
Abstract
Alzheimer’s disease (AD) is one of the progressive neurodegenerative disorders allied with genetic, lifestyle, and environmental factors and mainly affects the elderly population. Key pathological hallmarks of AD are extracellular depositions of beta-amyloid plaques, intracellular accumulation of neurofibrillary tangles (NFTs), and alterations in amyloid precursor protein (APP) gene, and it is clinically characterised by changes in cellular and molecular cascades of synaptic loss, cognitive dysfunctions, and neuronal death. Some evidences support the role of major biomarker candidates to induce AD, which are oxidative stress and metals, as brain is more prone to generate the reactive oxygen species (ROS) by different metal ions. Imbalance between free radicals and antioxidant defence system leads to cell apoptosis and dyshomeostasis of essential endogenous redox active metal ions in brain such as iron (Fe), copper (Cu), chromium (Cr), and cobalt (Co), and redox inactive metals like cadmium (Cd), arsenic (As), and lead (Pb) and inert metal zinc (Zn) show their toxic effects in Alzheimer’s disease. To overcome the toxic effects of metal-induced ROS, there must be enhancement of two major antioxidant enzymes, catalase and superoxide dismutase. The chapter emphasises the dual protagonists oxidative stress and metals and their relation in the progression of AD.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Alzheimer’s disease (AD) is a neurological condition characterised by progressive degeneration of nerve cells as it worsens, causing dementia and other brain abnormalities. It accounts for approximately 60–80% of cases and is mainly observed in the geriatric population as an age-related risk factor (Anonymous 2020). The changes in AD of the brain are structural changes like progressive brain atrophy and damaged neurons of the cerebral cortex and hippocampus regions causing functional changes like difficulty in thinking and concentration, delusions, mental decline, disorientation, forgetfulness, and inability to create new memories.
There are countless propositions to explain AD pathogenesis, involving the amyloid cascade theory, tau hyperphosphorylation theory, cellular excitotoxicity, metal-induced oxidative stress, and apoptosis (Fan et al. 2020). The excessive production of ROS and their further interactions with various metals can cause oxidative stress that alters different biological processes. Oxidative stress and metal-induced pathogenesis of AD are covered in this chapter.
2 Oxidative Stress
The difference between the amount of excessive oxidants (such as molecular oxygen and its derivatives, or ROS) produced and the antioxidants released to counteract them is known as oxidative stress. Because of the numerous intracellular metabolisms in the mitochondria, lipoxygenases, peroxisomes, and NADPH oxidase enzyme systems, oxidants are produced endogenously (Mattson 2004). In order to maintain physiological balance, an advanced enzymatic antioxidant system, such as catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPx), and a non-enzymatic antioxidant system, such as glutathione and vitamins A, C, and E, neutralise and regulate overall ROS levels (Akbar et al. 2016).
It should be emphasised that owing to the high concentration of polyunsaturated fatty acids, excitotoxicity, and a mixed-function oxidase system, brain cells are predominantly vulnerable to oxidative damage (Manisha et al. 2017). As a source of oxidative stress, ROS are largely formed in the nervous system and active in the brain and neurons. As glial cells (post-mitotic cells) are easily vulnerable or prone to free radicals and result in the degeneration of neurons, this stress primarily affects them. Some studies have reported that harmful or toxic effects of ROS on various cells may cause shrinkage of cytoplasm and condensation of the nucleus and nuclear envelope that lead to the formation of phosphatidylserine on the surface region of the cell, i.e. sign of apoptosis and necrosis (Moreira et al. 2009). It provides enough evidence that the aetiology of Alzheimer’s disease may be significantly influenced by the oxidative impairment triggered by free radicals (AD).
2.1 Oxidative Stress and Neurodegeneration
Neurodegeneration is a process that is characterised by abnormal functioning or complete loss of brain nerve cells commonly known as ataxia or dementia. Some factors like excitotoxicity, abnormal mitochondrial (Mt) functions, and finally programmed cell death are the main pathological changes in ageing and neurodegeneration of Alzheimer’s disease (AD). While considering the part oxidative stress plays in Alzheimer’s disease, some important points need to be discussed in short:
-
1.
Why nerve cells are especially most sensitive or more vulnerable to oxidative stress?
-
2.
What role do environmental and genetic factors play in the production of free radicals?
-
3.
How do the toxic ‘free radicals’ cause neuronal death or cell apoptosis?
-
4.
Oxygen is a key element in regulating various metabolic and physiological functions, and in normal physiology, it is exchanged for oxidative phosphorylation in mitochondria. Then how it becomes toxic to neurons?
Reactive oxygen species (ROS) are induced by the formation of free radicals associated with increased pro-oxidants and decreased antioxidants. It consists of nitric oxide (NO), hydrogen peroxide (H2O2), hydroxyl radicals (OH), and super-anions, which are highly reactive and damage mitochondrial activated microglial cells, which are present in the brain and turn as a reservoir of reactive oxygen species (ROS). A free radical is an electrically charged atom; one such free radical is superoxide, which is produced by the addition of an electron to an oxygen molecule (Fridovich 1999; Nordberg and Arnér 2001; Leigh 1990). These formed radicals do not get destroyed, so they enter various tissues, cells, cell organelles, organs, and organ systems; alter various cell signalling mechanisms, metabolic dysfunctions, and signal transduction pathways; and generate a phenomenon called oxidative stress. This production of reactive oxygen species (ROS) stimulates the mitogen-activated pathway (MAP), which in turn triggers a cascade of calcium excitotoxicity and apoptosis. Additionally, it was discovered that older rats’ front parietal cortex, cortical neurons, and white matter all had much higher levels of RNA oxidation (Liu et al. 2002). Extensive oxidative damage to nucleic acids that results in changes in DNA structure is another indicator of oxidative stress and metals associated with AD (Cioffi et al. 2019).
Humans are continuously exposed to free radicals that are produced by any chemical, physical, or biological pollutants that are generated from the industries and pollutants, which are present in the ecosystem. Some physical sources like ionising radiations, γ-rays, cosmic rays, and X-rays also generate free radical ions in the environment. Living organisms are always attacked by oxygen free radical (OFR), which is present in pollutants with oxygen. Environmental exposures, like air pollution, might increase an organism’s ability to create ROS; consequently, air pollution exposure may constitute a risk factor for AD by amplifying oxidative stress mechanisms capable of triggering physiological changes in the central nervous system (Yang and Omaye 2009). Air pollution has been found in clinical tests to impair cognitive performance and cause cerebrovascular damage (Moulton and Yang 2012). ROS-mediated damage seems to be complex in Alzheimer’s disease, with interactions between dysfunctional mitochondria, redox transition metals, and other variables.
2.2 Relation Between Oxygen and Oxidative Stress
Oxygen, oxidative stress, and hypoxia are some factors that affect the cell’s physiology. A constant supply of oxygen is indispensable for nerve survival and physiology. However, the role of oxygen and its associated mechanisms and other cascade processes in the nervous system is complex. As oxygen is essential for cells, whether neuronal or other cells that are involved in tissue formation, excessive oxygen level can be equally hazardous. During oxidative phosphorylation, the breakdown of glucose molecules takes place in the mitochondria (Mt) with the help of oxygen via ATP molecules. Mitochondria itself promotes oxidative stress and increases its intensity. Any genetic alterations like DNA mutation lead to dysfunction in the generation of ATP, leading to neuronal dysfunction and further leading to neuronal death. The primary reason of neuronal cell hypersensitivity to oxidative stress is related to structural and metabolic variables. The brain is predominantly sensitive to oxidative stress (Behl 2005), and glial cells of diverse types are involved in structural support and metabolic requirements. When compared to other endothelial cells in the body, endothelial cells are encircled by glial cells and are impervious to the absorption of different substances and defence cells such as macrophages. Furthermore, the brain requires greater oxygen and glucose intake to make continual ATP pooling in vivo for appropriate brain physiology since it always strives to maintain the other organs functioning, and it must be in normal physiology. Wherever the antioxidant levels are reduced in the aged brain, the consumption of oxygen is highly converted to free radicals and generates reactive oxygen species (ROS).
2.3 Mechanism of Generation of Reactive Oxygen Species (ROS)
The creation of free radicals is caused by oxygen molecules, which are hazardous to cells under certain conditions. Cells have regulatory mechanisms for the effective interaction of oxygen and metal ions, which results in reactive oxygen species (ROS) generation and oxidative stress. The metabolic route is useful for interacting with organic molecules in vivo since contact with oxygen is an important necessity owing to reactive oxygen species (ROS) and is actively inappropriate. In any form of reactive oxygen species (ROS) generation, molecular oxygen is needed to be actual and cellular or physiologically functioning, which is evolved in a wide range of metallo-enzymes that facilitate reactive oxygen species (ROS) generation upon the interaction of redox metals with oxygen using catalytic pathways:
The whole reaction of the pooled steps (3) is termed Haber-Weiss reaction:
In totalling the above reactions, the subsequent reactions may also take place:
Fenton reaction yields hydroxyl radical, which in turn rejoins with various biomolecules, including lipids, proteins, and DNA, and causes their oxidation.
2.4 Role of Amyloid Beta (Aβ)-Induced Oxidative Stress in Alzheimer’s Disease (AD)
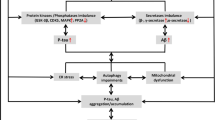
AD is characterised by the existence of disorientated plaques of amyloid proteins, hyperphosphorylation of neurofibrillary tangles of tau proteins, changes in amyloid precursor protein (APP) gene, and clinical symptoms like memory loss, motor deficiency, and cognitive impairment by inducing various mechanistic factors like ROS, neuroinflammation, cerebrovascular damage, Aβ accumulation, and neuronal loss. Stress can also cause vasculitis in the brain, making the brain more susceptible to dementia. Stressful life events have been linked to causing havoc to the brain, which could lead to AD. Numerous studies demonstrate that oxidation processes occur in the AD brain and that Aβ proteins can directly generate free radicals by activating the NADPH oxidase system (Shelat et al. 2008). The condition progresses as the reactive chemicals develop into pro-oxidants, which sustain an ongoing auto-destructive process (Praticò 2005) (Fig. 2.1).

Causative factors of Alzheimer’s disease (AD)
A fragment of the amyloid precursor protein (APP), which is a normal neuron membrane protein synthesised in the brain, aggregates to form extracellular amyloid plaques. Excessive synthesis of Aβ can be neurotoxic and can cause oxidative stress. Direct evidence shows increasing oxidation stress in AD:
-
1.
Generation of free radicals in neurons and ROS formation
-
2.
Decreased neurocytochromes like cytochrome C oxidase in the brain
-
3.
Elevation of oxidation of proteins and DNA in aged brain
-
4.
Increased per oxidation of lipids in AD ventricular fluid due to membrane damage by ROS
-
5.
Amyloid β peptide capable of generating free radicals
-
6.
Decreased energy metabolism due to alterations in metabolic pathways
-
7.
Decreased polyunsaturated fatty acids and increased aldehyde (4-hydroxynonenal) products during the peroxidation of lipids
-
8.
Advanced glycation end products (AGE), malondialdehyde, and SOD-1 neurofibrillary tangles
The enzymes glutamine synthetase and creatine kinase can both be rendered inactive by the direct production of ROS. ROS can control the JNK/stress-activated protein kinase pathways. The instigation of these cascades is related to the hyperphosphorylation of tau proteins and Aβ-induced cell death (Ferrer et al. 2005). Aβ is created when the amyloid precursor protein is successively broken down by the secretases, β-site amyloid precursor protein-cleaving enzyme 1, and γ-secretase protein of the amyloidogenic pathway (APP). BACE 1 initiates the amyloidogenic process by cleaving the APP at two secretase sites. As a result, the plasma membrane secretes APP, which is a long version of APP, as well as CTF-99 or CTF-89. Since Aβ42 is more likely to oligomerise than Aβ40 or Aβ38, which increases its capacity to exert cytotoxic effects, Aβ42 looks to be more neurotoxic than Aβ40 or Aβ38 (Dimitrov et al. 2013), causing oxidative mutilation to synaptic membranes and inducing hyperphosphorylation of tau proteins (Fan et al. 2020). These neurons internalised and degraded the Aβ peptide when exposed to soluble oligomers of it (AβOs), released extracellular vesicles containing the active enzyme catalase (CAT), and selectively secreted interleukins-6 and -10 and vascular endothelial growth factor (VEGF) into the medium. The enzymes glutamine synthetase and creatine kinase can be inactivated directly by ROS, which is produced by the production of Aβ fragment (de Godoy et al. 2018). As a result, the main metabolic variation in AD is the alteration of Aβ into hazardous compounds by ROS, which form senile plaques and promote apoptosis (Ribarič 2018). In fact, a growing body of research indicates that the primary toxic agents in AD rather than amyloid deposits are soluble Aβ oligomers, monomers, and protofibrils (Verma et al. 2015). Fyn is rendered inactive as a result of γ-secretase, with Aβ activating the enzyme (STEP) striatal enriched protein tyrosine phosphatase, which sets off a chain of subsequent events that result in dendritic spine collapse (Mairet-Coello et al. 2013). It is clear that the interaction between Aβ and oxidative stress has a significant role in the pathological changes experienced by AD patients.
2.5 Oxidative Damage-Induced Tau Neurofibrillary Tangles
Microtubule-associated protein (MAP), tau protein, is recurrently present in the cytosol as well as in axons of neurons. Overexpression of Tau protein causes the course of stabilisation and destabilisation of microtubules, protein interactions that augment the pathological effects of tau and inhibition of kinase-dependent transport of Golgi complex-derived vesicles into neuritis, transportation defects and APP trafficking into neuron cells causing mutations in genes (apoE4), neurodegenerative histological dysfunctions, increased metal levels, inflammation, and consequent loss of synapses and neurons. Through the triggering of the p38 mitogen-activated protein kinase (MAPK) and c-Jun amino-terminal kinase, oxidative stress enhances the production of β-secretase (Tamagno et al. 2008) and promotes deviant phosphorylated tau through the activation of glycogen synthase kinase 3-(GSK-3-β) (Fang et al. 2000); the inactivation of certain molecules by oxidants may be significant as well. Prolyl isomerase PIN1 was discovered using a proteomic technique to be especially vulnerable to oxidative stress, being significantly downregulated and oxidised in the hippocampus of AD patients (Pastorino et al. 2006). Tau proteins include approximately 30 possible phosphorylation sites because of the abundance of serine and threonine phosphate-accepting residues. One of the many post-translational alterations that tau may experience is phosphorylation by kinases such as JKN, AMPK, and GSK-3. It was discovered that tau-enriched neurofibrillary tangles indicate oxidative damage. Nitric oxide is produced in close vicinity to the tau that creates the neurofibrillary tangles, according to research on the location of the enzyme dimethyl arginase (MacAllister et al. 1996) and regulation of the activity of nitric oxide synthase in hippocampal tissue from AD patients. These conclusions are confirmed by the revelation that an antibody that recognises an HNE-lysine adduct co-localises with endogenously produced paired helical tau filaments from AD brains (Takeda et al. 2000). Furthermore, in AD patients’ brain, an aldehyde by-product of lipid peroxidation, i.e. acrolein, was found to co-localise with neurofibrillary tangles. Additionally, the antibody Alz50, which detects a conformational change in tau, corresponds with the antioxidant enzyme heme oxygenase-1 (HO-1), whose levels are noticeably raised in the AD and moderate cognitive impaired (MCI) brain (Carmel et al. 1996; Barone et al. 2012), and perhaps plays a critical role in the harmful development of dementia. Antioxidant therapy, for instance of nitrone and N-acetyl cysteine, inhibited the immune response to tau oligomers, demonstrating a further straight involvement of oxidative tension in tau assembly (Du et al. 2016).
3 ROS-Induced Disruption of Calcium Signalling in Alzheimer’s Disease
In the AD brain, calcium (Ca2+) dysregulation ensues preceding to the development of Aβ plaques plus neurofibrillary tangles, signifying so as disturbance in cytoplasmic Ca2+ may be one of the disease’s primary causes. Cellular Ca2+ homeostasis is a critical regulator of many aspects of neuronal physiology, including synaptic plasticity, growth and differentiation, action potential properties, knowledge, and memory. The pathogenesis of AD is influenced by plasma membrane Ca2+ channels, lysosomal Ca2+ signalling, and mitochondrial Ca2+ signalling and shows apoptosis, necrosis, degeneration, and poor autophagy, which are also facilitated by aberrant cellular Ca2+. Hydroxyl radicals, superoxide anion, and hydrogen peroxide are recognised ROS that control Ca2+ signalling pathways. The ROS generation/Ca2+ signalling paradigm was discovered as a consequence of functional impairment of membrane-bound receptors and channels that regulate Ca2+ influx or efflux brought on by oxidative stress-induced lipid peroxidation. The primary Ca2+ storage organelle in a cell is the ER, which can release Ca2+ in response to electrical and chemical cell stimulation (Bootman et al. 2001) through two types of Ca2+ release channels, the IP3R and the RyR. Ca2+ modulation is indirectly accomplished by causing membrane-damaging oxidative stress through voltage-dependent channels and ionotropic glutamate receptors, Ca2+ pumps, and increases Ca2+ inflow (Keller et al. 1997; Blanc et al. 1998; Mark et al. 1997). Aβ aggregates can inhibit Ca2+ signalling in a number of ways, including by activating the InsP3R and RyR, which release Ca2+ from ER storage, and by generating cation permeation pores on the plasma membrane, which enables Ca2+ entry. In response to agonists of phospholipase C (PLC)-coupled receptors, the IP3R releases Ca2+. When PLC is activated, PI (4,5) P2 is cleaved, releasing diacylglycerol and INS (1, 4, 5) P3 (IP3), which binds to IP3R. Calcium-induced calcium release by IP3R is a mechanism that further increases Ca2+ release through RyR (CICR). It has previously been demonstrated that IP3-evoked Ca2+ transients are amplified by the presence of the AD-linked presenilin 1 (PS1) mutation (Stutzmann et al. 2004). Exaggerated ER Ca2+ signals were shown to be caused by RyR activation brought on by Ca2+ released by IP3R. Increased intracellular Ca2+ overload, specifically its release from the ER, may cause an excessive amount of Ca2+ to be taken up by the mitochondria. The closeness of the ER and mitochondria on a physical level may contribute to this impact (Pinton et al. 1998; Csordás et al. 2006).
Increased ROS generation and decreased mitochondrial membrane potential are effects of excessive Ca2+ buildup in mitochondria. Cell death results from the large Ca2+ influx into mitochondria that causes the potential of the mitochondrial membrane to collapse (Duchen 2000). The classic store-operated Ca2+ entry (SOCE) mechanism is activated by the depletion of ER Ca2+, which then causes the stimulation of plasma membrane Ca2+ channels, which eventually stimulate a prolonged extracellular Ca2+ influx to the cytosol. By controlling DRD1 that is dopamine receptor D1 to activate CaMKII (which is a Ca2+/calmodulin-dependent protein kinase II) via the non-canonical Gaq-Ca2+ signalling pathway, GHSR1a contributes to hippocampus synaptic physiology and memory preservation (Kern et al. 2015; Hsu et al. 2018a; Seminara et al. 2018). In AD, tau is abnormally hyperphosphorylated, which disrupts axonal transport and results in the death of neuronal cells. Given that numerous kinases are triggered through Ca2+, disturbance of Ca2+ homeostasis caused via the PS mutation may greatly increase tau phosphorylation and cause neurofibrillary tangle development. Ca2+ fluctuations, mitochondrial function, gene expression, and apoptosis are just a few of the cellular activities that store-operated Ca2+ entry signalling in AD regulates. Some of the molecular targets include NMDR, which lessens cognitive impairment and behavioural outcomes in patients with moderate Alzheimer’s disease by preventing excessive Ca2+ influx and sustained glutamate release, which cause excitotoxicity of neurons, rescuing tau hyperphosphorylation and protecting synapse type A. Regulating sodium and calcium permeability, which improves memory and learning, type L VGCC diminishes the synthesis of A-42 and the neurotoxicity caused by A-25-33 in cortical neurons; several of these agents have good blood-brain barrier penetration; T-type voltage-gated calcium channels are controlled in their activation, which improves cognitive performance; RyR stabilises ER calcium release, prevents synaptic loss, and enhances cognitive function, whereas InsP3R guards cells by limiting excessive caspase-3 and calcium activity (Tong et al. 2018). Antioxidants and mechanisms that control Ca2+ homeostasis by preventing its release from ER may be effective therapeutic strategies for preventing AD-related neuronal death.
4 ROS-Induced Mitochondrial Dysfunction (Mitochondrial Cascade Hypothesis)
Mitochondria control both cellular metabolism and apoptosis. The onset of AD has been related to microglial mitochondrial oxidative stress. AD has been connected, via a number of mechanisms, to elevated reactive oxygen species (ROS) generation and decreased mitochondrial membrane potential. AD interacts with microglial receptors including TREM2, which opens up a cascade of reactions that harm mitochondria and amplify inflammatory and cytotoxic reactions. Microglia’s generation of mitochondrial ROS increases as a result of fibrillary Aβ stimulation of NADPH oxidase, which worsens the neurotoxic effects.
The pathophysiology of AD is influenced by the metals, microglia, TREM2, apoptosis, P2X7R, and mitochondrial dysfunction. Damage to the mitochondria impairs their ability to produce energy, causes oxidative stress, and produces mitochondrion-derived damage-associated molecular patterns that harm neurons and promote inflammation. The dynamics of mitochondrial fission and fusion are also out of balance in AD, which results in abnormal mitochondrial dispersion in neurons. Reactive oxygen species can be created when electrons decrease oxygen outside of the electron transport chain (ROS). By coordinating electron transfer with the pumping of protons across the inner mitochondrial membrane, mitochondria make ATP. Membrane potential (MP) and ROS production are related, and a high MP encourages more ROS production. High MP alters the redox potential of ETC carriers and lengthens the half-life of ubisemiquinone, both of which increase the generation of ROS. Additionally, any damage to ETC components could cause decreased intermediates to stall, increasing the chance of an electron sliding and lowering O2 to produce ROS. To maintain the functioning of neurons, mitochondria produce ATP using the electron transport chain (ETC). Reduced mtDNA copy number from altered mitochondrial dynamics may lead to problems in mitochondrial electron transport function (Readnower et al. 2011). mtDNA oxidation rises in comparison to nuclear DNA, leading to an age-dependent buildup of mtDNA mutations. These mutations would result in a general decrease in the number of copies of the mtDNA, which would lower oxidative phosphorylation. The development of AD is aided by the elevated levels of cyclophilin D and Aβ in synaptic mitochondria. Patients with AD have abnormal mitochondrial dynamics, and it has been found that mitochondrial fission occurs more regularly than fusion in AD. Evidence shows that there are fewer mitochondria in AD, which is consistent with larger mitochondria, which supports this observation (Hirai et al. 2001). Fis1 and other proteins linked to fusion (dynamin-like protein and OPA1) have been found to have higher protein levels in response to APP overexpression through the synthesis of Aβ. Aβ has also been demonstrated to harm Drp1 by oxidative damage, which leads to mitochondrial fission (Cho et al. 2009). There is growing indication from studies that mitochondrial dysfunction is a key influence in the onset and advancement of AD.
5 Oxidative Stress and Damage to Biomolecules (Lipids, Proteins, DNA/RNA)
Strong oxidants called reactive oxygen species, also known as peroxynitrite, are produced when nitric oxide and superoxide anion react, and they can harm lipids, proteins, and nucleic acids (DNA and RNA). The pre-existing oxidative stress-induced damage that precedes and may involve and/or contribute to the neurofibrillary deterioration of neurons in the Alzheimer’s disease brain may constitute the selective variation of a multitude of intracellular proteins, including key enzymes and structural proteins. Another sign of oxidative stress linked to AD is extensive oxidative injury to nucleic acids that results in modifications to DNA assembly. The nucleotide guanosine is oxidised in DNA/RNA oxidation to create 8-hydroxyguanosine (8-oxoG). High amounts of 8-oxoG were discovered in neurons in the hippocampus, subiculum, entorhinal cortex, and frontal, temporal, and occipital neocortex in apoptotic brain samples from AD patients. Additionally, it was discovered that RNA oxidation was much higher in various locations such as the front parietal cortex, white matter, cortical neurons, and hippocampus of old rats. According to certain reports, oxidative stress causes changes to the proteins tau and Aβ. Tau interacts dynamically with the generated microtubules to aid in microtubule structure, and their organisation’s intracellular dynamics were shown to be disturbed in AD patients (Weingarten et al. 1975; Heston and White 1978).
Lipids, which make up the majority of cellular membranes, are essential for preserving the structural integrity of cells. The physical characteristics of cellular membranes are changed by excessive lipid oxidation, which can also lead to the covalent alteration of proteins and nucleic acids (Gaschler and Stockwell 2017). Numerous biological settings result in the assembly of lipid peroxides, which can act as signalling molecules by post-translationally altering proteins through enzymes or non-enzymatic mechanisms to be used in the production of lipid peroxides such as 5-lipoxygenase, 12/15-lipoxygenases, and chemistry of the Fenton-type reactions. The amount of the chromophore produced by the reaction between MDA and thiobarbituric acid can be evaluated by measuring the absorbance. This chromophore is utilised to detect lipid peroxides and the by-products of their breakdown. Similar to this, the degree of protein carbonylation has been determined using the reaction between the aldehyde moiety of 4-HNE and 2,4-dinitrophenylhydrazine. The role of lipid peroxides in numerous diseases and cell death has also been made clear by the fact that they can produce hazardous secondary messengers. In some situations, it has been demonstrated that the lipid breakdown product 4-HNE can cause apoptosis (Dalleau et al. 2013). Lipid peroxides’ capacity to produce harmful secondary messengers has also served to emphasise their significance in a number of diseases and the part that cell death plays in the development and control of inflammation (Ackermann et al. 2017). Inhibiting the enzymes responsible for their synthesis, or using peroxidation inhibitors, is a very popular method for averting the progress of lipid peroxides.
DNA and RNA, which are made up of proteins involved in the disease, peaked early and remained increased. According to the study, nucleic acid oxidation is a common occurrence in neurodegeneration. Protein synthesis is slowed down or is abnormal when mRNA, rRNA, and tRNA are oxidised. Oxidative stress messes with the regulatory mechanisms of noncoding RNAs, particularly microRNAs, as well as this translational machinery (miRNAs). Oxidised miRNAs can mistakenly recognise target mRNAs. The vulnerable brain areas of AD contain three downregulated miRNAs that are miR-107, miR-210, and miR-485 as well as seven upregulated miRNAs that are miR-125b, miR-146a, miR-200c, miR-26b, miR-30e, miR-34a, and miR-34c, all of which are associated to oxidative stress (Nunomura and Perry 2020).
Transfer RNA (tRNA), microRNA, and ribosomal RNA (rRNA) susceptible to oxidative damage (miRNA): The stimulation of caspase-3 and subsequent apoptosis are caused, in part, by the preferential attachment of poly(C)-binding protein 1 (PCB1) to oxidised mRNA containing two contiguous 8-oxoGua residues (Ishii et al. 2018). Because it is available in the mitochondrial inter-membrane space, cytochrome c (cytc) can catalyse the oxidation of transfer RNA (tRNA), which primes to the formation of a cross-linking complex between tRNA and cytc and encourages its release from mitochondria, which then causes apoptosis (Tanaka et al. 2012). Even though Bcl-xL and Bcl-w are not their natural targets, oxidised miRNA-184 that contains 8-oxo-guanosine binds with them. The eventual apoptosis is caused by a subsequent decrease in Bcl-xL and Bcl-w (Wang et al. 2015). Better early intervention tactics may result from a deeper understanding of the effects and cellular conduct processes of the oxidatively changed RNAs, which may reveal information about the primary causes of neurodegenerative disorders.
DNA oxidative damage may be a significant factor in ageing and neurological illnesses like Alzheimer’s disease (AD). Reactive oxygen species, predominantly hydroxyl radicals, can harm DNA by breaking DNA strands, forming DNA-DNA and DNA-protein cross links, exchanging and translocating sister chromatids, and producing at least 20 oxidised base adducts. Altering DNA bases can result in mutation and altered protein synthesis (Markesbery and Lovell 2006). Mild cognitive weakening, the first form of AD, has elevated levels of 8-OHG, 8-OHA, and 5,6-diamino-5-formamidopyrimidine in nuclear and mitochondrial DNA, indicating that DNA oxidation is an early occurrence in AD. Additionally, the interaction of DNA bases with alpha- and beta-unsaturated aldehydic by-products of lipid peroxidation, such as 4-hydroxynonenal and acrolein, can result in the production of large exocyclic adducts (Lovell and Markesbery 2007). Antioxidants as treatments have engrossed a lot of attentiveness due to the role that biomolecule oxidation and overall oxidative stress play in the progress of Alzheimer’s disease.
6 Role of Metals in AD
Metals are part of the earth’s crust and are found in water, the atmosphere, and many other ecosystems. Copper (Cu), chromium, cobalt, magnesium, iron (Fe), lithium, manganese (Mn), selenium (Se), nickel (Ni), and zinc (Zn) are among the essential metals. Metals play a vital role in our daily lives since they are engaged in so many enzymatic activities and serve crucial roles in maintaining cell structure and controlling processes like neurotransmission, antioxidant response, and gene expression. Both endogenous and exogenous metal exposures are known to cause changes in oligo-element homeostasis and can harm the central nervous system (CNS), cause oxidative stress, interfere with mitochondrial function, and inhibit the activity of various enzymes that play crucial catalytic, structural, and regulatory roles in various proteins, transporters, and receptors. These systems may be circumvented by toxic metal compounds, or they may be trapped inside and endanger the BBB. Evidence suggests that metal-induced neurotoxicity may be brought about via chemically induced blood-brain interface damage (Chen et al. 2014). While roughly they are necessary in tiny amounts, undue concentrations in the humanoid body typically cause neurotoxicity. The most prevalent deficiencies linked to metal-induced toxicity embrace mitochondrial malfunction, oxidative stress, and protein misfolding when metals get hoarded in the brain system (Wright and Baccarelli 2007; Angeli et al. 2014; Zhang et al. 2013; Seo et al. 2013). Numerous antioxidant enzymes, which are crucial for the brain and other organ functions, depend heavily on metal homeostasis. Additionally, it has been proposed that AD is caused by changes in critical metal homeostasis, which result in the interaction of metals with proteins and their consequent activation of aggregate formation (Gunter et al. 2010).
In the biological processes of metalloproteins and in neural processes, metal ions in the AD brains can rise up to three times over those in healthy control brains physiologically (Malecki 2001). A positive feedback loop of greater oxidation and higher ROS production is produced when the cations Zn2+ and Cu2+ bind to the hydrophilic N-terminal ends of Aβ peptides. Here, they are able to undertake continuous redox reactions that generate significant levels of ROS (Bondy et al. 1998; Strong et al. 1996).
7 Zinc
The second most common transition metal after Fe, Zn is a crucial trace element needed by humans and several other living organisms. Over 300 enzymes and metalloproteins use it as a cofactor, which controls gene transcription and antioxidant response. The testes, muscle, liver, and brain contain the majority of the body’s Zn. Zn deficiency in humans impacts learning capacity as well as mental and physical development.
Researchers have looked into the unusually high Zn2+ content in the brains of AD patients to draw conclusions about the relationship between the pathogenesis of AD and an imbalance in Zn2+ homeostasis. Zn inhibits amyloid-induced neurotoxicity by preferentially precipitating aggregation intermediates at low doses (a few micromolars) (Garza-Lombó et al. 2018; Spiers et al. 2022).
Zinc’s contribution to the pathophysiology of AD is supported by two major lines of evidence: (1) zinc concentrations in the brain, blood serum, and cerebrospinal fluid are frequently used as AD biomarkers and (2) zinc concentrations in brain, blood serum, and cerebrospinal fluid are densely innervated by zinc-containing axons, whereas those less affected by pathology contain few zinc-containing terminals (Chen et al. 2012). The phenomenon known as ‘zinc fooding’, in which zinc-containing neurons abruptly release free zinc into the brain’s extracellular spaces, is a key aspect of this examination. This phenomenon can appear in response to a number of circumstances, including ischaemia, convulsions, and traumatic brain damage. However, an experimental investigation using a tau mouse model revealed that Zn supplementation exacerbated the behavioural and biochemical deficiencies brought on by tau proteins (Barnham et al. 2003).
The high concentration of Zn2+ ions binding to β-amyloid may enhance the formation of fibrillar β-amyloid aggregation in the synaptic cleft, where oxidised Aβ protein can build up and combines with the protein to produce a precipitate of poisonous Aβ peptides that are unable to leave the synapses leading to neurodegeneration (Cuajungco et al. 2000). The easier formation of hazardous oligomers and, ultimately, plaques is made possible by the binding of metal ions to Aβ monomers. Microtubule instability caused by intracellular Zn depletion triggers the release of tau, hyperphosphorylation, and development of neurofibrillary tangles. The release of Zn from metallothionines by intracellular Zn excess, which results from Aβ aggregation and ROS production, may disrupt mitochondrial function and trigger apoptosis.
Metal ions can also interact with tau proteins in a similar way. It has been shown that Zn2+ can bind tau and encourage its phosphorylation (Garai et al. 2007). We discovered that tau fibrillisation and formation of paired helical filaments are induced and accelerated by even micromolar concentrations of Zn2+ ions. This is accomplished by upregulating kinases that phosphorylate tau, such as GSK-3, and inhibiting kinases that de-phosphorylate tau proteins, such as protein phosphatase 2A.
Zn influences key pro-inflammatory signalling pathways by acting as an anti-inflammatory element. Zn inhibits the dissociation of nuclear factor k B (NF-kB) from the inhibitory protein that it is paired with, preventing NF-kB from translocating into the nucleus and thus suppressing inflammation. Zn also prevents STAT3 activation brought on by IL-6 (Suh et al. 2000). Zn levels that are excessively high induce the inhibition of Cu and Fe 83 absorption, an increase in the production of ROS in the mitochondria, and disruption of the activity of metabolic enzymes, all of which lead to the induction of apoptosis. By activating ERK1/2, which can phosphorylate beclin-1 and thereby facilitate the formation of the beclin-1-PI3K complex during the autophagic process, zinc promotes autophagy. Additionally, zinc can facilitate the degradation of mTOR, a negative regulator of autophagy, which results in cell autophagy.
8 Iron Toxicity
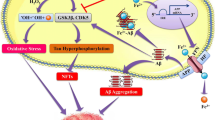
All living things require iron (Fe), a redox active metal, for healthy physiological functions. Fe is necessary for cell growth at the cellular level, but too much of it (Fe overload) results in oxidative stress and cell death. Fe homeostasis refers to the process of strictly controlling Fe levels. It is frequently found as heme-containing proteins, as a cofactor in proteins that contain Fe-sulphur clusters, and as proteins that contain Fe ions (Craven et al. 2018). A couple of examples of heme-containing proteins are catalases and peroxidases. Fe is very strongly associated with the pathophysiology of AD.
Although free Fe is more likely to form free radicals and exchange electrons with adjacent molecules than bound Fe, bound Fe is regarded as safe because it can lead to additional Fe release from proteins that contain Fe, such as heme proteins, ferritin, and Fe-sulphur clusters. A lack of iron can also make it easier for the body to absorb divalent elements including lead (Pb), cadmium (Cd), aluminium (Al), and manganese (Mn). Even in the lack of excess Mn in the brain, Fe shortage might promote Mn accumulation there (Atwood et al. 2003). There is an inverse link between Mn and Fe because Fe and Mn, along with other important metals, are regulated inside the basal ganglia by influx into the brain via transferrin and TfRs, as well as by DMT-1. Fe-induced oxidative stress is particularly hazardous (Naqvi et al. 2010). This is because it can create an intracellular positive feedback loop that worsens the toxic effects of brain Fe overload. Fe is taken up by the BBB in the brain. The capillary endothelial cell TfR absorbs Fe in the form of transferrin-Fe3+. Additionally, Fe3+ can attach to p-tau monomers to cause aggregation and creation of oligomers. NFTs can undergo continuous Fenton redox reactions as a result of the accumulation of Fe3+ ions, which produces large levels of ROS and intensifies intracellular oxidative stress. For iron to be absorbed from food, the DMT-1 is a crucial transporter. Intestinal DMT-1 levels are upregulated by Fe deprivation, and Mn absorption and neurotoxicity are also increased (Cristóvão et al. 2016).
9 Copper Toxicity
Copper is thought to be the metal ion that is most redox reactive. Redox regulation guards against various forms of oxidative stress and preserves ‘redox equilibrium’ by managing the redox state. In actuality, the redox characteristics of Cu ions are what cause the neurotoxic effects, increasing ROS generation in the CNS (Gammoh and Rink 2017). The brain is thought to be particularly vulnerable to the harmful effects of ROS due to its high metabolic rate and relatively low capacity for cellular control when compared to other organs. Cu is a necessary element and a crucial part of many enzymes, such as cytochrome c oxidase, dopamine hydroxylase, monoamine oxidase, and superoxide dismutase (Cu/Zn-SOD or SOD1), which are crucial for oxygen and electron transport, protein modification, and production of neurotransmitters (Mezzaroba et al. 2019). ROS manifestations and neurodegeneration have been linked to necessary metals, such as Cu. Cu in excess is neurotoxic and has been associated with the progression of AD.
Because of its weak albumin binding, it is likely for Cu ions to be liberated from the albumin-bound moiety at the BBB and then transported into the brain. The ATP-dependent transport of Cu across the BBB is carried out by two intracellular proteins called Cu-transporting P-type ATPases (ATP7A and ATP7B), which belong to a subclass of ATPases. However, it is unclear how these proteins are distributed within the BBB. The BBB is impermeable to Cu under typical physiological circumstances. Specific Cu transport systems are needed to move Cu between two fluid compartments across the BBB. The BBB-mediated variations in copper ion (Cu2+) levels have a significant impact on Aβ metabolism. Cu can only passively diffuse into the cerebrospinal fluid under specific pathological circumstances where BBB permeability is reduced.
Three steps can be used to describe how copper ions associate with Aβ. Copper is first bound to endogenous reductants, and then Cu (II) is reduced to copper. Copper’s reductive state causes molecular oxygen to also undergo reduction, ensuing in the generation of ROS. The toxicity of amyloid oligomers and plaques is exacerbated as a result of copper’s direct interactions with Aβ (Mills et al. 2010).
Copper interacts with amyloid and tau proteins to promote aberrant protein accumulation (Bader et al. 2011). Cu may promote amyloid precursor protein and amyloid peptide self-aggregation, and elevated levels of Cu in cerebral fluid have been observed in some AD patients (Mezzaroba et al. 2019).
Cu appears to accelerate the Aβ cascade by promoting Aβ production and accretion in AD plaques, and its deposition in the CNS. Intracellular tangles first form inside neurons when Fe and Cu bind to hyperphosphorylated tau protein. Free Fe2+ or Cu1+ species will cause harmful Fenton reactions, including the production of ROS and micro-inflammation (Smith et al. 1997). Cu’s potent affinity for Aβ, which promotes its aggregation and heightens oxidative stress via the Fenton reaction, has traditionally been thought to be its neurotoxic mechanism of action. Therefore, it has been proposed that Cu build-up mediates neurotoxicity and that its removal from the brain prevents or reverses the load of Aβ plaque. Recent research reveals that dyshomeostasis of Cu and its valency in the body, rather than acculturation and interaction with Aβ, are the primary determinants of either its neurotoxic or helpful effects as an essential metal. Aβ1–42 that has been stabilised by copper interacts with cell membranes to make them more permeable. Lysosomes, the organelles involved in autophagy, have shown an increase in copper. Elevated copper levels in the redox cycle of metal ions encourage autophagy and apoptosis in glioma cells through the activation of JNK and reactive oxygen species.
Deregulated copper ions may initiate and enhance tau hyperphosphorylation and formation of sheet-rich tau fibrils, which in turn lead to synaptic failure, neuronal death, and cognitive impairment found in AD patients.
The fact that genetic mutations on Cu transporters that cause loss of function cause severe neurological symptoms is another argument in AD pathology. Unique pathways of Cu neurotoxicity were postulated, mediated by non-neuronal cell lineages in the brain, like capillary endothelial cells, contributing to the development of AD neuropathology, along with its changed distribution (Acevedo et al. 2019).
10 Aluminium
Al is used in many different things, such as food packaging, preservative cans, cookware, automobiles, and vaccine adjuvants, to name a few. Oxidative stress and mitochondrial dysfunction are two potential reasons of the brain damage. In human AD, aluminium increases plaque and tangle pathology, impairing cholinergic neurotransmission and causing similar neural network destruction (Kawamata and Manfredi 2010). The prolonged retention percentage of aluminium in the brain suggests that it may accumulate to hazardous levels over time. The same neurons that form intracellular neurofibrillary clumps also develop aluminium deposits; however, Al hydroxide impairs long-term memory, increases anxiety, and kills neurons in the spinal cord and motor cortex. Aluminium salt produces localised neurodegenerative effects that resemble AD.
The growth of iron-driven oxidative stress events linked to numerous separate but functionally related gain-of-toxicity and loss-of-function processes is the postulated mechanism of aluminium-induced neurotoxicity. The inositol phosphate system and calcium control may be disrupted by aluminium deposition, which could lead to neurodegeneration. Aluminium promotes harmful redox reactions caused by iron, primarily by simultaneously activating superoxide dismutase and inhibiting catalase (Stutzmann et al. 2004). After chronic aluminium exposure, transgenic mice’s hippocampal tissues’ RNA expression can be examined to indicate redox stress patterns. Because the integrity of biological membranes is compromised by free radical attacks on proteins and lipids, unchecked redox reactions are devastating. By opening the mitochondrial permeability transition pore (MTP) and encouraging cytochrome c translocation into the cytoplasm, aluminium ions indirectly drive mitochondrial dysfunction. Effects of aluminium on biological systems, such as changes in protein accumulation, gene expression, and membrane disruption, have only been observed in quantity that is significantly higher than that found in people (Wang et al. 2020).
11 Manganese
Manganese (Mn) is a universally important trace element needed for healthy cellular homeostasis, growth, and development. The chemical forms of Mn include chelates, salts (sulphate and gluconate), and oxidation states (Mn2+, Mn3+, Mn4+, Mn6+, and Mn7+) (aspartate, fumarate, succinate) (Roos et al. 2006). In astrocytes, the abundant manganoprotein glutamine synthetase (GS) expresses itself mostly and produces glutamine by converting glutamate to glutamine. Low levels of Mn in the brain lower GS activity since it has been proposed that Mn regulates GS activity. Enhanced glutamate transport and glutamatergic oxidative stress, mitochondrial malfunction, dysregulation of autophagy, build-up of intracellular hazardous compounds, and apoptosis are some of the underlying mechanisms. Alzheimer’s disease (AD) and other neurological illnesses associated with ageing depend on mitochondria.
Being a necessary metal, manganese is mostly obtained through dietary means. However, inhaling large concentrations of the metal can result in brain manganese build-up and manganism, a condition that resembles neurodegeneration. Manganese from food passes across the blood-brain barrier (BBB), but it is absorbed through the olfactory transport channel from inhalation, causing a build-up of the metal in the brain. In animal models, too much manganese impairs MnSOD, which leads to oxidative stress and pathophysiology of AD, including Aβ build-up and tau phosphorylation. Although the potential relationship with clinical AD has not yet been proven, manganese binds Aβ and elevated manganese exposure is connected with cognitive losses in human epidemiologic research. A specific type of neurodegenerative disease called manganism is linked to extremely high manganese exposure. The amounts of additional critical metals affect how much manganese has an impact on the nervous system. Mn might contribute to the development of AD. According to reports, the development of senile plaques is linked to deregulated Mn metabolism in AD patients as well as a malfunctioning Mn-SOD scavenger system.
Patients with neuropathology-confirmed AD have brains with decreased mitochondrial MnSOD activity. Additionally, it has been shown that Fe regulates the transport of Mn across the BBB and that a disturbed distribution of Fe has been linked to the pathophysiology of AD (Ward et al. 2015). The majority of intracellular Mn is stored in mitochondria, and an increase in Mn levels in this organelle can directly affect oxidative phosphorylation, limiting the activity of F1-ATPase and, as a result, cellular ATP generation (Hsu et al. 2018b). The excessive ROS are then produced by oxidative stress caused by high intra-mitochondrial Mn levels, which leads to mitochondrial malfunction. Its pro-oxidant capability is increased by the transition from Mn+2 to Mn+3. Oxidative stress induced by Mn causes the mitochondrial transition to open.
12 Cadmium
Cadmium (Cd) is a proven human carcinogen and non-essential transition heavy metal. The main ways that people are exposed to cadmium are through their diet and cigarettes. Cadmium exposure through inhalation can pass past the olfactory bulb and blood-cerebrospinal fluid barrier and enter the brain. Cadmium induces oxidative stress, neuroinflammation, and neuronal death in animal models of the brain. By altering the BBB’s permeability, causing AD to aggravate thereby creating tau neurofibrillary tangles as cadmium causes neurotoxicity. Cadmium may be linked to clinical AD and reduced cognitive performance specifically in human ageing research (Yokel 2000). Given the uncertainties surrounding cadmium transport to the brain, the pathophysiologic connection between environmental cadmium exposure and AD is, however, rather tenuous.
A metal like cadmium that is redox inactive indirectly causes oxidative stress. For sulfhydryl groups of thiols like glutathione and metallothionine, cadmium exhibits a strong attraction. The antioxidant defence system is interfered by either short-term high-level exposure or long-term persistent low-level exposure (Nandi et al. 2019). In neural cells as well as brain endothelial cells, cadmium causes oxidative stress. Glutathione detoxification is activated at low cadmium levels. Glutathione depletion occurs at larger concentrations with ongoing oxidative stress.
Another putative cadmium-AD route is poisoning of cholinergic neurons. Acetylcholinesterase is altered, and basal forebrain cholinergic neurons degenerate as a result of cadmium exposure, which accelerates cell death on cholinergic neurons. Cadmium impairs neurodevelopment and induces oxidative stress-dependent neuroinflammation. Direct effects on neuronal cells through oxidative stress, neuroinflammation, and apoptosis are well understood. By altering the BBB’s permeability and combining with other neurotoxicants, cadmium may also cause neurotoxicity, aggregation of Aβ, and formation of tau neurofibrillary tangles (Lidsky 2014). Cadmium exposure causes pathogenic mechanisms that lead to cognitive dysfunction and AD pathogenesis.
13 Lead
Lead contamination is widespread as a result of recent and previous industrial usage. Lead enters the bloodstream through ingestion, inhalation, or cutaneous absorption before it can cross the blood-brain barrier (Deore et al. 2021). Lead is a strong neurotoxin that disrupts the brain in a generalised manner and leads to oxidative stress, mitochondrial damage, endoplasmic reticulum stress, excitotoxicity, altered homeostatic metal signalling, inflammation, and finally neuronal apoptosis (Du et al. 2017). Lead treatment results in memory problems and AD-related pathologies in animal models, including alterations in tau, APP, and Aβ.
Additionally, it disrupts Ca2+ homeostasis, prevents PKC115 from being phosphorylated, and lowers nitric oxide generation. Intelligence, executive functioning, memory, attention, processing speed, language, emotion, and motor and visuospatial skills are all negatively impacted by Pb exposure.
Microtubule-associated protein tau (MAPT): Transgenic mice treated with 0.2% lead acetate water during PNDs 1–20 had lead-related altered expression of MAPT and miR-34c, a miRNA that targets MAPT causing cytoskeleton stability impairment and neuronal dysfunction (Martinez-Finley et al. 2013). Rats of both sexes exposed to these conditions had increased tau protein in the forebrain and cerebellum and tau hyperphosphorylation. Mice and rats that were exposed to lead early in life had pathology that was related to Alzheimer’s disease (Bihaqi and Zawia 2013). Increased tau mRNA, tau protein, its transcriptional regulators (Sp1 and Sp3), and site-specific tau hyperphosphorylation were all linked to early-life lead exposure.
14 Conclusion
In conclusion, metal-induced oxidative stress has been identified as a significant contributor to the development and advancement of Alzheimer’s disease. Elevated levels of metals such as copper, aluminium, and iron have been found in the brains of individuals with Alzheimer’s disease, and these metals can generate reactive oxygen species and lead to oxidative damage to cells and tissues. This damage can result in the accumulation of amyloid beta protein and tau tangles, which are key pathological features of Alzheimer’s disease. Moreover, metal-induced oxidative stress can also lead to inflammation and cell death, which can further contribute to the deterioration of brain function in individuals with Alzheimer’s disease. For example, studies have shown that individuals with Alzheimer’s disease have higher levels of aluminium in their brains compared to healthy individuals. Additionally, researchers have found that increasing the levels of copper in the brains of mice leads to the accumulation of amyloid beta protein and the development of Alzheimer’s disease-like symptoms. Similarly, studies have demonstrated that iron accumulation in the brain is associated with the development of Alzheimer’s disease.
Targeting metal-induced oxidative stress may represent a promising strategy for the prevention and treatment of Alzheimer’s disease. For example, studies have shown that chelating agents, which bind to metals and remove them from the body, can reduce oxidative stress and improve cognitive function in individuals with Alzheimer’s disease. Additionally, dietary interventions, such as increasing the intake of antioxidants, may also help to mitigate metal-induced oxidative stress and prevent or delay the onset of Alzheimer’s disease.
References
Acevedo K, Masaldan S, Opazo CM, Bush AI (2019) Redox active metals in neurodegenerative diseases. J Biol Inorg Chem 24(8):1141–1157. https://doi.org/10.1007/s00775-019-01731-9
Ackermann JA, Hofheinz K, Zaiss MM, Krönke G (2017) The double-edged role of 12/15-lipoxygenase during inflammation and immunity. Biochim Biophy Acta Mol Cell Biol Lipids 1862(4):371–381. https://doi.org/10.1016/j.bbalip.2016.07.014
Akbar H, Duan X, Saleem S, Davis AK, Zheng Y (2016) RhoA and Rac1 GTPases differentially regulate agonist-receptor mediated reactive oxygen species generation in platelets. PLoS One 11(9):e0163227. https://doi.org/10.1371/journal.pone.0163227
Angeli S, Barhydt T, Jacobs R, Killilea DW, Lithgow GJ, Andersen JK (2014) Manganese disturbs metal and protein homeostasis in Caenorhabditis elegans. Metallomics 6(10):1816–1823. https://doi.org/10.1039/c4mt00168k
Anonymous (2020) 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. https://doi.org/10.1002/alz.12068
Atwood CS, Obrenovich ME, Liu T, Chan H, Perry G, Smith MA, Martins RN (2003) Amyloid-beta: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-beta. Brain Res Brain Res Rev 43(1):1–16. https://doi.org/10.1016/s0165-0173(03)00174-7
Bader B, Nübling G, Mehle A, Nobile S, Kretzschmar H, Giese A (2011) Single particle analysis of tau oligomer formation induced by metal ions and organic solvents. Biochem Biophys Res Commun 411(1):190–196. https://doi.org/10.1016/j.bbrc.2011.06.135
Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, Williamson NA, White AR, Hinds MG, Norton RS, Beyreuther K, Masters CL, Parker MW, Cappai R (2003) Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J Biol Chem 278(19):17401–17407. https://doi.org/10.1074/jbc.M300629200
Barone E, Di Domenico F, Sultana R, Coccia R, Mancuso C, Perluigi M, Butterfield DA (2012) Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic Biol Med 52(11–12):2292–2301. https://doi.org/10.1016/j.freeradbiomed.2012.03.020
Behl C (2005) Oxidative stress in Alzheimer’s disease: implications for prevention and therapy. Subcell Biochem 38:65–78. https://doi.org/10.1007/0-387-23226-5_3
Bihaqi SW, Zawia NH (2013) Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to lead (Pb). Neurotoxicology 39:95–101. https://doi.org/10.1016/j.neuro.2013.07.010
Blanc EM, Keller JN, Fernandez S, Mattson MP (1998) 4-hydroxynonenal, a lipid peroxidation product, impairs glutamate transport in cortical astrocytes. Glia 22(2):149–160. https://doi.org/10.1002/(sici)1098-1136(199802)22:2<149::aid-glia6>3.0.co;2-2
Bondy SC, Guo-Ross SX, Pien J (1998) Mechanisms underlying the aluminum-induced potentiation of the pro-oxidant properties of transition metals. Neurotoxicology 19(1):65–71
Bootman MD, Collins TJ, Peppiatt CM, Prothero LS, MacKenzie L, De Smet P, Travers M, Tovey SC, Seo JT, Berridge MJ, Ciccolini F, Lipp P (2001) Calcium signalling—an overview. Semin Cell Dev Biol 12(1):3–10. https://doi.org/10.1006/scdb.2000.0211
Carmel G, Mager EM, Binder LI, Kuret J (1996) The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem 271(51):32789–32795. https://doi.org/10.1074/jbc.271.51.32789
Chen X, Guo C, Kong J (2012) Oxidative stress in neurodegenerative diseases. Neural Regen Res 7(5):376–385. https://doi.org/10.3969/j.issn.1673-5374.2012.05.009
Chen P, Parmalee N, Aschner M (2014) Genetic factors and manganese-induced neurotoxicity. Front Genet 5:265. https://doi.org/10.3389/fgene.2014.00265
Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science (New York, N.Y.) 324(5923):102–105. https://doi.org/10.1126/science.1171091
Cioffi F, Adam RHI, Broersen K (2019) Molecular mechanisms and genetics of oxidative stress in Alzheimer’s disease. J Alzheimers Dis 72(4):981–1017. https://doi.org/10.3233/JAD-190863
Craven KM, Kochen WR, Hernandez CM, Flinn JM (2018) Zinc exacerbates tau pathology in a tau mouse model. J Alzheimers Dis 64(2):617–630. https://doi.org/10.3233/JAD-180151
Cristóvão JS, Santos R, Gomes CM (2016) Metals and neuronal metal binding proteins implicated in Alzheimer’s disease. Oxidative Med Cell Longev 2016:9812178. https://doi.org/10.1155/2016/9812178
Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnóczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174(7):915–921. https://doi.org/10.1083/jcb.200604016
Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, Huang X, Farrag YW, Perry G, Bush AI (2000) Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem 275(26):19439–19442. https://doi.org/10.1074/jbc.C000165200
Dalleau S, Baradat M, Guéraud F, Huc L (2013) Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ 20(12):1615–1630. https://doi.org/10.1038/cdd.2013.138
de Godoy MA, Saraiva LM, de Carvalho LRP, Vasconcelos-Dos-Santos A, Beiral HJV, Ramos AB, Silva LRP, Leal RB, Monteiro VHS, Braga CV, de Araujo-Silva CA, Sinis LC, Bodart-Santos V, Kasai-Brunswick TH, Alcantara CL, Lima APCA, da Cunha-E Silva NL, Galina A, Vieyra A, De Felice FG et al (2018) Mesenchymal stem cells and cell-derived extracellular vesicles protect hippocampal neurons from oxidative stress and synapse damage induced by amyloid-β oligomers. J Biol Chem 293(6):1957–1975. https://doi.org/10.1074/jbc.M117.807180
Deore MS, Keerthana S, Naqvi S, Kumar A, Flora SJS (2021) Alpha-lipoic acid protects co-exposure to Lead and zinc oxide nanoparticles induced neuro, Immuno and male reproductive toxicity in rats. Front Pharmacol 12:626238. https://doi.org/10.3389/fphar.2021.626238
Dimitrov M, Alattia JR, Lemmin T, Lehal R, Fligier A, Houacine J, Hussain I, Radtke F, Dal Peraro M, Beher D, Fraering PC (2013) Alzheimer’s disease mutations in APP but not γ-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat Commun 4:2246. https://doi.org/10.1038/ncomms3246
Du X, West MB, Cheng W, Ewert DL, Li W, Saunders D, Towner RA, Floyd RA, Kopke RD (2016) Ameliorative effects of antioxidants on the hippocampal accumulation of pathologic tau in a rat model of blast-induced traumatic brain injury. Oxidative Med Cell Longev 2016:4159357. https://doi.org/10.1155/2016/4159357
Du K, Liu M, Pan Y, Zhong X, Wei M (2017) Association of Serum Manganese Levels with Alzheimer’s disease and mild cognitive impairment: a systematic review and meta-analysis. Nutrients 9(3):231. https://doi.org/10.3390/nu9030231
Duchen MR (2000) Mitochondria and calcium: from cell signalling to cell death. J Physiol 529 Pt 1(Pt 1):57–68. https://doi.org/10.1111/j.1469-7793.2000.00057.x
Fan L, Mao C, Hu X, Zhang S, Yang Z, Hu Z, Sun H, Fan Y, Dong Y, Yang J, Shi C, Xu Y (2020) New insights into the pathogenesis of Alzheimer’s disease. Front Neurol 10:1312. https://doi.org/10.3389/fneur.2019.01312
Fang X, Yu SX, Lu Y, Bast RC Jr, Woodgett JR, Mills GB (2000) Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci U S A 97(22):11960–11965. https://doi.org/10.1073/pnas.220413597
Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribé E, Dalfó E, Avila J (2005) Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr Alzheimer Res 2(1):3–18. https://doi.org/10.2174/1567205052772713
Fridovich I (1999) Fundamental aspects of reactive oxygen species, or what’s the matter with oxygen? Ann N Y Acad Sci 893:13–18. https://doi.org/10.1111/j.1749-6632.1999.tb07814.x
Gammoh NZ, Rink L (2017) Zinc in infection and inflammation. Nutrients 9(6):624. https://doi.org/10.3390/nu9060624
Garai K, Sahoo B, Kaushalya SK, Desai R, Maiti S (2007) Zinc lowers amyloid-beta toxicity by selectively precipitating aggregation intermediates. Biochemistry 46(37):10655–10663. https://doi.org/10.1021/bi700798b
Garza-Lombó C, Posadas Y, Quintanar L, Gonsebatt ME, Franco R (2018) Neurotoxicity linked to dysfunctional metal ion homeostasis and xenobiotic metal exposure: redox signaling and oxidative stress. Antioxid Redox Signal 28(18):1669–1703. https://doi.org/10.1089/ars.2017.7272
Gaschler MM, Stockwell BR (2017) Lipid peroxidation in cell death. Biochem Biophys Res Commun 482(3):419–425. https://doi.org/10.1016/j.bbrc.2016.10.086
Gunter TE, Gerstner B, Lester T, Wojtovich AP, Malecki J, Swarts SG, Brookes PS, Gavin CE, Gunter KK (2010) An analysis of the effects of Mn2+ on oxidative phosphorylation in liver, brain, and heart mitochondria using state 3 oxidation rate assays. Toxicol Appl Pharmacol 249(1):65–75. https://doi.org/10.1016/j.taap.2010.08.018
Heston LL, White J (1978) Pedigrees of 30 families with Alzheimer disease: associations with defective organization of microfilaments and microtubules. Behav Genet 8(4):315–331. https://doi.org/10.1007/BF01067395
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA (2001) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21(9):3017–3023. https://doi.org/10.1523/JNEUROSCI.21-09-03017.2001
Hsu TM, Noble EE, Reiner DJ, Liu CM, Suarez AN, Konanur VR, Hayes MR, Kanoski SE (2018a) Hippocampus ghrelin receptor signaling promotes socially-mediated learned food preference. Neuropharmacology 131:487–496. https://doi.org/10.1016/j.neuropharm.2017.11.039
Hsu HW, Bondy SC, Kitazawa M (2018b) Environmental and dietary exposure to copper and its cellular mechanisms linking to Alzheimer’s disease. Toxicol Sci 163(2):338–345. https://doi.org/10.1093/toxsci/kfy025
Ishii T, Hayakawa H, Igawa T, Sekiguchi T, Sekiguchi M (2018) Specific binding of PCBP1 to heavily oxidized RNA to induce cell death. Proc Natl Acad Sci U S A 115(26):6715–6720. https://doi.org/10.1073/pnas.1806912115
Kawamata H, Manfredi G (2010) Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space. Antioxid Redox Signal 13(9):1375–1384. https://doi.org/10.1089/ars.2010.3212
Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, Mattson MP (1997) Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem 69(1):273–284. https://doi.org/10.1046/j.1471-4159.1997.69010273.x
Kern A, Mavrikaki M, Ullrich C, Albarran-Zeckler R, Brantley AF, Smith RG (2015) Hippocampal dopamine/DRD1 signaling dependent on the ghrelin receptor. Cell 163(5):1176–1190. https://doi.org/10.1016/j.cell.2015.10.062
Leigh GJ (1990) Nomenclature of inorganic chemistry. Recommendations. Blackwell Scientific Publications, Oxford
Lidsky TI (2014) Is the aluminum hypothesis dead? J Occup Environ Med 56(5 Suppl):S73–S79. https://doi.org/10.1097/JOM.0000000000000063
Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, Hagen TM, Cotman CW, Ames BN (2002) Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha-lipoic acid. Proc Natl Acad Sci U S A 99(4):2356–2361. https://doi.org/10.1073/pnas.261709299
Lovell MA, Markesbery WR (2007) Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res 35(22):7497–7504. https://doi.org/10.1093/nar/gkm821
MacAllister RJ, Parry H, Kimoto M, Ogawa T, Russell RJ, Hodson H, Whitley GS, Vallance P (1996) Regulation of nitric oxide synthesis by dimethylarginine dimethylaminohydrolase. Br J Pharmacol 119(8):1533–1540. https://doi.org/10.1111/j.1476-5381.1996.tb16069.x
Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F (2013) The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through tau phosphorylation. Neuron 78(1):94–108. https://doi.org/10.1016/j.neuron.2013.02.003
Malecki EA (2001) Manganese toxicity is associated with mitochondrial dysfunction and DNA fragmentation in rat primary striatal neurons. Brain Res Bull 55(2):225–228. https://doi.org/10.1016/s0361-9230(01)00456-7
Manisha WH, Rajak R, Jat D (2017) Oxidative stress and antioxidants: an overview. Int J Adv Res Rev 2(9):110–119. https://www.researchgate.net/publication/319468596
Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP (1997) Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci 17(3):1046–1054. https://doi.org/10.1523/JNEUROSCI.17-03-01046.1997
Markesbery WR, Lovell MA (2006) DNA oxidation in Alzheimer’s disease. Antioxid Redox Signal 8(11–12):2039–2045. https://doi.org/10.1089/ars.2006.8.2039
Martinez-Finley EJ, Gavin CE, Aschner M, Gunter TE (2013) Manganese neurotoxicity and the role of reactive oxygen species. Free Radic Biol Med 62:65–75. https://doi.org/10.1016/j.freeradbiomed.2013.01.032
Mattson MP (2004) Pathways towards and away from Alzheimer’s disease. Nature 430(7000):631–639. https://doi.org/10.1038/nature02621
Mezzaroba L, Alfieri DF, Colado Simão AN, Vissoci Reiche EM (2019) The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 74:230–241. https://doi.org/10.1016/j.neuro.2019.07.007
Mills E, Dong XP, Wang F, Xu H (2010) Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Future Med Chem 2(1):51–64. https://doi.org/10.4155/fmc.09.140
Moreira PI, Duarte AI, Santos MS, Rego AC, Oliveira CR (2009) An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J Alzheimers Dis 16(4):741–761. https://doi.org/10.3233/JAD-2009-0972
Moulton PV, Yang W (2012) Air pollution, oxidative stress, and Alzheimer’s disease. J Environ Public Health 2012:472751. https://doi.org/10.1155/2012/472751
Nandi A, Yan LJ, Jana CK, Das N (2019) Role of catalase in oxidative stress- and age-associated degenerative diseases. Oxidative Med Cell Longev 2019:9613090. https://doi.org/10.1155/2019/9613090
Naqvi S, Samim M, Abdin M, Ahmed FJ, Maitra A, Prashant C, Dinda AK (2010) Concentration-dependent toxicity of iron oxide nanoparticles mediated by increased oxidative stress. Int J Nanomedicine 5:983–989. https://doi.org/10.2147/IJN
Nordberg J, Arnér ES (2001) Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med 31(11):1287–1312. https://doi.org/10.1016/s0891-5849(01)00724-9
Nunomura A, Perry G (2020) RNA and oxidative stress in Alzheimer’s disease: focus on microRNAs. Oxidative Med Cell Longev 2020:2638130. https://doi.org/10.1155/2020/2638130
Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP (2006) The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 440(7083):528–534. https://doi.org/10.1038/nature04543
Pinton P, Pozzan T, Rizzuto R (1998) The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J 17(18):5298–5308. https://doi.org/10.1093/emboj/17.18.5298
Praticò D (2005) Peripheral biomarkers of oxidative damage in Alzheimer’s disease: the road ahead. Neurobiol Aging 26(5):581–583. https://doi.org/10.1016/j.neurobiolaging.2004.09.020
Readnower RD, Sauerbeck AD, Sullivan PG (2011) Mitochondria, amyloid β, and Alzheimer’s disease. Int J Alzheimers Dis 2011:104545. https://doi.org/10.4061/2011/104545
Ribarič S (2018) Peptides as potential therapeutics for Alzheimer’s disease. Molecules (Basel, Switzerland) 23(2):283. https://doi.org/10.3390/molecules23020283
Roos PM, Vesterberg O, Nordberg M (2006) Metals in motor neuron diseases. Exp Biol Med (Maywood, N.J.) 231(9):1481–1487. https://doi.org/10.1177/153537020623100906
Seminara RS, Jeet C, Biswas S, Kanwal B, Iftikhar W, Sakibuzzaman M, Rutkofsky IH (2018) The neurocognitive effects of ghrelin-induced signaling on the hippocampus: a promising approach to Alzheimer’s disease. Cureus 10(9):e3285. https://doi.org/10.7759/cureus.3285
Seo YA, Li Y, Wessling-Resnick M (2013) Iron depletion increases manganese uptake and potentiates apoptosis through ER stress. Neurotoxicology 38:67–73. https://doi.org/10.1016/j.neuro.2013.06.002
Shelat PB, Chalimoniuk M, Wang JH, Strosznajder JB, Lee JC, Sun AY, Simonyi A, Sun GY (2008) Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J Neurochem 106(1):45–55. https://doi.org/10.1111/j.1471-4159.2008.05347.x
Smith MA, Harris PL, Sayre LM, Perry G (1997) Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A 94(18):9866–9868. https://doi.org/10.1073/pnas.94.18.9866
Spiers JG, Cortina Chen HJ, Barry TL, Bourgognon JM, Steinert JR (2022) Redox stress and metal dys-homeostasis appear as hallmarks of early prion disease pathogenesis in mice. Free Radic Biol Med 192:182–190. https://doi.org/10.1016/j.freeradbiomed.2022.09.025
Strong MJ, Garruto RM, Joshi JG, Mundy WR, Shafer TJ (1996) Can the mechanisms of aluminum neurotoxicity be integrated into a unified scheme? J Toxicol Environ Health 48(6):599–613. https://doi.org/10.1080/009841096161096
Stutzmann GE, Caccamo A, LaFerla FM, Parker I (2004) Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci 24(2):508–513. https://doi.org/10.1523/JNEUROSCI.4386-03.2004
Suh SW, Jensen KB, Jensen MS, Silva DS, Kesslak PJ, Danscher G, Frederickson CJ (2000) Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res 852(2):274–278. https://doi.org/10.1016/s0006-8993(99)02096-x
Takeda A, Smith MA, Avilá J, Nunomura A, Siedlak SL, Zhu X, Perry G, Sayre LM (2000) In Alzheimer’s disease, heme oxygenase is coincident with Alz50, an epitope of tau induced by 4-hydroxy-2-nonenal modification. J Neurochem 75(3):1234–1241. https://doi.org/10.1046/j.1471-4159.2000.0751234.x
Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo DG, Mattson MP, Tabaton M (2008) Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem 104(3):683–695. https://doi.org/10.1111/j.1471-4159.2007.05072.x
Tanaka M, Jaruga P, Küpfer PA, Leumann CJ, Dizdaroglu M, Sonntag WE, Boon Chock P (2012) RNA oxidation catalyzed by cytochrome c leads to its depurination and cross-linking, which may facilitate cytochrome c release from mitochondria. Free Radic Biol Med 53(4):854–862. https://doi.org/10.1016/j.freeradbiomed.2012.05.044
Tong BC, Wu AJ, Li M, Cheung KH (2018) Calcium signaling in Alzheimer’s disease & therapies. Biochim Biophys Acta Mol Cell Res 1865(11 Pt B):1745–1760. https://doi.org/10.1016/j.bbamcr.2018.07.018
Verma M, Vats A, Taneja V (2015) Toxic species in amyloid disorders: oligomers or mature fibrils. Ann Indian Acad Neurol 18(2):138–145. https://doi.org/10.4103/0972-2327.144284
Wang JX, Gao J, Ding SL, Wang K, Jiao JQ, Wang Y, Sun T, Zhou LY, Long B, Zhang XJ, Li Q, Liu JP, Feng C, Liu J, Gong Y, Zhou Z, Li PF (2015) Oxidative modification of miR-184 enables it to target Bcl-xL and Bcl-w. Mol Cell 59(1):50–61. https://doi.org/10.1016/j.molcel.2015.05.003
Wang L, Yin YL, Liu XZ, Shen P, Zheng YG, Lan XR, Lu CB, Wang JZ (2020) Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl Neurodegener 9:10. https://doi.org/10.1186/s40035-020-00189-z
Ward RJ, Dexter DT, Crichton RR (2015) Ageing, neuroinflammation and neurodegeneration. Front Biosci (Schol Ed) 7(1):189–204. https://doi.org/10.2741/S433
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72(5):1858–1862. https://doi.org/10.1073/pnas.72.5.1858
Wright RO, Baccarelli A (2007) Metals and neurotoxicology. J Nutr 137(12):2809–2813. https://doi.org/10.1093/jn/137.12.2809
Yang W, Omaye ST (2009) Air pollutants, oxidative stress and human health. Mutat Res 674(1–2):45–54. https://doi.org/10.1016/j.mrgentox.2008.10.005
Yokel RA (2000) The toxicology of aluminum in the brain: a review. Neurotoxicology 21(5):813–828
Zhang J, Cao R, Cai T, Aschner M, Zhao F, Yao T, Chen Y, Cao Z, Luo W, Chen J (2013) The role of autophagy dysregulation in manganese-induced dopaminergic neurodegeneration. Neurotox Res 24(4):478–490. https://doi.org/10.1007/s12640-013-9392-5
Acknowledgments
The authors acknowledge the award of Research Fellowships and financial support by the Department of Pharmaceuticals, Ministry of Chemical and Fertilizer, Government of India. NIPER-R communication no. is 395.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
The authors declare no conflict of interest.
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Fathima, S.A., Maurya, R., Naqvi, S. (2023). Oxidative Stress and Metals in Alzheimer’s Disease. In: Sharma, A., Modi, G.P. (eds) Natural Product-based Synthetic Drug Molecules in Alzheimer's Disease. Springer, Singapore. https://doi.org/10.1007/978-981-99-6038-5_2
Download citation
DOI: https://doi.org/10.1007/978-981-99-6038-5_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-6037-8
Online ISBN: 978-981-99-6038-5
eBook Packages: MedicineMedicine (R0)