
Abstract
Aims/hypothesis
Lineage conversion of non-beta cells into insulin-producing cells has been proposed as a therapy for the cure of diabetes. Glucagon-like peptide-1 (GLP-1) and its derivatives can induce beta cell neogenesis in vitro and beta cell mass expansion in vivo, but GLP-1 signalling has not been shown to regulate cell fate decisions in vivo. We therefore tested the impact of GLP-1 receptor (GLP1R) expression on beta cell differentiation in vivo.
Methods
Mice overexpressing GLP1R in pancreatic exocrine cells were generated by Cre-mediated recombination in sex-determining region Y-box 9 (SOX9)-expressing cells and then treated with exendin-4 and/or gastrin. Histological analysis was performed to detect cellular reprogramming from the exocrine lineage into insulin-producing cells.
Results
Whereas no newly generated beta cells were detected in the mice treated with exendin-4 alone, treatment with gastrin only induced the conversion of exocrine cells into insulin-producing cells. Furthermore, the overexpression of GLP1R, together with gastrin and exendin-4, synergistically promoted beta cell neogenesis accompanied by the formation of islet-like clusters. These newly generated beta cells expressed beta cell specific transcription factors, such as pancreatic and duodenal homeobox 1 (PDX1), NK6 homeobox 1 (NKX6.1) and musculoaponeurotic fibrosarcoma oncogene family A (MafA). These mice showed no histological evidence of pancreatitis or pancreatic dysplasia in their acini and had normal plasma amylase levels.
Conclusions/interpretation
Activation of GLP-1 and gastrin signalling induces beta cell neogenesis in the exocrine lineage without any deleterious pancreatic changes, which may lead to a potential therapy to cure diabetes by generating surrogate beta cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To date, insulin-producing cells have been generated from various differentiated cell types in adult organs, such as the liver, gut and pancreas, as well as from embryonic stem cells and induced pluripotent stem cells, which may lead to future cell therapies for the cure of diabetes. Owing to their shared developmental origins, non-beta cells in the pancreas, such as acinar cells and alpha cells, have attracted significant attention as potential candidates that might be easily reprogrammed into beta cells [1–3].
Ectopic expression of pancreas-specific transcription factors, such as pancreatic and duodenal homeobox 1 (PDX1), neurogenin 3 (NEUROG3) and musculoaponeurotic fibrosarcoma oncogene family A (MafA), has been shown to efficiently induce the reprogramming of non-endocrine pancreatic cells into other cell types, including insulin-producing cells [1, 4, 5]. Gene transfer methods may not be practical for clinical use owing to safety concerns, such as immune responses and random integration of the transferred gene into the genome. However, alternative methods for inducing beta cell reprogramming using humoral factors, such as incretins and growth factors, have recently received a large amount of attention [6].
Among the many humoral factors that regulate pancreas development, glucagon-like peptide-1 (GLP-1) and its analogue, exendin-4, have been reported to affect beta cell differentiation under in vitro and ex vivo conditions [7–9]. In addition, previous in vivo studies showed that the co-activation of GLP-1 and gastrin signalling increased the number of beta cells in mouse models of diabetes [10–12]. Thus, although the activation of GLP-1 signalling plays a role in beta cell mass maintenance, it remains unclear whether or not the in vivo activation of GLP-1 signalling directly induces beta cell neogenesis. To address this question, we applied genetic strategies based on a Cre/loxP-based lineage tracing system and generated a mouse model overexpressing GLP-1 receptor (GLP1R). Whereas no newly generated beta cells were detected by the activation of GLP-1 signalling alone, GLP-1 was able to facilitate beta cell neogenesis initiated by gastrin, although the reprogramming efficiency in our model was not as high as that of other mouse models [1, 13]. It is noted that activation of GLP-1 signalling did not induce pancreatitis and pancreatic dysplasia, suggesting that these safety concerns, which have been reported in previous clinical cases and in vivo studies of incretin-based therapies [14–16], were abated in our models. The present findings point to possible future therapies for diabetes via these signalling pathways.
Methods
Animals
NEUROG3-Timer, Sox9-CreER and ROSA26-lacZ reporter mice (R26R) were generated as described previously [17–19]. The transgenic construct CAG-CAT-Glp1r was assembled using a 2.9-kb CAG-CAT fragment cleaved from pCAG-CAT-lacZ [20], a 1.4-kb mouse Glp1r sequence obtained from mouse pancreas cDNA, a 1.3-kb internal ribosomal entry site (IRES)-egfp sequence cleaved from pAAV-Syn-AlstR- IRES2-egfp (plasmid #14895, Addgene, Cambridge, MA, USA) and a 0.2-kb fragment of the bovine growth hormone cassette gene. The 5.8-kb fragment of the CAG-CAT-Glp1r transgene was purified by electrophoresis and injected into fertilised eggs of B6D2F1 (BDF1) mice (Japan SLC, Hamamatsu, Japan). A total of 16 lines of CAG-CAT-Glp1r mice were generated and four lines highly expressing enhanced green-fluorescent protein (eGFP) were selected for the subsequent studies described herein. All animal procedures were approved by the Ethics Review Committee for Animal Experimentation of Osaka University Graduate School of Medicine. Transgenic mice were used after genotyping without randomisation. Most experiments were not blind. See electronic supplementary material (ESM) Methods for further details.
Sorting endocrine progenitors
The methods of NEUROG3-Timer mice generation, pancreatic cell dispersion and flow cytometry were as previously described [19, 21]. Briefly, pancreases from E17.5 NEUROG3-Timer embryos were treated with 0.05% trypsin/0.53 mmol/l EDTA (Invitrogen, Carlsbad, CA, USA) at 37°C for 5 min and inactivated by the addition of FBS. A MoFlo cell sorter (Dako Cytomation, Carpinteria, CA, USA) was used for sorting endocrine progenitors and other cell populations.
Induction of exogenous GLP1R expression
Tamoxifen (Sigma-Aldrich, St Louis, MO, USA) was prepared at 20 mg/ml in corn oil. For lineage tracing, Sox9-CreER;ROSA26-lacZ reporter strains or Sox9-CreER;CAG-CAT-Glp1r mice were injected subcutaneously with tamoxifen at 4 mg/20 g body weight at the age of 6 weeks, five times over a 2 week period. Four weeks after the first tamoxifen injection, the mice were injected with exendin-4 (100 μg/kg; Sigma-Aldrich) and/or human gastrin (1 mg/kg; Abbiotec, San Diego, CA, USA) intraperitoneally daily for 2 weeks, and then killed for further analyses at the age of 12 weeks. To inhibit GLP-1 signalling continuously, exendin-9-39 (150 pmol/kg/min; Sigma-Aldrich) was infused continuously using a mini-osmotic pump (Alzet, model 2002; Durect, Cupertino, CA, USA), which delivers the solution for up to 14 days.
Histology and immunostaining
Tissues were fixed in 4% paraformaldehyde in PBS at 4°C then washed in PBS, immersed in sucrose solution and embedded in Tissue-Tek (OCT Compound, Sakura, Japan) or processed routinely for paraffin embedding. Frozen and paraffin blocks were sectioned at 6 μm thickness and immunostained as previously described [22]. Although we used some representative images for histology, all data were used for quantification. See ESM Methods for further details.
TaqMan real-time PCR
Total RNA was isolated using the RNeasy Mini Plus kit (Qiagen, Valencia, CA, USA) and reverse-transcribed at 42°C for 30 min using the Verso cDNA synthesis kit (Thermo Fisher, Rockford, IL, USA) according to the manufacturer’s protocol. The expression levels of Glp1r (Mm00445292_m1), chymotrypsin-like elastase family member 1 (Cela1) (Mm00712898_m1) and pancreas-specific transcription factor 1a (Ptf1a) (Mm00479622_m1) mRNAs were quantified by TaqMan Real-Time PCR Assays (Applied Biosystems, Foster City, CA, USA), and normalised to glucuronidase, beta (Gusb, Mm00446953_m1). Data are expressed as mean ± SE.
Measurement of cAMP activity
Pancreatic tissues were collected from the mutant mice and control littermates at the age of 12 weeks. After homogenisation by polytron (Kinematica, Lucerne, Switzerland) in 0.1 M HCl, cAMP concentration (in supernatant fractions) was measured using a cAMP Direct Immunoassay Kit (#ab65355, Abcam, Cambridge, MA, USA).
Amylase activity
Plasma samples were collected from 12- and 24-week-old transgenic mice and stored at −20°C until use. Plasma amylase activity was measured using an Abaxis VetScan VS2 chemistry analyser (Abaxis, Union City, CA, USA).
Statistical analyses
Statistical analyses were performed using the JMP Statistical Discovery Software 9.0 (SAS Institute, Cary, NC, USA). Comparisons of two samples were performed by unpaired two-tailed t tests. Multiple groups were analysed by one-way ANOVA with a multiple comparison test and the Tukey–Kramer’s post-hoc test was used to compare different treatments in two mouse strains. A value of p < 0.05 was considered to indicate a statistically significant difference between two groups. Data are presented as the mean ± SE.
Results
GLP-1 receptor is expressed in pancreatic endocrine progenitors
Since GLP-1 and/or its analogue has been shown to induce beta cell differentiation from non-beta cells [7, 8], we hypothesised that GLP1R functions in pancreatic progenitors as well as in beta cells. To investigate the expression levels of Glp1r mRNA in endocrine progenitors during development, NEUROG3-expressing endocrine progenitors were sorted by fluorescence-activated cell sorting (FACS) from NEUROG3-Timer embryos, in which upstream and downstream sequences of human NEUROG3 drive the reporter protein DsRed-E5 that shifts its fluorescence peak from green to red over time. This enables the separation of endocrine progenitors from the more differentiated endocrine cells within a 6-h time window [19]. Transcriptome analysis by FACS revealed that Glp1r mRNA is expressed in green-fluorescent endocrine progenitors as well as in red-fluorescent mature endocrine cells (Fig. 1a, b), although the expression level of Glp1r is lower in the early progenitors compared with the islet cells from 4-week-old C57BL/6 mice, which includes more differentiated, but not fully mature, beta cells. These data suggest that GLP1R signalling pathway may function during pancreas development before beta cell specification.

GLP1R is expressed in pancreatic endocrine progenitors. (a) Pancreases from NEUROG-Timer embryos were dissected at embryonic day (E) 17.5 and sorted by FACS into four populations in a time-dependent manner. (b) The sorted cell populations were analysed by real-time PCR for Glp1r. Isolated islets from C57BL/6 mice at the age of 4 weeks (W) were analysed as a control. Expression levels of Glp1r were normalised to that of β-glucuronidase (n = 3 in each group)
Generation of transgenic mice that overexpress GLP-1 receptor
Since the expression of GLP1R is restricted to only a proportion of exocrine cells at much lower levels than that in beta cells [23], and Glp1r is expressed in pancreatic endocrine progenitors (Fig. 1a, b) and in newly generated beta cells in embryonic pancreases [24], we hypothesised that increased GLP-1 signalling may be able to induce the reprogramming of pancreatic exocrine cells into beta cells. To investigate this hypothesis, we generated a transgenic mouse line (CAG-CAT-Glp1r) in which ectopic expression of Glp1r is highly induced following Cre-mediated recombination (Fig. 2a). The egfp reporter gene was placed downstream of Glp1r cDNA via an IRES element, so that the exogenous GLP1R-expressing cells are labelled as green-fluorescent cells and their cell fates can be traced. The CAG-CAT-Glp1r mice were bred with the Sox9-CreER mice to generate double mutant Sox9-CreER;CAG-CAT-Glp1r (exo-Glp1r) mice. As a control, we used a reporter mouse model (Sox9-CreER;Rosa26-lacZ, exo-lacZ mice), which were generated by crossing Sox9-CreER mice with ROSA26-lacZ reporter strains (Fig. 2a) [17, 18]. Because the transcription factor sex-determining region Y-box 9 (SOX9) is expressed in pancreatic ductal cells during adulthood, exo-Glp1r and exo-lacZ mice treated with tamoxifen during adulthood can mark duct cells and their descendant acinar cells as eGFP- and β-galactosidase (β-gal)-expressing cells, respectively, whereas endocrine cells are never labelled [18].

Generation of transgenic mice that overexpress GLP1R. (a) Schematic representation of the Sox9-CreER;CAG-CAT-Glp1r (exo-Glp1r) mouse line and the exogenous expression of Glp1r after Cre-mediated recombination. Sox9-CreER;Rosa26-lacZ (exo-lacZ) mice were used as a control. (b–m) Immunofluorescent staining for (b, j) eGFP, (f) β-gal, (c, g, k) insulin, (d, h) DAPI, (l) Dolichos biflorus agglutinin (DBA) and (e, i, m) merged images in exo-Glp1r and exo-lacZ mice showed that eGFP and β-gal were expressed in acinar and duct cells, but not in the islets 6 weeks after the first tamoxifen injection. The efficiency of Cre-mediated recombination was comparable between exo-Glp1r and exo-lacZ mice. (j, l, m) Arrows indicate pancreatic ducts positive for eGFP. Scale bars, 100 μm. (n) Expression levels of Glp1r mRNA in the pancreases of exo-lacZ and exo-Glp1r mice were quantified by TaqMan real-time PCR. *p < 0.05; n = 4. (o) cAMP concentrations in the pancreases of exo-lacZ and exo-Glp1r mice. *p < 0.05; n = 4. White bars, exo-lacZ; black bars, exo-Glp1r
When Cre-mediated recombination was induced with tamoxifen in exo-Glp1r and exo-lacZ mice from 6 weeks of age, and immunostaining against eGFP and β-gal was performed 6 weeks after the first tamoxifen treatment, GFP- and β-gal-expressing cells were observed in ductal and acinar cells. The efficiency of Cre-mediated recombination of exo-Glp1r mice was comparable with that of exo-lacZ mice (percentage of eGFP- and β-gal-expressing cells among acinar cells: 51.8% ± 1.6% and 53.2% ± 5.1%, respectively, p = 0.83; among ductal cells: 46.9% ± 2.6% and 47.6% ± 2.8%, respectively, p = 0.88; Fig. 2b–m). Consistent with the immunostaining results for eGFP, Glp1r mRNA levels and cAMP concentrations in whole pancreases were significantly higher in exo-Glp1r mice than in exo-lacZ mice (Fig. 2n, o).
No beta cell neogenesis was observed by the activation of GLP-1 signalling alone
Although several reports have shown that GLP-1 and its derivatives can induce the generation of insulin-producing cells from pancreatic non-endocrine cells in vitro [7, 10] or can increase beta cell mass in vivo [25, 26], there is no direct evidence using genetic lineage tracing demonstrating that beta cell neogenesis is induced by the activation of GLP-1 signalling alone. To investigate whether or not the activation of GLP-1 signalling can affect the conversion of exocrine cells into beta cells, exendin-4 or PBS was administered to exo-Glp1r and exo-lacZ mice daily for 2 weeks (Fig. 3a). Double immunostaining against insulin and β-gal for exo-lacZ could not detect insulin/β-gal double-positive cells in the acinar area (Fig. 3b–e), consistent with a previous report showing no beta cell neogenesis during adulthood [18]. Furthermore, treatment of exo-Glp1r mice with exendin-4 or PBS did not result in any insulin/eGFP double-positive cells in the acinar area (Fig. 3f–i). Thus, activation of GLP-1 signalling failed to induce beta cell neogenesis in these mouse models.

No beta cell neogenesis was observed by the activation of GLP-1 signalling on its own. (a) Schematic of the experimental design for detecting beta cell neogenesis based on the Cre-loxP system. (b–i) Immunofluorescent staining for (b) β-gal, (f) eGFP, (c, g) insulin, (d, h) DAPI and (e, i) merged images in exo-lacZ mice treated with PBS and exo-Glp1r mice treated with exendin-4. All β-gal- and eGFP-positive cells were negative for insulin. Scale bar, 50 μm
Gastrin initiates beta cell neogenesis and activation of GLP-1 signalling enhances its effect
Gastrin is known to be expressed in the developing pancreas whereas it is not expressed in adult pancreas [27, 28], suggesting its possible role in pancreatic endocrine development. To investigate whether or not gastrin can induce beta cell neogenesis in the adult pancreas, exo-lacZ and exo-Glp1r mice were treated with gastrin and exendin-4 using the same protocol as above (Fig. 4a). Treatment with exendin-4 and gastrin significantly increased the percentage of beta cells and beta cell mass without affecting pancreatic weights (Fig. 4b–d). Immunostaining against insulin and β-gal detected insulin/β-gal double-positive cells in the acinar area when exo-lacZ mice were treated with gastrin alone (Fig. 4e–h). These results suggest that gastrin administration on its own induced beta cell neogenesis, although the efficiency of neogenesis was too low to affect total beta cell mass. Furthermore, co-administration of gastrin and exendin-4 significantly increased the number of insulin/β-gal double-positive cells compared with administration of gastrin alone (3.81 ± 0.45 and 1.79 ± 0.60/cm2 pancreas, respectively, p < 0.05; Fig. 4i). However, exo-Glp1r mice treated with gastrin demonstrated a significantly larger number of insulin/eGFP double-positive cells than insulin/β-gal cells in exo-lacZ mice treated with gastrin (3.40 ± 0.41 and 1.79 ± 0.60/cm2 pancreas, respectively, p < 0.05; Fig. 4i). Furthermore, co-injection of exendin-4 with gastrin dramatically increased the number of newly generated beta cells in exo-Glp1r mice, to 4.4-times higher than that in the exo-lacZ mutants (16.7 ± 1.7 and 3.81 ± 0.45/cm2 pancreas, respectively, p < 0.01; Fig. 4i). In addition, injection of both gastrin and exendin-4 elicited a 5-times greater increase in newly generated beta cells compared with gastrin injection alone (16.7 ± 1.7 and 3.40 ± 0.41/cm2 pancreas, respectively, p < 0.01; Fig. 4i). It is noted that islet-like-clusters consisting of two or more insulin-producing cells were observed in exo-Glp1r mice treated with exendin-4 and gastrin (Fig. 4j–n), but not in the other treatment groups. However, continuous inhibition of GLP-1 signalling using the GLP1R antagonist exendin-9-39, decreased the number of newly generated beta cells in mice treated with gastrin (Fig. 4o), suggesting that gastrin induces beta cell neogenesis at least partially through the activation of GLP-1 signalling. Taken together, these data suggest that the endogenous and genetic activation of GLP-1 signalling in the presence of gastrin enhances the reprogramming of non-endocrine cells into insulin-expressing cells, and this effect is synergistically increased by pharmacological activation by exendin-4.

Gastrin initiates beta cell neogenesis and activation of GLP-1 signalling enhances its effect. (a) Schematic of experimental design for detecting beta cell neogenesis. (b–d) Measurement of (b) total beta cell (insulin-positive) area per pancreas (i.e. beta cell proportion), (c) pancreatic weights and (d) beta cell mass of exo-lacZ and exo-Glp1r mice that were treated with exendin-4 (Ex-4) and/or gastrin (Gast). *p < 0.05; n = 4–6. Beta cell mass is the product of the beta cell proportion and pancreatic weight. (e–h) Immunofluorescent staining for (e) β-gal, (f) insulin, (g) DAPI and (h) merged images in exo-lacZ mice treated with gastrin alone. Double-positive cells for β-gal and insulin were observed, suggesting that exocrine-to-beta reprogramming was induced. (i) The number of insulin/eGFP (or β-gal) double-positive cells (i.e. newly generated beta cells) in the pancreases of the indicated mice. ND, not detected; *p < 0.05; n = 4–6. (j) Islet-like clusters were observed only in exo-Glp1r mice treated with exendin-4 and gastrin. While 56.9% of insulin/eGFP double-positive cells remained as single cells, 25.0% and 18.1% formed clusters with 2 and ≥3 insulin-positive cells, respectively. (k–n) Representative figures of islet-like clusters in exo-Glp1r mice treated with exendin-4 and gastrin, stained with (k) eGFP, (l) insulin, (m) DAPI and (n) a merged image. (k) Arrowheads indicate newly formed beta cells stained for both insulin and eGFP; arrows indicate acinar cells negative for eGFP. (o, p) The number of newly generated beta cells in the pancreases of (o) exo-lacZ and (p) exo-Glp1r mice treated with gastrin, together with or without exendin-9-39. *p < 0.05; n = 3. Scale bar, 50 μm
Newly generated insulin-producing cells express beta cell specific transcription factors
To characterise newly differentiated insulin-expressing cells derived from exocrine cells, fluorescent immunostaining was performed with antibodies against pancreas-specific transcription factors. In the pancreas of exo-Glp1r mice treated with exendin-4 and gastrin, >80% of the insulin/eGFP double-positive cells expressed NK6 homeobox 1 (NKX6.1), PDX1 and MafA (85.1%, 86.4% and 84.1%, respectively), which are pancreas-specific transcription factors that play essential roles in beta cell differentiation and maturation (Fig. 5a–e′) [29–33]. These neogenic insulin-producing cells did not express other endocrine hormones (glucagon, somatostatin and pancreatic polypeptide) or an exocrine enzyme (amylase; data not shown). These findings suggest that the activation of GLP-1 and gastrin signalling induces not only insulin transcription but also a certain level of beta cell maturation.

Newly generated insulin-producing cells express beta cell specific transcription factors. Immunofluorescent staining for (a, f, k, p, u, a′) eGFP, (b, g, l, q, v, b′) insulin, (c, h) PDX1, (m, r) NKX6.1, (w, c′) MafA and (e, j, o, t, y, e′) merged images in the pancreases of exo-Glp1r mice. Insulin/eGFP double-positive cells expressed (a–e) PDX1, (k–o) NKX6.1 and (u–y) MafA. (f–j, p–t, a′–e′) Pancreatic islets are shown as positive controls. (a, k, u) The dashed lines outline the clusters of newly formed beta cells. Scale bars, 50 μm
Activation of incretin signalling does not induce pancreatitis or pancreatic dysplasia
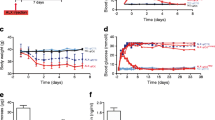
The activation of incretin signalling was reported to possibly be associated with pancreatitis and pancreatic dysplasia both in humans and animal models, although recent studies support the safety of incretin-based drugs [14–16, 34–36]. To test whether or not increased GLP-1 signalling in the pancreas results in undesired pathologies, pancreases from exo-Glp1r mice were examined microscopically. As shown in ESM Fig. 1a, there was no histological evidence of pancreatitis or pancreatic dysplasia in exendin-4 and gastrin-treated exo-Glp1r or exo-lacZ mice at 12 weeks of age. Immunostaining for Ki67 revealed comparable cell proliferation in the ducts and acini between the mutant mice and control littermates (ESM Fig. 1b, c). In addition, there was no significant increase in plasma amylase levels in the mutant mice (ESM Fig. 1d). Furthermore, TaqMan real-time PCR analysis showed no significant alterations in mRNA levels for elastase-1 (Cela1) and Ptf1a, a transcription factor that functions in acinar cells (ESM Fig. 1e, f) [37, 38]. In addition, pancreases of 24-week-old mutant mice revealed no abnormal findings in haematoxylin staining and no elevation of plasma amylase levels compared with C57BL/6 controls (data not shown). Thus, the activation of GLP-1 and gastrin signalling did not induce pancreatitis or pancreatic dysplasia, even with the ectopic activation of GLP-1 signalling in acinar cells.
Discussion
Although it has been reported that the activation of GLP-1 and gastrin signalling can induce beta cell neogenesis in vitro and increase beta cell mass in vivo [10, 25, 26, 39], there have been no direct in vivo studies of cell lineage analysing whether or not the activation of these pathways induces the cellular reprogramming of non-endocrine cells into insulin-producing cells. Here, we demonstrate direct in vivo evidence that gastrin administration alone initiates beta cell neogenesis within exocrine cells and that the concurrent activation of GLP-1 signalling synergistically enhances this effect, although the reprogramming efficiency in our model was lower than that of other reported strategies [1, 13].
GLP-1 and its derivatives, such as exendin-4 and liraglutide, have been shown in clinical studies to effectively improve glycaemic control with minimal risk of hypoglycaemia, and basic research has uncovered several mechanisms of their diabetes-protective effects on beta cells, such as the stimulation of insulin secretion, increase in beta cell replication and inhibition of beta cell apoptosis [40]. Based on the findings that GLP1R is detected in early endocrine progenitors of NEUROG3-Timer embryos (Fig. 1a, b), newly generated beta cells of Insulin-Timer embryos [24] and in acinar cells of the human adult pancreas [23], we hypothesised that GLP-1 may function in non-beta cells, including acinar and duct cells, and we activated the GLP-1 signalling pathway by the administration of exendin-4, a long-acting GLP1R agonist, and by overexpressing GLP1R in exocrine cells. Whereas in vitro studies have shown that GLP-1 and exendin-4 induce beta cell differentiation in the pancreatic tumour cell line AR42J [7, 9], our present study revealed that exendin-4 itself fails to initiate beta cell neogenesis in SOX9-lineage exocrine cells in vivo, even after the overexpression of GLP1R (Fig. 3f–i). SOX9-positive cells and their descendants are known to lose their capacity to differentiate into endocrine cells, even after in vivo regeneration experiments such as partial pancreatectomy and pancreatic duct ligation [18]. Thus, at least in our model, the activation of GLP-1 signalling alone is insufficient to reprogram SOX9-lineage cells into beta cells. Further optimisation of the dose and timing of administration of GLP-1 analogues is required for inducing beta cell neogenesis.
In contrast to the results of exendin-4 treatment, our mouse model directly demonstrated that beta cell neogenesis is induced by gastrin alone. Gastrin is expressed in the developing pancreas, particularly in endocrine populations [27, 28]. Although several reports have shown that combination therapy with gastrin and growth factors, such as EGF and TGF-α, increased beta cell mass [41–45], there was no evidence that gastrin could induce beta cell neogenesis as distinguished from beta cell replication. To our knowledge, this is the first report that a pharmacological dose of gastrin on its own initiated beta cell neogenesis independently of other humoral factors, such as EGF and GLP-1. Our results, taken together with previous reports, suggest that the activation of gastrin signalling combined with other humoral factors might provide a new therapeutic option that could be used to increase beta cell mass.
Recently, a clinical study of combination therapy with sitagliptin and lansoprazole in patients with type 1 diabetes failed to slow the progressive loss of insulin secretion [46]. This negative result might be due to an insufficient increase of GLP-1 or gastrin concentrations by these drugs. However, our direct approach with a GLP-1 analogue and gastrin induced the cellular reprogramming into beta cells, although the efficiency was not sufficient to cure streptozotocin-treated diabetic mice (data not shown). Further studies for optimising drug regimens could help to increase beta cell mass through beta cell neogenesis, which would lead to a cure for those diabetic patients who have lost most of their beta cells.
In the present study, Sox9-CreER mice were used to induce Cre-mediated recombination in SOX9-expressing cells, which resulted in the exogenous expression of GLP1R in duct cells and acinar cells [18]. Therefore, there are a number of possibilities for the origin of newly generated insulin-producing cells; namely, SOX9-expressing progenitors, differentiated duct cells or acinar cells. Several groups have demonstrated that acinar cells can be reprogrammed into beta cells under various conditions [1, 13, 47]. To clarify the origin of the newly generated insulin-producing cells, further studies should be performed using other Cre-expressing lines that induce Cre-mediated recombination specifically in acinar cells, such as Elastase1-CreER [48] and Ptf1a-CreER mice [47].
The mouse model overexpressing GLP1R helped us not only to investigate the beneficial effects of GLP-1 on beta cell differentiation, but also to evaluate possible pancreatic risks related to incretin therapy. The present data showed no obvious evidence of histological abnormalities or elevated amylase levels in GLP1R-overexpressing pancreases at the age of 24 weeks, 18 weeks after the induction of exogenous GLP1R in SOX9-lineage cells. Further careful observations for longer periods are required to apply our in vivo findings with this mouse model to human patients. In addition, it would be of interest to overexpress GLP1R in other tissues by crossing CAG-CAT-Glp1r mice with other Cre-expressing mouse lines, in order to test the physiological and/or pathological roles of GLP-1 signalling in various cell types in which its function remains controversial, such as pancreatic alpha cells [49] and thyroid C cells [50].
Abbreviations
- CELA1:
-
Chymotrypsin-like elastase family member 1
- eGFP:
-
Enhanced green fluorescent protein
- FACS:
-
Fluorescence-activated cell sorting
- β-Gal:
-
β-Galactosidase
- GLP-1:
-
Glucagon-like peptide-1
- GLP1R:
-
GLP-1 receptor
- IRES:
-
Internal ribosomal entry site
- MafA:
-
Musculoaponeurotic fibrosarcoma oncogene family A
- NEUROG3:
-
Neurogenin 3
- NKX6.1:
-
NK6 homeobox 1
- PDX1:
-
Pancreatic and duodenal homeobox 1
- PTF1A:
-
Pancreas-specific transcription factor 1a
- SOX9:
-
Sex-determining region Y-box 9
References
Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA (2008) In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature 455:627–632
Collombat P, Xu X, Ravassard P et al (2009) The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into α and subsequently β cells. Cell 138:449–462
Gu G, Dubauskaite J, Melton DA (2002) Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129:2447–2457
Gasa R, Mrejen C, Leachman N et al (2004) Proendocrine genes coordinate the pancreatic islet differentiation program in vitro. Proc Natl Acad Sci U S A 101:13245–13250
Miyatsuka T, Kaneto H, Shiraiwa T et al (2006) Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev 20:1435–1440
Pagliuca FW, Melton DA (2013) How to make a functional β-cell. Development 140:2472–2483
Zhou J, Wang X, Pineyro MA, Egan JM (1999) Glucagon-like peptide 1 and exendin-4 convert pancreatic AR42J cells into glucagon- and insulin-producing cells. Diabetes 48:2358–2366
Suzuki A, Nakauchi H, Taniguchi H (2003) Glucagon-like peptide 1 (1-37) converts intestinal epithelial cells into insulin-producing cells. Proc Natl Acad Sci U S A 100:5034–5039
Yew KH, Prasadan KL, Preuett BL et al (2004) Interplay of glucagon-like peptide-1 and transforming growth factor-beta signaling in insulin-positive differentiation of AR42J cells. Diabetes 53:2824–2835
Tamaki M, Fujitani Y, Uchida T, Hirose T, Kawamori R, Watada H (2010) Combination treatment of db/db mice with exendin-4 and gastrin preserves β-cell mass by stimulating β-cell growth and differentiation. J Diabetes Investig 1:172–183
Fosgerau K, Jessen L, Lind Tolborg J et al (2013) The novel GLP-1-gastrin dual agonist, ZP3022, increases β-cell mass and prevents diabetes in db/db mice. Diabetes Obes Metab 15:62–71
Suarez-Pinzon WL, Power RF, Yan Y, Wasserfall C, Atkinson M, Rabinovitch A (2008) Combination therapy with glucagon-like peptide-1 and gastrin restores normoglycemia in diabetic NOD mice. Diabetes 57:3281–3288
Baeyens L, Lemper M, Leuckx G et al (2014) Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol 32:76–83
Gier B, Matveyenko AV, Kirakossian D, Dawson D, Dry SM, Butler PC (2012) Chronic GLP-1 receptor activation by exendin-4 induces expansion of pancreatic duct glands in rats and accelerates formation of dysplastic lesions and chronic pancreatitis in the KrasG12D mouse model. Diabetes 61:1250–1262
Butler PC, Elashoff M, Elashoff R, Gale EA (2013) A critical analysis of the clinical use of incretin-based therapies: are the GLP-1 therapies safe? Diabetes Care 36:2118–2125
Li L, Shen J, Bala MM et al (2014) Incretin treatment and risk of pancreatitis in patients with type 2 diabetes mellitus: systematic review and meta-analysis of randomised and non-randomised studies. BMJ 348:g2366
Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21:70–71
Furuyama K, Kawaguchi Y, Akiyama H et al (2011) Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet 43:34–41
Miyatsuka T, Li Z, German MS (2009) Chronology of islet differentiation revealed by temporal cell labeling. Diabetes 58:1863–1868
Araki K, Araki M, Miyazaki J, Vassalli P (1995) Site-specific recombination of a transgene in fertilized eggs by transient expression of Cre recombinase. Proc Natl Acad Sci U S A 92:160–164
Miyatsuka T, Kosaka Y, Kim H, German MS (2011) Neurogenin3 inhibits proliferation in endocrine progenitors by inducing Cdkn1a. Proc Natl Acad Sci U S A 108:185–190
Schwitzgebel VM, Scheel DW, Conners JR et al (2000) Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 127:3533–3542
Pyke C, Heller RS, Kirk RK et al (2014) GLP-1 receptor localization in monkey and human tissue: novel distribution revealed with extensively validated monoclonal antibody. Endocrinology 155:1280–1290
Miyatsuka T, Matsuoka TA, Sasaki S et al (2014) Chronological analysis with fluorescent timer reveals unique features of newly generated β-cells. Diabetes 63:3388–3393
Xu G, Stoffers DA, Habener JF, Bonner-Weir S (1999) Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 48:2270–2276
Stoffers DA, Kieffer TJ, Hussain MA et al (2000) Insulinotropic glucagon-like peptide 1 agonists stimulate expression of homeodomain protein IDX-1 and increase islet size in mouse pancreas. Diabetes 49:741–748
Brand SJ, Fuller PJ (1988) Differential gastrin gene expression in rat gastrointestinal tract and pancreas during neonatal development. J Biol Chem 263:5341–5347
Suissa Y, Magenheim J, Stolovich-Rain M et al (2013) Gastrin: a distinct fate of neurogenin3 positive progenitor cells in the embryonic pancreas. PLoS One 8, e70397
Sander M, Sussel L, Conners J et al (2000) Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of β-cell formation in the pancreas. Development 127:5533–5540
Matsuoka TA, Artner I, Henderson E, Means A, Sander M, Stein R (2004) The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc Natl Acad Sci U S A 101:2930–2933
Olbrot M, Rud J, Moss LG, Sharma A (2002) Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U S A 99:6737–6742
Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H (2002) MafA is a glucose-regulated and pancreatic β-cell-specific transcriptional activator for the insulin gene. J Biol Chem 277:49903–49910
Matsuoka TA, Zhao L, Artner I et al (2003) Members of the large Maf transcription family regulate insulin gene transcription in islet β cells. Mol Cell Biol 23:6049–6062
Nyborg NC, Mølck AM, Madsen LW, Knudsen LB (2012) The human GLP-1 analog liraglutide and the pancreas: evidence for the absence of structural pancreatic changes in three species. Diabetes 61:1243–1249
Chadwick KD, Fletcher AM, Parrula MC et al (2014) Occurrence of spontaneous pancreatic lesions in normal and diabetic rats: a potential confounding factor in the nonclinical assessment of GLP-1-based therapies. Diabetes 63:1303–1314
Egan AG, Blind E, Dunder K et al (2014) Pancreatic safety of incretin-based drugs—FDA and EMA assessment. N Engl J Med 370:794–797
Cockell M, Stolarczyk D, Frutiger S, Hughes GJ, Hagenbüchle O, Wellauer PK (1995) Binding sites for hepatocyte nuclear factor 3β or 3γ and pancreas transcription factor 1 are required for efficient expression of the gene encoding pancreatic α-amylase. Mol Cell Biol 15:1933–1941
Rose SD, Swift GH, Peyton MJ, Hammer RE, MacDonald RJ (2001) The role of PTF1-P48 in pancreatic acinar gene expression. J Biol Chem 276:44018–44026
Rooman I, Lardon J, Bouwens L (2002) Gastrin stimulates β-cell neogenesis and increases islet mass from transdifferentiated but not from normal exocrine pancreas tissue. Diabetes 51:686–690
Campbell JE, Drucker DJ (2013) Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 17:819–837
Wang TC, Bonner-Weir S, Oates PS et al (1993) Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. J Clin Invest 92:1349–1356
Brand SJ, Tagerud S, Lambert P et al (2002) Pharmacological treatment of chronic diabetes by stimulating pancreatic β-cell regeneration with systemic co-administration of EGF and gastrin. Pharmacol Toxicol 91:414–420
Rooman I, Bouwens L (2004) Combined gastrin and epidermal growth factor treatment induces islet regeneration and restores normoglycaemia in C57Bl6/J mice treated with alloxan. Diabetologia 47:259–265
Suarez-Pinzon WL, Lakey JR, Brand SJ, Rabinovitch A (2005) Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet β-cells from pancreatic duct cells and an increase in functional β-cell mass. J Clin Endocrinol Metab 90:3401–3409
Suarez-Pinzon WL, Yan Y, Power R, Brand SJ, Rabinovitch A (2005) Combination therapy with epidermal growth factor and gastrin increases β-cell mass and reverses hyperglycemia in diabetic NOD mice. Diabetes 54:2596–2601
Griffin KJ, Thompson PA, Gottschalk M, Kyllo JH, Rabinovitch A (2014) Combination therapy with sitagliptin and lansoprazole in patients with recent-onset type 1 diabetes (REPAIR-T1D): 12-month results of a multicentre, randomised, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol 2:710–718
Pan FC, Bankaitis ED, Boyer D et al (2013) Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 140:751–764
Desai BM, Oliver-Krasinski J, de Leon DD et al (2007) Preexisting pancreatic acinar cells contribute to acinar cell, but not islet β cell, regeneration. J Clin Invest 117:971–977
De Marinis YZ, Salehi A, Ward CE et al (2010) GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab 11:543–553
Nauck MA, Friedrich N (2013) Do GLP-1-based therapies increase cancer risk? Diabetes Care 36(Suppl 2):S245–S252
Acknowledgements
We thank F. Lynn (The University of British Columbia, Vancouver, BC, Canada), H. Kim (Korea Advanced Institute of Science and Technology, Daejeon, South Korea) and A. Popiel (Tokyo Medical University, Tokyo, Japan) for their helpful advice and criticism, and S. Takebe (Osaka University Graduate School of Medicine) for her assistance with the experiments.
Funding
This work was supported by the JDRF Advanced Postdoctoral Fellowship award (10-2010-561), JSPS KAKENHI (No. 25461348), the Takeda Science Foundation, the Suzuken Memorial Foundation (all to TMi), the Lilly Incretin Basic Research Aid Program (to TMi and TMa), National Institutes of Health Grants R01 DK021344 and P30 DK63720 (to MSG) and the Nora Eccles Treadwell Foundation (to MSG).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
TMi and SS designed the whole project and wrote the manuscript. SS, MT, YY, and HA contributed to the acquisition and analysis of the data. TMa, TY, HK, YF, MSG, HW, and IS contributed to the interpretation of data. All authors revised the manuscript critically for important intellectual content and approved the final version of the manuscript. TMi is the guarantor of this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 102 kb)
ESM Fig. 1
(PDF 289 kb)
Rights and permissions
About this article

Cite this article
Sasaki, S., Miyatsuka, T., Matsuoka, Ta. et al. Activation of GLP-1 and gastrin signalling induces in vivo reprogramming of pancreatic exocrine cells into beta cells in mice. Diabetologia 58, 2582–2591 (2015). https://doi.org/10.1007/s00125-015-3728-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3728-z