
Abstract
Background and aim
IgA nephropathy is one of the leading causes of primary glomerulonephritis worldwide and an important etiology of renal disease in young adults. IgA nephropathy is considered an immune complex-mediated disease.
Methods
This review article summarizes recent evidence on the pathophysiology of IgA nephropathy.
Results
Current studies indicate an ordered sequence of multi-hits as fundamental to disease occurrence. Altered glycan structures in the hinge region of the heavy chains of IgA1 molecules act as auto-antigens, potentially triggering the production of glycan-specific autoantibodies. Recognition of novel epitopes by IgA and IgG antibodies leads to the formation of immune complexes galactose deficient-IgA1/anti-glycan IgG or IgA. Immune complexes of IgA combined with FcαRI/CD89 have also been implicated in disease exacerbation. These nephritogenic immune complexes are formed in the circulation and deposited in renal mesangium. Deposited immune complexes ultimately induce glomerular injury, through the release of pro-inflammatory cytokines, secretion of chemokines and the resultant migration of macrophages into the kidney. The TfR1/CD71 receptor has a pivotal role in mesangial cells. New signaling intracellular mechanisms have also been described.
Conclusion
The knowledge of the whole pathophysiology of this disease could provide the rational bases for developing novel approaches for diagnosis, for monitoring disease activity, and for disease-specific treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Immunoglobulin A nephropathy (IgAN) is a primary glomerulonephritis, first described by Berger and Hinglais in 1968 by the detection of mesangial immunodeposits of immunoglobulin A (IgA) and immunoglobulin G (IgG) on renal biopsies of patients with persistent microscopic hematuria. IgAN is one of the leading causes of glomerulonephritis worldwide. IgAN usually occurs in adolescents and young adults with predominance of male gender, being the male-to-female ratio 2:1 or 3:1 [1]. Although there are clear geographical and ethnic variances, IgAN is more common in Asians. IgAN was diagnosed in 40 % of native kidney biopsies in Asia, compared with 20 % in Europe, 5–10 % in the United States and less than 5 % in Central Africa [2]. It should be mentioned that differences in the guidelines for renal biopsy might, at least in part, explain the variability in the prevalence of IgA worldwide [3].
The commonest signs and symptoms of IgAN are hematuria and variable degrees of proteinuria. Gross hematuria is frequent in children after exposure to upper respiratory tract infections and may be the first symptom of the disease [4]. In adults, IgAN usually runs an asymptomatic course. Abnormal urine sediment and proteinuria are found by routine health examination. The most common clinical presentation is macroscopic hematuria accompanied by variable degree of proteinuria. Massive proteinuria or nephrotic syndrome is unusual at diagnosis. Moreover, IgAN is characterized by a highly variable course that evolves from a mild renal disease without renal function decline [5] to end-stage renal disease (ESRD) [6, 7]. According to a Japanese cohort with 1012 IgAN patients, renal survival was 84.3, 66.6 and 50.3 % in 10, 20 and 30 years, respectively [7]. In children, a reduction of 50 % in the estimative of glomerular filtration rate (GFR) was reported in 12–18 % of patients with 5–10 years of follow-up [8, 9]. Accordingly, renal survival was 87.6 % among Chinese children and 86 % among Swiss children followed for 4.6 and 10 years, respectively [8, 9]. There are several clinical and histology factors that help to predict the final outcome of patients with IgAN. Clinical predictors of worse outcome are hypertension, altered renal function at diagnosis, urinary protein excretion at the baseline and urinary protein excretion during follow-up [10]. Histology findings that indicate unfavorable outcomes are mesangial hypercellularity (M), endocapillary hypercellularity (E), segmental glomerulosclerosis (S) and interstitial fibrosis/tubular atrophy (T), according to the original studies that proposed the Oxford classification [11, 12]. The VALIGA (European Validation of the Oxford Classification of IgAN) study evaluated 1147 patients from 13 European countries and also provided an independent validation of the predictive value of the M, S and T lesions across a broader spectrum of the disease [1]. However, a systematic review and meta-analysis of 16 retrospective studies that validated the Oxford classification did not confirm the role of mesangial hypercellularity as a prognostic factor and added the presence of crescents to the other three factors (endocapillary hypercellularity, segmental glomerulosclerosis and interstitial fibrosis/tubular atrophy) as determinants of progression to ESRD [6].
The mesangial deposition of dominant or co-dominant IgA is what defines the disease. Concomitant deposits of IgG and IgM, C3 [13–16] and properdin [17] may also be seen. C4 and C4d, mannose-binding lectin (MBL) and C5b–C9 immunostaining can also be detected [18], while C1q is usually absent [14, 16, 18]. It should be pointed, however, that the immunostaining for C4 and C4d, mannose-binding lectin (MBL) and C5b–C9 in kidney biopsy is sometimes difficult to reproduce and to interpret precisely. These findings may suggest activation of alternative and lectin pathways of the complement system in the pathogenesis of IgAN [19]. However, mesangial granular IgA deposits have also been documented in healthy population without evidence of renal injury [20] and in patients with mesangioproliferative glomerulonephritis [21]. The aim of this review article is to summarize the main findings related to the pathophysiology of IgAN.
Physiological IgA structure and function
In humans, IgA exceeds the daily production of all other immunoglobulins (~70 versus ~22 mg/kg/day of IgG and ~7 mg/kg/day of IgM). The lower plasma level of IgA compared with that of IgG is due to shorter circulatory half-life of IgA (5 versus 21 days of IgG) and the fact that approximately two-thirds of produced IgA is selectively transported to external secretions [2]. IgA is found in serum and mucosal secretions in two distinct isotypes, IgA1 and IgA2. Both isotypes can form polymers. The multimeration is dependent on the J chain at the C-terminus of α-chains. In serum, IgA is mainly found as monomeric form (aound 80 % in healthy subjects). The predominant isotype is IgA1 (~85 % of total IgA), which is consistent with the dominance of IgA1-producing plasma cells in the bone marrow. On the other hand, polymeric IgA (dimers or tetramers) predominates in external secretions [22]. The IgA1/IgA2 ratio reflects differential distribution of IgA1 and IgA2 secreting cells in corresponding mucosal tissues. According to IgA1/IgA2 ratio, IgA1-secreting cells prevail in salivary and lacrimal glands, respiratory tract and upper gastrointestinal tract in comparison to a slight predominance of IgA2-secreting cells in the colon and female reproductive system [23].
The predominant form of both IgA1 and IgA2 in mucosal secretion is secretory IgA (S-IgA) that constitutes the first line of mucosal immunity [24]. To pass through the mucosal epithelium, IgA specifically binds at the epithelium basolateral surface to the polymeric immunoglobulin receptor (pIgR). IgA dimerization is critical for the interaction with the extracellular domain of pIgR. The resultant dimeric IgA (dIgA)/pIgR complex is internalized in membrane vesicles and transferred to the apical cell surface. dIgA is then released into mucosal secretions with a secretory component, the extracellular portion of the pIgR. Both components together form S-IgA [25]. S-IgA exists as two subclasses, S-IgA1 and S-IgA2, where S-IgA2 has a shorter hinge joining the Fab and Fc regions. Also, S-IgA2 is significantly nonplanar in its structure, in distinction to the near planar structure of S-IgA1. The shorter hinge region of IgA2 and the presence of secretory component appear to displace the four Fab regions out of the Fc plane in S-IgA2. Differences in the structure result in specific immune properties for S-IgA2 and S-IgA1 and may explain the differences of S-IgA1 and S-IgA2 in terms of interactions with antigens, susceptibility to proteases and effects on receptors [26].
IgA has been primarily viewed as an anti-inflammatory antibody. The presence of S-IgA at mucosal surfaces might warrant that inflammatory processes are kept under control. S-IgA prevents penetration of the mucosal wall by pathogenic microorganisms or foreign antigens, serving as an antiseptic barrier [27]. S-IgA can surround microorganisms and be repelled by mucosal surfaces, can agglutinate microbes and interfere with bacterial motility and can interact with and neutralize bacterial products such as enzymes and toxins [28]. Moreover, if antigens achieve the lamina propria, dIgA can interact and transport them back to the lumen via the pIgR route, before recognition by inflammatory cells. This process might be an effective way to clear the mucosa of undesired excessive immune complexes that otherwise may trigger immune response [29]. Many IgA-deficient patients do not suffer from serious complications but are more susceptible to allergies and autoimmune diseases, suggesting that the lack or low levels of S-IgA may lead to inappropriate immune response against food components or indigenous bacterial flora. This may also play a role in several mucosal disorders, such as gluten-sensitive enteropathy and inflammatory bowel diseases, since the frequencies of these diseases are increased in selective IgA-deficient patients [29].
On the other hand, dIgA is also considered as a very potent stimulus to initiate inflammatory process. Once microbial flora and food components in the intestinal tract reach the lamina propria through diffusion or transcytosis, they are opsonized with dimeric IgA without the secretory component. dIgA has also the ability to recruit neutrophils and other cells of the myeloid lineage by interacting with human IgA Fc receptor (FcαRI), also known as CD89. This mechanism functions as a second line of defense in mucosal areas.
In serum, IgA has also a dual function in immune responses, acting as pro- or anti-inflammatory substance. Naturally occurring serum monomeric IgA induces inhibitory signals through FcαRI/CD89, likely to dampen excessive inflammatory responses in serum. Several groups have demonstrated that, in the absence of antigens, serum IgA downregulates IgG-mediated phagocytosis, chemotaxis, bactericidal activity, oxidative burst activity, and cytokine release in human cells in vitro [30]. Instead, dimeric or multimeric IgA-opsonized pathogens lead to potent pro-inflammatory responses when binding to FcαRI in neutrophils and Kupffer cells to clear the infection [28]. Thus, FcαRI can act as a molecular switch, directing signals toward either an activating or an inhibitory function within the immune system. It should be mentioned that FcαRI can also be shed from cell membrane, being referred as soluble FcαRI. IgA-soluble FcαRI complexes have also an important role in IgAN, as detailed in next sections.
Heavy chains of IgA1 and IgA2 show marked homology in their primary structure (CH2, 99.3 % and CH3, 98 % homology) [2]. The main difference between these two subtypes is the hinge region (HR) between domains one and two of the heavy chain (CH1 and CH2), leading to diverse biological properties. IgA1 has duplicated insertion of amino acids in HR, while this structure is absent in IgA2 [24]. The extended HR of IgA1 may add sequential flexibility for the Fab fragment and, thereby, increases the antigen-binding capacity. However, IgA1 HR is highly susceptible to specific IgA1 proteases, such as serine and cysteine proteases. Most of these enzymes are specific for human IgA1 HR, considering the low homology of this region in other vertebrates [2]. These enzymes are produced by Haemophilus influenzae, Streptococcus pneumoniae, Neisseria menin gitidis, Neisseria gonorrhoeae, Streptococcus sanguis and other bacteria [24]. The antigen specificity is also distinct between the two subtypes of IgA. In general, antibodies specific for proteins and glycoproteins of microbial or food origin are present dominantly in the IgA1 isotype while antibodies to polysaccharides, lipopolysaccharides and lipoteichoic acid are associated mainly with the IgA2 isotype [31].
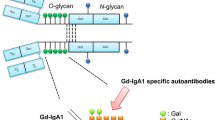
Figure 1 shows the structure of galactose-deficient IgA1. IgA1 contains a HR rich in proline, serine and threonine. Normal human IgA1 in the circulation has simple core O-glycosides basically composed by N-acetyl-galactosamine carbohydrate linked to an oxygen atom of serine or threonine in the HR. Attached to N-acetyl-galactosamine is galactose, forming O-glycans. Usually, no more than six among the nine residues of serine and threonine are glycosylated in each HR, acting as potential sites for O-glycan attachment. Four and five glycans attached to HR are the commonest form [32]. This O-glycan structure is lacking in IgA2. O-glycosylation is mediated by specific glycosyltransferases in the Golgi apparatus of IgA1-secreting cells [33]. The O-glycans’ binding regions consist of N-acetylgalactosamine (GalNAc) with β1,3-linked galactose (Gal), with or without attached sialic acid (N-acetylneuraminic acid—NeuNAc) [34]. The synthesis of the O-glycoside is initiated via enzymatic addition of GalNAc to serine or threonine residues by UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases (GalNAcTs). Gal is linked to the GalNAc by the enzyme β1,3-galactosyltransferase (C1GalT1). The stability of C1GalT1 is given by its specific chaperone, Core 1 β1,3-galactosyltransferase-specific chaperone (Cosmc). The sialic acid NeuNAc is linked to GalNAc by α2,6-sialyltransferase GalNAc I and II (ST6GalNAc I or ST6GalNAc II) or linked to Gal by α2,3-sialyltransferase [35].

Schematic view of the O-glycosylation of immunoglobulin A1 showing the heterogeneity of O-glycans in hinge region. CH, heavy chain; GalNAc, N-acetylgalactosamine; GalNAcTs, N-acetylgalactosyltransferases; Ser, serine; Thr, threonine; NeuAc, neuraminic acid; Gd-IgA1, galactosylated-deficient immunoglobulin A1; C1GalT1, enzyme β1,3-galactosyltransferase; Cosmc, Core 1 β1,3-galactosyltransferase-specific chaperone; ST3Gal, α2,3-N-sialyltransferase; ST6GalNAc II, α2,6-sialyltransferase N-acetylgalactosamine II; NeuNAc, N-acetylneuraminic acid
The composition of carbohydrates in normal serum IgA1 is quite variable. Most prevalent form includes GalNAc-Gal disaccharide (T antigen), with mono- and di-sialylated residues. Serum IgA1 usually presents few galactose-deficient O-glycosides (Gd-IgA1) [31]. Patients with IgAN have increased levels of circulating free O-glycoside IgA1 (Tn antigen). When sialylated, this structure is known as sialylated Tn antigen (STn) [36, 37]. The Tn antigen is part of the epitope recognized by IgG and IgA, forming immune complexes capable of inducing renal lesion [38, 39].
Brief overview of IgA nephropathy pathogenesis
The pathogenesis of IgAN is still not completely elucidated. Current studies indicate an ordered sequence of “four hits” as fundamental to the disease occurrence [2, 35, 40, 41]. Figure 2 shows a schematic view of the “four hits”. First, there is an anomalous production of galactosylated-deficient IgA1 O-glycoforms. Genetic stimuli and innate immune response, mainly via Toll-like receptors (TLRs), seem to be involved in this process, which also alters the crosstalk between mucosa and bone marrow [42]. Mucosal B lymphocytes, that produce galactosylated-deficient IgA1, are displaced to systemic sites, especially the bone marrow, raising the serum Gal-deficient IgA1 (Gd-IgA1). However, the presence of circulating aberrant IgA1 alone is not sufficient to cause kidney injury. The “second hit” in the pathogenesis of IgAN is the production of antibodies against the undergalactosylated hinge. These antibodies are naturally occurring in the plasma, but their production may be exacerbated following infections by bacteria and virus that express N-acetylgalactosamine as surface antigens. A cross-immune response thus happens between antigens of microorganisms and Gd-IgA1 [43]. Episodes of gross hematuria after infections of air or intestinal mucosa are described as clinical manifestations of IgAN [4]. IgAN is, therefore, considered an immune complex-mediated disease in which autoantibodies, IgG and IgA, are produced against the Gd-IgA1. Binding of glycan-specific IgG and IgA antibodies to aberrant IgA1 forms the immune complexes, which are the “third hit” in the pathogenesis of IgAN [44]. The formation of circulating Gd-IgA1 immune complexes induces an alteration in the interaction between IgA and FcαRI/CD89, the IgA receptor in myeloid cells [25]. As a consequence, cleavage of the extracellular domain of FcαRI is induced, leading to the formation of circulating Gd-IgA/FcαRI immune complexes. Immune complexes with soluble Gd-IgA/FcαRI are also implicated in the pathogenesis of IgAN [25]. Antibody binding to IgA1 hinge hampers the recognition of IgA1 by the liver, consequently reducing the serum clearance of this molecule. The trigger for renal injury is the deposition of immune complexes in the glomerular mesangium, defined as the “fourth hit” [45, 46]. These immune complexes are formed by Gd-IgA/FcαRI and Gd-IgA/IgG or IgA. The deposition of these immune complexes occurs mainly in mesangium with little tubular or epithelial deposits. There is evidence that immune complexes of higher sizes are more likely to induce kidney damage than smaller ones [39]. The transferrin receptor (TfR1/CD71) is a known mesangial receptor able to bind and internalize pIgA1 and immune complexes containing IgA1 [31]. Immune complexes containing Gd-IgA1 deposition are initial inducers of inflammation. By activating other glomerular and tubular cells, the inflammatory process, which is initially restricted to mesangial areas, progresses to glomerular and tubulointerstitial sites [41].

Schematic view of the “four hits” related to the pathogenesis of immunoglobulin A nephropathy (IgAN). B lymphocytes produce galactosylated-deficient immunoglobulin A1 (Gd-IgA1) that triggers the production of glycan-specific autoantibodies in circulation. The binding of these antibodies to Gd-IgA1 leads to the formation of immune complexes (IC). The liver is not able to remove IC. The immune complexes deposit in renal mesangium. B cells type B lymphocytes, Gd-IgA1 galactosylated-deficient Immunoglobulin A1, Anti-Gd-IgA1 antibodies against galactosylated-deficient Immunoglobulin A1; IC immune complexes
Insights from genetic studies
The cause of primary IgAN is unknown. While most IgAN patients are sporadic cases, familial aggregation of the disease has been well recognized [47–51]. Familial clustering of a disease often suggests a genetic effect. However, genome-wide association studies (GWAS) of sporadic cases of IgAN was considered the most promising approach to identify susceptibility genes. These studies were aimed at detecting variants at genomic loci that are associated with some diseases, as IgAN [52]. The first published GWAS for IgAN was performed in Europe [53] and was followed by three larger studies performed in Chinese cohorts [54, 55] and European and East Asian ancestry [56]. Several susceptibility loci were identified and candidate gene approach based on the disease pathogenesis pointed out the meaningful genes. The implicated pathways include antigen processing and presentation [major histocompatibility complex (MHC) region], the complement system (CFHR1/3 and ITGAM–ITGAX loci), regulation of mucosal IgA production (TNFSF13 and LIF/OSM loci), and innate immunity against pathogens (DEFA, CARD9, ITGAM–ITGAX, and VAV3 loci) [57]. Only 4–7 % of the disease variance can be due to these loci [53–55, 58].
Complex selective pressure is suggested by the close correlation between the IgA nephropathy risk allele frequencies and the variation of disease prevalence among different ethnic populations. Helminth infection could be a potential source of selection pressure, as the highest global burden of soil-transmitted helminthes infections occurs in Asia. The increased incidence of IgAN in Asia may represent an untoward consequence of protective adaptation to mucosal invasion by local pathogens. The enhanced immune response conferred by risk alleles would simultaneously explain the known association of mucosal infections as a trigger for IgAN [56].
Immune system and mucosa–bone marrow crosstalk
Classically, the immune system is divided into innate and adaptive immunity [59]. The innate response is rapid but nonspecific. Innate immunity includes recognition of pathogen-associated molecular patterns (PAMPs) by macrophages, dendritic cells, leukocytes, and other cells, followed by opsonization and phagocytosis. Receptors of PAMPs are the Toll-like receptors (TLRs). TLRs belong to a family of pattern-recognition receptors that link innate and acquired immune system [60]. Activation of TLRs induces the maturation of dendritic cells, cytokine release and recruitment of macrophages and lymphocytes. Mature dendritic cells interact with T cells, activating specific T cell response—the adaptive response [42].
The production of mucosal IgA occurs by the activation of follicular B cells in germinal centers. The antigen-presenting cells located below the specialized M cells take up antigens and present them to T helper cells. The interaction between B and T cells under cytokines stimulation, mainly transforming growth factor (TGF)-beta and interleukin (IL)-10, stimulates B cells to undergo class switching from IgM to IgA and somatic hypermutation for affinity maturation [61]. However, the stimulation of IgA production occurs also by a T cell-independent pathway. The TLR/MyD88 (TLR2, TLR4, TLR5, TLR9) activated by antigens may stimulate antigen-presenting cells in the lamina propria and stromal cells to release innate response signals (as BAFF, APRIL, TNF, TGF-beta, retinoic acid), which in turn promotes the conversion of IgM to IgA in B cells, regardless of T-lymphocytes [35].
It has been suggested that changes in the immune response of the mucosa cells leads to reduced antigen clearance with continuous antigen exposure, which may trigger the production of anomalous IgA1 [42]. In several experimental studies, antigens capable of enhancing serum altered IgA1 or inducing IgAN have been tested, such as Staphylococcus aureus [62], H. influenzae [63], Sendai virus [64, 65], gliadin [66]. On the other hand, no food or viral antigens were consistently found in mesangial deposits, suggesting a nonspecific alteration of IgA1 production via innate immune response [42]. The mechanisms of this unbalance are still unknown. There is evidence that TLRs are possible culprits, both in the increased production of abnormal IgA1, as well in the aberrant glycosylation [67–69]. TRL9 are hyperexpressed in plasmocytoid dendritic cells in tonsils of patients with IgAN [68]. Upregulation of TLR4 was found in patients with IgAN, particularly associated with proteinuria and microscopic hematuria [67]. Furthermore, reduced levels of the chaperone (Cosmc) were found to be involved in undergalactosylation. This process is promoted via activation of TLR4 by bacterial LPS in vitro [69].
Despite the association of respiratory and intestinal infections with IgAN exacerbation, it is well recognized that circulating Gd-IgA1 antibodies are primarily produced in the bone marrow [70]. Some researchers suggest that B cells previously encountered the antigens at other sites and, at that point, relocated them to bone marrow [42]. It has been hypothesized that there is a defect in the crosstalk between mucosa and bone marrow, induced by cytokines and adhesion molecules [70]. IgA-secreting cells have traffic pattern associated with their site of origin. Homing and differentiation of plasma cells in the lamina propria of the small gut is associated with upregulation of integrin α4β7, CCR9 and CCR10 receptors, whereas the expression of integrin α4β1 and CXCR4 is linked to the homing and differentiation of B cells in the bone marrow [71]. Batra and co-workers showed increased expression of α4β1 by CD3T cells in patients with IgAN [72].
Abnormal glycosylation of IgA1
Studies with IgAN patients showed alterations in glycosylation of circulating IgA1 [73, 74] and IgA1 from mesangial immunodeposits [75]. Typically, the hinge region of IgA1 contains O-glycans formed by N-acetylgalactosamine and the disaccharide galactose (GalNAc-Gal, also known as T antigen) and their sialylated forms (ST antigens) [30, 37, 76]. Some IgA1 molecules in patients with IgAN present O-glycosides with galactose deficiency in HR, which consist of only GalNAc (Tn antigen) or GalNAc terminal with sialylation (sTn antigen). This finding was inferred from the reduction of reactivity of IgA1 to specific T lectins antigens (jacalin) in patients with IgAN [73], and increased reactivity to GalNAc-specific lectins, such as that produced by Helix aspersa [36].
Using Epstein–Barr virus in circulating B cells that produce IgA, the under-galactosylation was seen only in patients with IgAN, compared to normal controls [43]. This change can be basically explained by the expression of key enzymes in the O-glycosylation pathway [36, 77, 78]. First of all, there are different subtypes of N-acetylgalactosyltransferases (GalNAcTs). GalNAcT2 is ubiquitous and appears to have a predominant role in IgA O-glycosylation. GalNAcT2 rarely attaches glycans to sites next to GalNAc regions of the HR. However, five GalNAcTs, rather than GalNAcT2, showed very weak and almost negligible activity toward HR, and their specificities were totally different from those of GalNAc-T2 [77]. Indeed, the link between isotypes of GalNAcT and O-glycosylation defects is still unclear. Probably, the increased expression and activity of sialyltransferase ST6GalNAc II may result in a premature sialylation, which, in turn, inhibits the attachment of galactose to N-acetylgalactosamine in IgA1 Tn antigens. Accordingly, Suzuki et al. observed an increased expression of ST6GalNAc II in B lymphocytes from patients with IgAN [36]. In addition, reduced expression and activity of galactosyltransferase C1GalT1 [36] and decreased expression of C1GalT1-specific chaperone Cosmc [78] have also been observed in peripheral B lymphocytes from patients with IgAN.
It should be mentioned that data concerning either intrinsic or extrinsic regulation of glycosylation of IgA1 in B cells are still limited. Among serum proteins with O-glycosylation, two have been studied in detail in patients with IgAN: the C1 esterase inhibitor and IgD. In both cases, there were no alterations to the O-galactosylation pattern in patients with IgAN [79]. The result indicates that patients with IgAN exhibit galactose-deficient O-glycans uniquely on IgA1 in the circulation, but not on other glycoproteins. These observations argue against the existence of a global deficit in O-glycosylation of serum proteins in IgAN, and suggest that this defect may be limited to a specific B lymphocyte population secreting Gd-IgA1. Moreover, only a fraction of B lymphocytes produces Gd-IgA1 in patients with IgAN [36]. It is now recognized that O-glycosylation defects happen in late stages of B cell development and maturation as a consequence of abnormal immune regulation induced by acquired stimuli [42]. Furthermore, external stimuli and cytokines influence the O-glycosylation of IgA [35]. Increased Th2 response can reduce the O-glycosylation of IgA [80]. IL-4 stimulation of B cell lines causes increased production of IgA and significant reduction in the levels of Cosmc mRNA and of C1GalT1 mRNA, leading to a defect in the galactosylation of HR [67]. Cosmc inhibition was also evidenced by LPS stimulation of B lymphocytes [69]. All these evidences point to an acquired defect of glycosylation whose stimulus comes from an abnormal immune response.
On the other hand, evidence of a genetic contribution for IgAN has come to light. Recently, researchers have found that microRNAs may play an important role in the pathogenesis of IgAN [81, 82]. MicroRNAs (miRNAs) are short, noncoding RNA molecules that regulate gene expression. Patients with IgAN exhibited lower C1GALT1 expression, which negatively correlated with miR-148b expression [81]. Also, GalNAcT2 is a potential target of the miRNA called let-7b. In IgAN patients, this miRNA is significantly upregulated compared with healthy blood donors. In ex vivo experiments, let-7b decreased GalNAcT2 levels in peripheral blood mononuclear cells of IgAN patients, whereas the loss of let-7b function in healthy blood donors led to an increase of GalNAcT2 mRNA and its protein level [82].
Recently, a new approach to hypogalactosylated structures is being analyzed based on microheterogeneity studies [34, 83]. This approach includes the direct determination of sites of attachment of the O-glycans as well as the characterization of microheterogeneity of glycans at each site. For example, in normal individuals, Thr228, Ser230, Ser232 and Thr233 residues are the most common sites attached to glycan, while Thr225 and Thr236 residues are predominantly hypogalactosylated. Indeed, for IgA1 myeloma protein, Thr225, Thr228 and Ser232 were predominantly attached to GalNAc-Gal disaccharide, whereas GalNAc-Gal deficient or the absence of glycan was determined at Ser230, Thr233 and Thr236 [33]. This microheterogeneity in HR rises a new questioning, extrapolated to IgAN. It is highly possible that the shapes of nephritogenic Gd-IgA1 arise not only from galactosylation insufficiency, but also from different binding sites of O-glycosides at amino acid residues in HR.
Anti-glycan antibodies and immune complexes
As a result of deficiency of galactose, residues of N-acetylgalactosamine in the truncated IgA1 hinges are exposed as novel epitopes [44]. Poorly galactosylated IgA1 O-glycoforms may act as auto-antigens, potentially triggering the production of glycan-specific autoantibodies. Recognition of novel epitopes by IgA and IgG antibodies leads to the formation of immune complexes Gd-IgA1/IgA and Gd-IgA1/IgG [44]. Virtually, all serum Gd-IgA1 is combined with other anti-glycan-specific antibody [19]. Anti-glycoside antibodies occur naturally in plasma. Some bacteria and virus express N-acetylgalactosamine as surface antigens. An infection by these agents could facilitate the production of anti-glycosides, which might cross-react with Gd-IgA, causing or exacerbating IgAN [45]. This may explain why urinary abnormalities are often intensified during episodes of upper airway infections in patients with IgAN [4].
Moreover, it seems that the anti-glycoside antibodies in patients with IgAN have a peculiarity in their primary structure with high affinity for Gd-IgA1, as shown by Suzuki et al. [43]. This research group has cloned EBV-immortalized lymphocytes that secreted IgG with specificity for Gd-IgA1. The analysis of the sequence of light (V L) and heavy (V H) chains of IgG showed unique features in complementarity-determining region three (CDR3) of V H. Serine was observed in the third position of CDR3 from six of seven patients with IgAN. On the other hand, alanine was detected in the same position from all six healthy controls. Furthermore, on reversing the residue serine to alanine through site-directed mutagenesis, there was a reduction of the affinity of recombinant IgG for Gd-IgA1 [43]. Currently, we still do not know if this change (Ala to Ser in CDR3) originates from genetic variation or somatic mutation during an active immune response [31].
The production of complexes with IgA reduces plasma clearance [84]. IgA is catabolized mainly by the liver, through the asialoglycoprotein receptor (ASGP-R), which recognizes sialylated glycoproteins [85, 86]. In the case of IgA, Gal and terminal GalNAc are recognized. However, binding of the anti-GalNAc glycoside-specific antibody prevents recognition of glycosides in HR by ASGP-R, thereby reducing catabolism. Also, immune complexes have difficulty to cross hepatic fenestrated capillaries up to the space of Disse, consequently reducing the contact with hepatocytes expressing ASGP-R [2].
Furthermore, the level of specific IgG anti-glycoside is increased in patients with IgAN and is associated with degree of proteinuria and with the amount of urinary IgA1–IgG immune complexes [43]. Increased antigen–antibody complexes of IgA1 were observed in acute episodes of gross hematuria [43]. Consequently, immune complexes not effectively metabolized by the liver are deposited in mesangial region due to high molecular weight [87]. This deposition stimulates mesangial proliferation and local cytokines release [88].
Immune complexes with soluble FcαRI/CD89 are also implicated in the pathogenesis of IgAN [25]. FcαRI is the only Fc receptor that specifically binds to IgA. Physiologically, FcαRI plays an essential anti-inflammatory role in immunity by allowing the transmission of inhibitory signals as a consequence of binding to serum monomeric IgA. This effect prevents the development of autoimmunity. Moreover, when combined to FcRγ, FcαRI can act as a molecular switch, directing signals toward either anti- or pro-inflammatory actions. FcRγ-associated FcαRI mediates either activating (with multimeric IgA) or inhibitory responses (with monomeric IgA), being a dual-function type receptor that operates differently according to the type of ligand [25]. In IgAN, the formation of circulating Gd-IgA1 immune complexes induces an alteration in the interaction between IgA and FcαRI (e.g., increased affinity or increased aggregation). As a consequence, cleavage of the extracellular domain of FcαRI is induced, leading to the formation of circulating IgA/FcαRI immune complexes, which are found in mesangial deposits. IgA/FcαRI immune complexes have been implicated in disease exacerbation through the release of pro-inflammatory cytokines, secretion of chemokines and the resultant migration of macrophages into the kidney [40, 89–91].
Mesangial deposition of immune complexes with Gd-IgA1, cell activation and glomerular injury
The deposition of immune complexes (IC) of Gd-IgA1/specific anti-glycan IgG in the mesangium plays a critical role in the pathogenesis of IgAN [45, 46]. Isolated deposition of Gd-IgA1 or isolated glycoside antibodies is not sufficient to trigger inflammatory response [39, 92]. The deposition is predominantly in the mesangium with limited amount of immune complexes in podocytes and tubular epithelial cells [41].
Studies with cultured human mesangial cells showed that immune complexes (Gd-IgA1/IgG) of high molecular weight (>800 kDa) had greater impact on mesangial proliferation and cytokine production. In contrast, Gd-IgA1, complexed or non-complexed into smaller immune complexes, was not able to induce mesangial proliferation [39, 93]. Of note is the fact that immune complexes with a definite molecular mass induced further mesangial proliferation in patients with IgAN compared to healthy controls [84]. Moreover, immune complexes containing higher amounts of Gd-IgA1 produced more intense mesangial proliferation [39]. On the other hand, no GalNAc epitopes were found in the mesangium, suggesting that specific IgG deposition is not directly targeted to the mesangium without formation of immune complexes in situ [43]. Such findings show how relevant Gd-IgA1 antigens and Gd-IgA1/specific IgG or IgA antibodies are to the formation of immune complexes and the essential role of passive deposition of circulating immune complexes containing Gd-IgA1 for the activation of mesangial cells.
The role of soluble FcαRI mesangial deposition in the pathogenesis of IgAN was further supported by findings obtained with transgenic mice expressing FcαRI. These animals spontaneously developed IgAN with IgA mesangial deposition, macrophage infiltration, hematuria, weak proteinuria, but without renal dysfunction [94]. After crossing mice overexpressing human IgA1 and human FcαRI mice, the offspring spontaneously presented a complete phenotype of human IgAN, with kidney inflammation, IgA1 mesangial deposition, hematuria, and proteinuria [95]. The double transgenic model confirmed that the IgA1/FcαRI interactions conferred to circulating immune complexes a nephritogenic behavior on the mesangium [25].
Signaling pathways of immune response in the kidney of patients with IgAN remain to be clarified. Indeed, some classic receptors for IgA (pIgR, FcαRI and Fc-α/μR) were not found in mesangial cells of some patients with IgAN [96]. Also, IgA1 receptors in podocytes and tubular epithelial cells are still unknown [41]. In this regard, Kaneko et al. identified integrin α1/β1 and α2/β1 heterodimer as a potential receptor for IgA1 in human glomerular mesangial cells in IgAN [97]. The combination of IgA1 with this receptor induced mesangial cell proliferation and collagen deposition [97]. The transferrin receptor (TfR1/CD71) is another well-known mesangial receptor able to bind and internalize pIgA1 and immune complexes containing IgA1 [31]. Moreover, the binding and internalization trigger a positive feedback loop, inducing higher expression of TfR1/CD71 in mesangial cells [98]. The IgA1/sFcαRI immune complexes have also the capacity to bind mesangial TfR1 [95]. IgA1 deposition involved a direct binding of sFcαRI to mesangial TfR1, resulting in TfR1 upregulation [40]. sFcαRI–TfR1 interaction induced mesangial surface expression of transglutaminase 2 (TGase2), which in turn upregulated TfR1 expression. Tissue TGase2 was considered an essential molecule in IgAN pathogenesis in mice as it controls mesangial deposition of IgA1 complexes that lead to renal dysfunction. TGase2 would be the factor responsible for TfR1 overexpression in primary cells implicated in IgA-related diseases and celiac disease. In the absence of transglutaminase 2, IgA1/sFcαRI deposits were dramatically impaired. Therefore, TGase2 may facilitate IgA1/sFcαRI deposition and mesangial cell activation, being considered a potential target for therapeutic intervention [95].
In IgAN, kidney injury begins with the activation of mesangial cells, which alters the profile of cytokines and other mediators. Novak et al. showed that there are two types of IgA immune complexes (stimulatory and inhibitory) capable of producing distinct patterns of intracellular phosphorylation. Stimulatory immune complexes have higher mass with elevated amounts of Gd-IgA1. These immune complexes exhibited strong stimulatory effect on expression of IL-6 and IL-8 genes [93]. Polymeric IgA1 interactions with TfR1 on human mesangial cells induced cell proliferation and secretion of IL-6 through activation of phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mechanistic target of rapamycin (MTOR) and the extracellular signal-regulated kinase (ERK) 1/2 kinase pathways [25]. Tamouza et al. showed activation of the MAPK/ERK pathway (identified by ERK1/2 phosphorylation) in a subgroup of IgAN patients presenting with high blood pressure and proteinuria higher than 1 g per day. Interestingly, IgAN patients with less pronounced proteinuria had no expression of mesangial p-ERK1/2 in spite of the presence of IgA deposits in the mesangium [99]. The link between mesangial ERK1/2 phosphorylation and proteinuria in IgAN patients points to a dysregulated mesangial–podocyte crosstalk, resulting in an altered filtration barrier (indirect podocyte injury through mesangial stimulation) [99].
The complement system also plays a significant role in the pathogenesis of IgAN. Complement activation occurs via three pathways: (1) classic pathway, activated primarily by immune complexes with IgG and IgM, but also by necrotic or apoptotic cells and acute-phase proteins such as C-reactive protein [100], (2) alternative pathway, initiated by spontaneous breaking of the C3 component, but with current evidence of the participation of properdin as initiator, in addition to its role in stabilizing the C3 convertase [101], and (3) lectin pathway, which, instead of using antibodies to recognize pathogens, acts through plasma proteins, such as mannose-binding lectins (MBL) and ficolins, to identify carbohydrate patterns found on the surface of microorganisms [100]. Regardless of the complement pathway, an amplification cascade is initiated in IgAN, which releases soluble pro-inflammatory molecules and membrane attack complex (C5b–9 complex), resulting in osmotic cell lysis. The complement system is highly regulated by short half-life activated factors and endogenous regulatory proteins [100]. In ~90 % of IgAN patients, C3 is detected in the glomerulus [13–16]. The activation of the alternative complement pathway is also suggested by: (1) the presence of properdin in most renal biopsies, co-localized with IgA deposits [17], (2) the absence of C1q [14, 16, 18] and (3) the elevation of C3 catabolites in serum of patients with IgAN [101]. However, recent findings also suggest the involvement of the lectin pathway in IgA nephropathy [102]. Mesangial deposits of MBL are seen in 25 % of patients with IgAN [18]. Mesangial deposits of MBL and C4 and/or C4 catabolites are potential markers for the progression of IgAN [103, 104]. In contrast, patients with MBL deficiency tend to show better clinical presentation and lower levels of serum creatinine and proteinuria than patients without MBL deficiency [105, 106]. N-glycans present on the heavy chains of secretory IgA1 are candidate ligands for lectin pathway activation [107]. However, further studies are essential to confirm the role of MBL ligands in the pathogenesis of IgAN.
Activation of mesangial cells by immune system has been considered the initial step of inflammation in the pathogenesis of IgAN [108]. Figure 3 shows the pathways related to renal injury in patients with IgAN. Activated mesangial cells could initiate three mechanisms that operate independently or synergistically to develop kidney injury in IgAN patients: (1) tubulointerstitial infiltration by monocytes and macrophages; (2) tubulointerstitial injury secondary to intraluminal exposure to albumin; and (3) glomerulus–podocyte–tubule crosstalk [41]. Mesangial cells activated by immune complexes proliferate and release extracellular matrix proteins, chemokines and cytokines. Such pro-inflammatory and pro-fibrotic molecules (TNF, IL-6, angiotensin II, platelet-derived growth factor) modify the gene expression of podocyte and stimulate the infiltration of inflammatory cells into the tubulointerstitial compartment [41]. The attracted inflammatory cells, in turn, release further cytokines that activate tubular epithelial cells, which amplify the inflammatory cascade by releasing more pro-inflammatory and pro-fibrotic mediators. A positive feedback loop is then activated, perpetuating the process. Inflammatory mediators released by mesangial cells may also change the slit diaphragm in podocytes, thus causing proteinuria. Proteinuria, by itself, stimulates chemotaxis and migration of immune cells in various glomerular diseases [109].

Schematic view of the pathways related to renal injury in patients with immunoglobulin A nephropathy (IgAN). The deposition of immune complexes (galactose-deficient IgA1 combined with anti-glycan IgG/IgA) in mesangium triggers complement activation, production of angiotensin II, recruitment of inflammatory cytokines, and stimulation of renal fibrosis. These mechanisms resulted in podocyte and renal tubular cell injury, leading to progressive renal dysfunction. TNF-α tumor necrosis factor alpha, TGF-β transforming growth factor beta, Ang II angiotensin II, PDGF platelet-derived growth factor, IL-6 interleukin 6, PAI-1 plasminogen activator inhibitor 1, EMT extracellular matrix, LP lectin pathway, AP alternative pathway, EGF epithelial growth factor, FGF fibroblast growth factor
Concluding remarks
IgA nephropathy is an immune complex-mediated disease with a multi-hit kinetics. The IgAN pathophysiology is not yet fully elucidated, as well as the role of IgA in the regulation of immune–inflammatory functions. However, knowledge of the pathophysiology of this immune complex disease has undoubtedly progressed with discoveries involving the structure of hypogalactosylated IgA, the formation of antibodies directed against these new epitopes, the formation and mesangial deposition of Gd-IgA/IgG or IgA and Gd-IgA/FcαRI immune complexes and the glomerulus–podocyte–tubule crosstalk. Studies on IgA receptors allowed advances even in the understanding of normal IgA immune response. The characterization of FcαRI/CD89 receptor highlighted the duality of IgA in promoting immune response, pro- or anti-inflammatory, according to their binders. Furthermore, it was demonstrated that the soluble FcαRI is capable of interacting with mesangial receptors, notably TfR1/CD71, and, thereby, triggering the activation of the mesangial cell. Some signaling mechanisms in mesangial cells have also been described, such as transglutaminase 2, activation of the MAPK/ERK pathway and activation of the alternative and lectin pathways. These, and further advances in the studies of the molecular basis and mechanisms of IgAN, will provide essential information for future earlier diagnosis, better monitoring of the clinical course or response to treatment and, ultimately, disease-specific therapy.
References
Coppo R, Troyanov S, Bellur S, Cattran D, Cook HT, Feehally J, Roberts ISD, Morando L, Camilla R, Tesar V, Lunberg S, Gesualdo L, Emma F, Rollino C, Amore A, Praga M, Feriozzi S, Segoloni G, Pani A, Cancarini G, Durlik M, Moggia E, Mazzucco G, Giannakakis C, Honsova E, Sundelin BB, Di Palma AM, Ferrario F, Gutierrez E, Asunis AM, Barratt J, Tardanico R, Perkowska-Ptasinska A. Validation of the Oxford classification of IgA nephropathy in cohorts with different presentations and treatments. Kidney Int. 2014;86(4):828–36.
Mestecky J, Raska M, Julian BA, Gharavi AG, Renfrow MB, Moldoveanu Z, Novak L, Matousovic K, Novak J. IgA nephropathy: molecular mechanisms of the disease. Annu Rev Pathol. 2013;8:217–40.
Levy M, Berger J. Worldwide perspective of IgA nephropathy. Am J Kidney Dis. 1988;12(5):340–7.
Wyatt RJ, Kritchevsky SB, Woodford SY, Miller PM, Roy S, Holland NH, Jackson E, Bishof NA. IgA nephropathy: long-term prognosis for pediatric patients. J Pediatr. 1995;127(6):913–9.
Gutiérrez E, Zamora I, Ballarín JA, Arce Y, Jiménez S, Quereda C, Olea T, Martínez-Ara J, Segarra A, Bernis C, García A, Goicoechea M, García de Vinuesa S, Rojas-Rivera J, Praga M. Long-term outcomes of IgA nephropathy presenting with minimal or no proteinuria. J Am Soc Nephrol. 2012;23(10):1753–60.
Lv J, Shi S, Xu D, Zhang H, Troyanov S, Cattran DC, Wang H. Evaluation of the Oxford classification of IgA nephropathy: a systematic review and meta-analysis. Am J Kidney Dis. 2013;62(5):891–9.
Moriyama T, Tanaka K, Iwasaki C, Oshima Y, Ochi A, Kataoka H, Itabashi M, Takei T, Uchida K, Nitta K. Prognosis in IgA nephropathy: 30-year analysis of 1,012 patients at a single center in Japan. PLoS One. 2014;9(3):e91756.
Le W, Zeng C-H, Liu Z, Liu D, Yang Q, Lin R-X, Xia Z-K, Fan Z-M, Zhu G, Wu Y, Xu H, Zhai Y, Ding Y, Yang X, Liang S, Chen H, Xu F, Huang Q, Shen H, Wang J, Fogo AB, Liu Z-H. Validation of the Oxford classification of IgA nephropathy for pediatric patients from China. BMC Nephrol. 2012;13(1):158.
Edström Halling S, Söderberg MP, Berg UB. Predictors of outcome in paediatric IgA nephropathy with regard to clinical and histopathological variables (Oxford classification). Nephrol Dial Transplant. 2012;27(2):715–22.
Bartosik LP, Lajoie G, Sugar L, Cattran DC. Predicting progression in IgA nephropathy. Am J Kidney Dis. 2001;38(4):728–35.
Cattran DC, Coppo R, Cook HT, Feehally J, Roberts ISD, Troyanov S, Alpers CE, Amore A, Barratt J, Berthoux F, Bonsib S, Bruijn JA, D’Agati V, D’Amico G, Emancipator S, Emma F, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene H-J, Haas M, Herzenberg AM, Hill PA, Hogg RJ, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Leung CB, Li L-S, Li PKT, Liu Z-H, Mackinnon B, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int. 2009;76(5):534–45.
Roberts ISD, Cook HT, Troyanov S, Alpers CE, Amore A, Barratt J, Berthoux F, Bonsib S, Bruijn JA, Cattran DC, Coppo R, D’Agati V, D’Amico G, Emancipator S, Emma F, Feehally J, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene H-J, Haas M, Herzenberg AM, Hill PA, Hogg RJ, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Li L-S, Li PKT, Liu Z-H, Mackinnon B, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. 2009;76(5):546–56.
Vangelista A, Frascà GM, Mondini S, Bonomini V. Idiopathic IgA mesangial nephropathy: immunohistological features. Contrib Nephrol. 1984;40:167–73.
Espinosa M, Ortega R, Gómez-Carrasco JM, López-Rubio F, López-Andreu M, López-Oliva MO, Aljama P. Mesangial C4d deposition: a new prognostic factor in IgA nephropathy. Nephrol Dial Transplant. 2009;24(3):886–91.
Sahin OZ, Yavas H, Taslı F, Gibyeli DG, Ersoy R, Uzum A, Cirit M. Prognostic value of glomerular C4d staining in patients with IgA nephritis. Int J Clin Exp Pathol. 2014;7(6):3299–304.
Espinosa M, Ortega R, Sánchez M, Segarra A, Salcedo MT, Gonza F, Camacho R, Espinosa M, Ortega R, Sa M, Pinedo F, Gutierrez E, Valera A, Leon M, Valdivia MA, Cabrera R, Lo K, Cobo MA, Rodriguez R, Balları J, Arce Y, Garcı B. Association of C4d deposition with clinical outcomes in IgA nephropathy. Clin J Am Soc Nephrol. 2014;9(4):897–904.
Rauterberg EW, Lieberknecht HM, Wingen AM, Ritz E. Complement membrane attack (MAC) in idiopathic IgA-glomerulonephritis. Kidney Int. 1987;31(3):820–9.
Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, Stahl GL, Matsushita M, Fujita T, van Kooten C, Daha MR. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol. 2006;17(6):1724–34.
Wyatt RJ, Julian BA. IgA Nephropathy. N Engl J Med. 2013;368(25):2402–14.
Waldherr R, Rambausek M, Duncker WD, Ritz E. Frequency of mesangial IgA deposits in a non-selected autopsy series. Nephrol Dial Transplant. 1989;4(11):943–6.
Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. 2003;63(6):2286–94.
Woof JM, Mestecky J. Mucosal immunoglobulins. Immunol Rev. 2005;206:64–82.
Brandtzaeg P, Johansen F-E. Mucosal B cells: phenotypic characteristics, transcriptional regulation, and homing properties. Immunol Rev. 2005;206:32–63.
Woof JM, Russell MW. Structure and function relationships in IgA. Mucosal Immunol. 2011;4(6):590–7.
Robert T, Berthelot L, Cambier A, Rondeau E, Monteiro RC. Molecular insights into the pathogenesis of IgA nephropathy. Trends Mol Med. 2015;21(12):762–75.
Bonner A, Almogren A, Furtado PB, Kerr MA, Perkins SJ. The nonplanar secretory IgA2 and near planar secretory IgA1 solution structures rationalize their different mucosal immune responses. J Biol Chem. 2009;284(8):5077–87.
Mathias A, Pais B, Favre L, Benyacoub J, Corth B. Role of secretory IgA in the mucosal sensing of commensal bacteria. Gut Microbes. 2014;5(6):688–95.
Aleyd E, Heineke MH, Van Egmond M. The era of the immunoglobulin A Fc receptor Fc a RI; its function and potential as target in disease. Immunol Rev. 2015;268(1):123–38.
Yel L. Selective IgA deficiency. J Clin Immunol. 2010;30(1):10–6.
Kerr MA. The structure and function of human IgA. Biochem J. 1990;271(2):285–96.
Novak J, Julian BA, Mestecky J, Renfrow MB. Glycosylation of IgA1 and pathogenesis of IgA nephropathy. Semin. Immunopathol. 2012;34(3):365–82.
Tarelli E, Smith AC, Hendry BM, Challacombe SJ. Human serum IgA1 is substituted with up to six O-glycans as shown by matrix assisted laser desorption ionisation time-of-flight mass spectrometry. Carbohydr Res. 2004;13(339):2329–35.
Takahashi K, Smith AD, Poulsen K, Kilian M, Julian BA, Mestecky J, Novak J, Renfrow MB. Naturally occurring structural isomers in serum IgA1 O-glycosylation. J Proteome Res. 2012;11(2):692–702.
Takahashi K, Wall SB, Suzuki H, Smith AD, Hall S, Poulsen K, Kilian M, Mobley JA, Julian BA, Mestecky J, Novak J, Renfrow MB. Clustered O-glycans of IgA1: defining macro- and microheterogeneity by use of electron capture/transfer dissociation. Mol Cell Proteom. 2010;9(11):2545–57.
Yu H-H, Chu K-H, Yang Y-H, Lee J-H, Wang L-C, Lin Y-T, Chiang B-L. Genetics and immunopathogenesis of IgA nephropathy. Clin Rev Allergy Immunol. 2011;41(2):198–213.
Suzuki H, Moldoveanu Z, Hall S, Brown R, Vu HL, Novak L, Julian BA, Tomana M, Wyatt RJ, Edberg JC, Alarcón GS, Kimberly RP, Tomino Y, Mestecky J, Novak J. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest. 2008;118(2):629–39.
Hiki Y, Horii A, Iwase H, Tanaka A, Toda Y, Hotta K, Kobayashi Y. O-linked oligosaccharide on IgA1 hinge region in IgA nephropathy. Fundamental study for precise structure and possible role. Contrib Nephrol. 1995;111:73–84.
Wang Y, Zhao M-H, Zhang Y-K, Li X-M, Wang H-Y. Binding capacity and pathophysiological effects of IgA1 from patients with IgA nephropathy on human glomerular mesangial cells. Clin Exp Immunol. 2004;136(1):168–75.
Novak J, Tomana M, Matousovic K, Brown R, Hall S, Novak L, Julian BA, Wyatt RJ, Mestecky J. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005;67(2):504–13.
Floege J, Moura IC, Daha MR. New insights into the pathogenesis of IgA nephropathy. Semin Immunopathol. 2014;36(4):431–42.
Lai KN. Pathogenesis of IgA nephropathy. Nat Rev Nephrol. 2012;8(5):275–83.
Coppo R, Amore A, Peruzzi L, Vergano L, Camilla R. Innate immunity and IgA nephropathy. J Nephrol. 2010;23(6):626–32.
Suzuki H, Fan R, Zhang Z, Brown R, Hall S, Julian BA, Chatham WW, Suzuki Y, Wyatt RJ, Moldoveanu Z, Lee JY, Robinson J, Tomana M, Tomino Y, Mestecky J, Novak J. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest. 2009;119(6):1668–77.
Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. 1999;104(1):73–81.
Barratt J, Eitner F, Feehally J, Floege J. Immune complex formation in IgA nephropathy: a case of the ‘right’ antibodies in the ‘wrong’ place at the ‘wrong’ time? Nephrol Dial Transplant. 2009;24(12):3620–3.
Schlöndorff D, Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol. 2009;20(6):1179–87.
Levy M. Familial cases of Berger’s disease and anaphylactoid purpura: more frequent than previously thought. Am J Med. 1989;87(2):246–8.
Scolari F, Amoroso A, Savoldi S, Mazzola G, Prati E, Valzorio B, Viola BF, Nicola B, Movilli E, Sandrini M, Campanini M, Maiorca R. Familial clustering of IgA nephropathy: further evidence in an Italian population. Am J Kidney Dis. 1999;33(5):857–65.
Schena FP, Scivittaro V, Ranieri E. IgA nephropathy: pros and cons for a familial disease. Contrib Nephrol. 1993;104:36–45.
Rambausek M, Hartz G, Waldherr R, Andrassy K, Ritz E. Familial glomerulonephritis. Pediatr Nephrol. 1987;1(3):416–8.
Johnston PA, Brown JS, Braumholtz DA, Davison AM. Clinico-pathological correlations and long-term follow-up of 253 United Kingdom patients with IgA nephropathy. A report from the MRC glomerulonephritis registry. Q J Med. 1992;84(304):619–27.
Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24.
Feehally J, Farrall M, Boland A, Gale DP, Gut I, Heath S, Kumar A, Peden JF, Maxwell PH, Morris DL, Padmanabhan S, Vyse TJ, Zawadzka A, Rees AJ, Lathrop M, Ratcliffe PJ. HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol. 2010;21(10):1791–7.
Yu X-Q, Li M, Zhang H, Low H-Q, Wei X, Wang J-Q, Sun L-D, Sim K-S, Li Y, Foo J-N, Wang W, Li Z-J, Yin X-Y, Tang X-Q, Fan L, Chen J, Li R-S, Wan J-X, Liu Z-S, Lou T-Q, Zhu L, Huang X-J, Zhang X-J, Liu Z-H, Liu J-J. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet. 2012;44(2):178–82.
Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, Sanna-Cherchi S, Men CJ, Julian BA, Wyatt RJ, Novak J, He JC, Wang H, Lv J, Zhu L, Wang W, Wang Z, Yasuno K, Gunel M, Mane S, Umlauf S, Tikhonova I, Beerman I, Savoldi S, Magistroni R, Ghiggeri GM, Bodria M, Lugani F, Ravani P, Ponticelli C, Allegri L, Boscutti G, Frasca G, Amore A, Peruzzi L, Coppo R, Izzi C, Viola BF, Prati E, Salvadori M, Mignani R, Gesualdo L, Bertinetto F, Mesiano P, Amoroso A, Scolari F, Chen N, Zhang H, Lifton RP. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43(4):321–7.
Genet N. HHS public xccess. 2015;46(11):1187–96.
Magistroni R, DAgati VD, Appel GB, Kiryluk K. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015;89(1):167–75.
Kiryluk K, Novak J. The genetics and immunobiology of IgA nephropathy. J Clin Invest. 2014;124(6):2325–32.
Rabb H. The T cell as a bridge between innate and adaptive immune systems: implications for the kidney. Kidney Int. 2002;61(6):1935–46.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–84.
Cerutti A. The regulation of IgA class switching. Nat Rev Immunol. 2008;8(6):421–34.
S Sharmin, Y Shimizu, M Hagiwara, K Hirayama, A Koyama. Staphylococcus aureus antigens induce IgA-type glomerulonephritis in Balb/c mice. J Nephrol. 2004;17(4):504–11.
Yamamoto C, Suzuki S, Kimura H, Yoshida H, Gejyo F. Experimental nephropathy induced by Haemophilus parainfluenzae antigens. Nephron. 2002;90(3):320–7.
Amore A, Coppo R, Nedrud JG, Sigmund N, Lamm ME, Emancipator SN. The role of nasal tolerance in a model of IgA nephropathy induced in mice by Sendai virus. Clin Immunol. 2004;113(1):101–8.
Jessen RH, Emancipator SN, Jacobs GH, Nedrud JG. Experimental IgA-IgG nephropathy induced by a viral respiratory pathogen. Dependence on antigen form and immune status. Lab Invest. 1992;67(3):379–86.
Coppo R, Mazzucco G, Martina G, Roccatello D, Amore A, Novara R, Bargoni A, Piccoli G, Sena LM. Gluten-induced experimental IgA glomerulopathy. Lab Invest. 1989;60(4):499–506.
Coppo R, Camilla R, Amore A, Peruzzi L, Daprà V, Loiacono E, Vatrano S, Rollino C, Sepe V, Rampino T, Dal Canton A. Toll-like receptor 4 expression is increased in circulating mononuclear cells of patients with immunoglobulin A nephropathy. Clin Exp Immunol. 2010;159(1):73–81.
Suzuki H, Suzuki Y, Narita I, Aizawa M, Kihara M, Yamanaka T, Kanou T, Tsukaguchi H, Novak J, Horikoshi S, Tomino Y. Toll-like receptor 9 affects severity of IgA nephropathy. J Am Soc Nephrol. 2008;19(12):2384–95.
Qin W, Zhong X, Fan JM, Zhang YJ, Liu XR, Ma XY. External suppression causes the low expression of the Cosmc gene in IgA nephropathy. Nephrol Dial Transplant. 2008;23(5):1608–14.
Buck KS, Smith AC, Molyneux K, El-Barbary H, Feehally J, Barratt J. B-cell O-galactosyltransferase activity, and expression of O-glycosylation genes in bone marrow in IgA nephropathy. Kidney Int. 2008;73(10):1128–36.
Kunkel EJ, Butcher EC. Plasma-cell homing. Nat Rev Immunol. 2003;3(10):822–9.
Batra A, Smith AC, Feehally J, Barratt J. T-cell homing receptor expression in IgA nephropathy. Nephrol Dial Transplant. 2007;22(9):2540–8.
Andre PM, Le Pogamp P, Chevet D. Impairment of jacalin binding to serum IgA in IgA nephropathy. J Clin Lab Anal. 1990;4(2):115–9.
Tomana M, Matousovic K, Julian BA, Radl J, Konecny K, Mestecky J. Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney Int. 1997;52(2):509–16.
Hiki Y, Odani H, Takahashi M, Yasuda Y, Nishimoto A, Iwase H, Shinzato T, Kobayashi Y, Maeda K. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001;59(3):1077–85.
Coppo R, Amore A. Aberrant glycosylation in IgA nephropathy (IgAN). Kidney Int. 2004;65(5):1544–7.
Iwasaki H, Zhang Y, Tachibana K, Gotoh M, Kikuchi N, Kwon Y-D, Togayachi A, Kudo T, Kubota T, Narimatsu H. Initiation of O-glycan synthesis in IgA1 hinge region is determined by a single enzyme, UDP-N-acetyl-alpha-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase 2. J Biol Chem. 2003;278(8):5613–21.
Qin W, Zhou Q, Yang L-C, Li Z, Su B-H, Luo H, Fan J-M. Peripheral B lymphocyte beta1,3-galactosyltransferase and chaperone expression in immunoglobulin A nephropathy. J Intern Med. 2005;258(5):467–77.
Smith AC, de Wolff JF, Molyneux K, Feehally J, Barratt J. O-glycosylation of serum IgD in IgA nephropathy. J Am Soc Nephrol. 2006;17(4):1192–9.
Chintalacharuvu SR, Nagy NU, Sigmund N, Nedrud JG, Amm ME, Emancipator SN. T cell cytokines determine the severity of experimental IgA nephropathy by regulating IgA glycosylation. Clin Exp Immunol. 2001;126(2):326–33.
Serino G, Sallustio F, Cox SN, Pesce F, Schena FP. Abnormal miR-148b expression promotes aberrant glycosylation of IgA1 in IgA nephropathy. J Am Soc Nephrol. 2012;23(5):814–24.
Serino G, Sallustio F, Curci C, Cox SN, Pesce F, De Palma G. Role of let-7b in the regulation of N-acetylgalactosaminyltransferase 2 in IgA nephropathy. Nephrol Dial Transplant. 2015;30(7):1132–9.
Franc V, Řehulka P, Raus M, Stulík J, Novak J, Renfrow MB, Šebela M. Elucidating heterogeneity of IgA1 hinge-region O-glycosylation by use of MALDI-TOF/TOF mass spectrometry: role of cysteine alkylation during sample processing. J Proteom. 2013;92:299–312.
Novak J, Vu HL, Novak L, Julian BA, Mestecky J, Tomana M. Interactions of human mesangial cells with IgA and IgA-containing immune complexes. Kidney Int. 2002;62(2):465–75.
Stockert RJ, Kressner MS, Collins JC, Sternlieb I, Morell AG. IgA interaction with the asialoglycoprotein receptor. Proc Natl Acad Sci USA. 1982;79(20):6229–31.
Tomana M, Kulhavy R, Mestecky J. Receptor-mediated binding and uptake of immunoglobulin A by human liver. Gastroenterology. 1988;94(3):762–70.
Novak J, Julian BA, Tomana M, Mestecky J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol. 2008;28(1):78–87.
Nieuwhof C, Kruytzer M, Frederiks P, van Breda Vriesman PJ. Chronicity index and mesangial IgG deposition are risk factors for hypertension and renal failure in early IgA nephropathy. Am J Kidney Dis. 1998;31(6):962–70.
Berthelot L, Robert T, Vuiblet V, Tabary T, Braconnier A, Dramé M, Toupance O, Rieu P, Monteiro RC. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. 2015;88(4):815–22.
Vuong MT, Hahn-Zoric M, Lundberg S, Gunnarsson I, van Kooten C, Wramner L, Seddighzadeh M, Fernström A, Hanson LÅ, Do LT, Jacobson SH. Association of soluble CD89 levels with disease progression but not susceptibility in IgA nephropathy. Kidney Int. 2010;78(12):1281–7.
Boyd JK, Barratt J. Immune complex formation in IgA nephropathy: CD89 a ‘saint’ or a ‘sinner’? Kidney Int. 2010;78(12):1211–3.
Novak J, Moldoveanu Z, Renfrow MB, Yanagihara T, Suzuki H, Raska M, Hall S, Brown R, Huang W-Q, Goepfert A, Kilian M, Poulsen K, Tomana M, Wyatt RJ, Julian BA, Mestecky J. IgA nephropathy and Henoch–Schoenlein purpura nephritis: aberrant glycosylation of IgA1, formation of IgA1-containing immune complexes, and activation of mesangial cells. Contrib Nephrol. 2007;157:134–8.
Novak J, Raskova Kafkova L, Suzuki H, Tomana M, Matousovic K, Brown R, Hall S, Sanders JT, Eison TM, Moldoveanu Z, Novak L, Novak Z, Mayne R, Julian BA, Mestecky J, Wyatt RJ. IgA1 immune complexes from pediatric patients with IgA nephropathy activate cultured human mesangial cells. Nephrol Dial Transplant. 2011;26(11):3451–7.
Launay BP, Grossetête B, Arcos-fajardo M, Gaudin E, Torres SP, Beaudoin L, De Serre NP, Lehuen A, Monteiro RC. Fc-alpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’ s disease): evidence for pathogenic soluble receptor—IgA complexes in patients and CD89 ransgenic mice. J Exp Med. 2000;191(11):1999–2009.
Berthelot L, Papista C, Maciel TT, Biarnes-Pelicot M, Tissandie E, Wang PHM, Tamouza H, Jamin A, Bex-Coudrat J, Gestin A, Boumediene A, Arcos-Fajardo M, England P, Pillebout E, Walker F, Daugas E, Vrtosvnik F, Flamant M, Benhamou M, Cogné M, Moura IC, Monteiro RC. Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J Exp Med. 2012;209(4):793–806.
Leung JC, Tsang AW, Chan DT, Lai KN. Absence of CD89, polymeric immunoglobulin receptor, and asialoglycoprotein receptor on human mesangial cells. J Am Soc Nephrol. 2000;11(2):241–9.
Kaneko Y, Otsuka T, Tsuchida Y, Gejyo F, Narita I. Integrin alpha1/beta1 and alpha2/beta1 as a receptor for IgA1 in human glomerular mesangial cells in IgA nephropathy. Int Immunol. 2012;24(4):219–32.
Moura IC, Arcos-Fajardo M, Gdoura A, Leroy V, Sadaka C, Mahlaoui N, Lepelletier Y, Vrtovsnik F, Haddad E, Benhamou M, Monteiro RC. Engagement of transferrin receptor by polymeric IgA1: evidence for a positive feedback loop involving increased receptor expression and mesangial cell proliferation in IgA nephropathy. J Am Soc Nephrol. 2005;16(9):2667–76.
Tamouza H, Chemouny JM, Raskova Kafkova L, Berthelot L, Flamant M, Demion M, Mesnard L, Paubelle E, Walker F, Julian BA, Tissandié E, Tiwari MK, Camara NOS, Vrtovsnik F, Benhamou M, Novak J, Monteiro RC, Moura IC. The IgA1 immune complex-mediated activation of the MAPK/ERK kinase pathway in mesangial cells is associated with glomerular damage in IgA nephropathy. Kidney Int. 2012;82(12):1284–96.
Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33(6):479–92.
Wyatt RJ, Julian BA. Activation of complement in IgA nephropathy. Am J Kidney Dis. 1988;12(5):437–42.
Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, Novak J. Current understanding of the role of complement in IgA nephropathy. J Am Soc Nephrol. 2015;26(7):1503–12.
Nasri H, Ahmadi A, Rafieian-Kopaei M, Bashardoust B, Nasri P, Mubarak M. Association of glomerular C4d deposition with various demographic data in IgA nephropathy patients; a preliminary study. J Nephropathol. 2015;4(1):19–23.
Espinosa M, Ortega R, Sánchez M, Segarra A, Salcedo MT, González F, Camacho R, Valdivia MA, Cabrera R, López K, Pinedo F, Gutierrez E, Valera A, Leon M, Cobo MA, Rodriguez R, Ballarín J, Arce Y, García B, Muñoz MD, Praga M. Association of C4d deposition with clinical outcomes in IgA nephropathy. Clin J Am Soc Nephrol. 2014;9(5):897–904.
Ohsawa I, Ishii M, Ohi H, Tomino Y. Pathological scenario with the mannose-binding lectin in patients with IgA nephropathy. J Biomed Biotechnol. 2012;2012:476739.
Liu L-L, Liu N, Chen Y, Wang L-N, Jiang Y, Wang J, Li X-L, Yao L, Fan Q-L. Glomerular mannose-binding lectin deposition is a useful prognostic predictor in immunoglobulin A nephropathy. Clin Exp Immunol. 2013;174(1):152–60.
Ohsawa I, Ishii M, Ohi H, Tomino Y. Pathological scenario with the mannose-binding lectin in patients with IgA nephropathy. J Biomed Biotechnol. 2012;2012(Figure 1):476739.
Moura IC, Benhamou M, Launay P, Vrtovsnik F, Blank U, Monteiro RC. The glomerular response to IgA deposition in IgA nephropathy. Semin Nephrol. 2008;28(1):88–95.
Lai KN, Leung JCK, Chan LYY, Guo H, Tang SCW. Interaction between proximal tubular epithelial cells and infiltrating monocytes/T cells in the proteinuric state. Kidney Int. 2007;71(6):526–38.
Acknowledgments
This study was partially supported by CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior), CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil) and FAPEMIG (Fundação de Amparo à Pesquisa do Estado de Minas Gerais, Brazil).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None declared.
Additional information
Responsible Editor: Graham R. Wallace.
Rights and permissions
About this article

Cite this article
Fabiano, R.C.G., Pinheiro, S.V.B. & Simões e Silva, A.C. Immunoglobulin A nephropathy: a pathophysiology view. Inflamm. Res. 65, 757–770 (2016). https://doi.org/10.1007/s00011-016-0962-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-016-0962-x