
Abstract
Immunoglobulin A nephropathy (IgAN) or Berger’s disease is the most common form of primary glomerulonephritis in the world and one of the first causes of end-stage renal failure. IgAN is characterized by the accumulation of immune complexes containing polymeric IgA1 in mesangial areas. The pathogenesis of this disease involves the deposition of polymeric and hypogalactosylated IgA1 (Gd-IgA1) in the mesangium. Quantitative and structural changes of Gd-IgA1 play a key role in the development of the disease due to functional abnormalities of two IgA receptors: the FcαRI (CD89) expressed by blood myeloid cells and the transferrin receptor (CD71) on mesangial cells. Abnormal Gd-IgA1 induces release of soluble CD89, which participates in the formation of circulating IgA1 complexes. These complexes are trapped by CD71 that is overexpressed on mesangial cells in IgAN patients together with the crosslinking enzyme transglutaminase 2 allowing pathogenic IgA complex formation in situ and mesangial cell activation. A humanized mouse model expressing IgA1 and CD89 develops IgAN in a similar manner as patients. In this model, a food antigen, the gliadin, was shown to be crucial for circulating IgA1 complex formation and deposition, which could be prevented by a gluten-free diet. Identification of these new partners opens new therapeutic prospects for IgAN treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Immunoglobulin A (IgA)
IgA is a relatively “unique” antibody isotype in humans due, on one hand, to its heterogeneity of molecular forms, subclasses and glycosylation and, on the other, to the unexpected poor participation of serum IgA in systemic immune responses [1]. IgA, differentially distributed between the systemic and mucosal immune system, is by far the most abundantly produced isotype in humans. More IgA is produced per day (66 mg/kg/day) than all other classes combined [2], notably at epithelial sites where most is lost daily within exocrine secretions as mucosal surface encompasses more than 400 m2. Serum IgA, the second most abundant isotype in the circulation, mainly consists of monomers derived from bone marrow plasma cells [1], whereas secretory IgA is synthesized as dimers by local plasma cells before being transported to mucosal surfaces through epithelial cells by the polymeric Ig receptor [3]. IgA displays a T-shaped structure, which differs from the common Y-shape of other Ig [4]. IgA is divided into closely related subclasses, IgA1 and IgA2, which basically differ by the absence of a 13-amino acid sequence in the hinge region of the IgA2 molecule [5]. This difference explains the resistance of IgA2 to the action of bacterial proteases (i.e., from Streptococcus mutants, Neisseria meningitidis and Haemophilus influenzae) and may underlie the predominance of IgA2 in mucosal secretions [5]. Interestingly, the IgA system differs substantially in the species studied, i.e., man vs. mice and rats. While two IgA subclasses are recognized in humans, only one class exists in mice and rats [6]. Serum IgA is mostly monomeric in humans, and polymeric in mice. Clearance via the hepatobiliary route plays an important role in mice, but not in humans [1]. In serum, IgA (mainly monomeric and of the IgA1 subclass) constitutes one-fifth of the total Ig pool due to a rapid catabolism (half-life: 3–6 days). IgA is the most glycosylated form of Ig, with carbohydrates representing about 6 % of its content. Unlike IgA2, the heavy chains of IgA1 molecules contain a unique insertion in the hinge-region segment between the first and second constant region domains. This hinge region has a high content of serine and threonine residues, which are the sites of attachment of up to five O-linked glycan chains consisting of N-acetylgalactosamine with a β1,3-linked galactose and sialic acids [7]. Sialic acid can also be attached to N-acetylgalactosamine by an α2,6 linkage. It is noteworthy, however, that the carbohydrate composition of these O-linked glycans in the hinge region of serum IgA1 is heterogeneous, the commonest forms including N-acetylgalactosamine-galactose disaccharide, and its mono- and di-sialylated variants.
Role of IgA receptors in homeostasis and immune protection
Serum monomeric IgA (mIgA) is a poor player in systemic immune responses. It plays rather a paradoxical role in immunity with its involvement in immune inhibition but also as a pathogenic actor in inflammatory diseases depending on the type and structure of IgA. The major role of serum mIgA in physiology is as a powerful anti-inflammatory effector towards the immune system. It has been demonstrated by several groups for more than two decades that in the absence of antigen, serum IgA down-regulates IgG-mediated phagocytosis, chemotaxis, bactericidal activity, oxidative burst activity, and cytokine release [8–14]. The most striking supporting evidence for the inhibitory role of serum mIgA comes from patients with selective IgA deficiency [15]. In these patients, both IgA1 and IgA2 are usually markedly reduced or absent. In addition to increased infections of the respiratory and gastrointestinal tract, these patients show increased susceptibility to autoimmune and allergic disorders including celiac disease, lupus, rheumatoid arthritis, autoimmune endocrinopathies, chronic active hepatitis, ulcerative colitis, Crohn’s disease, and autoimmune hematologic disorders [16].
The anti-inflammatory action of serum mIgA was enigmatic until the discovery of IgA Fc receptors. Fc receptors are defined by their specificity for the Fc fragment of immunoglobulin isotypes, and receptors for IgA are referred to as FcαR [6]. Although they are not structurally related, there are several types of IgA receptors. Three of them are considered bona fide FcαR. The first one is designated FcαRI (or CD89), and is a receptor specific for IgA, capable of binding both human IgA1 and IgA2 subclasses. The other FcαRs are the polymeric Ig receptor (PIgR), involved in Ig transport across epithelial barriers, and Fcα/µR, both having the common property of binding IgA and IgM. The four alternative IgA receptors are the asialoglycoprotein receptor, the transferrin receptor, FcRL4 and DCSIGN/SIGNR1 [6, 17].
Among these IgA receptors, CD89 plays an essential anti-inflammatory role in immunity allowing transmission of inhibitory signals following binding of serum mIgA. CD89 is the only IgA Fc receptor expressed on blood myeloid cells including monocytes/macrophages, dendritic cells, Kupffer cells, neutrophils and eosinophils [6]. Interestingly, like IgA, CD89 differs substantially between species since mice fail to express a CD89 homolog. CD89 can bind IgA1 and IgA2 with moderate affinity (Ka~106 M−1) in the boundaries of CHα2 and CHα3 domains. While pIgA and IgA immune complexes (IC) bind with a greater avidity [18], mIgA binding to CD89 was found to be transient in vitro using surface plasmon resonance approaches. It seems reasonable however to assume that, in the presence of high concentrations of circulating mIgA (~2 mg/ml), CD89 may be constantly occupied. Supporting this statement, there is indeed evidence in the literature that IgA can be detected on the surface of monocytes and neutrophils—this was initially described as “cytophilic” IgA [19, 20]. Recently, we demonstrated for the first time that monovalent targeting of CD89 either by mIgA or anti-CD89 Fab inhibits IgG-mediated phagocytosis in human monocytes [21, 22], explaining previously described inhibitory functions of serum IgA [8–14]. More importantly, we were able to show that CD89-mediated inhibition was not associated with conventional inhibitory motifs such as the immunoreceptor tyrosine-based inhibition motif (ITIM). Indeed CD89 does not contain any ITIM in its cytoplasmic tail, but it can be expressed with or without physical association to the FcRγ adaptor [23]. While γ-less CD89 recycles monomeric IgA playing an essential role in mIgA homeostasis, FcRγ-associated CD89 mediates either activating or inhibitory responses [21], defining this receptor as a dual function receptor which operates depending on the type of the ligand: multimers or monomers. Both opposite functions were mediated by the FcRγ adaptor which contains an immunoreceptor tyrosine-based activation motif (ITAM) in its cytoplasmic tail. These data might explain why CD89 is considered a unique member of the FcR family. Analysis of CD89’s 3-dimensional structure reveals that it binds IgA in a manner different from other Ig isotypes. Its two Ig-like domains are oriented at right angles and two CD89 molecules can bind one IgA molecule within the EC1 domain, at a site completely different from the other FcRs, yielding a 2:1 stoichiometry as compared to the 1:1 stoichiometry of IgE and IgG to their FcRs [24]. The CD89 gene is not located in the FcR gene cluster but in chromosome 19, inside the leukocyte receptor cluster (LRC). CD89 is distantly related to other FcRs, being more homologous to LRC-encoded activating and inhibitory receptors [25].
Taken together, our findings on the functional regulation of CD89 allow the definition of a new inhibitory motif: the inhibitory ITAM, or ITAMi [26]. ITAMi signaling results from an inhibitory configuration of the ITAM present within an activating receptor [26]. These configurations (activating vs. inhibitory) are dependent on the way the ITAM-containing receptor interacts with its ligand. In the case of CD89, while highly multimeric interaction induces an activating signal, interactions of the receptor with ligands binding at low valency such as mIgA or anti-CD89 Fab fragments produce an inhibitory response. Indeed, the 2:1 stoichiometry of CD89:mIgA interaction can lead to receptor dimerization. This dimeric interaction generates an inhibitory signal, in contrast to the signal induced by multimeric receptor aggregation. The inhibitory signal is also generated with anti-CD89 Fab fragments, which in principle should target the receptor in a monomeric fashion.
Specific targeting of the ITAM-containing receptor with a weakly binding ligand (low affinity, low avidity, low valency) induces only scant ITAM phosphorylation thereby favoring the recruitment of signaling effectors such as the phosphatase SHP-1 with inhibitory potential [21, 27]. This is in contrast to highly multimeric ligation, which leads to a strong Src kinase-mediated phosphorylation of FcRγ and recruitment of Syk [28, 29]. Monovalent targeting of CD89 by Fab anti-CD89 or by mIgA prevented asthma, nephritis or inflammatory arthritis development in CD89 Tg mice [21, 22, 27].
Role of IgA receptors as pathogenic actors in IgA nephropathy
IgAN is the most common IgA-associated disease. The cause of IgAN, which is variable as regards the severity of its presentation, remains unknown. It occurs as a primary disease but can be associated with Henoch-Schönlein purpura, celiac disease or inflammatory bowel disease. In IgAN, IgA molecules deposited in the kidney are primarily pIgA1 [30]. There are many reports indicating that circulating IgA in patients with the disease is abnormal in terms of its concentrations, molecular size or aggregation state. However, IgA concentrations may not be an essential pathogenic factor since in IgA myeloma patients, IgA deposition in the kidney is not correlated to serum IgA levels, indicating that there is a particularity in the composition of nephritogenic IgA1 complexes in IgAN. Moreover, increased serum levels of IgA are often found in other IgA-associated diseases such as Henoch-Schönlein purpura, ankylosing spondylitis, Sjögren’s syndrome, alcoholic liver cirrhosis, celiac disease, inflammatory bowel disease, and dermatitis herpetiformis [31, 32]. Interestingly, a study has shown that the Fcα fragments of serum dimeric and trimeric but not mIgA1 aggregated to form multimers resistant to disruption in sodium-dodecyl-sulfate [33]. This might explain the propensity of pIgA to aggregate, facilitating interaction with its receptors at leukocyte and tissue levels. There is also evidence that mesangial IgA1 exhibits a negative net charge [30] and decreased glycosylation [34] in IgAN, suggesting that this abnormal structural feature could underlie its pathogenic potential. The O-linked carbohydrate chains present in the hinge region of IgA1 in IgAN patients appear to be different from those in normal individuals [35]. This lack of specific galactose residues could have marked effects on the properties of the molecule and create new epitopes that in turn could generate an immune response with IgG antibodies against hypogalactosylated (Gd) IgA1 [36, 37]. The trigger for this abnormality is unknown, but recent studies have shown that IgG anti-Gd-IgA1 antibodies are good markers of progression or recurrence after renal transplantation [38]. In addition, altered glycosylation favors self-aggregation of IgA1 [39].
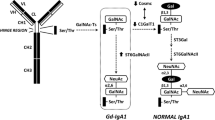
Increased levels of IgA, are associated with aberrantly decreased expression of CD89 [20, 40–42]. Studies in IgAN patients and patients with other IgA-associated diseases including human immunodeficiency virus (HIV) infection, alcoholic liver cirrhosis, and spondyloarthropathies indicate reduced CD89 expression levels on circulating monocytes and, to a lesser degree, on neutrophils [20, 40–42]. Addition of IgA has a negative effect on CD89 expression [20], possibly due to shedding of the CD89 extracellular domain [43] (Fig. 1a). Indeed, production of soluble CD89 was induced in vitro by pIgA incubated with CD89-transfected cells. The demonstration of soluble CD89 in serum of IgAN patients and not in serum from healthy controls supports this hypothesis [44]. However, in contrast to the increased circulating levels of IgA1-IgG complexes observed in severe IgAN patients, levels of IgA-sCD89 complexes were decreased in these patients (or transplanted patients with recurrence of IgA1 deposits) suggesting that CD89-containing complexes could be selectively trapped in the mesangium, aggravating the disease [45].

Proposed molecular mechanism involving IgA receptors in IgA nephropathy. a The pathogenic IgA1 complexes containing polymeric hypogalactosylated IgA1 (Gd-IgA1) alone or complexed with IgG anti-Gd-IgA1 antibodies may lead to enhanced IgA binding to CD89 on blood monocytes. This would generate soluble CD89/Gd-IgA1 complexes upon cleavage of FcRγ-less CD89. b Soluble CD89/Gd-IgA1 (and/or eventually soluble CD89/Gd-IgA1-IgG) may become deposited in the mesangium through binding of IgA1 and sCD89 to the CD71 receptor. This is a hypothetical representation, as the two binding sites of these molecules to CD71 remain unknown. The interaction of macromolecular IgA1 complexes with the CD71 receptor induces transglutaminase 2 expression at the mesangial cell surface, which will lead to CD71 activation, initiating an inflammatory feedback loop by inducing its enhanced expression together with cell proliferation and cytokine/chemokine production. The latter will further increase the inflammatory response and disease development
CD89 transgenic mice, in which the transgene is driven by a myeloid-specific promoter (human CD11b) conferring high CD89 expression on monocytes/macrophages, have been shown to spontaneously develop IgA nephropathy [43]. The mechanism in this model was shown to involve binding of mouse IgA dimers to human CD89 with subsequent receptor shedding and release of soluble CD89:IgA complexes that deposit in the kidney. However, these animals only developed mouse IgA deposits after 24 weeks of age due to the low affinity between mouse IgA and human CD89 and failed to show renal function alterations. The pathogenic role of sCD89 was recently demonstrated using a double transgenic mouse model expressing both human CD89 and human IgA1 (α1KI-CD89Tg mice) [46]. Circulating IgA1-sCD89 complexes in these mice induced mesangial deposits, hematuria and proteinuria in contrast to mice expressing human IgA1 alone. Moreover, sCD89 was detected in mesangial deposits in biopsies of patients with IgAN. This model also revealed that pathogenesis involves at least four elements: the interaction of IgA1-sCD89 complexes with another IgA1 receptor, with the transferrin receptor 1 (CD71) and with the transglutaminase 2 (TGase 2), a cross-linking enzyme that may play a role in amplification of in situ pathogenic complex deposition in the mesangium.
The mechanism responsible for deposition of IgA1 complexes in the mesangium in IgAN remained enigmatic for several years as patient mesangial cells fail to express CD89, Fcα/μR, ASGP-R and poly-IgR [47], until the identification of the CD71 as a mesangial IgA1 receptor in patients with IgAN [48]. It has been demonstrated that IgA1 complexes and hypogalactosylated IgA1 have a higher affinity for CD71 and trigger inflammation, via the release from mesangial cells of pro-inflammatory cytokines [such as interleukin (IL)-1, IL-6 and tumor necrosis factor (TNF)-α], and mesangial cell proliferation, with the potential to promote mesangial expansion and chemotactic activity towards leukocytes [49, 50]. Moreover, CD71 overexpression was associated with in situ ERK phosphorylation in biopsies of patients with severe disease [51]. However, lessons from transgenic animal studies suggest that in situ crosslinking of mesangial CD71 by both IgA and sCD89 in glomeruli is a necessary event. Soluble CD89 can bind CD71 and induce TGase 2, which in turn is translocated to the mesangial plasma membrane allowing cell activation by IgA1-sCD89 complexes. Thus, CD89 and CD71 may cooperate with abnormal IgA1 to account for initiation, progression and chronicity of the disease (Fig. 1b).
Common IgA and IgA receptor dysfunctions in celiac disease (CD) and IgAN
IgA plays an important role in various inflammatory diseases, such as CD and IgAN. Two main questions arise: (a) Is there a correlation between these two diseases? (b) How is IgA involved in this possible common pathogenic mechanism? Various studies have shown that IgAN occurs as a primary disease but it can be associated with Henoch-Schönlein purpura, CD or inflammatory bowel disease [52]. Indeed, the prevalence of CD in the general population is 0.5–1 % depending on the geographical region, and this percentage rises to 4 % in patients with IgAN [53]. Inversely, CD may be a risk factor for renal disease, especially for IgAN. Glomerular mesangial deposits of IgA occur frequently in untreated CD and they are associated with circulating IC containing IgA [54]. However, in this situation IgA seems to be deposited without being able to induce clinically overt glomerulonephritis.
Coppo et al. have shown the presence of IgA anti-gliadin antibodies, associated with elevated IgA levels, in the serum of IgAN patients [55]. Moreover, IgA and IgG anti-endomysium antibodies were detected in the serum of IgAN patients [56]. These data suggest that dietary components (e.g., gliadin) may play a role in IgAN by promoting IgA IC formation and perhaps favoring mesangial localization via lectinic interactions; thus they propose a possible association between IgAN and CD [57]. This is supported by the finding that treatment with a gluten-free diet in IgAN patients [58] and animal models [55] decreased IgA IC and IgA anti-gliadin antibody levels in the serum, and ameliorated clinical symptoms of the disease, such as proteinuria and hematuria. Nevertheless, there was relentless progression to renal failure.
Concerning the mucosal immunity in IgAN, Kovács et al. observed that IgAN patients compared to controls presented significantly higher intestinal permeability, related to increased proteinuria, microhematuria and serum IgA levels [59]. Thus, elevated intestinal permeability in IgAN patients may play a role in the pathogenesis of the disease and adversely influence its progression. Another study demonstrated the presence of a rectal mucosal sensitivity to gluten in one-third of IgAN patients, but without any signs of CD, suggesting that such sub-clinical inflammation to gluten might be involved in the pathogenesis of IgAN in a subgroup of patients [60].
The pathogenic mechanism that links IgAN and CD is not yet clear [61]. It is known that lectins, particularly gliadins, can bind pIgA1 containing Gal and GalNac residues [57]. This reaction leads to the formation of macromolecular IgA1 IC. Interestingly, patients with CD display increased levels of Gd-IgA1 in the circulation [62]. Moreover, gliadin can also bind mesangial cells via lectinic bonds favoring the bridging of pIgA1 and IgA1 IC to these cells, and thus enhancing both IgA1 mesangial trapping and in situ IgA1 deposit formation. Gliadin binding on mesangial cells also modulates the production of immunological mediators and hemodynamic factors (increase of TNF-α and inhibition of prostaglandin E2 production) [57]. These changes might stimulate mesangial cell growth and mesangial matrix production, contributing to IgAN pathogenesis. Recent data from our laboratory show that gliadin can bind CD89 and induce nephrogenic sCD89-IgA1 complexes in α1KI-CD89Tg mice [63]. Mice were rendered gluten-sensitive by being administered a gluten-free diet for at least three generations. This led to a reduction in IgA1 mesangial deposition, a reduction in mesangial CD71 and TGase 2, reduced glomerular inflammatory-cell infiltration (CD11b+ and CD3+ cells), a reduction in hematuria, and reduced IgA1–sCD89 complexes in the serum and kidney eluates. Exposure to gluten in these sensitized mice led to intestinal injury, demonstrated by inflammation and villous atrophy, increased circulating IgA1–sCD89 complexes, mesangial IgA1 deposition, and elevated serum IgA1 anti-gliadin antibodies, that correlated with the level of proteinuria. Interestingly, a correlation was also found between anti-gliadin antibody levels in IgAN patients and proteinuria. Pertinently, early introduction of a gluten-free diet in α1KI-CD89Tg mice at 3 weeks of age prevented the typical development of mesangial IgA1 deposition and hematuria [63]. This study provides novel mechanistic insights into the interactions required for IgA deposition and opens up prospects for further examination of the link between IgAN, gliadin and IgA1-sCD89 complexes in large cohorts of IgAN patients with unexplained gastrointestinal symptoms.
The possible role of oral tolerance breakdown in the pathophysiology of IgAN has been proposed, but there is still no clear evidence for the triggering factor of this process. It seems that the breakdown of oral tolerance can be favored by perturbations of epithelial cell function, resulting in abnormal processing of dietary antigens, such as gliadin, which renders them immunogenic rather than tolerogenic. In this case, the cytokines produced by the immune system activation influence the epithelial cell secretory component and class II antigen presentation. These modifications lead to intestinal permeability changes, allowing increased antigen uptake and presentation with stimulation of the mucosa immune system, resulting in mucosal inflammatory diseases, like CD. Furthermore, we have previously shown that in CD patients, the gliadin peptide 31–49 crosses the intestinal barrier without being degraded by the lysosomal pathway, through a mechanism that bears some similarities to that observed in IgAN (overexpression of CD71 at the apical surface of enterocytes) [64]. To conclude, the efficacy of a gluten-free diet, together with the observed increase in intestinal permeability and in serum IgA reactivity to gliadin, and the increased levels of salivary and serum secretory IgA, have prompted the notion that the mucosal immune system plays an important role in IgAN pathophysiology.
Conclusions
IgA–IgA receptor interactions play a significant role in the pathophysiology of IgAN. Understanding the molecular events involved in the pathological process will help to identify new partners, opening up new prospects for the treatment of this disease.
References
Kerr MA (1990) The structure and function of human IgA. Biochem J 271:285–296
Solomon A (1980) Monoclonal immunoglobulins as biomarkers of cancer. In: Sell S (ed) Cancer markers. Humana Press, New York, pp 57–87
Phalipon A, Corthésy B (2003) Novel functions of the polymeric Ig receptor: well beyond transport of immunoglobulins. Trends Immunol 24:55–58
Boehm MK, Woof JM, Kerr MA, Perkins SJ (1999) The fab and fc fragments of IgA1 exhibit a different arrangement from that in IgG: a study by X-ray and neutron solution scattering and homology modelling1. J Mol Biol 286:1421–1447. doi:10.1006/jmbi.1998.2556
Woof JM, Kerr MA (2006) The function of immunoglobulin A in immunity. J Pathol 208:270–282. doi:10.1002/path.1877
Monteiro RC, Van De Winkel JGJ (2003) IgA Fc receptors. Annu Rev Immunol 21:177–204. doi:10.1146/annurev.immunol.21.120601.141011
Mestecky J, Tomana M, Moldoveanu Z et al (2008) Role of aberrant glycosylation of IgA1 molecules in the pathogenesis of IgA nephropathy. Kidney Blood Press Res 31:29–37. doi:10.1159/000112922
Van Epps DE, Williams RC (1976) Suppression of leukocyte chemotaxis by human IgA myeloma components. J Exp Med 144:1227–1242. doi:10.1084/jem.144.5.1227
Van Epps DE, Reed K, Williams RC (1978) Suppression of human PMN bactericidal activity by human IgA paraproteins. Cell Immunol 36:363–376
Wilton JMA (1978) Suppression by IgA of IgG-mediated phagocytosis by human polymorphonuclear leucocytes. Clin Exp Immunol 34:423–428
Wolf HM, Fischer MB, Puhringer H et al (1994) Human serum IgA downregulates the release of inflammatory cytokines (tumor necrosis factor-alpha, interleukin-6) in human monocytes. Blood 83:1278–1288
Nikolova EB, Russell MW (1995) Dual function of human IgA antibodies: inhibition of phagocytosis in circulating neutrophils and enhancement of responses in IL-8-stimulated cells. J Leukoc Biol 57:875–882
Wolf HM, Hauber I, Gulle H et al (1996) Anti-inflammatory properties of human serum IgA: induction of IL-1 receptor antagonist and FcαR (CD89)-mediated down-regulation of tumour necrosis factor-alpha (TNF-α) and IL-6 in human monocytes. Clin Exp Immunol 105:537–543. doi:10.1046/j.1365-2249.1996.d01-793.x
Olas K, Butterweck H, Teschner W et al (2005) Immunomodulatory properties of human serum immunoglobulin A: anti-inflammatory and pro-inflammatory activities in human monocytes and peripheral blood mononuclear cells. Clin Exp Immunol 140:478–490. doi:10.1111/j.1365-2249.2005.02779.x
Jacob CMA, Pastorino AC, Fahl K et al (2008) Autoimmunity in IgA deficiency: revisiting the role of IgA as a silent housekeeper. J Clin Immunol 28(Suppl 1):S56–S61. doi:10.1007/s10875-007-9163-2
Ludvigsson JF, Neovius M, Hammarström L (2014) Association between IgA deficiency and other autoimmune conditions: a population-based matched cohort study. J Clin Immunol 34:444–451. doi:10.1007/s10875-014-0009-4
Diana J, Moura IC, Vaugier C et al (2013) Secretory IgA induces tolerogenic dendritic cells through SIGNR1 dampening autoimmunity in mice. J Immunol 191:2335–2343. doi:10.4049/jimmunol.1300864
Wines BD, Sardjono CT, Trist HM et al (2001) The interaction of FcαRI with IgA and its implications for ligand binding by immunoreceptors of the leukocyte receptor cluster. J Immunol 166:1781–1789. doi:10.4049/jimmunol.166.3.1781
Lawrence DA, Weigle WO, Spiegelberg HL (1975) Immunoglobulins cytophilic for human lymphocytes, monocytes, and neutrophils. J Clin Invest 55:368–376
Grossetête B, Launay P, Lehuen A et al (1998) Down-regulation of Fc alpha receptors on blood cells of IgA nephropathy patients: evidence for a negative regulatory role of serum IgA. Kidney Int 53:1321–1335. doi:10.1046/j.1523-1755.1998.00885.x
Pasquier B, Launay P, Kanamaru Y et al (2005) Identification of FcαRI as an inhibitory receptor that controls inflammation: dual role of FcRγ ITAM. Immunity 22:31–42. doi:10.1016/j.immuni.2004.11.017
Rossato E, Ben Mkaddem S, Kanamaru Y et al (2015) Reversal of arthritis by human monomeric IgA through the receptor-mediated SH2 domain-containing phosphatase 1 inhibitory pathway. Arthritis Rheumatol 67:1766–1777. doi:10.1002/art.39142
Launay P, Patry C, Lehuen A et al (1999) Alternative endocytic pathway for immunoglobulin A Fc receptors (CD89) depends on the lack of FcRgamma association and protects against degradation of bound ligand. J Biol Chem 274:7216–7225
Herr AB, Ballister ER, Bjorkman PJ (2003) Insights into IgA-mediated immune responses from the crystal structures of human FcαRI and its complex with IgA1-Fc. Nature 423:614–620. doi:10.1038/nature01685
Davis RS, Dennis G Jr, Odom MR et al (2002) Fc receptor homologs: newest members of a remarkably diverse Fc receptor gene family. Immunol Rev 190:123–136. doi:10.1034/j.1600-065X.2002.19009.x
Blank U, Launay P, Benhamou M, Monteiro RC (2009) Inhibitory ITAMs as novel regulators of immunity. Immunol Rev 232:59–71. doi:10.1111/j.1600-065X.2009.00832.x
Kanamaru Y, Pfirsch S, Aloulou M et al (2008) Inhibitory ITAM signaling by FcαRI-FcRγ chain controls multiple activating responses and prevents renal inflammation. J Immunol 180:2669–2678. doi:10.4049/jimmunol.180.4.2669
Launay P, Lehuen A, Kawakami T et al (1998) IgA Fc receptor (CD89) activation enables coupling to syk and Btk tyrosine kinase pathways: differential signaling after IFN-gamma or phorbol ester stimulation. J Leukoc Biol 63:636–642
Kanamaru Y, Arcos-Fajardo M, Moura IC et al (2007) Fcα receptor I activation induces leukocyte recruitment and promotes aggravation of glomerulonephritis through the FcRγ adaptor. Eur J Immunol 37:1116–1128. doi:10.1002/eji.200636826
Monteiro RC, Halbwachs-Mecarelli L, Roque-Barreira MC et al (1985) Charge and size of mesangial IgA in IgA nephropathy. Kidney Int 28:666–671
Delacroix DL, Elkom KB, Geubel AP et al (1983) Changes in size, subclass, and metabolic properties of serum immunoglobulin A in liver diseases and in other diseases with high serum immunoglobulin A. J Clin Invest 71:358–367
Montenegro V, Monteiro RC (1999) Elevation of serum IgA in spondyloarthropathies and IgA nephropathy and its pathogenic role. Curr Opin Rheumatol 11:265–272
Almogren A, Kerr MA (2008) Irreversible aggregation of the Fc fragment derived from polymeric but not monomeric serum IgA1—Implications in IgA-mediated disease. Mol Immunol 45:87–94. doi:10.1016/j.molimm.2007.05.002
Allen AC, Bailey EM, Brenchley PEC et al (2001) Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: observations in three patients. Kidney Int 60:969–973. doi:10.1046/j.1523-1755.2001.060003969.x
Novak J, Julian BA, Tomana M, Mestecky J (2008) IgA glycosylation and IgA immune complexes in the pathogenesis of IgA Nephropathy. Semin Nephrol 28:78–87. doi:10.1016/j.semnephrol.2007.10.009
Tomana M, Novak J, Julian BA et al (1999) Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest 104:73–81
Suzuki H, Moldoveanu Z, Hall S et al (2008) IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest 118:629–639. doi:10.1172/JCI33189
Berthoux F, Suzuki H, Thibaudin L et al (2012) Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J Am Soc Nephrol 23:1579–1587. doi:10.1681/ASN.2012010053
Kokubo T, Hiki Y, Iwase H et al (1997) Evidence for involvement of IgA1 hinge glycopeptide in the IgA1-IgA1 interaction in IgA nephropathy. J Am Soc Nephrol 8:915–919
Grossetête B, Viard JP, Lehuen A et al (1995) Impaired Fc alpha receptor expression is linked to increased immunoglobulin A levels and disease progression in HIV-1-infected patients. AIDS Lond Engl 9:229–234
Silvain C, Patry C, Launay P et al (1995) Altered expression of monocyte IgA Fc receptors is associated with defective endocytosis in patients with alcoholic cirrhosis. Potential role for IFN-gamma. J Immunol 155:1606–1618
Montenegro V, Chiamolera M, Launay P et al (2000) Impaired expression of IgA Fc receptors (CD89) by blood phagocytic cells in ankylosing spondylitis. J Rheumatol 27:411–417
Launay P, Grossetête B, Arcos-Fajardo M et al (2000) Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J Exp Med 191:1999–2009
Berthelot L, Robert T, Vuiblet V et al (2015) Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. doi:10.1038/ki.2015.158
Monteiro RC, Moura IC, Launay P et al (2002) Pathogenic significance of IgA receptor interactions in IgA nephropathy. Trends Mol Med 8:464–468
Berthelot L, Papista C, Maciel TT et al (2012) Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J Exp Med 209:793–806. doi:10.1084/jem.20112005
Leung JCK, Tsang AWL, Chan DTM, Lai KN (2000) Absence of CD89, polymeric immunoglobulin receptor, and asialoglycoprotein receptor on human mesangial cells. J Am Soc Nephrol 11:241–249
Moura IC, Centelles MN, Arcos-Fajardo M et al (2001) Identification of the transferrin receptor as a novel immunoglobulin (Ig)a1 receptor and its enhanced expression on mesangial cells in Iga nephropathy. J Exp Med 194:417–426. doi:10.1084/jem.194.4.417
Moura IC, Arcos-Fajardo M, Sadaka C et al (2004) Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J Am Soc Nephrol JASN 15:622–634
Moura IC, Arcos-Fajardo M, Gdoura A et al (2005) Engagement of transferrin receptor by polymeric IgA1: evidence for a positive feedback loop involving increased receptor expression and mesangial cell proliferation in IgA nephropathy. J Am Soc Nephrol JASN 16:2667–2676. doi:10.1681/ASN.2004111006
Tamouza H, Chemouny JM, Raskova Kafkova L et al (2012) The IgA1 immune complex-mediated activation of the MAPK/ERK kinase pathway in mesangial cells is associated with glomerular damage in IgA nephropathy. Kidney Int 82:1284–1296. doi:10.1038/ki.2012.192
Monteiro RC (2010) Role of IgA and IgA Fc receptors in inflammation. J Clin Immunol 30:1–9. doi:10.1007/s10875-009-9338-0
Collin P, Syrjänen J, Partanen J et al (2002) Celiac disease and HLA DQ in patients with IgA nephropathy. Am J Gastroenterol 97:2572–2576. doi:10.1111/j.1572-0241.2002.06025.x
Pasternack A, Collin P, Mustonen J et al (1990) Glomerular IgA deposits in patients with celiac disease. Clin Nephrol 34:56–60
Coppo R, Amore A, Roccatello D (1992) Dietary antigens and primary immunoglobulin A nephropathy. J Am Soc Nephrol 2:S173
Pierucci A, Fofi C, Bartoli B et al (2002) Antiendomysial antibodies in Berger’s disease. Am J Kidney Dis 39:1176–1182. doi:10.1053/ajkd.2002.33387
Amore A, Emancipator SN, Roccatello D et al (1994) Functional consequences of the binding of gliadin to cultured rat mesangial cells: bridging immunoglobulin A to cells and modulation of eicosanoid synthesis and altered cytokine production. Am J Kidney Dis Off J Natl Kidney Found 23:290–301
Coppo R, Roccatello D, Amore A et al (1990) Effects of a gluten-free diet in primary IgA nephropathy. Clin Nephrol 33:72–86
Kovács T, Kun L, Schmelczer M et al (1996) Do intestinal hyperpermeability and the related food antigens play a role in the progression of IgA nephropathy? I. Study of intestinal permeability. Am J Nephrol 16:500–505
Smerud HK, Fellström B, Hällgren R et al (2009) Gluten sensitivity in patients with IgA nephropathy. Nephrol Dial Transplant 24:2476–2481. doi:10.1093/ndt/gfp133
Papista C, Berthelot L, Monteiro RC (2011) Dysfunctions of the Iga system: a common link between intestinal and renal diseases. Cell Mol Immunol 8:126–134. doi:10.1038/cmi.2010.69
Lebreton C, Ménard S, Abed J et al (2012) Interactions among secretory immunoglobulin A, CD71, and transglutaminase-2 affect permeability of intestinal epithelial cells to gliadin peptides. Gastroenterology 143(698–707):e4. doi:10.1053/j.gastro.2012.05.051
Papista C, Lechner S, Ben Mkaddem S et al (2015) Gluten exacerbates IgA nephropathy in humanized mice through gliadin–CD89 interaction. Kidney Int. doi:10.1038/ki.2015.94
Matysiak-Budnik T, Moura IC, Arcos-Fajardo M et al (2008) Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med 205:143–154. doi:10.1084/jem.20071204
Acknowledgments
This work was supported by grants from ANR, FRM, INFLAMEX, INSERM, CNRS and Paris Diderot University. Sebastian M. Lechner PhD fellowship was supported by CORDDIM
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no financial conflicts of interest.
Rights and permissions
About this article

Cite this article
Lechner, S.M., Papista, C., Chemouny, J.M. et al. Role of IgA receptors in the pathogenesis of IgA nephropathy. J Nephrol 29, 5–11 (2016). https://doi.org/10.1007/s40620-015-0246-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-015-0246-5