
Abstract
NANOG is important for the early stages of embryogenesis and embryonic stem cell maintenance. It is also involved in the nuclear reprogramming necessary for forming induced pluripotent stem cells, thus offering enormous potential for cell therapies. Here, the expression and purification of human NANOG protein from the soluble fraction of bacterial cell lysate are reported. To produce fusion tagged transducible version of NANOG protein, we fused the human NANOG cDNA sequence to different fusion tags for affinity purification and efficient cell and nuclear delivery. Subsequently, the optimal expression conditions were identified, and the recombinant fusion protein was purified to homogeneity with a retained secondary structure. Additionally, NANOG is linked to cellular multipotency, which plays crucial roles in stemness, tumor progression and migration, thus highlighting its potential role as a therapeutic target for a myriad of malignancies such as cervical cancer. Therefore, we investigated the effect of application of the HTN-NANOG fusion protein on HeLa cells. Upon protein transduction in HeLa cells, the cell migration rate was augmented in the presence of NANOG. Furthermore, we observed the downregulation of p27 in HeLa cells in the presence of NANOG at mRNA and protein levels. From a future perspective, this bioactive form of human NANOG protein can be used to understand the molecular roles of NANOG in several cellular processes associated with cancer and induced pluripotent stem cells.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Homeobox (homeodomain) protein NANOG is a transcription factor of the ANTP (Antennapedia) class and is conserved in vertebrates [1, 2]. NANOG expression is observed during the initial stages of mouse embryonic development and in primordial germ cells [3,4,5,6,7,8]. During mouse embryonic development, lack of NANOG expression in the inner cell mass failed to produce epiblast, and instead, formed parietal endoderm-like cells [4]. Thus, the expression of NANOG is crucial for the epiblast formation [4, 9, 10]. Furthermore, the mRNA level of NANOG is downregulated after implantation [3]. High expression of NANOG holds epiblast essence, whereas low expression promotes differentiation into primitive endoderm. In embryonic stem cells (ESCs), NANOG is a vital player in the transcriptional pluripotency regulatory network along with OCT4 and SOX2 [11,12,13]. Moreover, the role of NANOG in ESCs is to maintain self-renewal and pluripotency in a cytokine-independent fashion [3, 4]. Intriguingly, the deletion of NANOG did not disrupt the chimera formation potential of ESCs [3].
Furthermore, ESCs are identical to induced pluripotent stem cells (iPSCs), and NANOG holds the potential for reprogramming due to the presence of the homeodomain, which is functionally preserved among all vertebrates [2]. The dose of NANOG is critical in cell-fusion-mediated reprogramming, and its overexpression improved the efficiency of reprogramming by 200-fold [14]. Several studies have reported that NANOG overexpression can enhance and hasten iPSC generation [9, 15]. Additionally, the role of NANOG in reprogramming is critical for reaching the pluripotent ground state by enabling the conversion of pre-induced pluripotent stem cells to bonafide iPSCs [9, 16]. Moreover, NANOG not only induces pluripotency but can also overcome reprogramming obstacles [17]. Inconsistent with these studies, a few studies have reported that including NANOG in the reprogramming cocktail does not improve reprogramming efficiency and is thus not critical for iPSC generation [18, 19]. Despite the deletion of the NANOG gene from somatic cells, these cells can be reprogrammed to form iPSCs and form teratomas and chimeras [19], indicating their dispensability in the reprogramming process. Another study reported that, although the elimination of NANOG decreased reprogramming efficiency, it was not required for iPSC generation [20]. Thus, these studies indicate that other members of the transcriptional network of pluripotency can substitute for NANOG loss. However, the same studies have suggested that NANOG is essential during the final stage of the reprogramming process and that its depletion affects reprogramming efficiency [19, 20]. Numerous reports have demonstrated the generation of human iPSCs using NANOG via lentiviral transduction [21,22,23,24]. In addition, the derivation of mouse and human iPSCs has also been reported using retroviral vectors, which include NANOG in the reprogramming cocktail [25, 26]. It is noteworthy that the use of viral-based approaches showed high reprogramming efficiency but induces genomic alteration. To circumvent these limitations of including viral components, several other approaches, such as recombinant proteins, mRNA, Sendai virus and miRNAs, are favored [27, 28]. Among all the approaches for iPSC generation, the recombinant protein-based method is the safest [29].
Recombinant proteins are of great importance for industrial applications and clinical research. The benefits of using bacterial systems, such as Escherichia coli (E. coli), for the production of recombinant proteins are easy genetic manipulation and low production costs. However, this system also has several unavoidable blockages, such as codon usage bias, weak expression, insoluble expression, complicated purification protocols, protein misfolding and so forth. Thus, the expression of recombinant proteins opens an avenue for the generation of iPSCs. These cells can serve as patient-specific cells for cell therapies. Herein, the generation of a recombinant human NANOG fusion protein along with its secondary structure determination is reported, which possesses cytoplasmic and nuclear translocation ability, and is demonstrated to be biologically active. This recombinant version of human NANOG can potentially be used in the cocktail of transcription factors for iPSC generation.
In addition to the role of NANOG in iPSCs generation, anomalous NANOG expression has been shown in different types of human malignancies. Furthermore, ectopic expression of NANOG caused augmented proliferation and downregulation of tumor suppressors, however, it did not result in tumor formation. Therefore, unlike other reprogramming factors, NANOG cannot be considered an oncogene [30]. In stark contrast to earlier finding, several studies have reported NANOG expression in different tissues, including the breast, cervix and kidney [31,32,33]. Several such studies will provide a deeper understanding of molecular networks in cancer and help in prognosis and treatment. Thus, the recombinant version of human NANOG generated in this study can further be helpful to understand the biological role of NANOG in cancer.
2 Materials and Methods
2.1 Plasmids, Strains, Reagents and Cell Lines
The personalized plasmids (pUC-HTN-GOI and pUC-GOI-NTH) were procured from GenScript. E. coli BL21 (DE3) cells were utilized as host for recombinant protein expression. Isopropyl β-D-1-thiogalactopyranoside (IPTG), Luria–Bertani broth, terrific broth, kanamycin, sodium phosphate mono/dibasic, sodium chloride and imidazole was purchased from HiMedia. Bradford reagent was purchased from Bio-Rad. Dulbecco's Modified Eagle medium (DMEM), fetal bovine serum (FBS) and penicillin–streptomycin solution (P/S) were purchased from Invitrogen.
Apart from this, two human cell lines, HeLa (NCCS, India) and human dermal fibroblasts [HDFs; HiMedia CL011-2XT25)], were cultured in a complete growth medium [DMEM, FBS (5%) and P/S (1%)].
2.2 Construction of Expression Vector and Optimization for Soluble NANOG Expression
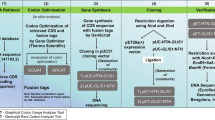
A schematic figure detailing the stepwise process from retrieval of gene sequence [codon-optimized coding sequence of NANOG along with a set of fusion tags (Table 1) (gene inserts)] to cloning to confirmation of cloning of NANOG in desired vector is shown in Fig. 1. Furthermore, pET28a(+)-HTN-NANOG and pET28a(+)-NANOG-NTH were transformed into BL21(DE3) cells using CaCl2 method. To identify the optimal expression, the same approach was used as designated earlier [34, 35]. The cells were lysed using the lysis buffer mentioned in Table 2.

Diagram representation of the codon optimization and cloning strategy used in this study. H, Histidine; T, TAT, Trans-activator of transcription; N, NLS, Nuclear localization sequence/signal
2.3 One-Step Purification of NANOG Via Metal Affinity Chromatography
To purify HTN-NANOG fusion protein, 1.2 L of lysate of the recombinant E. coli BL21(DE3) cells harboring pET28a(+)-HTN-NANOG were established as mentioned above. The resulting lysate was loaded onto the pre-equilibrated nickel-NTA column, followed by complete washing with buffers and then eluted with elution buffer. The composition of all buffers for purification purpose is mentioned in Table 2.
After purification, size-exclusion chromatography of the eluted fractions was performed as previously described [34]. The collected protein fractions were quantified using the Bradford assay [36].
2.4 SDS-PAGE, Coomassie Staining and Western Blotting (Immunoblotting) Analysis
SDS-PAGE, Coomassie staining and Western blotting (immunoblotting) was performed as previously described [35]. The primary antibodies [anti-His (BioBharati, BB-AB0010; 1:5000), anti-NANOG (Merck Millipore, AB9220; 1:5000)], anti-GAPDH (BioBharati, BB-AB0060; 1:5000), anti-H3 (BioBharati, BB-AB0055; 1:5000), anti-p27 (Cell Signaling Technology, D69C12; 1:4000), β-actin (BioBharati, BB-AB0024); 1:5000], and secondary antibodies [anti-rabbit IgG antibody (Invitrogen, 31,460; 1:5000)] were used in the immunoblot analysis.
2.5 Circular Dichroism (CD) Spectroscopy
The full-length HTN-NANOG protein secondary structure was determined by CD spectroscopy [J-1500 spectropolarimeter (Jasco, MD, USA)] and further scrutinized by online tool BeStSel (Beta Structure Selection) [37, 38] as previously described [34, 35].
2.6 Stability of HTN-NANOG Protein
Stability was performed as described previously [39] and clarified samples were then evaluated by immunoblotting using the NANOG antibody.
2.7 Subcellular Fractionation
HDFs cells (1 × 105) were seeded in a T-25 flask and incubated overnight with vehicle control or HTN-NANOG protein (400 nM). Subcellular fractionation was carried out as previously described [40] and analyzed by immunoblotting with specific antibodies.
2.8 Cell Proliferation Assay
HDFs (1 × 104 cells/well) were seeded in 24-well plates and were treated with HTN-NANOG (200 nM) or vehicle control. After reaching approximately 70–80% confluency, the cells were trypsinized. Cell counting was performed using a hemocytometer [41]. A cell suspension was intermixed with exclusion dye solution (Bio-Rad) and loaded into the cell counting chamber. Later, the live cells within a specific area were counted and recorded. Because the chamber volume is well-defined (commonly 0.1 mm3), the number of cells calculated per area multiplied by the dilution factor determines the number of live cells per mL. The data are presented with a graph of the cumulative cell number against days.
2.9 Cell Migration (Scratch) Assay
HeLa cells (0.6 × 105) were seeded in a 24-well plate in a growth medium. Scratch assay was performed as earlier [42]. The rate of cell migration was calculated as previously described [43].
2.10 RT-qPCR Analysis
HeLa cells (1 × 105 cells per well) were seeded in a six‐well plate and treated with vehicle control or HTN-NANOG protein (200 nM) for 3 consecutive days. RNA isolation, synthesis of complementary DNA and RT-qPCR were performed and analyzed as described previously [44]. The primers used in this study are mentioned in Table 3.
2.11 Statistical Analysis
Data analysis (unpaired student’s t-test) were analyzed using GraphPad Prism 8 software and presented as mean ± standard deviation (SD) of three independent experiments. Values of p < 0.05 (*p < 0.05; ***p < 0.001) were with statistical significance.
3 Results
3.1 Cloning and Expression Parameter Optimization
The coding (codon-optimized) sequence of human NANOG was fused with three fusion tags as shown in Table 1, Fig. 1. The fused gene insert was cloned in pET28a(+) to generate two constructs as shown in Fig. 1. These genetic constructs were verified using restriction analysis using enzymes (Fig. 2) and DNA sequencing. These genetic constructs were transformed in E. coli strain BL21(DE3) for expression. Next, expression parameters were identified by screening different values (Table 4) for maximal soluble expression.

Cloning of gene inserts, HTN-NANOG and NANOG-NTH, in pET28a(+) expression vector. The GOI was fused as per the schematic diagram (top) with human NANOG cDNA sequence to generate pET28a(+)-HTN-NANOG and pET28a(+)-NANOG-NTH. The resulting plasmids were then confirmed by restriction digestion using various restriction enzymes. GOI, Gene of interest; H, His; T, TAT and N, NLS
Based on these observations, maximal soluble expression of NANOG fusion proteins was observed with the gene constructs (HTN-NANOG and NANOG-NTH) induced at 18 °C (Fig. 3). However, no soluble expression of HTN-NANOG was induced at 37 °C, and many truncations were observed for NANOG-NTH (37 °C) (Fig. 3). Hence, these constructs were excluded from further analyses. Notably, in the case of NANOG, only N-terminally tagged NANOG (HTN-NANOG) induced at 18 °C showed the maximal soluble expression with no truncations. Hence, this gene construct (HTN-NANOG), induced at 18 °C with a post-induction incubation time of 12 h, was selected for further experiments (Fig. 3). These results demonstrate that the solubility of the protein was improved by reducing the temperature of the induced culture. Several studies have demonstrated the expression and purification of full-length and truncated recombinant NANOG proteins from several mammalian species [12, 45,46,47,48,49,50,51]. However, these studies only showed the interaction of NANOG with its consensus DNA binding sequence [12, 45, 49,50,51], however, the comprehensive demonstration of bioactivity of full-length human NANOG is still obscure. Thus, this study mainly focuses on the exploration of the bioactivity of full-length recombinant NANOG protein.

Identification of the optimal induction temperature to achieve maximal soluble expression of recombinant human NANOG fusion protein. E. coli BL21(DE3) strain was transformed with pET28a(+) vectors harboring the fusion gene inserts and the expression of both N-terminally tagged and C-terminally tagged recombinant NANOG protein was at two different temperatures: 37 °C (post-incubation time of 2 h) and 18 °C (post-incubation time of 12 h). Then, the harvested cells were lysed to obtain the total cell lysate (L) fraction and further centrifuged to obtain a soluble/supernatant (S) cell fraction and an insoluble/pellet (P). Protein samples (20 µg) were loaded on 12% SDS-PAGE gel and further verified by immunoblotting (bottom). M, Marker; UI, uninduced (total cell lysate); L, cell lysate; P, pellet portion; S, supernatant portion; kDa, kilodaltons; α, antibody. (n = 4) * Truncation of fusion proteins
3.2 Purification and Secondary Structure Estimation of HTN-NANOG Protein
After screening the parameters for soluble expression analysis, HTN-NANOG was expressed in soluble form when induced at 18 °C. Hence, we aimed to purify this protein using a simple and straightforward affinity chromatography-based purification procedure. The HTN-NANOG protein band (~55 kDa) was observed in the elution fractions (Fig. 4). The purified HTN-NANOG protein was identified using SDS-PAGE (Fig. 4; top) and immunoblotting using a histidine antibody (Fig. 4; middle) and NANOG antibody (Fig. 4; bottom). Both antibodies detected all protein fragments, proving that there was no bacterial protein. Thus, we have demonstrated simple and straightforward native purification of the NANOG fusion protein.

SDS-PAGE and immunoblot analysis of NANOG fusion protein purification by metal affinity chromatography. HTN-NANOG protein. M, Marker; L, Lysate; S, Supernatant portion; FT, Flow-through portion; W (1–3), Wash buffer (1–3); E, Elution portion; kDa, Kilodalton; α, Antibody
The far-ultraviolet CD spectroscopic method is widely used to unveil the folding features of certain proteins where the secondary structure is obscure [52, 53]. Thus, the secondary structural conformation of purified recombinant HTN-NANOG was evaluated using CD (Fig. 5a). CD spectrum analysis using the BeStSel tool suggested that this fusion protein consisted primarily of random coils (~46%), followed by β‐sheets (~21%), turns (~18%) and α‐helices (~15%) (Fig. 5b). The results account that purified recombinant HTN-NANOG protein has a secondary structure retained post-purification.

Determination of the secondary structure of the NANOG protein using far-UV CD spectroscopy. The purified NANOG protein was analyzed for its secondary structures using far-UV CD spectroscopy. a The CD spectra were represented as delta epsilon (M − 1 cm − 1; Y-axis) vs wavelength (nm; X-axis). b CD spectra were evaluated with the BeStSel online tool, and the resulting structural configuration (α-helix, β-sheets, turn and others) is represented using bar graphs (n = 3)
3.3 Stability and Transduction Ability of HTN-NANOG Fusion Protein
Further, the protein stability of the purified NANOG protein was studied. This fusion protein was found to be stable for at least 24 h, which was identified using immunoblotting with the NANOG antibody (Fig. 6a, b).

Stability and localization of HTN-NANOG protein determined with a subcellular fractionation assay. a Stability of purified recombinant proteins under standard cell culture conditions was analyzed by immunoblotting. b Densitometric analysis of data from (a) and (c) representative immunoblot showing cytoplasmic (C) and nuclear (N) fractions of NANOG fusion protein-treated or untreated (vehicle control) HDFs cells using NANOG, GAPDH and Histone H3 antibodies. M, Marker; C, Cytosolic portion; N, Nuclear portion
Next, we demonstrated the transduction ability of HTN-NANOG fusion protein in HDFs using subcellular fractionation. HDFs have no endogenous expression of NANOG; therefore, these cells were used in this study. Similar to immunofluorescence staining, subcellular fractionation can also be used to know the protein localization in a cell [40, 54]. Moreover, subcellular localization is vital for proper protein function. The fractions (nuclear and cytoplasmic) of the human cell lines were separated by centrifugation. These fractions were further analyzed by immunoblotting. In this study, GAPDH (Glyceraldehyde-3-Phosphate Dehydrogenase) and Histone H3 were used as internal loading controls for the cytoplasmic (GAPDH) and nuclear (H3) fractions (Fig. 6c), respectively. Immunoblotting analysis exhibited that the majority of the HTN-NANOG fusion proteins were present in the nuclear fraction of cells treated with NANOG. This implied that the fusion tag nuclear localization signal (NLS) is responsible for the efficient nuclear translocation of the recombinant fusion protein. The homeodomain in human NANOG tends to be localized in the nucleus, implying that it has a NLS [55]. The presence of a stretch of six amino acids [YKQVKT (136–141 aa)] in the homeobox is responsible for nuclear localization [56]. Adding an extra NLS to NANOG sequence has further enabled efficient nuclear delivery of the purified recombinant NANOG protein.
3.4 Effect of the Recombinant HTN-NANOG Protein on Cell Proliferation of HDFs
Several studies have reported that exogenous expression of NANOG in ESCs and fibroblasts resulted in increased cell proliferation [57, 58]. In view of this, we also explored the effect of NANOG on human fibroblast (HDF) cells. To investigate cell proliferation, we counted the cells using a hemocytometer at different time intervals and observed augmented cell proliferation of HDFs. The data was analyzed and are shown in Fig. 7. The results of our study are in concordant with prior studies [57, 58]. Hence, our study provides evidence that NANOG plays a proliferative role in HDFs and is bioactive.

Effect of purified HTN-NANOG protein on cell proliferation. Cell proliferation was assessed via cell counting. Y-axis signifies the cumulative cell number, and the X-axis signifies days of treatment (n = 3)
3.5 Effect of the Recombinant HTN-NANOG Protein on the Migration of HeLa Cells
The oncogenic potential of NANOG is well established and its presence has been detected in cervical cancer [33, 59, 60]. Forced NANOG expression has been reported to result in increased migration, invasion and tumorigenesis in HeLa cells [61]. Thus, to confirm that the purified HTN-NANOG fusion protein was biologically active, the migration rate of HeLa cells was evaluated using an in vitro scratch assay. Based on the observation of the effect of HTN-NANOG protein transduction in HeLa cells, NANOG protein-treated wells showed faster cell migration compared to the wells treated with the vehicle control (Fig. 8a, b; ***p < 0.001). These results were consistent with previously published results [61]. An earlier study used NANOG mRNA, but we used NANOG as a recombinant protein in our study for functional investigation. Thus, this study demonstrated that the HTN-NANOG fusion protein facilitated the migration of HeLa cells and was bioactive.

Effect of purified HTN-NANOG protein on the migration of HeLa cells. a Scratch assay of HeLa cells. Cell migration was induced in the presence of HTN-NANOG. Scale bar: 100 μm. b Graphical representation of the changes in migration rate following treatment with vehicle control or NANOG protein. Quantitative data are expressed as the mean ± SD (n = 3). ***p < 0.0001
3.6 Effect of the Recombinant HTN-NANOG Protein on the Cell Cycle Inhibitor P27 Gene
To further explore the functional characteristics of NANOG, we investigated the effect of NANOG on cell cycle factor p27 (CDKN1B). Furthermore, p27Kip1 is a tumor suppressor and inhibitor of Cyclin/Cyclin-Dependent Kinase (CDK) complexes. Thus, p27 plays a vital role in the regulation of the cell cycle. Transient activation of NANOG in fibroblasts is linked to the downregulation of p27 [62]. Hence, we also attempted to examine the effect of HTN-NANOG on p27 mRNA and protein levels in HeLa cells. Cells were harvested for RNA isolation and complementary DNA (cDNA) synthesis was carried out from total RNA. Quantitative real-time PCR (RT-qPCR) was performed using GAPDH as a reference. The data indicated a significant decrease in the p27 at RNA level (Fig. 9a; *p < 0.05). To further validate this at the protein level, we performed immunoblotting using β-actin as a loading control for normalization (Fig. 9b). Immunoblotting analysis showed that the expression of p27 was downregulated at the protein level in the presence of HTN-NANOG. These data demonstrate that the downregulation of the p27 (at RNA and protein level) in the presence of protein is in line with an earlier reported study [62]. Hence, this confirms that the purified recombinant HTN-NANOG protein is bioactive.

Effect of the purified HTN-NANOG fusion protein on p27 levels in HeLa cells. a Relative expression of the p27 mRNA in vehicle control and HTN-NANOG analyzed by RT-qPCR. Quantitative statistics are shown as the mean ± SD (n = 3). *p < 0.05. b Immunoblot analysis of p27 protein expression in the presence of the vehicle control and NANOG protein (n = 3)
4 Discussion
Human NANOG (915 bp) contains four exons and three introns. The human NANOG protein is a long chain of 305 amino acids and has N-terminal, homeobox domain and C-terminal region. The N-terminal region (94 aa) is abundant in serine, proline and threonine and is responsible for governing the transcription of NANOG. The homeobox domain (60 aa) binds to the DNA core region. The C-terminal region (151 aa) is tryptophan-rich, and investigations have shown that this region is responsible for homodimerization. Thus, NANOG acts as a transcription factor by controlling transcription through interactions with the promoters of myriad genes via these domains [55, 56, 63]. NANOG is a transcriptional activator that binds to the 5’–TAAT–3’ essential regions of the DNA sequence [64, 65]. A triad of transcription factors, including OCT4, SOX2 and NANOG, appear to be at the heart of pluripotency [11]. Unlike OCT4 and SOX2, solitary NANOG cells conserve pluripotency in the absence of pluripotency maintenance signals. Knockdown studies have revealed the crucial role of NANOG in embryogenesis staging. NANOG is important for the maintenance of self-renewal and pluripotency. These properties prevent the ESCs and iPSCs from differentiation [11].
Additionally, NANOG facilitates pre-iPS cells to attain ground-state pluripotency (iPSCs), which is crucial in the process of molecular reprogramming [9]. During the reprogramming of somatic cells, NANOG plays a critical role in inducing pluripotency [9]. NANOG boosts the activation of STAT3; hence, the synergistic action between them promotes the establishment of naive pluripotency in ESCs and during the initial stages of reprogramming [66]. iPSCs that are deficient in NANOG are identical to wild-type iPSCs in transcriptional status; and upon further exploration, teratomas and chimeric formation are underpinned. Hence, this study demonstrates that NANOG is dispensable for iPSC formation [19]. A recent study has demonstrated the distinctive ability of human NANOG to initiate prion-like clusters. These assemblies could act as a means for delivering DNA elements for their interactions with others, which is essential for the re-organization of chromatin structures and activation of ground-state pluripotency in a dose-dependent manner [67].
We successfully purified HTN-NANOG from a soluble fraction of E. coli cultures using affinity chromatography. Based on these observations, the soluble protein expression was relatively low (purity >90%) and might be due to its aggregation property when induced at larger volumes. Additionally, another study reported the purification of human NANOG, in which, among all the domains, the C-terminal domain has limited solubility. In contrast, the homeodomain and N-terminal domains are favorably soluble [51]. Strikingly, another study also showed the expression of full-length human NANOG protein along with 17 kDa protein (Skp) chaperone [51]. The role of Skp is to aid the folding of outer membrane proteins and their entry into membranes. Furthermore, Skp is well established to enhance the folding of recombinant proteins and further, directs the cargo to the periplasm of E. coli [68]. In agreement with the preceding evidence, the wild-type NANOG-GB1 fusion protein was also expressed and found to be soluble when co-expressed with the Skp chaperone. This study reasoned that the Skp chaperone is equipped with a hydrophobic cage that encases wild-type NANOG and further co-operates with tryptophan residues upon exposure [67]. In addition, human NANOG, by virtue of its C-terminal domain, is apt for aggregation, which accounts for the limited biophysical characterization of the protein and its individual domains [67]. Therefore, we aimed to determine the secondary structure of the full-length NANOG fusion protein.
The crystal structure of homeodomain NANOG has been documented as a structural scaffold formed by three helices in both mice and humans [45, 69]. A recent study showed that the CD spectra of each domain individually, the N-terminal domain of human NANOG, comprises intrinsically disordered regions responsible for displaying a random-coil signature in NMR spectra and are further found in line with the computational prediction [67]. In contrast, the far-UV CD spectra of the C-terminal domain revealed β-sheets and resulted in a reduced number of β-sheets in the mutated version of human NANOG [67]. According to available data, our study showed that the secondary structure of the full-length HTN-NANOG protein is retained and is likely to be bioactive.
A multitude of studies has been reported on the generation of reprogramming factors, namely, OCT4, SOX2, NANOG, PDX1, NGN3, GLIS1, GATA4, TBX5, HAND2, ETS2, MEF2C and MESP1, in E. coli using similar approaches [35, 40, 42, 44, 58, 62, 70,71,72,73,74,75,76,77,78]. These investigations also revealed the utility of NLS in proteins that facilitate the delivery of recombinant proteins to the nucleus in human cells. Several reports have observed that the fusion tags [post protein delivery into cells (via TAT) and their nucleus (via NLS)] do not hamper the bioactivity of recombinant proteins [35, 39, 40, 44, 58, 62, 70,71,72,73, 78]. Thus, cytoplasmic and nuclear transport of reprogramming factors can be attained via TAT and NLS fusion without requiring additional transduction reagents. For this, we demonstrated that the protein is stable under standard cell culture conditions and has the ability to enter the nucleus using a subnuclear fractionation assay.
Additionally, our study highlights the proliferative effect of recombinant NANOG on human dermal fibroblasts, emphasizing its role in malignancy. Next, we analyzed the impact of exogenously delivered NANOG fusion proteins on HeLa cell migration. Thus, we revealed that the stem cell-linked factor NANOG possesses tumorigenic properties in cervical cancer. Furthermore, we observed the downregulation of p27 in HeLa cells in the presence of NANOG at the mRNA and protein levels. This study provides evidence that tagging the protein at either terminal does not hinder its bioactivity. The generation of biologically active NANOG might trigger the endogenous expression of NANOG and may also participate in the auto- and inter-regulatory loops of pluripotency during the induction of iPSCs. The proliferative effect of NANOG on cells may offer an excellent platform for the expansion of mature cells in vitro. From a cancer perspective, it may act as a molecular marker for prognosis and cure.
References
Booth HAF, Holland PWH (2004) Eleven daughters of NANOG. Genomics 84(2):229–238. https://doi.org/10.1016/j.ygeno.2004.02.014
Theunissen TW, Costa Y, Radzisheuskaya A, van Oosten AL, Lavial F, Pain B et al (2011) Reprogramming capacity of Nanog is functionally conserved in vertebrates and resides in a unique homeodomain. Development 138(22):4853–4865. https://doi.org/10.1242/dev.068775
Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A (2003) Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 113(5):643–655. https://doi.org/10.1016/s0092-8674(03)00392-1
Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K et al (2003) The homeoprotein nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113(5):631–642. https://doi.org/10.1016/S0092-8674(03)00393-3
Komatsu K, Fujimori T (2015) Multiple phases in regulation of Nanog expression during pre-implantation development. Dev Growth Differ 57(9):648–656. https://doi.org/10.1111/dgd.12244
Chambers I, Silva J, Colby D, Nichol J, Nijmeijer B, Robertson M et al (2007) Nanog safeguards pluripotency and mediates germline development. Nature 450(7173):1230–1234. https://doi.org/10.1038/nature06403
Yamaguchi S, Kimura H, Tada M, Nakatsuji N, Tada T (2005) Nanog expression in mouse germ cell development. Gene Expr Patterns 5(5):639–646. https://doi.org/10.1016/j.modgep.2005.03.001
Lavial F, Acloque H, Bertocchini F, MacLeod DJ, Boast S, Bachelard E et al (2007) The Oct4 homologue PouV and Nanog regulate pluripotency in chicken embryonic stem cells. Development 134(19):3549–3563. https://doi.org/10.1242/dev.006569
Silva J, Nichols J, Theunissen TW, Guo G, van Oosten AL, Barrandon O et al (2009) Nanog is the gateway to the pluripotent ground state. Cell 138(4):722–737. https://doi.org/10.1016/j.cell.2009.07.039
Messerschmidt DM, Kemler R (2010) Nanog is required for primitive endoderm formation through a non-cell autonomous mechanism. Dev Biol 344(1):129–137. https://doi.org/10.1016/j.ydbio.2010.04.020
Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP et al (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122(6):947–956. https://doi.org/10.1016/j.cell.2005.08.020
Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X et al (2006) The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38(4):431–440. https://doi.org/10.1038/ng1760
Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, Orkin SH (2006) A protein interaction network for pluripotency of embryonic stem cells. Nature 444(7117):364–368. https://doi.org/10.1038/nature05284
Silva J, Chambers I, Pollard S, Smith A (2006) Nanog promotes transfer of pluripotency after cell fusion. Nature 441(7096):997–1001. https://doi.org/10.1038/nature04914
Hanna J, Saha K, Pando B, Van Zon J, Lengner CJ, Creyghton MP et al (2009) Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 462(7273):595–601. https://doi.org/10.1038/nature08592
Gingold JA, Fidalgo M, Guallar D, Lau Z, Sun Z, Zhou H et al (2014) A genome-wide RNAi screen identifies opposing functions of snai1 and snai2 on the nanog dependency in reprogramming. Mol Cell 56(1):140–152. https://doi.org/10.1016/j.molcel.2014.08.014
Theunissen TW, Van Oosten AL, Castelo-Branco G, Hall J, Smith A, Silva JCR (2011) Nanog overcomes reprogramming barriers and induces pluripotency in minimal conditions. Curr Biol 21(1):65–71. https://doi.org/10.1016/j.cub.2010.11.074
Zhao Y, Yin X, Qin H, Zhu F, Liu H, Yang Wet al (2008) Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell 3(5):475–479. https://doi.org/10.1016/j.stem.2008.10.002
Schwarz BA, Bar-Nur O, Silva JCR, Hochedlinger K (2014) Nanog is dispensable for the generation of induced pluripotent stem cells. Curr Biol 24(3):347–350. https://doi.org/10.1016/j.cub.2013.12.050
Carter AC, Davis-Dusenbery BN, Koszka K, Ichida JK, Eggan K (2014) Nanog-independent reprogramming to iPSCs with canonical factors. Stem Cell Rep 2(2):119–126. https://doi.org/10.1016/j.stemcr.2013.12.010
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318(5858):1917–1920. https://doi.org/10.1126/science.1151526
Maherali N, Ahfeldt T, Rigamonti A, Utikal J, Cowan C, Hochedlinger K (2008) A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 3(3):340–345. https://doi.org/10.1016/j.stem.2008.08.003
Haase A, Olmer R, Schwanke K, Wunderlich S, Merkert S, Hess C et al (2009) Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell 5(4):434–441. https://doi.org/10.1016/j.stem.2009.08.021
Li Y, Zhao H, Lan F, Lee A, Chen L, Lin C et al (2010) Generation of human-induced pluripotent stem cells from gut mesentery-derived cells by ectopic expression of OCT4/SOX2/NANOG. Cell Reprogramming (Formerly” Cloning and Stem Cells”), 12(3):237–247. https://doi.org/10.1089/cell.2009.0103
Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R et al (2008) Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci 105(8):2883–2888. https://doi.org/10.1073/pnas.0711983105
Moon J-H, Yun W, Kim J, Hyeon S, Kang PJ, Park G et al (2013) Reprogramming of mouse fibroblasts into induced pluripotent stem cells with Nanog. Biochem Biophys Res Commun 431(3):444–449. https://doi.org/10.1016/j.bbrc.2012.12.149
Borgohain MP, Haridhasapavalan KK, Dey C, Adhikari P, Thummer RP (2019) An insight into DNA-free reprogramming approaches to generate integration-free induced pluripotent stem cells for prospective biomedical applications. Stem Cell Rev Rep 15(2):286–313. https://doi.org/10.1007/s12015-018-9861-6
Haridhasapavalan KK, Borgohain MP, Dey C, Saha B, Narayan G, Kumar S, Thummer RP (2019) An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene 686:146–159. https://doi.org/10.1016/j.gene.2018.11.069
Dey C, Raina K, Haridhasapavalan KK, Thool M, Sundaravadivelu PK, Adhikari P et al (2021) An overview of reprogramming approaches to derive integration-free induced pluripotent stem cells for prospective biomedical applications. Recent Adv iPSC Technol 231–287. https://doi.org/10.1016/B978-0-12-822231-7.00011-4
Fischedick G, Wu G, Adachi K, Araúzo-Bravo MJ, Greber B, Radstaak M et al (2014) Nanog induces hyperplasia without initiating tumors. Stem Cell Res 13(2):300–315. https://doi.org/10.1016/j.scr.2014.08.001
Ezeh UI, Turek PJ, Reijo RA, Clark AT (2005) Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer: Interdiscip Int J Am Cancer Soc 104(10):2255–2265. https://doi.org/10.1002/cncr.21432
Bussolati B, Bruno S, Grange C, Ferrando U, Camussi G (2008) Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J 22(10):3696–3705. https://doi.org/10.1096/fj.08-102590
Ye F, Zhou C, Cheng Q, Shen J, Chen H (2008) Stem-cell-abundant proteins Nanog, Nucleostemin and Musashi1 are highly expressed in malignant cervical epithelial cells. BMC Cancer 8(1):1–5. https://doi.org/10.1186/1471-2407-8-108
Thool M, Dey C, Bhattacharyya S, Sudhagar S, Thummer RP (2021) Generation of a recombinant stem cell-specific human SOX2 protein from Escherichia coli under native conditions. Mol Biotechnol 63(4):327–338. https://doi.org/10.1007/s12033-021-00305-y
Haridhasapavalan KK, Sundaravadivelu PK, Bhattacharyya S, Ranjan SH, Raina K, Thummer RP (2021) Generation of cell-permeant recombinant human transcription factor GATA4 from E. coli. Bioprocess Biosyst Eng 44(6):1131–1146. https://doi.org/10.1007/s00449-021-02516-8
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72(1–2):248–254. https://doi.org/10.1006/abio.1976.9999
Micsonai A, Wien F, Bulyáki É, Kun J, Moussong É, Lee Y-H et al (2018) BeStSel: a web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res 46(W1):W315–W322. https://doi.org/10.1093/nar/gky497
Micsonai A, Bulyáki É, Kardos J (2021) BeStSel: from secondary structure analysis to protein fold prediction by circular dichroism spectroscopy. In: Structural genomics. Springer, Berlin, pp 175–189. https://doi.org/10.1007/978-1-0716-0892-0_11
Dey C, Thool M, Bhattacharyya S, Sudhagar S, Thummer RP (2021) Generation of biologically active recombinant human OCT4 protein from E. coli. 3 Biotech 11(5):1–16. https://doi.org/10.1007/s13205-021-02758-z
Narayan G, Agrawal A, Joshi N, Gogoi R, Nagotu S, Thummer RP (2021) Protein production and purification of a codon-optimized human NGN3 transcription factor from E. coli. Protein J 40(6):891–906. https://doi.org/10.1007/s10930-021-10020-x
Patel TB, Bertics PJ (2006) Epidermal growth factor: methods and protocols, vol 327. Springer Science & Business Media
Dey C, Venkatesan V, Thummer RP (2022) Identification of optimal expression parameters and purification of a codon-optimized human GLIS1 transcription factor from Escherichia coli. Mol Biotechnol 64(1):42–56. https://doi.org/10.1007/s12033-021-00390-z
Grada A, Otero-Vinas M, Prieto-Castrillo F, Obagi Z, Falanga V (2017) Research techniques made simple: analysis of collective cell migration using the wound healing assay. J Investig Dermatology 137(2):e11–e16. https://doi.org/10.1016/j.jid.2016.11.020
Haridhasapavalan KK, Das NJ, Thummer RP (2022) Generation of a transducible version of a bioactive recombinant human TBX5 transcription factor from E. Coli. Curr Res Biotechnol 4:66–77
Jauch R, Ng CKL, Saikatendu KS, Stevens RC, Kolatkar PR (2008) Crystal structure and DNA binding of the homeodomain of the stem cell transcription factor Nanog. J Mol Biol 376(3):758–770. https://doi.org/10.1016/j.jmb.2007.11.091
Hu PF, Guan WJ, Li XC, Ma YH (2012) Construction of recombinant proteins for reprogramming of endangered Luxi cattle fibroblast cells. Mol Biol Rep 39(6):7175–7182. https://doi.org/10.1007/s11033-012-1549-4
Yu M, Lian S, Han H, Yu K, Li G, Lian Z, Li N (2013) Four recombinant pluripotency transcriptional factors containing a protein transduction domain maintained the in vitro pluripotency of chicken embryonic stem cells. Sci China Life Sci 56(1):40–50. https://doi.org/10.1007/s11427-012-4426-4
Zhang H, Ma Y, Gu J, Liao B, Li J, Wong J, Jin Y (2012) Reprogramming of somatic cells via TAT-mediated protein transduction of recombinant factors. Biomaterials 33(20):5047–5055. https://doi.org/10.1016/j.biomaterials.2012.03.061
Yang WC, Patel KG, Lee J, Ghebremariam YT, Wong HE, Cooke JP, Swartz JR (2009) Cell-free production of transducible transcription factors for nuclear reprogramming. Biotechnol Bioeng 104(6):1047–1058. https://doi.org/10.1002/bit.22517
Yang WC, Welsh JP, Lee J, Cooke JP, Swartz JR (2011) Solubility partner IF2 Domain I enables high yield synthesis of transducible transcription factors in Escherichia coli. Protein Expr Purif 80(1):145–151. https://doi.org/10.1016/j.pep.2011.06.017
Ha SC, Pereira JH, Jeong JH, Huh JH, Kim SH (2009) Purification of human transcription factors Nanog and Sox2, each in complex with Skp, an Escherichia coli periplasmic chaperone. Protein Expr Purif 67(2):164–168. https://doi.org/10.1016/j.pep.2009.05.003
Kelly SM, Jess TJ, Price NC (2005) How to study proteins by circular dichroism. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1751(2):119–139. https://doi.org/10.1016/j.bbapap.2005.06.005
Micsonai A, Wien F, Kernya L, Lee Y-H, Goto Y, Réfrégiers M, Kardos J (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Natl Acad Sci 112(24):E3095–E3103. https://doi.org/10.1073/pnas.1500851112
Yu Z, Huang Z, Lung ML (2013) Subcellular fractionation of cultured human cell lines. Bio-Protoc 3(9):e754–e754
Do HJ, Lim HY, Kim JH, Song H, Chung HM, Kim JH (2007) An intact homeobox domain is required for complete nuclear localization of human Nanog. Biochem Biophys Res Commun 353(3):770–775. https://doi.org/10.1016/j.bbrc.2006.12.100
Chang DF, Tsai SC, Wang XC, Xia P, Senadheera D, Lutzko C (2009) Molecular characterization of the human NANOG protein. Stem Cells 27(4):812–821. https://doi.org/10.1634/stemcells.2008-0657
Zhang J, Wang X, Chen B, Suo G, Zhao Y, Duan Z, Dai J (2005) Expression of Nanog gene promotes NIH3T3 cell proliferation. Biochem Biophys Res Commun 338(2):1098–1102. https://doi.org/10.1016/j.bbrc.2005.10.071
Peitz M, Münst B, Thummer RP, Helfen M, Edenhofer F (2014) Cell-permeant recombinant Nanog protein promotes pluripotency by inhibiting endodermal specification. Stem cell Res 12(3):680–689. https://doi.org/10.1016/j.scr.2014.02.006
Gu TT, Liu SY, Zheng PS (2012) Cytoplasmic NANOG-positive stromal cells promote human cervical cancer progression. Am J Pathol 181(2):652–661. https://doi.org/10.1016/j.ajpath.2012.04.008
Ding Y, Yu AQ, Li CL, Fang J, Zeng Y, Li DS (2014) TALEN-mediated Nanog disruption results in less invasiveness, more chemosensitivity and reversal of EMT in Hela cells. Oncotarget 5(18):8393–8401. https://doi.org/10.18632/oncotarget.2298
Ding Y, Yu AQ, Wang XL, Guo XR, Yuan YH, Li DS (2016) Forced expression of Nanog with mRNA synthesized in vitro to evaluate the malignancy of HeLa cells through acquiring cancer stem cell phenotypes. Oncol Rep 35(5):2643–2650. https://doi.org/10.3892/or.2016.4639
Münst B, Thier MC, Winnemöller D, Helfen M, Thummer RP, Edenhofer F (2016) Nanog induces suppression of senescence through downregulation of p27KIP1 expression. J Cell Sci 129(5):912–920. https://doi.org/10.1242/jcs.167932
Oh JH, Do HJ, Yang HM, Moon SY, Cha KY, Chung HM, Kim JH (2005) Identification of a putative transactivation domain in human Nanog. Exp Mol Med 37(3):250–254. https://doi.org/10.1038/emm.2005.33
Jeter CR, Yang T, Wang J, Chao HP, Tang DG (2015) Concise review: NANOG in cancer stem cells and tumor development: an update and outstanding questions. Stem Cells 33(8):2381–2390. https://doi.org/10.1002/stem.2007
Patra SK, Vemulawada C, Soren MM, Sundaray JK, Panda MK, Barman HK (2018) Molecular characterization and expression patterns of Nanog gene validating its involvement in the embryonic development and maintenance of spermatogonial stem cells of farmed carp, Labeo rohita. J Anim Sci Biotechnol 9(1):1–17. https://doi.org/10.1186/s40104-018-0260-2
Stuart HT, Van Oosten AL, Radzisheuskaya A, Martello G, Miller A, Dietmann S et al (2014) NANOG amplifies STAT3 activation and they synergistically induce the naive pluripotent program. Curr Biol 24(3):340–346. https://doi.org/10.1016/j.cub.2013.12.040
Choi KJ, Quan MD, Qi C, Lee JH, Tsoi PS, Zahabiyon M et al (2022) NANOG prion-like assembly mediates DNA bridging to facilitate chromatin reorganization and activation of pluripotency. Nature Cell Biol 24(5):737–747. https://doi.org/10.1038/s41556-022-00896-x
Bothmann H, Plückthun A (1998) Selection for a periplasmic factor improving phage display and functional periplasmic expression. Nat Biotechnol 16(4):376–380. https://doi.org/10.1038/nbt0498-376
Hayashi Y, Caboni L, Das D, Yumoto F, Clayton T, Deller MC et al (2015) Structure-based discovery of NANOG variant with enhanced properties to promote self-renewal and reprogramming of pluripotent stem cells. Proc Natl Acad Sci U S A 112(15):4666–4671. https://doi.org/10.1073/pnas.1502855112
Bosnali M, Edenhofer F (2008) Generation of transducible versions of transcription factors Oct4 and Sox2. Biol Chem 389(7):851–861. https://doi.org/10.1515/BC.2008.106
Stock K, Nolden L, Edenhofer F, Quandel T, Brüstle O (2010) Transcription factor-based modulation of neural stem cell differentiation using direct protein transduction. Cell Mol Life Sci 67(14):2439–2449. https://doi.org/10.1007/s00018-010-0347-1
Thier M, Münst B, Edenhofer F (2011) Exploring refined conditions for reprogramming cells by recombinant Oct4 protein. Int J Dev Biol 54(11–12):1713–1721. https://doi.org/10.1387/ijdb.103193mt
Thier M, Münst B, Mielke S, Edenhofer F (2012) Cellular reprogramming employing recombinant Sox2 protein. Stem Cells Int. https://doi.org/10.1155/2012/549846
Narayan G, Sundaravadivelu PK, Agrawal A, Gogoi R, Nagotu S, Thummer RP (2021) Soluble expression, purification, and secondary structure determination of human PDX1 transcription factor. Protein Expr Purif 180:105807. https://doi.org/10.1016/j.pep.2020.105807
Haridhasapavalan KK, Sundaravadivelu PK, Voorkara U, Kaveeshwar V, Thummer RP (2022) Generation of the recombinant version of a bioactive human MEF2C transcription factor from E. coli. In: Healthcare research and related technologies-proceedings of NERC. Springer Nature
Haridhasapavalan KK, Sundaravadivelu PK, Thummer RP (2020) Codon optimization, cloning, expression, purification, and secondary structure determination of human ETS2 transcription factor. Mol Biotechnol 62(10):485–494. https://doi.org/10.1007/s12033-020-00266-8
Haridhasapavalan KK, Ranjan SH, Bhattacharyya S, Thummer RP (2021) Soluble expression, purification, and secondary structure determination of human MESP1 transcription factor. Appl Microbiol Biotechnol 105(6):2363–2376. https://doi.org/10.1007/s00253-021-11194-1
Haridhasapavalan KK, Sundaravadivelu PK, Joshi N, Das NJ, Mohapatra A, Voorkara U et al (2022) Generation of a recombinant version of a biologically active cell-permeant human HAND2 transcription factor from E. coli. Sci Rep 12(1):16129. https://doi.org/10.1038/s41598-022-19745-w
Acknowledgements
North Eastern Region—Biotechnology Programme Management Cell (BT/PR16655/NER/95/132/2015), DBT, Govt. of India funded this study.
Author information
Authors and Affiliations
Contributions
MT designed and performed the experiments, assembled and analyzed the data, wrote the manuscript; SS analyzed and interpreted the data; and RPT conceptualized and designed the study, analyzed the data, supervised the experiments, wrote the manuscript, and provided financial support. The final draft of the manuscript was approved by all the authors for publication.
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
None.
Conclusion
The purification of HTN-NANOG fusion protein from E. coli with a retained secondary structure and its bioactivity in human cells is demonstrated in this study. This study reported that human NANOG fusion protein enhanced proliferation, migration and downregulated p27 gene expression in HeLa cells, demonstrating its bioactivity. This biologically active human NANOG protein can be utilized to elucidate the biological function of NANOG in various cellular processes associated with cancer and induced pluripotent stem cells.
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Thool, M., Sudhagar, S., Thummer, R.P. (2023). Soluble Expression and Purification of Biologically Active Human NANOG from Escherichia coli. In: Pandey, L.M., Gupta, R., Thummer, R.P., Kar, R.K. (eds) Healthcare Research and Related Technologies. NERC 2022. Springer, Singapore. https://doi.org/10.1007/978-981-99-4056-1_6
Download citation
DOI: https://doi.org/10.1007/978-981-99-4056-1_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-4055-4
Online ISBN: 978-981-99-4056-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)