
Abstract
Emissions of greenhouse gases from industrial activity, traffic, and solid waste landfills contribute directly to the air pollution crisis. To keep the quantities of carbon dioxide in the atmosphere at a safe level, air pollution must be reduced. Many studies have reported techniques for converting the main greenhouse gas, carbon dioxide, into viable fuels as a method of reducing air pollution. The trending topic of photocatalytic conversion of CO2 to fuels has shown high potential. This process is environment-friendly due to its capability to be performed at ambient temperature and pressure and hence utilize less energy. This review focuses on the thermodynamic and reaction kinetics of photocatalytic CO2 reduction. Various types of photocatalysts used for CO2 reduction are discussed excessively by considering metallic, non-metallic, and composite systems. In addition, necessary experimental and product analysis parameters are covered and mentioned in detail.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
A significant amount of carbon dioxide (CO2) has been produced and released into the atmosphere as a result of the development of industries. CO2 levels are expected to rise roughly up to twice their current quantity [1–5]. CO2 exhaust, being a highly disruptive greenhouse gas, can unbalance the atmosphere and the biosphere accordingly. As a result, encouraging CO2 reduction and usage limitations is critical. Because CO2 is a linear molecule with high ionization energy and a low electron affinity, it is easier to reduce than to oxidize [1–5]. To reduce CO2 and enhance the reaction progress, various approaches have been used. Chemical reduction, photochemical reduction, electrochemical reduction, and biological transformations, for example, are investigated. The CO2 photoreduction reaction is the most promising among these approaches.
Photosynthesis combines the creation of O2 and carbohydrates with the fixation of CO2 using solar light energy. This process creates a readily available carbon source as well as an aerobic environment that can support practically all life forms on the planet [2–5]. With an extra input of energy from photosystem I, photosystem II photoinduces water oxidation, which supplies a key supply of reducing equivalents (water-derived electrons and protons) to transform CO2 into biomass, food, and fuel [1].
For more than 30 years, the idea of imitating the overall natural photosynthetic cycle of chemical conversion of CO2 into hydrocarbon fuels has piqued interest. Including biological conversion [1], thermochemical conversion [2], electrochemical conversion [3], and photocatalytic reduction of CO2 [4]. Due to CO2’s thermodynamic stability, a large amount of energy is required to break the C = O bond during the conversion process [5]. Compared to other technologies, photocatalytic CO2 reduction into hydrocarbon fuels is a difficult but promising path. It can be a source of a sustainable alternative to traditional fossil fuels, according to the following benefits: (i) It can be done under relatively mild settings, such as at ambient temperature and pressure; (ii) This process starts with a mass of abandoned CO2 and is powered by unlimited and pure solar energy; (iii) CO2 photoreduction may directly generate short-chain hydrocarbon fuels like CH4, CH3OH, C2H6, and so on, [3–5] alleviating the world's growing energy crisis; (iv) the commercialization of this technique will allow CO2 to replace fossil fuels as a source of carbon in the chemical industry [5].
In this review, we focus on the reduction of CO2 using irradiated photons and spotlighting different drawbacks as well. Section 2 depicts the fundamental mechanisms of photocatalytic CO2 reduction by deeply exploiting the thermodynamics and kinetics of photocatalytic CO2 reduction (PCR). In Sect. 3, the experimental parameters affecting the process are discussed in detail. Photocatalysts types, selection, and preparations are discussed in Sect. 4. Then, product characterization, analysis, and selectivity are explained in Sect. 5. Section 6 states the main challenges facing PCR and the promising opportunities of this topic ahead. Finally, the conclusion in Sect. 7 sums up the main points and declares the critical parameters to overcome the previously mentioned obstacles facing this research work.
2 Fundamentals, Mechanisms, and Kinetics of PCR
2.1 Thermodynamics of PCR
Table 1 illustrated the standard redox potential ΔE0 and the Gibb free energy ΔG0 of the multi-electron water splitting and CO2 reduction, respectively [6]. The positive ΔG0 substantiated that the CO2 reduction process is the endothermic one and it is a high challenge to carry out at room temperature. It also proved that the CO2 reduction reaction can archive much more energy than the water splitting reaction [6, 7].
The multi-electronic processes are more highly captivating than the mono-electronic process because the required energy for electron transfer is smaller. Besides that, the reaction by one electron possesses a bigger reduction potential of −1.9 V versus normal hydrogen electrode (NHE). Therefore, it calls for larger kinetics (overvoltage) [7, 9, 10].
A Latimer-Frost diagram shown in Fig. 1 depicted the multi-proton and multi-electron reduction of CO2 in a water solution at pH 7 [11]. Table 1 also listed a summary of the standard reduction potentials of CO2 for the half-cell reactions (at pH 7 in water solution versus NHE) [7, 9, 10, 12–14]. From Table 2 and Fig. 1, we can observe that the thermodynamic barrier was lowered significantly by a multi-electron and proton-assisted approach to the reduction of CO2 [11]. On the other hand, the conversion from CO2 to CO, then to H2CO, and then to alcohols or hydrocarbons also has a lower kinetic barrier if it is collated into the mono-electron process [11, 15]. Thus, the proton-assisted multi-electron transfer is a promising candidate to reduce CO2.

(Copyright 2014, Springer. License Number 5311300338819)
Latimer–Frost photograph of the multi-proton and multi-electron processes of CO2 in water solution at pH = 7.
Via the multi-electron transfer process, several compounds like CH3OH, HCOOH, and HCHO can be produced from the reduction of H2CO3 and the carbonate ions in the solution. The potentials of both H2CO3 and CO32− mentioned in Table 2 are conclusive that the pathway of methanol formation from H2CO3 or CO32− is more thermodynamically favorable than that from CO2 [12, 13].
The thermodynamics of the CO2 chemical change hindering increases due to the low energy class and high stability of CO2. Consequently, the targeted conversions need much more energy. Furthermore, due to its inertness, the utilization of catalysts is a considerable requirement [10, 15]. Because the reduction of CO2 is sophisticated and the transformation of CO2 into hydrocarbon fuels utilizing assisted catalyst has been expansively reported with different aspects. Some of these approaches are thermochemical, electrochemical, photo-electrochemical (PEC), and photochemical reductions [15, 16]. The lack of releasing more greenhouse effect gas, eco-friendly and economical reduction of CO2 into value chemicals will become a trending topic if the renewable energies are used widely. Meanwhile, the new routes of the artificial photosynthetic system (APS) e.g. PEC or photochemical reduction of CO2 into solar fuel, are the urgent aims in the reduction of CO2 [17]. For instance, Halmann was a pioneer to explore the p-type of GaP which was able to transform CO2 to CH3OH via the photo-electrochemical process in 1978 []. Then, Inoue and coworkers reported several chemical products such as HCHO, CH3OH, and HCOOH by PCR utilizing TiO2, ZnO, and CdS, GaP, and SiC aqueous sedimentations [14]. Thanks to APS systems, humans can use solar light or CO2 as chemical energy. In the past decade, photoreduction of CO2 was intensively studied because of the urgency to find out solutions for environmental pollution problems and their high applicability.
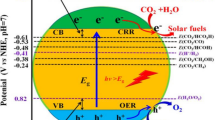
According to the semiconductor materials the production of the photogenerated (PG) charge carriers (electrons and holes), as shown in Fig. 2, are created by the absorption of photons with energy larger than or equal to its bandgap (Eg). The bandgap (energy difference between the valence band (VB) and the conduction band (CB)) plays a crucial key in forming photocatalytic behaviors of the semiconductors [18, 19]. On the surface of the semiconductors, the PG holes diffused from the VB react with water to induce either O2 or hydroxyl radicals (•OH). Then, •OH radicals oxidize the surrounding organic contaminants on the semiconductors’ surface [18, 19]. On the other hand, the electrons of the CB take part in the reduction process, which reacts either with water to yield H2 or with CO2 to fabricate hydrocarbon fuels [18–20]. To attain CO2 photoreduction, there are many requirements for an elite photo-catalyst. The PCR have to be positioned more positively than the lowermost CB of the photocatalyst. The oxidation potentials of H2O to O2 ought to be located at a more negative potential than the uppermost VB. The redox reaction can be carried out under irradiation at a specific energy, which is equal to or greater than the Eg of the photocatalysts [21]. In 1979, the first published article in which the CB of the semiconductor approaches significantly more negative potential compared with the redox potential to perform a certain reaction of CO2 reduction. Hence, the efficiency of obtaining products from CO2 reduction escalated [14].

(Copyright 2014, Springer. License Number 5311300338819)
Bandgap of some semiconductor photocatalysts and the redox potential of PCR at pH 7 in water solution.
Many promising semiconductors shown in Fig. 3 such as TiO2 [22, 23], ZnO [14, 24–27], ZnS [17, 28–31], SrTiO3 [32–35], SiC [14, 36–38], Cu2O [39–42], CdS [14, 43–48], GaP [14, 49], TaON [50–53], C3N4 [32, 54–56], BiVO4 [57–61] and Ta3N5 [62–65] are appropriate for PCR. Among them, TiO2 is a highly highlighted material, which was studied severally. TiO2 attracts attention due to its many excellent properties such as being non-toxic and cheap, made up of abundant elements, and resistant to photo-corrosion. Even possessing a lot of good behaviors, TiO2 is poor visible light absorption because of a wide bandgap, so it ought to be enhanced. Conversely, Cu2O, CdS, GaP, TaON, C3N4, and Ta3N5 are promising materials for PCR under visible light irradiation. But, they are quite sensitive because of the weak photostability, so they also should be improved. From Fig. 3, on the right side, many photocatalysts are possessing more negative CB levels suitable for the PCR.

Steps containing in PCR on a heterogeneous photo-catalyst
2.1.1 Processes of PCR
Not only do the appropriate Eg and CB potential play a salient role, but variously several other factors also significantly affect the yield of the PCR, for example, the photocatalytic process and CO2 reduction kinetics. According to a typical route of PCR over a semiconductor photo-catalyst, 4 main steps are composing the excitation, the transport, the segregation, and the electro-catalytic reduction of CO2 and the water oxidation. In detail, the initial process (1) in Fig. 3, which indicates the formation of electron–hole pairs as the semiconductor absorbs the photon energy larger than or equal to the bandgap of the material, is the excitation. Hence, the materials with a narrow bandgap (Eg < 3.0 eV or λ > 415 nm) are high potential candidates for rising the excitation yields of electron–hole pairs by visible light. From these fundamentals, many new novels to synthesize the innovative photocatalysts utilizing the visible light region were intensively studied.
The second process (2) in Fig. 3 describes the segregation of the PG charge carriers and their relocation on the surface for specific chemical reactions. In the third process (3) in Fig. 3, the number of excited charge carriers is decreased dramatically because the holes and electrons recombine and form heat, as a consequence, the efficiency is decreased. This process can call by the deactivation one. To avoid these unwanted phenomena and improve the yield, the transfer of the PG charge carriers to the surface necessitates enhancing and prohibiting the recombination in the bulk plays a vital key. The segregation and the recombination process are dominated by structural and electronic behaviors or photocatalysts. Hence, several beneficial strategies are studied intensively to maximize the segregation and minimize the recombination such as the fabrication of semiconductor/nano-carbon heterojunctions or nanostructured semiconductors or semiconductor heterojunctions.
After the PG electrons come to the surface, the fourth process (4) in Fig. 3 or the electro-catalytic reduction of CO2, which is frequently a multi-electron and multi-step process related to a cascade of reactions. Electron and photon transfer, C–O bond breaking, C–H/C–C bone creation, and numerous chemical compounds, will be taken place by the PG electrons trapped in the CO2 reduction co-catalysts (CRC) or the surface active sites [66–71]. Typically, to result in a high yield, the surface of semiconductors ought to be fulfilled by the CRC. While the photocatalytic reduction of CO2 occurs, does the formation of some kinds of stable products be to need a minimum of two electrons due to some intermediates being easily changed or hard to find out and enumerate. Due to the complicated multi-step mechanism, any approach which enhances CO2 reduction kinetics ought to be a probable route to improve the yield. The synthesis of mesoporous photocatalysts loading CRC is a good example for such a purpose [9].
Simultaneously with the fourth process (4), the electro-catalytic oxidation of water via PG holes caught in the water oxidation co-catalysts (or the surface active sites) will happen once the PG holes approach the surface and are referred to as the fifth process (5). Enhancing water oxidation can encourage the segregation of PG charge carriers on the surface of semiconductors, hence inducing developed activity of CO2 photoreduction. Additionally, the sixth step (6) in Fig. 3 shows the surface charge recombination process of the holes and electrons. The surface charge recombination and the efficiency of the photocatalytic CO2 reduction process are inversely proportional. To reduce recombination, surface trapping ought to be enhanced by increasing the surface properties of the photocatalysts [9].
Besides that, the seventh process (7) and the eighth process (8) in Fig. 3 show the electro-catalytic H2 evolution and reduction products of the electro-catalytic oxidation, respectively. In the seventh process (7), the consumption rate of PG electrons for CO2 reduction will be decreased steadily by the H2 evolution effect. In the eighth process (8), the oxidation of the CO2 reduction products by the PG holes is detriment for both water oxidation and CO2 reduction. The quantum yield will be unwantedly affected by these aforementioned two processes. Hence, the efficient approaches to enhance the PCR yield need to comprehensively consider such unfavorable factors [9].
2.2 Possible Mechanisms of PCR
In photocatalytic CO2 reduction, there are many reduction products formed with the joining of several protons and electrons, thus, the photocatalysts need to meet extraordinarily specific requirements. Table 3 depicts several related reactions containing variable numbers of protons and electrons and their recorded reduction potentials [14, 18].
Furthermore, the adsorption of CO2 on the photocatalysts as well as various reaction pathways also will affect the final products significantly. Nevertheless, existing of some ambiguous intermediate species has resulted in some challenges in the reaction mechanism study. Figure 4a and b show two possible CO2 reduction routes through formaldehyde and carbene pathways [73].

(Copyright 2020, Royal Society Chemistry License Number 1222250–1)
Schematic demonstration for the a formaldehyde pathway and b carbene pathway for CO2 reduction. (Reproduced with permission [73].
In detail, the formaldehyde pathway is the fast hydrogenation pathway related to the simultaneous hydrogenation and deoxygenation reactions, namely displayed by the route: CO2− → HCOOH → HCHO → CH3OH → CH4. Whereas, HCOOH is generated by the combination of CO2−, protons, and electrons. Then, they are transformed into HCHO, dihydroxymethyl, two protons, and CH4 in the final step. This pathway is evidenced to be thermodynamically feasible [73]. On the other hand, the CO2 reduction reaction, which is the fast deoxygenation, can also occur following the carbene pathway (CO2− → CO → C → CH3 → CH3OH/CH4). Namely, there is the deoxygenation reaction in the initial step and then the hydrogenation reaction in the second one [8, 26].
In the carbene pathway, the CO intermediate usually is transformed easily into the final products because of the weak affinity of the CO products and the surface of the photocatalysts. The CO intermediate can interact with the protons or electrons to generate the CH3OH or CH4, the surface of the photo-catalyst possesses a CO strong adsorption capacity. According to the two possible mechanisms, C–O is attacked by protons in both routes. On the other hand, there are some highlighted feature characterizations belonging to each mechanism. For example, in the carbene pathway, the carbon or mixed coordination mode is probably to break the C–O linkage. Likewise, in the formaldehyde pathway, the CO2− will adapt to oxygen coordination mode [24, 27].
2.3 Kinetics of PCR
Based on the various mechanisms, many types of kinetic equations were generated. For modeling the photocatalytic reduction of carbonate by the TiO2 under UV light (carried out in an aqueous solution), a Langmuir–Hinshelwood (L–H) equation was expressed. The results show that the photocatalytic reduction rate of carbonate is adsorption-controlled [74]. Whereas, a one-site L–H equation was used further to simulate the reduction rate of carbon dioxide to carbon monoxide (or hydrogen) and methane utilizing water over TiO2 [74–77]. Additionally, the mechanism of Anpo, in which carbon monoxide is defined as the primary intermediate, was strongly supported by this model [74, 75, 77].
In conclusion, because of the complicated processes of PCR and their kinetics, the yield of PCR on the photo-catalyst surface is so low. The reduction potentials of carbon dioxide conversion to hydrogen are less feasible than to methane or/and methanol in order of thermodynamic aspects. Nevertheless, because of the multi-electron reduction process leading to the kinetics of CO2 reduction, it is unfavorable. Thus, the enhancement reduction kinetics of CO2 makes a significant contribution to improving the efficiency. Whereas there are many complicated processes in PCR. Water oxidation, CO2 activation, and CO2 reduction kinetics processes also contribute important roles in the overall efficiency. Hence, to improve the overall efficiency, the modification of the kinetics and photocatalytic processes of CO2 reduction is critical. These can be achieved by the synthesis of high-efficient photocatalysts with significant surface areas [21, 78, 79]. Figure 5 demonstrates all of the critical factors which can influence the overall PCR efficiency.

Keys influencing photocatalytic yield and corresponding design strategies
3 Experimental Parameters Consideration
Photocatalytic conversion of CO2 into value‐added hydrocarbon fuels or useful chemical products has been the focus of active research. The photocatalytic conversion rate of CO2 to methanol depends on the photocatalyst used, the photoreactor design, and experimental parameters. Here, we introduce several considerations for photocatalytic CO2 reduction experiments. Preparation of catalysts, light source, and type of photoreactors as the PCR experiments preparation. Experimental parameters include reaction temperature, CO2 pressure, and presence of impurities adsorbed on the photocatalyst.
3.1 Preparation of Catalysts
There are numerous articles have been reported, including pure, doped, metal–organic framework based and composite photocatalysts, synthesized by using various methods, and used for the photocatalytic conversion of CO2 into fuels [80–87]. They are discussed excessively in section four.
3.2 Source of Light
Among the important criteria for photocatalytic CO2 reduction are light intensity and irradiation nature [88]. The number of photons falling on a unit area in a unit of time determines the intensity of an illumination source [89]. According to the literature, most photocatalysts work better in the UV region, hence they can only capture a small portion of the solar spectrum irradiation [90]. To get over this limitation, semiconductors are being modified to harvest a wider range of sunlight spectrum.
Aside from that, light can be focused and diverted to increase irradiation photon flow [91]. By focusing light over TiO2 and Pt/TiO2, Han et al. were able to reduce CO2. The authors tested CO2 photoreduction with various concentrating ratios (CRs). CRs are defined as the ratio of concentrated light flux (amount of energy per unit time per unit area) on the photocatalyst surface to ambient flux (under non-concentrated conditions).
By varying the distance between the Fresnel lens (placed between the light source and the photocatalyst) and the photocatalyst surface, the light irradiation is concentrated and modulated, resulting in variable light intensities with different light concentrated focal regions. The optimum concentration ratio (CR) increased dramatically, according to their findings [91]. Based on these findings, better light interaction with the photocatalyst under ideally concentrated light will result in a significant improvement in the yield [92]. Employing a greater intensity radiation source, the geometrical design of the photoreactor, the lamp to photoreactor distance, and the use of fiber optics are all appropriate techniques for maximizing the radiation intensity on the reaction medium [89].
3.3 Geometry and Design of Photoreactors
Photo-reactor geometry and design also encourage maximum photon flux distribution, allowing for a large active surface area with a high mass transfer rate and minimal light diffusion effects. However, in most reactor geometries when light impinges on the photocatalyst's surface from the center or side, a shadow is created on the other side, preventing a significant amount of the photocatalyst from being activated. Fabricating suitable reactor designs to achieve a consistent distribution of light and greater photocatalyst dispersion could improve light-photocatalyst contact [93]. A variety of ways have been documented in the literature to achieve this, including the use of various reactor geometries and catalyst supports [93–95].
Monoliths have been used as a photocatalyst support material in innovative photoreactors, attempting to overcome mass transfer restrictions and limited light distribution efficiency seen in immobilized photocatalysts. Monoliths have consistent structures, supported with parallel channels that come in a variety of shapes and sizes depending on how they are extruded. These materials have a large surface area per unit volume and other appealing characteristics such as minimal pressure drop, high mechanical strength, and thermal stability, making them superior to traditional catalyst arrangements (powders and pellets). By comparing the performance of TiO2 coated micro channel monolith and cell type support, Tahir et al. investigated the effect of photocatalyst dispersion (dispersed as a single layer over stainless steel cell). The TiO2-coated monolith showed a considerable increase in CO output in their research. This increase was mostly due to a larger exposed photocatalyst surface that was available for photocatalytic activity. This improvement was attributed to the monolith's increased illuminated surface allowing for more efficient photon usage [97].
Fiber optic reactors have an advantage over packed bed reactors in terms of photocatalyst dispersion and light spreading across a vast surface area. Nguyen et al. investigated the yield of photocatalytic CO2 reduction using photocatalyst coated on optical fiber against photocatalyst coated on a glass plate. For the same amount of photocatalysts, their research showed 15.2 times CH4 yield and increased C2H4 yield 11.6 times more. The synergistic effects of catalyst dispersion and effective light utilization could explain this [97].
Wang et al. used a fiber optic reactor to perform CO2 photoreduction and ascribed the increased output to the progressive and uniform distribution of light during irradiation. Optical fibers have the advantages of catalyst support and effective light distribution, but they also have the disadvantages of limited reactor capacity utilization and shorter light transit distance from the point of incidence. They occupy 20–30% of the reactor capacity, but the effective use of incident light is limited because of the limited catalyst-coated area [99]. Ola et al. fabricated an internally illuminated monolith reactor by combining the mutual effects of greater monolith surface area and effective light distribution of fiber optics and compared the CO2 reduction performance of this system to that of a slurry reactor. Due to the higher surface area of the monolith and the equal dispersion of light by optical fibers, it was discovered that internal illumination of the monolith reactor by optical fibers increased quantum efficiency by 23 times [97]. Optical fibers constructed of carved polymethylmethacrylate were placed into a NiO/InTaO4 coated monolith (honeycomb structure) by Liou et al. When used for photocatalytic CO2 reduction, this reactor increased product yield (methanol and acetaldehyde). Increased surface area, better photocatalyst loading, and effective light use are all factors that contribute to a higher yield [99].
3.4 Effect of Temperature Variation
Due to long-wavelength irradiation, concentrating solar light raises the temperature depending on the CR [12, 28, 29]. Photocatalytic CO2 reduction at high temperatures is promising because it bypasses the thermal barriers that cause slow reaction rates and low yields [96]. The effectiveness of temperature rise in photoreaction can be demonstrated by increased effective collisions between photogenerated charges and reactants, which are directly proportional to the reaction rate [97]. Furthermore, increasing temperature increases the desorption of products, allowing CO2 to adsorb on unoccupied sites, resulting in a faster reaction rate [98].
The reaction temperature influences photocatalytic CO2 to methanol conversion. Thus, determining the ideal temperature is difficult since temperature affects the methanol production rate in four different ways, as shown in Table 4. First, the amounts of CO2 that can dissolve in water is affected by temperature, therefore decreasing the temperature increases CO2 solubility in water. The increasing samount of dissolved CO2 in water can speed up the synthesis of methanol. When water is chilled from 25 to 0 °C, the solubility of CO2 in water increases by around 2.5 times, resulting in a 2.5-fold increase in dissolved CO2 [89, 97, 98]. Second, the temperature affects the ease with which reactants adhere to the surface of the catalyst.
Because there is less thermal agitation at low temperatures, reactants soak more quickly onto the catalytic surface. The photocatalytic reaction rate naturally increases with increasing the amounts of reactants adsorbed on the catalyst surface. Third, temperature influences the rate of product desorption, affecting catalyst poisoning and, as a result, catalyst availability. Catalyst poisoning occurs when reactive intermediates and products are more likely to remain adsorbed on the catalyst surface at lower temperatures, preventing further catalytic activity. As a result of the scarcity of vacant catalytic adsorption sites on the surface, fresh reactants are unable to adsorb on the surface, slowing the photocatalytic CO2 reduction [89, 97, 98]. Fourth, the decreased temperature has an undesirable influence on the photocatalytic CO2 reduction process because it reduces the diffusion rates and collision frequencies of the reactants, resulting in a lower reaction rate constant for methanol generation. These four temperature impacts of the photocatalytic reaction show that the negative effects of high and low temperatures can be avoided without sacrificing the favorable effects [89]. When the temperature was raised from 25 to 75 ℃, Wang et al. discovered that the production rate nearly doubled [99]. However, the reaction temperature should not rise too high, as this may cause the CO2 to desorb, slowing down the photoreduction process.
3.5 Flowing CO2 Gas Pressure
The CO2 gas pressure in the reaction chamber, like reaction temperature, plays an essential role in controlling the rate of CO2 reduction in product production. Increased CO2 solubility in water with pressure causes an increase in reaction rate and, as a result, an increase in reaction product generation rate. Because cooling down the solvent can lower the product desorption rate, clog the catalyst surface, and slow down the reduction process, boosting CO2 solubility by raising pressure is preferred over reducing the temperature for the goal of enhancing photocatalytic CO2 reduction. Aside from enhanced reaction products, increasing CO2 concentration caused by increased CO2 pressure in aqueous media has also been shown to improve product selectivity. The increase in methanol formation rate, on the other hand, does not increase endlessly with pressure. It increases to a maximum value at optimum pressure and then begins to drop as pressure is increased further. Mizuno et al. investigated the effect of CO2 gas pressure on the rate of reaction product formation and found that methanol was produced at ambient pressure [100]. However, as CO2 pressure increased, the rate of methanol formation increased sharply, peaking at 1.0 MPa, and then declining significantly with further CO2 pressure increases. When choosing CO2 reduction reactor designs [101], the expense of producing advanced high-pressure systems must be taken into consideration.
3.6 Effect of Contaminant
Organic contaminants in the catalyst or on the catalyst surface might cause a falsely positive result since they are often reduced more efficiently than CO2 to create diverse reduction products. This is especially true when the concentration of products obtained from photocatalytic CO2 reduction is low, as it has been in almost all photocatalysis experiments.
Small particle size and wide surface area of the TiO2 photocatalyst (and this also applies to other photocatalysts) are crucial elements in adsorbing airborne organic pollutants, which are more reactive than even CO2, according to Neatu et al. [102]. As a result, they are more easily reduced than CO2 and become the source of photoproducts, which are sometimes mistaken for CO2 reduction reaction products [103]. When the photocatalyst amount is large but the produced yield is low this can be attributed to the adsorbed contaminants. This condition can be avoided by calcining the photocatalyst before the photocatalytic experiment to remove any carbon-containing impurities that may be present on its surface. Because CO2 is more stable than other carbon-containing organic contaminants, organic impurities linked to photocatalyst surfaces degrade more quickly, resulting in an overestimation of photocatalytic activity and yield. In summary, photocatalyst contamination can lead to false results, hence rigorous surface cleaning is required to obtain accurate and precise results.
4 Photocatalyst Types, Selection, and Preparation
4.1 Metal System
CO2 photocatalysts are divided into five types: metal, mixed oxides, metal sulfides, polymeric materials, metal–organic framework, and others. Metal and mixed oxides, metal sulfides, and the metal–organic framework are included in the metallic system, which reduces CO2 into several forms, such as carbon monoxide, methane, methanol, and formic acid for fuel. Some examples included in the metal and mixed oxides are titanium oxide, iron oxide, tantalum oxide, copper oxide, niobium oxide, and strontium oxide. Furthermore, some examples of metal sulfides included are cadmium sulfide, zinc sulfide, MoS2, SnS2, Bi2S3, In2S3, and Znln2S4. Meanwhile, MIL 101, PMOF-AI, Co-ZIF9, and MOF 525-Co are examples of metals included in the metal–organic framework (MOF) group [104]. Activity and selectivity are essential properties in the selection of photocatalysts. To obtain a photocatalyst with high activity and selectivity, several things must be considered, including band structure, surface state, and photoreaction conditions. Increasing the activity and selectivity of photocatalysts can be done by optimizing carbon dioxide absorption, optimizing light harvesting, charge separation effectiveness, and synergistic effects [104].
4.1.1 Metal and Mixed Oxides
As previously described, TiO2 is the most widely used CO2 photocatalyst of metal oxides and mixed oxides. This is due to its non-toxicity, stability, slightly corrosive nature, and low cost. This metal is widely used to convert CO2 to methane and carbon monoxide. However, TiO2 is known to have a large bandgap (3.2 eV); due to this large bandgap, TiO2 only shows photocatalytic activity under UV radiation. In contrast, sunlight as a source of radiation is still not qualified. Therefore, the development of this photocatalyst to have a narrower bandgap is still being carried out [104]. Several strategies to increase the ability of TiO2 are doping using Ag and Cu or using co-doping [104, 105]. This doping can reduce bandgap energy, improve interfacial charge transfer, trap electrons, and allow the use of visible light [104, 105]. Another strategy is to use heterostructured crystal growth. The next modification is to insert the defect chemistry into the forbidden gap on the catalyst surface by thermal treatment [104, 105]. Modification through the formation of nanomaterials on TiO2 also shows advantages in the catalytic behavior. Modifying the nanostructure can increase the diffusion rate and surface area to increase the catalytic activity [105]. Furthermore, to increase the efficiency of the excitation process, modifications using a dye sensitizer can be made by increasing the wavelength so that the catalytic activity increases [104, 105]. There are several methods for TiO2 synthetizing including sol–gel, hydrothermal, impregnation, one-pot, and co-precipitation. Sol–gel process, hydrothermal synthesis, and one-pot synthesis were implemented using TiO2 and multi-walled carbon nanotube composites to improve the photocatalytic performance [104, 105]. A co-precipitation method can narrow the bandgap to absorb large amounts of energy from visible light. Synthesis using the impregnation method was performed by increasing irradiation and doping with metal oxides such as CuO, CoO, and Fe2O3 [105] (Fig. 6).

TiO2 synthesis methods [105]
Silver, rhodium, gold, palladium, and platinum are metals that are often used as co-catalysts with TiO2 to increase TiO2 occupation. The rate of methane formation is reported to be increased due to the use of noble metal co-catalysts. One of the noble metals, Palladium, acts as a co-catalyst on the TiO2 surface and provides active sites for CO2 adsorption and activity [106]. In addition, the size of metal nanoparticles is a critical factor to determine the activity and the rate of methane formation [106].
Copper is a transition metal that is often used as a TiO2 co-catalyst. This is because they are abundant, non-toxic, low in cost, and potential alternatives to the use of noble metals. The use of this co-catalyst showed an increase in the formation of methane. In addition, the use of Cu doping on TiO2 under UV-rich illumination can convert CO2 into formic acid [106]. The use of Cu-TiO2 was also reported to increase the efficiency of light harvesting under UV conditions to produce carbon monoxide and methane from CO2. Copper has also recently been reported to depress hydrogen development in CO2 photoreduction, thereby selectively producing CO [106].
Subsequently, the use of binary co-catalysts (Cu2O-Pt/TiO2 and MgO-Pt/ TiO2 systems) was investigated. Platinum in TiO2 is intended to promote electron capture and inhibit charge pair recombination. However, the use of Platinum increased H2, so it takes Cu2O or MgO to suppress the formation of H2. As a result, CH4 will be obtained with high selectivity. Recently, multi-heterojunctions were fabricated on TiO2-MnOx-Pt films. This multi-heterojunction can efficiently separate charged pairs to produce three times higher CH4 and methanol than pure TiO2 [106].
Au shows the effect of surface plasmon resonance on TiO2. Synergistic mixing of the plasmonic effects of Au and Pt nanoparticles as electron absorbers with TiO2 nanofibers was reported to increase visible light harvesting and inhibit the recombination of photoexcited TiO2. The reduction of CO2 under UV light and visible light for the Au-Cu alloy on TiO2 as a photocatalyst showed excellent performance. The electron selectivity for CH4 evolution can reach 97% under visible light irradiation. This high light harvesting ability comes from the plasmonic effect of Au [106].
Cu is one of the most commonly used metals for CO2 reduction photocatalyst. This is because Cu has low bandgap energy and a high conductive band. CuO and Cu2O are reported to have small band gaps of 1.7 and 2.2 eV. CuO nanomaterials are known to absorb visible light effectively and produce photogenerated electrons and holes. Cu-based photocatalysts have three pathways for CO2 reduction. There is the formaldehyde, carbinol, and glyoxal pathway. The Formaldehyde pathway can produce formic acid, methane, and methanol from Cu-based photocatalysts for CO2 reduction. The carbene pathway is usually used to produce methane and methanol. While the glyoxal pathway is usually used to produce formic acid [107].
The reduction activity and selectivity of CO2 can be affected by the morphology, particle size, and dispersibility of Cu when different methods are used. CuO which has a high density with poor dispersion will affect light absorption due to masking between particles. To address this problem, it is possible to utilize CuO thin film to enhance the catalytic effect. In addition, the use of glass fiber mesh coating CuO can also be done to increase the production of CH4. The photocatalytic activity and selectivity of CuO products can also be increased through size modification or create quantum dots of CuO. Modification of the size of CuO to be smaller will increase light harvesting and charge transfer separation due to the risen uniform distribution [106, 107]. The modification with CuO quantum dots plays a significant role in the CO2 adsorption and activation [107]. There are several methods to synthesize CuO. Among them, the solvothermal method and impregnation are quite famous [107].
The use of pure Cu is also carried out to minimize semiconductor energy bandgap. This reduced band gap energy can make full use of visible light to improve light utilization and encourage the practical application of photocatalysis. Cu can be prepared by microwave hydrothermal method and secondary calcination. Cu2O was studied as a photocatalyst to reduce CO2. This metal oxide is known to have high photocatalytic activity. Doping using Cu2O is known to improve its energy band structure and increase photocatalytic activity. Cu2O can be prepared by hydrothermal deposition-reduction method and microwave-assisted in situ reduction chemistry [107] (Fig. 7).

Cu synthesis methods
Perovskite is a metal oxide that is also used as a photocatalyst for CO2 reduction [108]. Perovskite is used as a photocatalyst because it provides a broad spectrum for CO2 conversion. In addition, perovskite also has good stability, flexible composition, efficient catalytic activity, long charge diffusion, low cost, and easy preparation [108]. Perovskite converts CO2 through light harvesting, which then separates electrons and transfers them from VB to CB. Then charge photogeneration is carried out before the redox reaction occurs on the catalyst surface until finally the product is formed [108]. There are several classifications of perovskite oxides, including ABO3, halide, layered, ruddlesden-popper phase, aurivillius phase, (110) layered, (111) layered, Dion-Jacobson phase, and oxynitrides [108].
ABO3 perovskite consists of alkali metal cations at site A and transition metal cations at site B. These transition metal cations regulate the perovskite catalytic activity and electron mobility. One way to narrow the perovskite band gap can be done by forming a solid solution. This is because the solid solution has a lower CB with a higher VB. In addition, the formation of this solid solution can also reduce the CO2 induced due to some perovskites having a band gap not suitable for CO2 reduction under visible light. This solid solution formation also offers advantages such as band gap control, charge transfer, and chemical stability [108].
Perovskite halides (ABX3) are another type of perovskite group where the cation at site A is usually Cs+ or Rb+, while the cation at site B is Pb, Ge, or Sn and the halide is located at site X. Perovskite halides are usually used to produce CH4 and CO at the surface of the catalyst. There is also layered perovskite which has flexibility in its structure, effectiveness in charge transport, and attractive optoelectronic characteristics [108].
The Aurivillius phase is a perovskite with cations such as Na, K, Ca, Sr, Ba, and Bi located at site A while, cations such as Fe, Cr, Ti, Ga, Nb, V, Mo, and W are located at site B. On the other hand, the Dion-Jacobson phase (An1BnO3n+1) is a perovskite with cations such as Rb, K, Ag, and Cs at site A while Pr, Sm, Nd, and La at site B. This perovskite has a band gap of about 3.8–4.3 eV which allows it to work under UV light. Ruddlesden-Popper phase (A2An−1BnO3n+1) is a perovskite consisting of alkali metals at site A and transition metals at site B. Finally, perovskite oxynitrides (ABO2−xN1+x) is a perovskite consisting of alkali metals at site A with transition metal at site B [108] (Fig. 8).

(Copyright Elsevier License Number 5326340974814)
Perovskite classification [108].
Besides being used as the main catalyst, several metal oxides can also be used as co-catalysts including Cu2O, CuO, NiO, MgO, CO3O4, and Fe2O3. The use of copper oxide as a co-catalyst can intensify CO2 adsorption on the surface and suppress charge recombination. Cu is also able to increase the electron density at the active site when it is excited and facilitates CO2RR which requires multi-electron transfer. Photocatalyst activity can be increased through the formation of oxygen vacancies at the active sites of Cu2O. Not only that, but this formation is also able to reduce the rate of charge recombination. CuO is usually used as a co-catalyst together with SiC, TiO2, K2Ti6O13, and NaTaO3 as the main catalyst. Magnesium oxide itself is usually used as a co-catalyst to increase the activation and adsorption of CO2 because of its ability to interact with CO2 strongly. This metal oxide is usually used with TiO2 to catalyze the reduction of CO2. Meanwhile, nickel oxide is usually used as a co-catalyst to inhibit the reverse reaction and increase charge carrier separation. This co-catalyst can increase the production of methanol, methane, and carbon monoxide. Nickel oxide is often used as a co-catalyst along with InTaO4, InNbO4, KTaO3, K2Ti6O13, and InVO4 as the main catalyst. Last but not least, is the use of cobalt oxide as a co-catalyst for the promotion of the oxygen evolution reaction and suppressing corrosiveness due to the buildup of photogenerated holes [109].
4.1.2 Metal Sulfides
On the other hand, metal sulfides such as ZnS and CdS have also been reported. Similar to TiO2, ZnS only absorbs visible light due to its large bandgap (3.66 eV). In contrast to CdS which has a smaller bandgap (2.4 eV), this metal is preferred for CO2 reduction. The catalytic performance of metal sulfide can be improved by combining metal sulfide photocatalysts with other photocatalysts. Incorporating metal sulfide photocatalysts with different photocatalysts will provide more advantages in bandgap regulation. The use of metal sulfides as photocatalysts is known to have great research potential. Unfortunately, metal sulfides have less stability during photocatalytic processes leading to structural damage [104].
Molybdenum disulfide (MoS2) is a layered binary sulfide widely applied in photocatalytic due to its excellent optical/electrical properties and flexible electronic band structure. The layer on MoS2 has a high d electron density, so it has potential in gas–solid CO2 photoreduction systems for methanol production. Bismuth sulfide (Bi2S3) is known to have a narrow bandgap (≈1.3 eV), so it is also getting attention in its role as a photocatalyst. In addition, this photocatalyst is also known to have low toxicity, high biocompatibility, good performance in reducing CO2, and an absorption coefficient in the visible light region (>105 cm−1). Like Bi2S3, In2S3 is also widely used for CO2 reduction. This metal has an expansive light response range due to its narrow bandgap (2.0–2.3 eV), and low toxicity [72] (Fig. 9).

Metal-sulfide band gap
One method that can be used to synthesize ZnS is heat-free synthesis with Cu+ and Cd2+ co-catalysts doping to maximize light utilization [72]. There is also the synthesis of ZnS with zinc hydroxide and thiourea using the surface of Ru nanoparticles to form formic acid, which is known to provide high product selectivity [72]. Metal sulfide type MoS2 can be synthesized by hydrothermal method and Chemical vapor deposition (CVD) synthesis. Likewise, with the synthesis of In2S3 through the hydrothermal cationic exchange method [72]. SnS2/SnO2 type metal sulfide photocatalyst can be synthesized by hydrothermal method and sequential template to maximize CO formation [72]. The ion-exchange method between Bi and Cd2S in ethylene glycol solution can be carried out to synthesize Bi2S3/CdS/FeTCPP photocatalysts [72] (Fig. 10).

Metal-sulfide synthesis
Besides being used as the main catalyst, metal sulfide can also be used as a co-catalyst because it can increase charge separation and add more active sites. The co-catalysts commonly used in this group are MoS2 and NiS2 [9, 109]. MoS2 is usually used together with Bi2WO6 under visible light to produce both ethanol and methanol. One method that can be used for the preparation of this co-catalyst is impregnation-calcination method. In addition, MoS2 is also used with TiO2 to produce methanol using the in situ grown method [109]. On the other hand, NiS2 is used as a co-catalyst with graphite carbon nitride (g-C3N4) to produce CO due to its ability to accelerate photogenerated electron–hole pair separation. In addition, NiS2 can also be used with ZnO to produce CO and CH4. However, the use of metal sulfide as a co-catalyst is also reported to have drawbacks due to its poor stability [109].
4.1.3 Metal–Organic Framework
Another metal system that is widely used is MOF. MOF can be used as co-catalysts as well as stand-alone photocatalysts. The MOF structure is potentially a photocatalyst because the network is porous, regular, and heterogeneous so that ions and CO2 can move freely into the matrix. MOF is also known to increase the reactivity of other catalysts. However, as with metal sulfides, MOFs have poor stability and problems in economic viability on an industrial scale. The use of MOFs as photocatalysts can still be improved by modifying the structure and adjusting the reactive functional groups. The metal-added MOF photocatalyst has a bandgap from 1.52–2.4 eV. Meanwhile, if MOF is used as a composite, the bandgap variation is 1.6–3.1 eV [104].
MOF can be synthesized through solvothermal, slow evaporation, microwave assistance, electrochemistry, sonochemistry, and mechanochemistry. These five methods generally combine three main components: metal salts, ligands, and solvents. The solvothermal method is the most frequently used method for MOF synthesis by involving high boiling point heating between organic linking ligands and metal salts in a solvent. Slow evaporation synthesis is preferred because it does not require external energy and uses only room temperature, even though it takes a long time. Microwave-assisted synthesis involves heating a solution with microwaves to produce nano-sized crystals. The electrochemical method is carried out by adjusting the pH of the solvent at room temperature without metal salts and consisting only of a mixture of organic linkages and electrolytes to provide metal ions. Synthesis using the sonochemical method is based on molecular changes due to ultrasonic wave radiation to produce fine crystalline materials. Finally, the mechanochemical synthesis method is based on applying a mechanical force without a solvent to form a porous MOF [110].
Metal systems have several drawbacks, including some metals toxicity, being not environmentally friendly, and have low selectivity. However, metal can still be used, such as doping carbon material as a photocatalyst. In addition, the use of bimetallic MOF is also known to be more efficient than the monometals (Fig. 11).

(Copyright Elsevier License Number 5326341140976)
MOF synthesis [110].
Besides being used as the main catalyst, MOF can also be used as a co-catalyst for CO2 reduction in nanocomposites. MOF acts as a kinetic process driver in the catalytic reaction and increases CO2 adsorption while other components act as light harvesters. For example the use of Co-Zif-9 as a co-catalyst together with [Ru(bpy)3]Cl2.6H2O as a photosensitizer, and TEOA as an electron donor under 380–700 nm irradiation. Although the use of this co-catalyst is known to have high catalytic efficiency, the selectivity of each product is quite low. Not only that, but photobleaching also causes a decrease in catalytic activity. So, it is necessary for coupling with semiconductors such as g-C3N4 and CdS as light harvesters. In addition, MOF is also used as a co-catalyst with TiO2. This system shows that the photocatalytic activity for methane production increases up to five times [111].
Homogeneous metal complexes have been widely used for CO2 reduction photocatalysts. However, this metal complex does not have long-term stability, is difficult to separate from the reaction mixture, the product will be contaminated, and it is difficult to recycle. Therefore, the use of MOF as a host to support homogeneous metal complexes is considered the right choice. This is based on the nature of the MOF which has a high surface area and uniform pores, so it can be adapted for the diffusion of reactants. MOFs also have well-defined and isolated sites for anchoring catalytic species, so they can be used to construct single-site catalysts [111].
4.1.4 Metal Complex
Transition metal complex ions have been widely used as photocatalysts. Some examples of metal complexes used as photocatalysts include complexes of ruthenium, osmium, rhenium, cyclometallation iridium, metalloporphyrins, and organic dyes. This photocatalyst has several properties, including selective light absorption, relatively inefficient reverse electron transfer rate, fast decomposition, sufficient reducing power for electron transfer, stability in ground and excited states, and reacts selectively to CO2. The metal complex-based homogeneous photocatalytic system consists of a photosensitizer and a CO2 reduction catalyst. The Ru(II)-Re(I) photocatalyst is reported to reduce CO2 efficiently when installed on solid materials. Photocatalysts made with photosensitizers and metal complex catalysts showed efficient, selective, and long-lasting results. Building a photocatalyst with a hybrid system can be done by connecting a photosensitizer and a catalyst to a solid material so that electron transfer will take place faster [111].
The Re dynamic complex has CO2 reduction activity and high product selectivity. The Re(bpy)(CO)3Cl complex has a stable and adjustable structure so that it has the potential as a CO2RR photocatalyst. When photocatalytic CO2RR is carried out in a mixed solution of DMF/TEOA, the addition of TEOA can capture CO2 even at the atmospheric level. Re(bpy-)(CO)3 has also been reported to react with CO2 in the dark. In addition, the use of a photosensitizer compatible with the Re complex can also increase the efficiency and durability of the catalyst [112]. Unlike the Re complex, which only reduces CO2 to CO, the Ru photocatalyst can catalyze the conversion of CO2 to CO and HCOOH. This reaction occurs through the capture of one CO2 molecule by Ru(bpy−)2(CO) to form Ru(bpy−)2(CO)(CO2) so that one H+ can be added to create the complex [Ru(bpy−)2(CO)(COOH)]+. This complex will then produce HCOOH through the acceptance of two electrons. However, it doesn't stop at this point, the [Ru(bpy−)2(CO)(COOH)]+ complex will then be protonated in the CO cycle so that water molecules are released and form CO molecules [112].
4.1.5 MXenes
MXenes are transition metal carbides, nitrides, or carbon nitrides that have great potential as photocatalysts. These metals consist of transition metals such as Scandium, Titanium, and Vanadium, elements from groups III A and IV A, and carbon or nitride elements. The general formula for MXenes is Mn+1Xn or Mn+1XnTx where T is a surface functional group such as oxygen, fluorine, and hydroxyl. Despite their potential as photocatalysts, MXenes cannot be used directly due to their non-semiconducting nature. Therefore, most of the MXenes are used as co-catalysts. MXene is commonly used in CO2 reduction to improve photogeneration of charge carriers, photogenerated species separation, photo-corrosion inhibition, enhance CO2 adsorption and activation, enhance light absorption, and photothermal effects. MXene can be applied in reactions of gas and liquid phase systems [114].
The photocatalytic activity and photostability of the semiconductor can be enhanced through the combination of the metallic conductivity of MXene with a suitable band structure. This combination allows for efficient electron migration from the semiconductor to the MXene. In addition, the regulation of the surface functional group of MXene can maximize CO2 adsorption and activation. This is because the surface functional groups act as active sites for the photocatalytic reactions. MXenes can also improve light and photothermal harvesting for metal nanoparticles. MXenes as co-catalysts have a significant role in regulating product selectivity so diesel fuel production can be enhanced [114].
MXene as a co-catalyst is usually used with nitrides, metal oxides, metal salts, perovskite, and MOF as the main catalyst. One example of the use of an MXene/Nitride catalyst is the use of g-C3N4/Ti3C2. This heterojunction is reported to improve CO2 adsorption, photogenerated charge carrier separation, and hybrid stability. There is also the use of MXene with metal oxides such as TiO2 which is known to accelerate the efficiency of electron–hole separation. This combination will enhance the photocatalytic reduction reaction with 3.7 times higher methane production than commercial TiO2. Other combinations such as perovskite CsPbBr3 with MXene Ti3C2 were reported to produce higher CO and CH4 than CsPbBr3 NCs. This is due to MXene's ability to increase the photocatalytic activity [115] (Fig. 12).

MXene synthesis
Generally, the basic synthesis of MXenes goes through the etching process. There are three etching approaches, namely fluorination etching, fluorine-free etching, and electrochemical etching. In the fluorination etching process, materials derived from hydrofluoric acid (HF) or salt solutions containing fluorine are used. Meanwhile, in the fluorine-free etchant method, Ti3C2Tx is fluorine-free in NaOH or KOH solutions. This method is considered to have a higher capacity for electrochemical properties. The electrochemical etching method is carried out through a redox reaction between the anode and cathode in an electrolytic cell through the application of voltage. Wet etching using HF is a method that is often used to synthesize MXenes. This is because HF-based etching has high selectivity and can break M-A metal bonds [115].
The MXenes as a photocatalytic co-catalyst synthesis is divided into three methods, namely mechanical mixing, self-assembly, and in situ oxidation. Mechanical mixing is a simple method for catalyst synthesis. This method includes advantages such as saving energy and low cost. Mechanical mixing is carried out through the process of grinding solid powder and mixing the substances in solution to deposit MXene on the surface of the photocatalyst. The self-assembly method is considered to have advantages over the mechanical mixing method, due to its ability to have closer contact and more uniform dispersion. Lastly, there is the direct in situ oxidation method of MXene. This method produces MXene in the form of MO/MXene or MO/MXene/C, where C is an amorphous carbon that can be used as a co-catalyst. Unfortunately, this method can only be used if the MO is a semiconductor [116] (Fig. 13).

(Copyright Elsevier License Number 5326340653842)
MXenes synthesis schematic [116].
4.1.6 Metal Phosphide
Metal phosphides are widely used because of their abundance, low prices, and high electrical conductivity. Some examples of catalysts belonging to this group include CoxP, Ni2P, Fe2P, Cu3P, WP, and InP. The presence of this P group moderates the strength of the metal phosphide bond for the catalytic product conversion process, in which the active site facilitates product desorption. One example of a metal phosphide catalyst for CO2 conversion is InP which has a band gap of 1.35 eV. Due to this large band gap, some modifications need to be made. One of them is to minimize the InP particle size to the quantum dot (QD) scale. The advantage of this QD modification is an increase in the redox potential so that the photocatalytic activity will also increase. This modification is also able to provide a more active site for the photocatalytic reaction [117].
In addition, metal phosphides are also commonly used as co-catalysts. This material can create sufficient and strong contact with the photocatalyst to effectively separate the electron–hole pairs. Metal phosphides are also considered to have an electronic structure like Pt so that they can be used as a substitute for precious metal co-catalysts. The bond between metal and phosphide can induce a small ligand effect so that the product will separate from the catalyst surface. Not only that, but this bond is also able to capture reaction intermediates so that the catalytic activity will increase [109].
Some examples of metal phosphide co-catalysts commonly used are FeP with polymeric carbon nitride as the main catalyst and WP with g-C3N4. The use of FeP co-catalyst with carbon nitride polymer was reported to be able to produce five times higher CO than the use of pure carbon nitride. The use of metal phosphide-based co-catalysts also has good stability. In addition, there was a narrowing in the band gap from 2.77 to 2.40 eV due to a change in the position of the VB resulting in increased light absorption under UV light. WP which was used as a co-catalyst with g-C3N4 also showed satisfactory results. CO production is reported to have increased considerably due to the use of this co-catalyst. Life cycle stability has also been reported to improve. This is due to the promotion of electron transfer due to the presence of P-N bonds at the WP and g-C3N4 interfaces [109].
4.1.7 Bismuth-Based p-Block Semiconductors
Most photocatalysts such as TiO2 and ZnO use a d-block semiconductor. In this discussion, p-block elements will be used to construct a CO2 reduction photocatalyst. This is because p-blocks have the potential to increase their photocatalytic activity in visible light. The p electrons in the hybridized state can decrease the CB state and increase the VB state so that the band gap becomes narrower. Not only that, another advantage that can also be obtained is the mobility of the photoexcited charge carriers and the charge separation will be increased [118].
Research conducted by Cui et al. [118] discusses the use of bismuth as a semiconductor by utilizing p electrons as a CO2 reduction photocatalyst. According to them, there are three strategies to increase photoreduction activity, constituent adjustment, vacancy engineering, and heterostructure construction. The adjustment of the constituents is carried out through the adjustment of the halogen component and the ratio of Bi:O:X. Meanwhile, vacancy engineering is carried out through the engineering of oxygen, bismuth, and halogen vacancies. Finally, the heterostructure construction was carried out through the BixOyXz construction of nanomaterial heterostructures and 2D semiconductors. Through this strategy, it is hoped that the band structure can be controlled so that the catalytic reduction conditions are met along with the introduction of high-level active sites on the photocatalyst surface. Improvements in energy conversion efficiency are also expected through the promotion of charge separation and transportation [118].
4.2 Non-metallic Photocatalyst
Photocatalysts in application to reduce CO2 by using non-metallic materials exhibit rapid growth for photoconversion of CO2. In addition, this material also shows tremendous potential as an alternative to photocatalysts owing to its abundance on earth, cost-effectiveness, high electrical conductivity, as well as environmental friendliness. Moreover, they also exhibit outstanding catalytic activity, durability, and exceptional selectivity in transforming CO2 into solar fuel. In this section, we will explain and elaborate the non-metallic photocatalysts for CO2 reduction [119].
4.2.1 Graphene
Graphene has been studied in-depth and has shown considerable interest in its application in photocatalytic CO2 reduction. In addition, many experiments and studies have tried to design, develop, and apply graphene as a photocatalyst to reduce CO2 [119]. The structure of graphene-based catalyst is unique. Aside from its large specific surface area, graphene also has many other desirable properties, including excellent electron transfer, transparency, high resistance to temperature changes, flexibility, and good CO2 adsorption, which have made it ideal to be used as CO2 photocatalysts [120]. The separation of charge carriers in photocatalytic CO2 reduction can also be facilitated by graphene. In addition, graphene has a large contact area and interacts strongly with other catalysts, so the photocatalytic reaction and performance for reducing CO2 can be promoted and improved by graphene [121].
Some common graphene includes pure graphene, graphene oxide (GO), and reduced graphene oxide. The three materials possess different functional groups, which gives them different properties as well [122]. A modified Hummers’ method is the common method to synthesize these materials using graphite as their raw material. The synthesis process with this technique begins with the chemical oxidation of graphite. The next step for preparing GO is exfoliation. GO typically has abundant oxygen-carrying functional groups, such as carboxyl, epoxy, hydroxyl, and carbonyl groups. The bandgap value of GO is about 2.2 eV while graphene pristine has a bandgap of 0 eV and does not have surface functional groups. The reduction of graphene oxide will produce rGO [123]. The reduction techniques are thermal treatment and chemical methods. The bandgap of rGO is variable, depending on the degree of reduction, from ~1.00 to 1.69 eV. The surface functional groups of rGO are the same as GO. The performance of graphene oxide can be improved by surface modification, doping, morphology control, and defect introduction. Tan et al. [124] investigated TiO2 doped GO. The wet chemical impregnation technique is the technique used to combine GO with TiO2 in this experiment. A modified Hummers’ technique was used to prepare graphite oxide powder. Followed by the exfoliation process through ultrasonication for 1.5 h and separation of the graphite layer. Within 8 h of reaction time, GO-TiO2 successfully yielded 3.45 mol g−1 h−1 of CH4. This performance represents a 14-fold performance increase over commercially available TiO2 P25 [124].
The reaction scheme of the CO2 reduction through GO-TiO2 is shown in Fig. 14. The photocatalytic CO2 reduction mechanism is suggested followed Langmuir–Hinshelwood (L–H) model. First, reactant molecules, in the form of carbon dioxide and water molecules, adsorb onto the photocatalyst surface. The next process is the surface reaction. At this stage where ∙CO2− radicals are generated from photogenerated electrons. Then, these ∙CO2− radicals will be transferred to the adsorbed CO2. Meanwhile, on the other side, the hole reacts with the adsorbed H2O molecules. The product of this reaction is ∙OH radicals and H+ ions. Then, it will form O2. To produce intermediate radicals and hydrocarbon products, carbon radicals will react with H radicals formed by proton reduction on the photocatalyst surface [124].

(Copyright Elsevier License Number 5321211075597)
The Langmuir–Hinshelwood mechanism [124].
4.2.2 g-C3N4
Graphite carbon nitride is a layered semiconductor material. g-C3N4 is a non-metallic material in the form of a polymer material and is composed of a polymer combination of tri-s-triazine/heptazine (C6N7) or triazine (C3N3) [120]. According to its wavelength of 460–430 nm, the g-C3N4 band gap value is narrow, ~2.7–2.9 eV. g-C3N4 also shows a CBat a negative position of about 1.1 V versus NHE. This property meets the thermodynamic requirements of photocatalytic CO2 reduction reactions [125]. Therefore, under visible light and sunlight CO2 will be activated and reduced successfully. The easy fabrication process, metal-free composition, durability, low cost, environmental friendliness, and good chemical and thermal stability are the superiority as a semiconductor material for the photocatalyst of g-C3N4. Furthermore, the conduction band of 1.14 eV and the valence band of 1.57 eV, make it a polymeric semiconductor suitable for visible light. However, g-C3N4 has drawbacks namely low exposed surface area, insufficient use of visible light, and fast photo-induced electron–hole pair recombination [126, 127].
Song et al. [128] developed the use of g-C3N4 as a photocatalyst in CO2 reduction applications. They obtained g-CN as a catalyst. g-CN was incorporated with C and O simultaneously. The donor–acceptor structure was developed using hexa-methyl melamine (HMM) as a doping agent that provides C and O. Simple thermal condensation method of melamine was used to fabricate g-CN-coupled Samples C and O with HMM at elevated temperatures. The catalyst exhibits efficient photocatalytic CO2 reduction activity and durability. Their experiment resulted in a production rate of CO at 34.97 µmol·gcat−1 in a 4-h-reaction-time. This result is 4.3-fold greater than that obtained by pristine g-CN [128] (Fig. 15).

(Copyright Elsevier License Number 5321220110079)
Photocatalytic reduction of CO2 mechanism through g-C3N4 [128].
4.2.3 Boron Nitride (BN)-Based Photocatalyst
Boron nitride has similarities to graphene, in particular the boron nitride with a hexagonal structure or h-BN. h-BN is a 2D material whose structure imitates a honeycomb, consisting of boron (B) and nitrogen (N) atoms with the same amounts and has excellent thermal conductivity and a large surface area. This material shows great potential in catalysis applications, especially in photocatalytic CO2 reduction. h-BN has a broad bandgap of 5.2–5.5 eV. Similar to h-BN, porous BN also has a wide bandgap (~5.5 eV) [131].
Cao et al. [130] successfully fabricated the h-BN (O/BN) nanosheets. This nanosheet is modified with ultra-thin oxygen-containing a single bond between the B atom and O atom. When O is associated with the B atom, it has a greater electronegativity, so the introduction of the O atom into the h-BN aims to form a single bond between the B atom and the O atom. These nanosheets exhibit a great performance for CO2 conversion to H2 at a production rate of 3.3 µmol·gcat−1·h−1 and CO at 12.5 µmol·g−1·h−1 [130].
4.2.4 Black Phosphorus (BP)
Black phosphorus, a 2D layered material, is an emerging remarkable photocatalytic semiconductor, especially for CO2 reduction. BP nanosheets have advantages such as a tailorable band gap value of about 0.3–2.0 eV as the layer decreases from bulk to monolayer, suitable electronic structure, excellent optical properties, and high mobility of charge carriers, which is about 1000 cm2·V−1·s−1 [131].
For example, a previous study succeeded in making a CO2 reduction photocatalyst from the fabrication of a monolayer of black phosphorus with a modified hydroxyl functional group, also known as M-BP-OH. In situ preparation of the photocatalyst can be achieved through the green exfoliation technique. In the synthesis using this technique, nitrogen is needed. From this research, the M-BP-OH exhibits an excellent photocatalytic activity performance. 112.6 mol·g−1·h−1 of carbon monoxide (CO) can be produced by this photocatalyst by reducing CO2. Furthermore, the M-BP-OH exhibit excellent stability under humid condition (90% humidity) for 24 h, ambient conditions for a month, and cycling test using a Xe lamp at 300 W for 60 h [132].
Recently, a hybrid material constructed of BP QDs and g-C3N4 was prepared to convert CO2 to CO. As a result of the research, the material can catalyze the CO2 photochemical conversion reaction to produce CO. The CO product produced at high production levels was 6.54 mol·g−1·h−1 [125]. Compared to whole g-C3N4 which was only able to produce 2.65 μmol per gram of catalyst used for a 1-h basis, the yield achieved was much higher. Chen et al. (2021) fabricated a heterojunction with an S-scheme in which a stable ambient-based 2D/2D material (Pt/BP-Bi2WO6) for photocatalytic CO2 reduction to syngas. The Pt/BP-OvMBWO heterojunction is electrostatically assembled. Pt/BP nanosheets are negatively charged and the OvMBWO nanosheets are positively charged. These two nanosheets were electrostatically assembled to obtain the heterojunction. As a result, this material can generate CO and H2 as high as 20.5 µmol·g−1·h−1 and 16.8 µmol·g−1·h−1, respectively [131].
4.2.5 Silicon Carbide (SiC)
Silicon carbide is a kind of environmentally friendly semiconductor. Great chemical and thermal stability are other advantages of SiC. The band gap of SiC is wide, about 2.4–3.2 eV, and has a conduction band potential of −1.40 eV. Due to these properties, SiC has become a promising semiconductor material as a photocatalyst for CO2 reduction [119]. Compared to the CO2 reduction potential of some products, the potential of SiC is more negative. Owing to its properties, SiC is a suitable material for the reduction of CO2 that requires a high potential. SiC has about 250 polytypes. However, in general, there are 2 kinds of SiC polytype, β-SiC, and α-SiC. Cubic 3C-SiC, which belongs to the β-SiC polytype is SiC. Meanwhile, the hexagonal SiC, such as 2H-, 4H-, 6H-SiC, and others belong to the α-SiC polytype [133]. Among these polytypes, the polytype that has the highest bandgap value of 2H-SiC is 3.3 eV while the lowest band gap of 3C-SiC is 2.4 eV [134].
Wang et al. [135] developed a β-SiC polytype of SiC. The developed SiC is a hollow-Sphere with an open mouth and is synthesized using environmentally friendly techniques. Glucose is used as a carbon source in this fabrication. In the application of photocatalytic CO2 reduction with purified water, the prepared SiC showed a very active performance for converting CO2 to methane due to the uniqueness of its electronic structure. Besides that, it is also supported by its hollow structure and its large BET surface area (28 m2/g). The performance of β-SiC Hollow-Sphere with Pt charge is greatly improved. Under sunlight, 2.0 wt% of Pt showed that the conversion of CO2 to CH4 reached 67.2 mol·g−1. While the results with bare SiC only reached 28.1 mol/g, which means the results using β-SiC Hollow-Sphere with Pt loading were 2 times higher. Moreover, for the evolution of CH4 from CO2, the performance shown by this material is also more than that of many metal oxides. Figure 16 illustrates the charge transfer process for reducing CO2 with water to produce methane under simulated sunlight in a Pt/SiC hollow sphere [135].

(Copyright Elsevier License Number 5321211504925)
Photocatalytic CO2 reduction mechanism in the Pt/SiC hollow spheres [135].
The development of polytype β-SiC, in the form of 3C-SiC, was also carried out by Li and Sun [136] on a gram scale. 3C-SiC is produced in the form of nanoparticles by using the ball milling method with a top-down approach. A cheap 3C-SiC crystalline powder is used for the fabrication of 3C-SiC nanoparticles. Ball-milling machine with high energy is used to grow nanoparticles by chemical vapor deposition. A 420 nm wavelength filter light from a Xe lamp with a power of 300 W is used in photocatalytic CO2 reduction. 3C-SiC nanoparticles were dispersed into a glass reactor for the reduction of CO2. 3C-SiC is capable of harvesting visible light. The band gap value is relatively small, about 2.36 eV, so 3C-SiC has an ideal band position straddling CO2 reduction. In addition, 3C-SiC has high stability, thermal conductivity, and relatively low price. The achieved selectivity for reducing CO2 to CH4 is as high as 90% combined with efficient water oxidation. Therefore, 3C-SiC has great potential and deserves consideration in photocatalytic CO2 reduction applications [136].
Liu et al. [133] fabricated a nanocage with a heterophase junction of 2H/3C-SiC. They prepared this nanocage by a simple magnesiothermic reduction technique mediated by low-temperature molten salt. SiO2 was used as a template. Uniform heterophase junctions in the SiC nanocage will be formed by the 2H and 3C crystalline phases. This accelerates photogenerated electron transfer effectively and is very important in improving the performance to generate CO through the photocatalytic reaction of CO2 reduction. CO produced by SiC nanocage was recorded to be 4.68 mol·g−1·h−1. This result is 3.25-fold more than commercially available SiC [133].
4.2.6 Covalent Organic Frameworks (COFs)
Known as COFs, covalent organic frameworks contain crystalline organic pores with orderly crystal structures connected with organic compounds by covalent bonds. COFs are polymeric materials. COF is built by elements that tend to be lightweight, such as carbon, hydrogen, oxygen, nitrogen, boron, and silica. As a new material class, COFs have some advantages. The advantages of COF include tailorable pore size, large specific area, tailored functionalities, and good thermal stability [119].
For example, Lu and coworkers designed a sequence of COFs in the form of crystalline 2D porphyrin-tetrathiafulvalente, TTCOF-M. The notation M indicates other elements used in the material, such as 2H, zinc (Zn), nickel (Ni), and copper (Cu). This material was synthesized as a photocatalyst for artificial photosynthesis including reducing CO2. In this study, no sacrificial agents, precious metal co-catalysts, or supplementary photosensitizers were added to the photocatalyst system. Schiff base condensation technique between 5,10,15,20-tetrakis (4-aminophenyl)-porphinato] (TAPP-M) which is metalized is the technique used in this study for the fabrication of TTCOF- M. Meanwhile, the solvothermal method was used for the preparation of 2,3,6,7-tetra (4-formylphenyl)-tetrathiafulvalene [137].
The mechanism of the CO2 reduction reaction process with H2O oxidation is shown in Fig. 17b. Visible light irradiation encourages the CO2 reduction process with H2O oxidation. Before the photo-induced electron transfer process from the tetrathiafulvalene (TTF) part to the TAPP part, the photons will be absorbed first. After the electrons are excited, they travel to the active catalytic site in TAPP, which is Zn/Cu. The excited electrons that have been transferred are used to reduce CO2. Meanwhile, TTF can generate O2 through H2O oxidation by the act of its photogenerated holes, in which electrons are obtained by the photocatalytic system from H2O to maintain the charge balance. This research was able to reduce CO2 with TTCOF-Zn/Cu COF using water as an electron donor. The highest performance is shown by TTCOF-Zn with CO production as high as 12.33 mmol and with high selectivity which is also combined with excellent durability [137].

(Copyright John Wiley and sons License Number 5319200443008)
a TTCOF-M synthesis, b CO2 reduction reaction process on TTCOF-M with H2O oxidation [137].
Fu et al. [138] successfully fabricated a crystalline bipyridine-containing sp2c-COF as a photocatalyst to reduce CO2. Rhenium complex [Re(CO)5Cl] was used to modify the COF and form a homogeneous catalyst Re-Bpy-sp2c-COF or [Re(bpy)(CO)3Cl]. This COF photocatalyst showed outstanding photocatalytic performance to convert CO2 into CO with a production rate of CO 1040 mol·g−1·h−1 and selectivity of 81% (H2) during 17.5 h of illumination. With dye sensitization, the photocatalytic performance was increased to reach 84% selectivity for more than 5 h of illumination and produce CO as much as 1400 mol·g−1·h−1 and 86% selectivity (CO/H2) [138].
Recently, Cui et al. [139] developed a ketoenamine-linked 2D COF, also known as TpBb-COF. TpBb-COF was prepared under solvothermal conditions by an acid-catalyzed Schiff-base condensation (Fig. 18a). There are two reactants used in this condensation. The first reactant is 2,6-diaminobenzo[1,2-d:4,5-d’]bisthiazole or Bb-NH2, the second reactant is 1,3,5-triformylphloroglucinol or Tp-CHO. The reactant is in the binary solvent mesitylene and dioxane. TpBb-COF was used for photocatalytic reaction to convert CO2 to produce CO. This material has a bandgap value of 1.72 eV. As a result, TpBb-COF showed excellent photocatalytic performance where as much as 52.8 mol·g−1·h−1 CO was able to be generated in pure CO2. Meanwhile, in conditions of 30% CO2 at 80 °C, the amount of CO that can be produced is 89.9 mol·g−1·h−1 [139].

(Copyright Royal society of chemistry License Number 1226810–1)
a TpBb-COF Synthesis b Stacking mode of TpBb-COF from the top and side view [139].
4.3 Composites System
Low et al. [140] designed nanoparticles of TiO2 on the MXene Ti3C2 for photocatalytic CO2 reduction. The nanoparticles grew in situ on MXene Ti3C2 through a simple calcination technique. This composite has a unique structure like rice crust. Due to the unique multilayer structure of TiO2 to Ti3C2, there is a large specific surface area which in turn provides a large active site for photocatalytic reduction. By using the optimized TiO2/Ti3C2 composite, the performance to reduce CO2 in photocatalytic reaction to produce methane was 3.7-fold more than commercial TiO2 or about 0.22 μmol·h−1. Ti3C2 has superior conductivity to facilitate the photogenerated electron transfer. Another consequence is that the photogenerated holes will also be suppressed by the effect of this property. TiO2 has a conduction band of about −0.25 eV against a normal hydrogen electrode. The composite band gap value is −0.04 eV concerning normal hydrogen electrodes, a more negative value than the Fermi Ti3C2 level (Fig. 19a) [140].

(Copyright Elsevier License Number 501735419)
a The migration of charge carrier on TiO2/Ti3C2 composite scheme b Photocatalytic CO2 reduction mechanism in TiO2/Ti3C2 composites [140].
Figure 19b illustrates the photocatalytic process to reduce CO2 in TiO2/Ti3C2 composites. First, with an interesting morphology in which a surface with a large specific area will increase the ability of this composite to adsorb CO2. Second, every component involved contributes to the outstanding performance. The black color properties of Ti3C2 increase the light absorption of the composite and create a photothermal effect. Third, on conductive Ti3C2 2D, a heterogeneous interface will be formed from in situ growth, where this interface can change the properties of the original material. The tight and strong contact facilitates the migration of charge carriers so that electron holes separate efficiently. In addition, photogenerated electrons also accumulate in Ti3C2 so it is very supportive of the multi-electron CO2 reduction reaction [140].
Wang et al. [126] developed a photocatalyst to reduce CO2 in the form of a nanocomposite z scheme between g-C3N4 and α-Fe2O3. The two materials are linked by Al-O. The first step to make this photocatalyst is to make a g-C3N4/α-Fe2O3 nanocomposite with an optimal ratio. The hydrolysis-solvothermal method with phase separation was used in the synthesis of nanocrystalline α-Fe2O3. To prepare g-C3N4, urea was heated in an alumina combustion vessel. The mass ratio of the two components was adjusted to obtain the optimal nanocomposite. Then, the photocatalyst composite of g-C3N4 and α-Fe2O3 which is linked by the Al-O bridge was fabricated by a two-step wet chemical technique. As the source of the Al-O bridge in the nanocomposite, an aqueous solution of AlCl3 was used. Compared to using pristine g-C3N4 and pristine Fe2O3, nanocomposite photocatalysts exhibit higher photocatalytic activity for enhanced CO2 conversion. After irradiation for 1 h, α-Fe2O3 showed low photocatalytic activity in reducing CO2 to several products, such as CO, CH4, and O2 with production rates of ~ 5.7 mol·g−1·h−1, ~ 0.3 mol·g−1·h−1, and ~ 6.2 mol·g−1·h−1 respectively. However, the addition of a large amount of g-C3N4, especially 15CN-F, give a positive contribution to the photocatalytic activity where the production rate for each product increased to ~ 15.8 mol CO g−1·h−1, ~ 3.1 mol CH4 g−1·h−1, and ~ 18.5 mol O2 g−1·h−1. This photocatalytic activity performance can be further enhanced by building a link or bridge between g-C3N4 and α-Fe2O3 (15CN-6Al-F) on 15CN-F nanocomposite in the form of Al-O. As a result, the amount of CO produced is 4 times higher than α-Fe2O3, or about 24 mol·g−1·h−1. The charge transfers and schematic of the photo-induced charge separation process for a g-C3N4/α-Fe2O3 nanocomposite linked by an Al-O bridge are illustrated in Fig. 20. Initially, the photoinduced electrons of α-Fe2O3 will join the g-C3N4 photoinduction holes in the g-C3N4/α-Fe2O3 nanocomposite when these two materials are excited simultaneously under a wavelength of 460 nm. In this case, a redox reaction will be induced because the holes in α-Fe2O3 and electrons in g-C3N4 are spatially separated and have thermodynamically sufficient energy. This reaction will result in a promoted charge separation which will increase the photocatalytic activity. Al-O bridges in nanocomposites will promote charge transfer from the α-Fe2O3 side to the g-C3N4 side and lead to increased photocatalytic performance for CO2 reduction. Photo-induction of electrons in g-C3N4 stimulates a reduction reaction with the H2O component to produce hydrogen atoms. The resulting hydrogen atoms attack the CO2 molecules to produce products in the form of CO and CH4. Meanwhile, another product, namely the radical hydroxyl group, could evolve O2 through the photoinduced hole reaction of α-Fe2O3 with H2O [126].

(Copyright Elsevier License Number 501735385)
Mechanism of photoinduction charge transfer and separation in g-C3N4/α-Fe2O3 nanocomposites linked by Al-O bridges [126].
Photocatalyst made from g-C3N4 was also recently fabricated by Xu and coworkers. In their research, they developed a photocatalyst nanocomposite with a ternary system between g-C3N4, Ag2CrO4, and graphene oxide having a Z-scheme mechanism. This nanocomposite photocatalyst system with g-C3N4 was developed because its ability to absorb visible light is low and its charge recombination is so fast. In this photocatalyst system, silver chromate (Ag2CrO4) nanoparticles are used which act as photosensitizers to drive charge transfer and harvest light. Silver chromate (Ag2CrO4) was used as a photosensitizer due to its narrow bandgap (-1.8 V). This is favorable for the g-C3N4 light absorption so that the amount of absorbed light by g-C3N4 can be increased. In addition, Ag2CrO4 may also pair with g-C3N4 to form binary heterojunctions to promote photogeneration charge separation. Graphene oxide (GO) was used as a co-catalyst. Apart from being a co-catalyst, GO is also an electron collector and provides a reactive site for CO2 conversion. A composite photocatalyst with a ternary system was recently prepared to reduce CO2 in photocatalytic reactions under simulated light irradiation to produce methane and methanol. The precipitation self-assembly technique was used for the fabrication of Ag2CrO4/g-C3N4/GO composites (Fig. 21) [141].

(Copyright Elsevier License Number 501735474)
Schematic of Ag2CrO4/g-C3N4/GO composite fabrication [141].
Under full spectrum sunlight simulation, the photocatalytic activity of the ternary composite Ag2CrO4/g-C3N4/GO in reducing CO2 was very good, as high as 1.03 mol·g−1. This result is 2.3-fold more than using the whole g-C3N4. The direct Z-scheme mechanism is a charge transfer process that occurs between Ag2CrO4 and g-C3N4 (Fig. 22). Apart from promoting charge separation, this charge transfer process enhances the reduction–oxidation capability of the photocatalytic system as well [141].

(Copyright Elsevier License Number 501735474)
Photocatalytic process in Ag2CrO4/g-C3N4/GO ternary composite through Z-scheme [141].
Crake et al. [142] developed a composite photocatalyst of inorganic and organic materials for CO2 reduction applications. In this study, they used titanium oxide and carbon nitride. Carbon nitride is used in the form of nanosheets (CNNS). The two materials, namely TiO2 and CNNS were prepared by hydrothermal in situ growth technique (Fig. 5a). This photocatalyst system does not require an expensive co-catalyst and CO2 reduction takes place in a single process. First, the preformed carbon nitride was exfoliated by sonication in water to form carbon nitride (CNNS) nanosheets. This CNNS was used to obtain TiO2 and CNNS composites. In the PTFE autoclave, titanium butoxide, hydrofluoric acid, and CNNS were added and mixed. The process was continued by heating at 180 °C for 24 h. The addition of hydrofluoric acid aims to control the formation of crystal facets. This photocatalyst exhibits an excellent photocatalytic CO2 reduction performance compared to its constituent materials, approximately an increase of more than tenfold. In addition, this composite also increases the capacity to adsorb CO2 [142] (Fig. 23).

(Copyright Elsevier License Number 5321210450514)
TiO2/Ti3C2 composite a Schematic of the fabrication process of TiO2/CNNS composites b Photocatalytic CO2 reduction reaction pathway [142].
4.3.1 TiO2-MOF Composite
The combination of TiO2 metal with MOF covers the shortcomings of each material as a CO2 reduction photocatalyst. These metals have their respective roles; TiO2 acts as a primary photocatalyst. Meanwhile, MOF acts as a photosensitizer for efficient light harvesting and electron transfer. Combining the two improves light absorption, electron–hole pair separation, and photocatalytic efficiency. The results obtained can be 5 times higher than pure metal TiO2 even though CO2 adsorption is reduced [143].
TiO2-MOF composites with TiO2 particles graphed onto (Cu3(BTC)2) microcrystals (TiO2 in HKUST-1) were found to have a bandgap of 1.6–3.1 eV under UV light. The use of this composite reduces the adsorption capacity of CO2, but the production of CH4 is five times higher than the use of pure TiO2 due to electron–hole separation [144].
Crake et al. [145] performed the synthesis of TiO2 through the solvothermal method. Then the in situ method was used to produce TiO2-MOF composites under UV–Vis irradiation. This composite prevents TiO2 aggregation by maintaining a high surface area. Furthermore, the porosity of the MOF results in a sizeable CO2 adsorption capacity so that the amount of adsorbed CO2 on the surface increases. Wang et al. [143] explain four types to produce TiO2-MOF composites. The first type uses organic ligands and metal templates of different morphology to form MOFs. The second type uses a precursor to produce TiO2 into a prepared MOF. The third type is the in situ method, as done by Crake et al. [145], and the last one uses a partial organic linker from MOF.
4.3.2 Graphene-MOF Composite
Another MOF combination, graphene, makes up for the lack of electrical conductivity in MOF and improves the photothermal stability. This composite can also reduce aggregation at high-temperature calcination and increase the catalytic activity of the active sites. The MOF in the composite acts as a substrate for the diffusion of the substrate because of its porous structure so that there is more room for additional components. In addition, MOF also has high crystallinity and stability during the photocatalytic process. Graphene is a conductor for light harvesting and electron transfer [110].
Briefly, there are two methods for making MOF-Graphene composites. The first way is physical mixing with pH adjustment to increase electrostatic adsorption. The second method is in situ growth to increase the interaction between graphene and MOF. The performance of these two methods can be improved in several ways, namely MOF aminase, polydopamine, heteroatom doping, use of graphene aerogel, and constructing heterogeneous structures [146].
4.3.3 Titanium-Based Perovskites
Modification of TiO2 catalyst with perovskite as host material was used to adjust optical properties and induce visible light absorption. This composite is also intended to form a catalyst that has good photo-corrosion resistance and thermal stability. In addition, this composite is applied as a candidate for producing hydrogen and hydrocarbons as well as water separation processes through photocatalytic reduction of CO2 [108].
Strontium titanate oxide (SrTiO3) and calcium titanate oxide (CaTiO3) are the most widely used perovskite oxides. SrTiO3 perovskite composites with TiO2 are known to have the ability to selectively separate CO2 reduction reactions and enhance charge transfer synergism. In addition, this perovskite can escalate the selectivity of CH4 and CO production. The addition of Pt and Pd nanoparticles to the composite material increased the photoconversion efficiency of CO2 to CH4. As a result, the production of CH4 and CO increased by 4.7 and 1.6 times, respectively, compared to ordinary composites. This is because Pt contributes to increase the efficiency of charge separation [108] (Fig. 24).

(Copyright Elsevier License Number 5326340974814)
Photocatalytic Mechanism over SrTiO3/TiO2 Composite. Reproduced with permission [108].
4.3.4 Graphene-Based Perovskites
Graphene is one of the most widely used photocatalysts for CO2 reduction. However, this material has a cypher bandgap which makes it incapable to perform charge separation. However, graphene offers high charge mobility and electron–hole pair separation through its interface. The use of composites with graphene enables increasing its light absorption, charge mobility, and catalytic activity in visible light through their synergistic effect [108].
The use of perovskite Bi2MoO6 composite with graphene for photocatalytic CO2 reduction was carried out to produce CH3OH and C2H5OH. Bi2MoO6 electrons will migrate from VB to CB, then the Mo atoms in the perovskite will trap photogenerated electrons to attract CO2. These electrons will then move towards graphene which has an electron-rich interface. Finally, CO2 absorption occurs due to the large surface area of graphene [147].
4.3.5 Graphitic Carbon Nitride-Based Perovskites
KNbO3 is a perovskite used as a composite with g-C3N4 and Pt. g-C3N4 functions as a sensitizer while Pt acts as a co-catalyst. The electrons will be migrated to the perovskite which is then switched to the Pt co-catalyst to reduce CO2 into CH4. The use of this composite system produces four times higher methane product than the use of pure g-C3N4, under visible light radiation. In addition, the use of perovskite as a composite material is proficient to increase the catalytic activity along with the increase in the concentration of perovskite. In another study, the use of perovskite NaNbO3 composites with g-C3N4 was able to increase methane production eightfold compared to the use of pure g-C3N4-Pt or NaNbO3. This is because there is a faster separation and charge transfer so that the photocatalytic activity also increases. In addition, g-C3N4 CB (−1.13 eV) was located at more negative potential than that of NaNbO3 (−0.77 eV) [108] (Fig. 25).

(Copyright Elsevier License Number 5326340974814)
Photocatalytic Mechanism over g-C3N4/ KNbO3 Composite. Reproduced with permission [108].
4.3.6 MXenes-Based Perovskites
MXenes are transition metal carbides/nitrides and carbonitrides that have the general formula Mn+1Xn, where M is a transition metal such as scandium, titanium, zirconium, hafnium, vanadium, niobium, tantalum, and molybdenum while X is carbon or nitrogen. Perovskite Bi2WO6 composite with MXene Ti3C2 yielded 4.6 times higher CH4 and CH3OH than the use of pure Bi2WO6 perovskite. This is because CB perovskite is more negative than Ti3C2 so there is a sudden electron migration as a result of photo induction to the heterojunction interface. In another study, CsPbBr3 NC/MXene perovskite nanocomposites were able to selectively produce CO and CH4 [148].
5 Product Characterization, Analysis, and Selectivity
5.1 Product Characterization and Analysis
CO2 photoreduction involves multi-electron reactions that can result in a wide range of products in the gas phase, including CO, CH4, and higher hydrocarbons, as well as numerous, oxygenates in the liquid phase, including alcohols, aldehydes, and carboxylic acids. Table 3 depicts a few reaction examples that result in gas and liquid products. According to a recent analysis by Izumi, certain reduction products with reasonably high yields that are not thoroughly verified may not be entirely made from CO2. While our study is focused on producing effective photocatalyst systems, there are a few critical challenges that need to be addressed in terms of CO2 reduction product analysis: (1) Is it possible to reliably detect the most likely reduction products? After accurate quantification of the products and elimination of the contribution from carbon contamination, a reliable conclusion on photocatalytic activity can be reached. (2) Will organic additions have an impact on the product analysis? Organic additives are frequently utilized in homogenous molecular systems as sacrificial reagents, solvents, photocatalysts, or photosensitizers. It is necessary to evaluate their impact on the analysis of CO2 reduction products. (3) Is it true that the measured products are the result of CO2 reduction? Verification techniques include 13C nuclear magnetic resonance (NMR) and gas chromatography-mass spectrometry (GC–MS).
5.1.1 Analysis of Gas Phase Products
The principal gaseous products of CO2 photoreduction are CH4 and CO, while H2 and/or O2 can also be formed as byproducts of water splitting [149]. Varghese et al. also found higher alkanes as photoreduction products of CO2 in their study [150]. Aside from the products, CO2 must be quantified as the primary species in the gas phase due to limited conversion to estimate the reaction progress and/or the extent of CO2 dissolution in the liquid phase [151]. The use of infrared spectroscopy (IR) or diffuse reflectance infrared Fourier transform spectroscopy (DRIFT) to check CO2 consumption and generation has been done on occasion [152]. The most widely used method for quantifying these gas species is gas chromatography (GC) [152]. GC–MS was also employed for routine CH4 and CO analysis in addition to GC [153] or to confirm the source of carbon [154].
5.1.2 Analysis of Liquid Phase Products
Although GC with TCD/FID is still the most commonly used technology for analyzing various forms of liquid oxygenates (mainly alcohols) [155], Other approaches have been used, such as MS, high-performance liquid chromatography (HPLC) [156], ion-exchange chromatography (IEC) [157], ultraviolet–visible (UV–Vis) spectroscopy (colorimetric assay) after reacting with chromotropic acid [158] or Nash reagent [159] and NMR [160]. Table5 summarizes the substances that can be evaluated and their detection limits utilizing various techniques, as well as the accompanying constraints.
5.2 Product Selectivity
Photocatalytic CO2 reduction can directly convert CO2 and H2O into hydrocarbon solar fuels utilizing endless solar energy as the only source of energy [161]. CO2, as the highest oxidation state of carbon, can be reduced into a variety of products by obtaining different numbers of electrons and protons, such as CO, HCOOH, HCHO, CH3OH, and CH4 [162]. Besides these C1 products, some C2 products (C2H4, C2H6, CH3CH2OH) derived from the C–C coupling reaction also can be detected in the photocatalytic CO2 reduction reactions [163].
In thermodynamics and kinetics, Fig. 26 summarizes some key influencing factors. To begin, light must be absorbed by a semiconductor photocatalyst to produce effective photogenerated electrons and holes. Photon energy and light intensity are the most common light-excitation attributes. The former determines whether a semiconductor can absorb excited photons and thus influence the product selectivity of thermodynamic reactions. The latter impacts the number of photogenerated electrons and holes generated by excitation, and subsequently affects the reaction rate and product selectivity of multiple electron processes kinetically.

(Copyright Elsevier License Number 5327490662508)
Important thermodynamic and kinetic influence factors in product selectivity of photocatalytic CO2 reduction reactions [163].
5.2.1 Product Pathways and Selectivity
Figure 27 depicts the various routes of complex photocatalytic CO2 reduction processes, which include electron and proton transfer, hydrogenation and deoxygenation, C–O bond breaking, and C–H bond formation [165]. CO2 adsorption on the surface of the photocatalyst initiates photocatalytic CO2 reduction processes. CO2 adsorption mechanisms in general include carbon and oxygen coordination with photocatalyst surface atoms [166]. The adsorption/desorption of reactants/intermediates, as well as the sequencing of hydrogenation and deoxygenation processes, are all affected by the C, H, and O affinities of photocatalysts, resulting in diverse product paths and selectivity [164].

(Copyright Elsevier License Number 5327490662508)
Possible pathways of photocatalytic CO2 reduction to CH4 on the surface of photocatalysts [164].
5.2.2 Main Strategies for Controlling Product Selectivity
Complex multi-step mechanisms are involved in photocatalytic CO2 reduction reactions. In terms of reaction activity and product selectivity, each procedure is critical. The photocatalytic CO2 reduction process can be broken down into the following phases in general: (1) Photocatalysts are activated by the right amount of sunlight and produce a large number of electrons and holes. (2) Charge carriers generated by photocatalysts separate and transfer from the bulk to the surface. (3) On the surface of photocatalysts, reactants (CO2 and H2O) are simultaneously captured and activated. (4) Photogenerated holes are consumed by H2O or additional sacrificial reagents to form oxidation products, whereas effective photogenerated electrons reach the surface active sites and catalytic reduction processes occur. (5) The resulting products desorb from the photocatalyst surface, and the re-exposed surface active sites engage in the subsequent catalytic events.
By adjusting the key phases of the photocatalytic CO2 reduction process, the product selectivity of photocatalytic CO2 reduction reactions can be controlled. The primary engineering strategies for enhancing photocatalytic CO2 reduction product activity and selectivity are given in Fig. 28.

(Copyright Elsevier License Number 5327490662508)
Main factors influencing the product selectivity of photocatalytic CO2 reduction reactions.
5.2.3 Light-Excitation Attributes
In general, the wavelength requirement of incident light that can excite semiconductors is determined by the band gaps of classic semiconductor photocatalysts. By altering the light-excitation properties, it is difficult to manage the product selectivity of CO2 reduction processes. Plasmonic photocatalysts have different modes of light absorption and excitation than standard semiconductor-based photocatalysts. The collective free electron oscillations induced by light irradiation, known as localized surface plasmon resonances (LSPRs), can produce strong light absorption in plasmonic nanoparticles (NPs) [167].
Adsorbed reactants on the surface of plasmonic NPs can be activated by the energetic (hot) electrons obtained. Excess hot electrons stored on the surface can participate in CO2 reduction processes with the help of an effective hole scavenger, and oxidation products result from the oxidation of the hole scavenger. The distribution of photoexcited hot electrons is highly influenced by the energy and density of photons, resulting in distinct reaction paths and rates. It should be possible to tune light-excitation properties to alter the product selectivity of plasmonic photocatalysts.
5.2.4 Band Structure of Photocatalysts
The redox abilities of photogenerated electrons and holes are highly dependent on the locations of photocatalysts’ CB and VB during photocatalytic processes. CO2 conversion to various reduction products necessitates certain reduction potentials of photogenerated electrons, as shown in Fig. 29. Regulating the band structure of photocatalysts to regulate the CB positions allows efficient management of product selectivity with respect to thermodynamics by obtaining designated reduction potentials of electrons.

(Copyright Elsevier License Number 5327490662508)
Reduction potentials of photocatalytic CO2 reduction to different products (versus SHE, pH = 7) [168].
5.2.5 Separation of Photogenerated Electrons and Holes
CO2 reductions by photocatalysis are multi-electron processes. The density of photogenerated electrons on photocatalyst surfaces can alter the rate of surface catalytic reactions, which in turn affects the overall product selectivity of the reaction. The separation efficiency of photogenerated electrons and holes in photocatalysts determines the density of surface photogenerated electrons [169]. As a result, increasing the effectiveness of charge separation of photocatalysts should be a key technique for controlling product selectivity [170].
5.2.6 Adsorption and Activation of Reactants
In a catalytic reaction, the adsorption and activation of reactant molecules is a crucial step. The only two reactant molecules in photocatalytic CO2 reduction processes are H2O and CO2. Their adsorption and activation capabilities on the photocatalyst surface have a significant impact on activity and product selectivity. CO2 is a nonpolar linear molecule that has a bond energy of 799 kJ·mol−1. CO2 is difficult to activate and diminish due to these properties [171]. CO2 adsorption on photocatalyst surfaces can change their linear molecular conformation, allowing the CO2 molecule to be activated for photocatalytic reduction processes [172]. CO2 adsorption and activation are usually synchronized. CO2 adsorption on the surface of photocatalysts is indicated by typical carbon coordination, oxygen coordination, and mixed coordination modes.
A partially charged species CO2 radical dot will arise as a result of the interaction between CO2 and the surface atoms of photocatalysts, which is the typical activation intermediate. The process of further reduction by acquiring electrons can occur smoothly after activation. The surface atomic states and characteristics of photocatalysts influence the coordination modes of CO2 radical dots. Better CO2 adsorption and activation increase CO2 usage and lower the reaction barrier, affecting the product selectivity of photocatalytic CO2 reduction processes [173].
H2O is a key reactant in the photocatalytic CO2 reduction process [174]. To complete the oxidation half-reaction, H2O consumes photogenerated holes. H2O also serves as a proton source for the CO2 hydrogenation process. The adsorption and dissociation of H2O to protons on the surface of photocatalysts can boost the photocatalytic CO2 reduction processes by further reducing the hydrocarbon products. Pt is a good candidate for dissociating H2O to H adatoms, and it is beneficial in the treatment of HER. Hydrocarbon compounds with a high reducing state can be obtained by further hydrogenation of CO2 with H adatoms obtained through H2O dissociation.
5.2.7 Control Reaction Active Sites
The product selectivity of photocatalytic CO2 reduction processes is influenced by the quantity and species of surface reactive sites. Different active sites are appropriate for various surface catalytic processes, yielding various products. High-index facets can bring more surface low-coordinated atoms than low-index facets, which can serve as good active sites for CO2 activation and catalytic processes.
The product selectivity of CO2 photocatalytic reduction was heavily impacted by the size of Pt NPs. The smaller Pt NPs resulted in better H2O reduction selectivity for H2 production. The bigger Pt NP-modified HTSO can achieve a better CH4 selectivity. Surface terrace sites were more common in bigger Pt NPs, while low-coordinated locations (corner and edge) were more common in smaller Pt NPs (Fig. 30a). The DFT simulations also revealed that terrace sites can operate as active sites for CH4 production, while low-coordinated areas are better for converting H2O to H2.

(Copyright Elsevier License Number 5327490662508)
a Relationship between selectivity of CH4, the ratio of terrace/surface, and size of Pt particles. b Process of partial CO-modified 1.8PHTSO by CO stepwise adsorption/desorption. c CO-TPD results of 1.8PHTSO after CO pulse adsorption and partial CO-modified 1.8PHTSO by He flow desorption at 280 °C. d Photocatalytic CO2 reduction performance of 1.8PHTSO and CO-1.8PHTSO [164].
The partial CO-modified 1.8PHTSO (1.8 nm Pt modified HTSO) may cover the corner and edge sites of Pt particles, as shown in Fig. 30b, c. The photocatalytic performance of 1.8PHTSO and CO-1.8PHTSO is compared in Fig. 30d, demonstrating that the terrace sites were favored for CH4 production. The size of Pt modified the geometric feature's surface site percentage, which varied the product selectivity of photocatalytic CO2 reduction processes [164].
5.2.8 Intermediates/Side Products of Adsorption and Desorption
Good reactant adsorption and simple product desorption are required to yield targeted products with high selectivity. Many different intermediates or side products can be generated during the photocatalytic CO2 reduction process. Intermediate and side products differ in their strong interactions with photocatalysts. The species are difficult to desorb from the surface of the photocatalyst if the contact was too strong. It is more likely that a further hydrogenation reduction reaction will be carried out [9, 164]. Intermediaries is the name given to these species. The weak interaction, on the other hand, will make desorption and release from photocatalyst surfaces easier, and these species act as key side products. In this regard, the adsorption and desorption of intermediates/side products can have a significant impact on photocatalytic CO2 reduction reaction product selectivity [9, 164, 173].
6 Challenges and Opportunities
Energy scarcity and environmental issues are becoming increasingly serious global issues and major challenges for humanity in the twenty-first century. Solar-driven CO2 reduction to value-added chemical fuels is a viable option for influencing the global carbon balance, with tremendous potential rewards. The current goal is to create photoactive materials that can chemically tie these light-driven redox processes together, achieving conversion efficiencies and selectivity that are superior to natural photosynthesis. Thermodynamically upward, multi-electron, multihole, and multi-proton reactions occur on a multicomponent photocatalyst in the photoconversion of CO2. In the fields of catalysis, energy science, semiconductor physics and engineering, and green chemistry, long-standing difficulties are posed. Several key considerations must be balanced to develop effective CO2 fixation and conversion, including:
-
(i)
A thorough understanding of the processes that take place on the surface of photocatalysts during CO2 reduction, such as CO2 and intermediate product adsorption and desorption, as well as the role of adsorbed water. In this regard, the research would combine experimental and computational methods to assess catalyst activity for CO2 photoreduction, advancing fundamental knowledge in the fields of material science, surface chemistry, photocatalysis, and nanotechnology. STM would be used to determine CO2 adsorption locations and explore molecular orbitals and transfer of charge. The local density of states, lowest-energy molecule structure, and vibrational modes would theoretically be clarified using density functional theory (DFT) computations. STM and DFT working together would give a natural framework for future research targeted at gaining an atomic-scale knowledge of CO2 photoreduction.
-
(ii)
A significant increase in the lifespan of the charge-separated state could result in a significant increase in CO2 photoreduction efficiency. This electron–hole recombination is two to three orders of magnitude faster than other electron transfer processes in terms of time scale. As a result, any approach that prevents electron–hole recombination will considerably improve CO2 photoreduction efficiency and rates. To decrease electron–hole pair recombination, the photocatalyst's electrical conductivity and diffusion length should be as high as possible. Charge-carrier transfer to surface reaction sites for reductive chemistry may be aided by ultrathin nanostructures. The electron–hole recombination can be decreased by spatial separation of photoexcited electrons and holes, which can be done via co-catalysts, Z-schemes, or heterostructures linking two semiconductors with properly aligned band structures.
-
(iii)
Light absorption by the photocatalyst affects the kinetics of CO2 photoreduction. Many optical approaches, such as structuring for multiple light scattering to increase the effective optical path length and up-conversion to convert nonabsorbed infrared light to absorbed visible light, could be useful for harvesting light to improve efficiency.
-
(iv)
To maximize the adsorption, transport, and desorption of reactants, intermediates, and products, a large surface area and porosity are necessary.
-
(v)
Because defects like oxygen vacancies regulate the majority of the chemistry at many metal oxide surfaces, oxygen vacancies are thought to play a critical role in electron trapping and CO2 activation. These defects can be discovered by in situ electron paramagnetic resonance spectroscopy, ultraviolet photoemission spectroscopy, and metastable impact electron spectroscopy, among other techniques.
-
(vi)
A high-efficiency process must be based on photoactive materials that are abundant on the planet, non-toxic, light-stable, scalable, and affordable in cost. Furthermore, with prolonged irradiation, the efficiency of photocatalytic CO2 reduction may be deactivated.
7 Conclusion
It may be argued that environmental issues are no longer localized issues, but have evolved into global issues concerning climate change. We are still far from having a superb as well as a cost-effective photocatalyst for photocatalytic CO2 reduction with H2O to overcome these challenges. As previously stated, photocatalytic processes are quite intricate in terms of both physical mechanism and product distribution, which limits their usefulness. The current state of research in this field is perplexing, and evaluating the efficiency of various photocatalysts is challenging due to the wide range of influencing factors and reaction circumstances. Many of the studies neglected some details like mass balance (moles of CO2 transformed into the specified product), product distribution, and the amount of reducing agent, as well as time requirements, solution pH, temperature, CO2 pressure, light power, and activity decay with time. Comprehensive research into this method is required before it can be put into practice on a large scale. A single, uniform and widely agreed standard format should be created.
The major challenges responsible for the current significantly low rate of average productivity in photocatalytic reduction of CO2 with H2O are low photocatalytic efficiency, low response to sunlight, inefficient electron transport between reduction and oxidation catalysts, and a high recombination rate of photogenerated species. The short lifetimes of one electron-reduced species and the photoexcited state in the presence of O2 produced by H2O oxidation are considered also more drawbacks. Even though UV light has more energy than visible light, visible light harvesting photocatalysts are the most desirable for this process due to the plentiful visible light from natural sunlight. Plasmonic photocatalysts, which are still in their inception, have already shown promise in overcoming the first two flaws listed above. Because the boundaries of these extremely effective noble metal photocatalysts are quickly growing, it is reasonable to expect plasmonic photocatalysts to play a substantial role in future environmental solutions.
Fabrication of ideal structured visible light-responsive photocatalysts with a wide bandgap, high rate of photogenerated electron–hole transport, high rate of photogenerated electron–hole transport, and low rate of recombination, to increase the possibility for practical application of photocatalytic CO2 reduction with H2O should be the focus of future research.
References
Bard AJ, Fox MA (1995) Acc Chem Res 28:141
Aresta M, Dibenedetto A (2007) Dalton Trans 2975
Halmann M, Fox MA (1978) Nature 275:115
Inoue T, Fujishima A et al (1979) Nature 277:637
Meduri A, Fuoco T et al (2014) Macromolecules 47:534–543
Bolton JR (1978) Solar Fuels Sci 202:705–711
Lehn JM, Ziessel R (1982) Proc Natl Acad Sci USA 79:701–704
Hori Y, Vayenas CG, White RE, Gamboa-Aldeco ME (2008) Modern aspects of electrochemistry. Modern aspects of electrochemistry. Springer, New York, 89–189
Li X, Wen J, Low J, Fang Y, Yu J (2014) Sci China Mater 57:70–100
Fujita E (1999) Coordin Chem Rev 185:373–384
Morris AJ, Meyer GJ, Fujita E (2009) Acc Chem Res 42:1983–1994
Bard AJ, Parsons R, Jordan J (1985) Standard potentials in aqueous solution. Routledge, New York, pp 195–197
Yahaya AH, Gondal MA, Hameed A (2004) Chem Phys Lett 400:206–212
Benson EE, Kubiak CP, Sathrum AJ, Smieja JM (2009) Chem Soc Rev 38:89–99
Song C (2006) Catal Today 115:2–32
Kumar B, Llorente M, Froehlich J et al (2012) Annu Rev Phys Chem 63:541–569
Kanemoto M, Shiragami T, Pac C, Yanagida S (1992) J Phys Chem 96:3521–3526
Linsebigler A, Lu G, Yates JJ (1995) Chem Rev 95:735–758
Nakata K, Fujishima A (2012) J Photochem Photobiol C 13:169–189
Chen X, Shen S, Guo L, Mao SS (2012) Chem Rev 110:6503–6570
Maeda K, Domen K (2007) J Phys Chem C 111:7851–7861
A. Dhakshinamoorthy, S. Navalon, A. Corma A, H. Garcia, Energy Environ Sci 5 (2012) 9217–9233.
Indrakanti VP, Kubicki JD, Schobert HH (2009) Energy Environ Sci 2:745–758
Guan GQ, Kida T, Harada T, Isayama M, Yoshida A (2003) Appl Catal A-Gen 249(1):1–18
Mahmodi G, Sharifnia S, Rahimpour F, Hosseini SN (2013) Sol Energ Mat Sol C 111:31–40
Nunez J, O’Shea VAD, Jana P, Coronado JM, Serrano DP (2013) Catal Today 209:21–27
Xi GX, Ouyang SX, Ye JH (2011) Chem Eur J 17:9057–9061
Fujiwara H, Hosokawa H, Murakoshi K, Wada Y, Yanagida S (1998) Langmuir 14:5154–5159
Inoue H, Moriwaki H, Maeda K, Yoneyama H (1995) J Photoch Photobio A 86:191–196
Koci KK, Reli M, Kozak O et al (2011) Catal Today 176:212–214
Kuwabata S, Nishida K, Tsuda R, Inoue H, Yoneyama H (1994) J Electrochem Soc 141:1498–1503
Aurian-Blajeni B, Halmann M, Manassen J (1980) Sol Energy 25:165–170
Lee WH, Liao CH, Tsai MF, Huang CW et al (2013) J Appl Catal B-Environ 132:445–451
Sui DD, Yin XH, Dong HZ et al (2012) Catal Lett 142:1202–1210
Zhou H, Guo JJ, Li P et al (2013) Sci Rep 3:1667
Li H, Lei Y, Huang Y et al (2011) J Nat Gas Chem 20:145–150
Yamamura S, Kojima H, Iyoda J, Kawai W (1987) Electroanal Chem 225:287–290
Jamshidi K, Hyon S-H, Ikada Y (1988) Polymer 29(12)
Bessekhouad Y, Robert D, Weber JV (2005) Catal Today 101:315–321
Li T, Wang WN, Zhan ZL et al (2010) Appl Catal B-Environ 100:386–392
Robert D (2007) Catal Today 122:20–26
Tseng IH, Chang WC, Wu JCS (2002) Appl Catal B-Environ 37:37–48
Chaudhary YS, Woolerton TW, Allen CS et al (2012) 48:58–60
Fujiwara H, Hosokawa H, Murakoshi K et al (1997) J Phys Chem B 101:8270–8278
Li X, Chen J, Li H et al (2011) J Nat Gas Chem 20:413–417
Li X, Liu HL, Luo DL et al (2012) Chem Eng J 180:151–215
Liu BJ, Torimoto T, Yoneyama H (1998) J Photoch Photobio A 113:93–97
Praus P, Kozak O, Koci K, Panacek A, Dvorsky R (2011) J Colloid Interf Sci 360:574–579
Barton EE, Rampulla DM, Bocarsly AB (2008) J Am Chem Soc 130:6342–6344
Hara M, Nunoshige J, Takata T, Kondo JN, Domen K (2003), Chem Commun 3000–3001
Kim ES, Nishimura N, Magesh G et al (2013) J Am Chem Soc 135:5375–5383
Maeda K, Higashi M, Lu DL, Abe R, Domen K (2010) J Am Chem Soc 132:5858–5868
Sekizawa K, Maeda K, Domen K, Koike K, Ishitani O (2013) J Am Chem Soc 135:4596–4599
Mao J, Peng T, Zhang X et al (2013) Catal Sci Technol 3:1253–1260
Wang X, Maeda K, Thomas A et al (2009) Nat Mater 8:76–80
Zhang JS, Chen XF, Takanabe K et al (2010) Angew Chem Int Ed 49:441–444
Jiang H, Dai H, Meng X et al (2011) Appl Catal B-Environ 105:326–334
Jiang H, Meng X, Dai H et al (2012) J Hazard Mater 217–218:92–99
Kudo A, Omori K, Kato J (1999) J Am Chem Soc 121:11459–11467
Kudo A, Ueda K, Kato H, Mikami I (1998) Catal Lett 53:229–230
Tokunaga S, Kato H, Kudo A (2001) Chem Mater 13:4624–4628
Chun WJ, Ishikawa A, Fujisawa H et al (2003) J Phys Chem B 107:1798–1803
Higashi M, Domen K, Abe R (2011) Energy Environ Sci 4:4138–4147
Li Y, Takata T, Cha D et al (2012) Adv Mater 25:125–131
Ma SSK, Hisatomi T, Maeda K, Moriya Y, Domen K (2012) J Am Chem Soc 134:9993–19996
Anpo M, Yamashita H, Ichihashi Y, Ehara S (1995) Electroanal Chem 396:21–26
He H, Zapol P, Curtiss LA (2012) Energy Environ Sci 5:6196–6205
Liu G, Hoivik N, Wang K, Jakobsen H (2012) Sol Energ Mat Sol C 105:53–68
Pipornpong W, Wanbayor R, Ruangpornvisuti V (2011) Appl Surf Sci 257:10322–10328
Sasirekha N, Basha SJS, Shanthi K (2006) Appl Catal B-Environ 62:69–180
Subrahmanyam M, Kaneco S, Alonso-Vante N (1999) Appl Catal B-Environ 23:169–174
Wang J, Lin S, Tian N, Ma T, Zhang Y, Huang H (2021) Adv Funct Mater 31:2008008
Kong T, Jiang Y, Xiong Y (2020) Chem Soc Rev 49:6579–6591
Ku Y, Lee W, Wang W (2004) J Mol Catal A-Chem 212:191–196
Lo CC, Hung CH, Yuan CS, Wu JF (2007) Sol Energ Mat Sol C 91:1765–1774
Tahir M, Amin NS (2013) Appl Catal A-Gen 467:483–496
Tan SS, Zou L, Hu E (2008) Catal Today 131:125–129
Wasielewski MR (2009) Acc Chem Res 42:1910–1921
Zhou H, Qu Y, Zeid T, Duan X (2012) Energy Environ Sci 5:6732–6743
Mahmoud Idris A et al (2022) J Colloid Interface Sci 607(2):1180–1188
Li N et al (2022) ACS Nano 16(2):3332–3340
Wang F et al (2022) ACS Nano 16(3):4517–4527
Usman M et al (2022) J Envir Chem Engin 10(3)
Tahir M, Tahir B (2022) J Mat Sci Techno 106:195–210
Pachaiappan R et al (2022) Chem Engin Res Des 177:304–320
Guo K et al (2023) J Envir Sci 125:290–308
Chen P et al (2022) ACS Catal 12(8):4560–4570
Camera-Roda GF, Santarelli CA (2005) Martin. Sol Ener 79(4):343–352
Lais A et al (2018) Int J Ener Res 42(6):2031–2049
Paulino et al (2016) Appl Catal B: Envir 185:362–370
Han S et al (2017) J Ener Chem 26(4):743–749
Ali S et al (2019) Catalysts 9(9)
Kočí K et al (2011) Catal Today 176(1):212–214
Ola O et al (2012) Appl Catal B: Envir 126:172–179
Liou et al (2011) Ener Envir Sci 4(4)
Saladin et al (1997) Chem Soc Faraday Trans 93:4159–4163
Tan et al (2017) Chem Engin J 308:248–255
Zhang et al (2009) Catal Today 148(3–4):335–340
Wang W, Ku Y (2003) J Photochem Photobio A Chem 159(1):47–59
Mizuno T et al (1996) J Photochem Photobiol A Chem 98(1–2):87–90
Ola et al (2015) J Photochem Photobio C Photochem Rev 24:16–42
Neatu et al (2014) Int J Mol Sci 15(4):5246–5262
Yui T et al (2011) ACS Appl Mater Interfac 3(7):2594–2600
Ochedi et al (2021) Envir Chem Lett 19:941–967
Rehman ZU et al (2022) Molecules 27(2069):1–30
Nguyen TP et al (2020) Nanomaterials 10(337):1–24
Wang W, Wang L, Su W, Xing Y (2022) J CO2 Util 61:1–21
Madi M, Tahir M, Tasleem S (2021) Advances in structural modification of perovskite semiconductors for visible light assisted photocatalytic CO2 reduction to renewable solar fuels: a review. J Envir Chem Engi 9:1–39
You J, Xiao M, Wang Z, Wang L (2022) J CO2 Util 55:1–22
Ikreedeegh RR, Tahir M (2021) A critical review in recent development of metal-organic frameworks (MOFs) with band engineering alteration for photocatalytic CO2 reduction to solar fuels. J CO2 Util 43:1–41
Chen Y, Wang D, Deng X, Li Z (2013) J Name 00:1–3
Kumagai H, Tamaki Y, Ishitani O (2021) Acc Chem Res 55:978–990
Luo et al (2019) Coord Chem Rev 390:86–126
Shen J, Wu Z et al (2021) Flat Chem 28:1–11
Zhao Y, Que M, Chen J, Yang C (2020) J Mat Chem C 1–53
Li K, Zhang S, Li Y, Fan J, Lv K (2021) MXenes as noble-metal-alternative co-catalysts in photocatalysis. Chi J Catal 42:3–14
Do KH, Kumar et al (2020) Chem Cat Chem 12(18):1–9
Cui D, Wang et al (2018) ASC 6(12):15936–15953
Shen H, Peppel T, Strunk J, Sun Z (2020) Solar RRL 4(8):1900546
Ochedi FO, Liu D, Yu J, Hussain A, Liu Y (2021) Envir Chem Lett 19(2):941–967
Hasani A, Teklagne MA et al (2020) Carbon Energy 2(2):158–175
Vu NN et al (2019) Adv Funct Mat 29(31):1901825
Mondal A, Prabhakaran A, Gupta S, Subramanian VR (2021) ACS Omega 6(13):8734–8743
Tan LL, Ong WJ, Chai SP, Mohamed AR (2017) Chem Engin J 308:248–255
Han C, Li J, Ma Z et al (2018) Sci Chi Mat 61(9):1159–1166
Wang J, Qin C, Wang H et al (2018) Appl Catal B Envir 221:459–466
Li G, Lian Z, Wang W, Zhang D, Li H (2016) Nano Energy 19:446–454
Song X, Li X, Zhang X, Wu Y, Ma C, Huo P, Yan Y (2020) Fabricating C and O co-doped carbon nitride with intramolecular donor-acceptor systems for efficient photoreduction of CO2 to CO. Appl Catal B Envir 268:118736
Shankar R, Sachs M, Francàs L et al (2019) J Mat Chem A 7(41):23931–23940
Cao Y, Zhang R, Zhou T, Jin S, Huang J, Ye L, Huang Z, Wang F, Zhou Y (2020) ACS Appl Mat Interf 12(8):9935–9943
Chen C, Hu J, Yang X, Yang T, Qu J, Guo C, Li CM (2021) ACS Appl Mat Interf 13(17):20162–20173
Zhu X, Huang S, Yu Q, She Y, Yang J, Zhou G, Li Q, She X, Deng J, Li H, Xu H (2020) Appl Catal B Envir 269:118760
Liu Y, Wang B, Li D, Shen J, Zhang Z, Wang X (2022) J Coll Interf Sci 622:31–39
Wu R, Zhou K, Yue CY, Wei J, Pan Y (2015) Progr Mat Sci 72:1–60
Wang Y, Zhang L, Zhang X et al (2017) Appl Catal B Envir 206:158–167
Li H, Sun J (2021) ACS Appl Mat Interf 13(4):5073–5078
Lu M, Liu J, Li Q, Zhang M, Liu M et al (2019) Angew Chem Int Ed 58(36):12392–12397
Fu Z, Wang X, Gardner AM, Wang X et al (2020) Chem Sci 11(2):543–550
Cui J et al (2021) J Mat Chem A 9(44):24895–24902
Low J, Zhang L, Tong T, Shen B, Yu J (2018) J Catal 361:255–266
Xu D, Cheng B, Wang W, Jiang C, Yu J (2018) Appl Catal B Envir 231:368–380
Crake A, Christoforidis KC, Godin R, Moss B, Kafizas A, Zafeiratos S, Durrant JR, Petit C (2019) Titanium dioxide/carbon nitride nanosheet nanocomposites for gas phase CO2 photoreduction under UV-visible irradiation. Appl Catal B Envir 242:369–378
Wang CC, Wang X, Liu W (2019) Chem Engin J
Li R et al (2014) Adv Mater 26(28):4783–4788
Crake A, Christoforidis KC, Kafizas A, Zafeiratos S, Petit C (2017) Appl Catal B Envir 210:131–140
Wang Z, Huang J, Mao J, Guo Q, Chen Z, Lai Y (2020) J Mat Chem A 1–30
Zhang W, Mohamed AR, Ong WJ (2020) Angew Chem Int Ed 59(51):22894–22915
Cao S, Shen B, Tong T, Fu J, Yu J (2018) Bioorg Med Chem Lett 28(21):2109–2115
Lizuka K, Wato T, Miseki Y, Saito K, Kudo A (2011) J Am Chem Soc 133:20863–20868
Varghese O, Paulose M, LaTempa T, Grimes C (2009) Nano Lett 9:731–737
Peng YP, Yeh YT, Shah SI, Huang C (2012) Appl Catal B 414–423
Lin W, Han H, Frei H (2004) J Phys Chem B 108:18269–18273
Asi MA, He C, Su M, Xia D, Lin L, Deng H, Xiong Y, Qiu R, Li X (2011) Catal Today 175:256–263
Teramura K, Iguchi S, Mizuno Y, Shishido T, Tanaka T (2012) Angew Chem Int Ed 51:8008–8011
Nguyen TV, Wu JCS (2008) Sol Energy Mater Sol Cells 92:864–872
Matsuoka S, Yamamoto K, Ogata T, Kusaba M, Nakashima N, Fujita E, Yanagida S (1993) J Am Chem Soc 115:601–609
Yanagida S, Kanemoto M, Ishihara K, Wada Y, Sakata T, Mori H (1997) Bull Chem Soc Jpn 70:2063–2070
Koci K, Matejka V, Kovar P, Lacny Z, Obalova L (2011) Catal Today 161:105–109
Rakibuddin M, Kim HY (2019) Beilstein J Nanotechnol 10:448–458
Irvine J, Eggins B, Grimshaw J (1990) Sol Energy 45:27–33
Li K et al (2016) ACS Catal 6:7485–7527
Fu J et al (2017) Small 13:1603938
Fu J, Jiang K, Qiu X, Yu J, Liu M (2020) Product selectivity of photocatalytic CO2 reduction reactions. Mater Today 32:223
Vasileff A et al (2018) Chem 4:1809–1831
Low J et al (2018) J Catal 361:255–266
Chang X et al (2016) Energy Environ Sci 9:2177–2196
Hattori Y et al (2018) Mater Today 21:590–593
Ran J et al (2018) Adv Mater 30:1704649
Xu F et al (2018) Appl Catal B 230:194–202
Shi G et al (2018) Appl Surf Sci 427:1165–1173
Zheng Y et al (2017) Nano Energy 40:512–539
D’Alessandro DM et al (2010) Angew Chem Int Ed 49:6058–6082
Han B et al (2018) Angew Chem Int Ed 57:16811–16815
Karamian E, Sharifnia S (2016) J CO2 Util 16:194–203
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Gemeda, T.N. et al. (2023). A Recent Review on Photocatalytic CO2 Reduction in Generating Sustainable Carbon-Based Fuels. In: Abdullah, H. (eds) Photocatalytic Activities for Environmental Remediation and Energy Conversion. Green Energy and Technology. Springer, Singapore. https://doi.org/10.1007/978-981-19-6748-1_4
Download citation
DOI: https://doi.org/10.1007/978-981-19-6748-1_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-6747-4
Online ISBN: 978-981-19-6748-1
eBook Packages: EnergyEnergy (R0)