, Doty T et al (2003) Dipeptidyl peptidase IV inhibitor treatment stimulates beta-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes 52:741–750
Duality of interest
All authors except M. J. Charron are or were (S. L. Conarello) employees of Merck & Co.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. L. Conarello, G. Jiang and J. Mu contributed equally to this work.
Rights and permissions
About this article
Cite this article
Conarello, S.L., Jiang, G., Mu, J. et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 50, 142–150 (2007). https://doi.org/10.1007/s00125-006-0481-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0481-3