
Abstract
The rhizosphere is a composite ecosystem which supports multiple bacterial populations that nourishes the terrestrial biosphere and plays a crucial role in the continuous recycling of minerals, nutrients, and organic matter through the soil. Diverse varieties of molecular tools based on immediate isolation and analysis of various compounds from environmental samples such as lipids, nucleic acids, and peptides have been discovered which have provided structural and functional data about microbial communities present in rhizospheric soil. With the advent of next-generation sequencing technologies (NGS), it has become possible to delve deeper into the rhizosphere microbiome to understand the unknown aspects of it. This has resulted in a shift from traditional approaches to the modern omics-based approach based on NGS sequencing technologies for discovering and distinguishing the vast microbial diversity to understand their interactions with different environmental factors. The major objective of this chapter is to provide insights on structural and functional rhizospheric microbial diversity analysis by the application of cutting-edge biotechnological tools. We have first glanced through the basic concepts of rhizosphere and its importance in plant system, the common rhizospheric microbial population, and looked at the plant–microbe interactions which are of prime importance in the rhizosphere ecosystem. Next, we come to the molecular tools used for rhizospheric microscopic diversity analysis—a detailed view into a few of the traditional approaches used for diversity approaches before proceeding to the rapidly emerging and more popular omics-based approaches used for rhizosphere microbial diversity analysis. We have also identified the merits and demerits, future opportunities of omics-based approaches in rhizosphere microbiology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
9.1 Introduction
Plant-associated microbes have been known to function as nitrogen fixers, zinc solubilizers, phosphorous solubilizers, potassium solubilizers, absorbers, accumulators, conversion of essential nutrients into a form that the plant can uptake, source of antibiotic and antifungal agents, source of commercially viable enzymes, inducer for production of plant hormones, the key to developing a tolerance for biotic and abiotic stress such as salinity, acidity, alkalinity, excess soil moisture, drought conditions, and extremely high or extremely low temperatures (Ali et al. 2018; Tanim et al. 2019). These microbes influence plant growth, and the growth of the plant and environmental stressors influence the structural and functional dynamics of such microbial niches. Majority of the plant-associated microbe population is formed by the microbiota colonizing the rhizosphere of the plant. The microbes show enforced as an ecological consequence of the influence of root exudates on which they thrive. This microbiota is extremely influential and diverse has immense applications in the field of biotechnology with significant agricultural and commercial importance.
The multiform microbiome carries out and regulates a variety of processes occurring in the rhizosphere of the plant that are relevant to plant proliferation. Betaproteobacteria act as nitrogen fixers by denitrification and nitrate reduction. Acinetobacter is known for the bioremediation of xenobiotic compounds. Gammaproteobacteria adapt for suitable growth in a rhizospheric environment requiring resistance to metal toxicity and metal reduction to get rid of the contaminant for bioremediation and enriching the soil to make it fertile. Pseudomonas and Bacillus species are known to act as heavy metal detoxifiers and participate in phosphorous solubilization. Clostridia exhibits the ability to reduce heavy metals through hydrogen metabolism and fermentation (Ghosh et al. 2019).
The increase in industrial activities over the past few decades and the intensive agricultural activities undertaken to meet heightened demand for crop production has resulted in pertinent inorganic and organic pollution. Metal pollution has led to excessive degradation of soil quality and adversely affected plant health as well as microbial population composition (Benidire et al. 2020). As a consequence of this, the use of biofertilizers involving the application of the rhizosphere-associated biome for assisting its growth by improving soil conditions to benefit the environment has been championed as an alternative for chemical fertilizers. Certain bacterial colonies in the plant roots act as bio-inoculants increasing the fertility of the soil via their metabolism and can be isolated for bioremediation purposes. The application of biofertilizers is also encouraged owing to the increasing popularity of organic farming. Some plant species are hyperaccumulators, with the rhizospheric communities regulating the uptake of metal ions contaminating the soil. Microbial diversity analysis is essential for determining the contaminant degradation ability of the various species in a microbial population. Microbial diversity can also help understand the biological impacts of certain farming practices on crop production (Wang et al. 2019), verify the presence of antibiotic and antifungal agent producing strains (Ali et al. 2018), or to isolate any other strain from the concerning rhizosphere responsible for the production of specific enzyme or metabolite.
Many molecular tools have been developed such as the traditionally employed techniques Amplified Ribosomal DNA Restriction Analysis (ARDRA) and Denaturing Gradient Gel Electrophoresis (DGGE) for microbial diversity analysis to facilitate understanding of the community dynamics and the microbial metabolic processes that occur in the rhizosphere. Both cultivation-based approach and DNA-based approach have been employed for this purpose. With the recent advancements in technological high-throughput sequencing tools with a reduced expense, time consumption, and hassle have been developed. These sequencing tools are next-generation sequencers that are utilized in the study of the entire genetic material present in a microbial community referred to as metagenomics. The sequencing tools are also employed in metatranscriptomics which is the study of gene expression and interaction, metaproteomics which refers to the characterization analysis of the protein expressed in the microbial, and metabolomics which refers to the study of the significant metabolites and exudates present in an ecological system. Metagenomics, metatranscriptomics, metaproteomics, and metabolomics studies can be and have been applied in numerous plant-related researches to understand and define the rhizospheric microbiota of the given plant and the specificity of the plant–microbe interactions. The significance of the rhizospheric biota for plant proliferation has been well acknowledged and analyzing their microbial diversity is important for characterizing their relevant functional traits (Singh et al. 2020).
9.2 What is Rhizosphere?
The rhizosphere is the region of the soil that can be defined as the zone that is immediately surrounding the root of a plant that houses a diverse range of microbial colonies (Ali et al. 2018). Different plant species have complex and unique microflora associated with them. This microflora may compose of bacteria, fungi, viruses, and archaea. Along with the host plant, these microbes form a delicately balanced ecosystem, and they may or may not benefit from the presence of these microbes that are found on their leaves, flowers, stems, and roots. The rhizosphere is inhabited by the root-associated microbes which maintain either a symbiotic or non-symbiotic relationship with the plant. These microbes thrive on the root exudates of the host plant and therefore the rhizosphere forms an agreeable niche for the microbial population. These microbes, being present at the soil–root interface of the plant exerts a significant influence over the rhizospheric processes that take place (de los Reyes et al. 2020). These microbial interactions are very distinctive based upon the microbial species. The evolution of the soil microflora of an individual plant has occurred simultaneously with the evolution of the plant itself over time (Ghosh et al. 2019). Therefore, the microbiome of the rhizosphere is unique to the specific host plant.
9.3 Rhizospheric Diversity
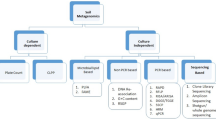
Any soil sample when taken and analyzed for microbial diversity shows the soil biota representing a huge number of microbial species. The diversity which is shown by a microbial community heavily depends upon the physicochemical properties of the soil which they inhabit. At the root–soil interface, the soil habitat shows an increase in favorable conditions to promote microbial colonization, and a higher density and diversity of microbes is thus seen in rhizospheric soil when compared to the bulk soil. These microbes vary in their function from site to site and species to species (Ghosh et al. 2019). The rhizosphere of a plant houses a plethora of culture-dependent and culture-independent microbial species. Microbial colonies found in the rhizospheric niches of the plant vary from species to species and influence crop production, productivity, and plant sustainability (Fig. 9.1). An abundance is witnessed in the number and highly diverse variety of microbial species occupying the rhizospheric niche.

Microbial interaction in the plant rhizosphere for nitrogen fixation, production of biologically active compounds and phytoremediation
9.3.1 Common Rhizospheric Population
Azobacter, Azospirillium, Azolla, Rhizobium, and Cyanobacteria species are common microbes generally that are known for their nitrogen-fixing abilities. These symbiotic microbes are Gram-negative, aerobic bacteria. These nitrogen-fixing bacteria act as natural fertilizers impacting the plant metabolism, production of antibiotics, and plant growth hormones, root development and allowing for enhanced nutrient uptake. Pseudomonas, Bacillus, Actinomyces, Agrobacterium, and Acetobacter are known to be phosphorous-solubilizing bacteria. Apart from partaking in photosynthesis, energy transfer, and other plant processes, phosphorus is an important element because it is known to limit the fixation of nitrogen. Presence of the aforementioned bacterial species in the rhizosphere is not only limited to solubilizing phosphorus for plant uptake to regulate the metabolic processes of the plant but also enhance the nitrogen fixation process. Pseudomonas and Bacillus species also commonly facilitate the solubilization of potassium. Various Pseudomonas, Bacillus, Rhizobium, and Azospirillum species have been classified as plant growth-promoting rhizobacteria (PGPR). They are important for plant proliferation with their ability to serve as biocontrol. These species have also been reported as being capable of solubilizing zinc (Reddy et al. 2020).
9.3.2 Plant-Microbe Interaction
The evolution of the microbial strains is majorly affected by environmental conditions and plant species. The microbes evolve as to facilitate growth-promoting activities, heightened nitrogen-fixing ability, act as a metal detoxifier, synthesis of biologically active compounds, and combat other the abiotic and biotic stresses its native plant species is subjected to. The plant–microbe interaction is governed by complex physiological and biochemical activities that facilitate communication.
The rhizospheric microbial interactions are very defined and specific in nature since the structural and functional diversity of the bacterial population is greatly influenced by the soil environment around the rhizosphere and the requirement of the native plant. A relative increase in microbial density and microbial activity is seen in rhizospheric soil when compared to non-rhizospheric soil, where the presence of such dense and hyperactive microbial niches is not observed (Shu et al. 2012). The growth stage of a plant has been reported to result in a marked change in the microbial community composition and dynamics. The plant species is a major determining factor for the community structure of the rhizosphere. Rhizobium harboring legumes is one such example (de los Reyes et al. 2020).
The plant–microbe interaction has made phytoremediation as a popular choice for bioremediation to keep in check contaminants that are found as a result of industrial activities and intensive farming methods. The class of Alphaproteobacteria, associated with the rhizosphere of the O. basilicum plant, has been known not only to tolerate high levels of polychlorobiphenyl in soil but also to exhibit major polychlorobiphenyl degradation activities. It has been reported that the rhizosphere has a significant effect on the bacterial genus isolated from the roots of a plant and the metabolic process of the said bacteria (Sánchez-Pérez et al. 2020). Therefore, the rhizospheric microflora ensures the efficiency of the phytoremediation process, by enhancing the contaminant removal potential.
9.4 Molecular Tools for Rhizospheric Microbial Diversity Analysis
9.4.1 Traditional Molecular Tools
Microbial community diversity analysis was conducted at the sites mentioned in the following table using traditional molecular tools approach which includes techniques such as ARDRA (amplified ribosomal DNA restriction analysis), RFLP (restriction fragment length polymorphism), DGGE (denaturing gradient gel electrophoresis), TGGE (temperature gradient gel electrophoresis), and RISA (ribosomal intergenic spacer analysis). The common bacterium phyla that were reported to be present predominantly were Proteobacteria, Firmicutes, and Actinobacteria. The analysis of microbial diversity at different sites shows that the dominant species in the rhizospheric microbial population of various plants were mostly Pseudomonas, Bacillus, and Rhizobium (Fig. 9.2; Table 9.1).

Molecular tools for characterizing rhizospheric microbial diversity
9.4.1.1 ARDRA
Amplified ribosomal DNA restriction analysis (ARDRA) is a molecular technique that mirrors the RFLP technique except that it applies to the 16s ribosomal subunit of culture-independent bacteria. The steps involved are amplification, digestion, and gel electrophoresis. The PCR amplified fragment with restriction endonuclease is resolved with electrophoresis gel and a pattern is obtained. The methods used to analyze RAPD patterns are also used to analyze ARDRA patterns. NT-SYS and PAST are used for information about whether bands are present or not (1’s and 0’s for presence and absence, respectively). These patterns can be used to create phylograms or phylogenetic trees which describe the restriction pattern and give a relationship between organisms. The most common softwares used are GelCompar II and BioNumerics. Clones are amplified with primers and digested by restriction endonucleases and the resultant fragments are separated by acrylamide gels. The resultant profiles can be used for community clustering in genotyping or strain typing (Ying et al. 2012). The advantages of ARDRA are quick analysis of variations, multiple strains, and species. The drawback of this method is the inability to provide information about the sample microorganisms and the requirement of large quantities of DNA. Similar to the outcome of ribotyping, this method is faster but is not much sensitive.
9.4.1.2 DGGE
Denaturing gradient gel electrophoresis (DGGE) is another culture-independent method for analysis of microbial community which yields a visual fingerprint representation of the microbial community. The axis of a polyacrylamide gel is exposed to a denaturing gradient. Primers are used for PCR amplification after which the DNA undergoes gel electrophoresis by the denatured gel. Change in melting temperatures due to variations cause different migration ultimately leading to separation. The DNA separates due to the melting domains which are changed from the denaturation whose branching patterns are analyzed. The change in denaturation concentration leads to a change in the migration rate which in turn leads to a banding pattern. By comparing these patterns and known sequences, the various species in the sample can be identified and analyzed (Nimnoi et al. 2011). Digital image analysis can be used to interpret the DGGE profiles. By comparing distances between the migrated and reference strains, species can be identified and classified. This analysis technique poses one drawback as its bands only generate partial sequences. As a molecular tool, this method is often mentioned as a pair with temperature gradient gel electrophoresis (TGGE). Around 95–99% of microbial diversity in a community can be identified through this method.
9.4.1.3 RISA
Ribosomal intergenic spacer analysis (RISA) is an analysis method of community fingerprinting. It involves PCR amplification, electrophoresis, and staining. The region between the two subunits, namely 16S and 23S, is called the intergenic spacer region. The PCR amplification of this region is the main step in RISA (Srivastava et al. 2016). RISA fragments are obtained from specific regions in both the subunits which have been targeted with oligonucleotide primers. Based on the microbial species, tRNAs are encoded by elements of the intergenic spacer region. For the most effective length and sequence heterogeneity, the ISR length ranges from 150 to 500 base pairs. Multiple dominant community members result in a mixed PCR product. This is followed by electrophoresis by polyacrylamide gel. Visualization of the resultant DNA is possible after staining. A complex banding pattern consisting of DNA bands that correspond to a bacterial population gives a community-specific profile. It has been used for soil sustainability studies, species population studies, etc.
9.4.2 Omics-Based Rhizospheric Microbial Diversity
Omics-based approaches involve the use of data available from multiple omics fields including genomics (DNA, genomes), transcriptomics (gene transcripts or mRNA expressions), proteomics (proteins, proteomes), and metabolomics (metabolites, metabolome) for the purpose of microbial diversity analysis for an environmental sample. The following table (Table 9.2) provides a summary of sites where omics-based technologies such as metagenomics, functional metagenomics, metatranscriptomics, metaproteomics, and metabolomics have been used to perform microbial diversity analysis for the rhizosphere. The most common bacteria phyla reported to be present predominantly in these studies are Proteobacteria, Acidobacteria, and Firmicutes.
9.4.2.1 Metagenomics (Using all NGS platforms)
Metagenomics is the study of genetic materials collected immediately from an environment. It is one of the most commonly used culture-independent method and first of the omics methods to be developed for the study of microbial diversity including unculturable soil microbial community. It involved sequencing of genes available directly from environmental samples and analysis of the sequence generated which can then be used for various data analysis and predictions. Sequencing of data is done by the various platforms of NGS, most popular of them being Illumina sequencing followed by 454 sequencing and pyrosequencing. Metagenomics approach has been widely used to characterize microbial communities of any environmental habitat (Andreote et al. 2012; Puranik et al. 2016; Wei et al. 2017). Metagenomic analysis has been used in the study of microbiome present in soil contaminations and other aspects related to it (Kumar et al. 2018; Ridl et al. 2016). Whole metagenome sequencing has been used to study the role of rice rhizosphere in the metabolism of methane and nitrogen (Bhattacharyya et al. 2016) and effect of a specific strain of bacillus on lettuce rhizosphere microbial community (Kröber et al. 2014). Studies have also been conducted on bacterial diversity of specific crop species (Knief et al. 2012; Uroz et al. 2010) as well as utilization of compound present in rhizosphere soil (Unno and Shinano 2012) thereby revealing the wide use of this method in diverse fields of microbiology.
9.4.2.2 Functional Metagenomics
Functional metagenomic approach is a type of metagenomic approach which focuses on the studying of gene function from a mixed population of DNA. It involves construction and screening of metagenomic libraries which help in annotations of gene function. It involves the identification of functional gene during screening without using previously retrieved sequenced genes enabling the identification of both novel and known genes (Mirete et al. 2015). This approach has been widely used to study functional traits in rhizosphere community selection, functional potentials of various ecosystems along with determining the community diversity (Alzubaidy et al. 2016; Bai et al. 2014; Mendes et al. 2014; Yan et al. 2017). This approach has been used for discovery of resistance genes in a particular ecological habitat (Mirete et al. 2015; Willms et al. 2019) and study of the effect on pollutant removal in ecological wastewater (Bai et al. 2017).
9.4.2.3 Metatranscriptomics
Metatranscriptomics studies gene expression of microbes (RNA expressions or gene transcripts) within natural environments, i.e., metatranscriptome. Metatranscriptome provides information about the active metabolic processes of the microbiome in a given condition in an environment (Kothari et al. 2017). Metatranscriptomics can retrieve and sequence mRNAs from an environmental microbial community without any previous idea of the genes that might be expressed by the community thereby making it advantageous and less biased (Poretsky et al. 2009). NGS is the preferred technique for sequencing in metatranscriptomics with Illumina sequencing and 454 sequencing being the most popular platforms. Metatranscriptome analysis has been used for studying microbial diversity and host–microbiome interactions of various plant species (Cao et al. 2015; Rampadarath et al. 2018). Effects on the rhizosphere due to various factors of soil, soil contamination, developmental stages of plants have been investigated through this approach (Chaparro et al. 2014; Yergeau et al. 2018). Comparative studies based on metatranscriptomics have been performed among various samples to reveal the variations and effects of changing factors such as plant species, day timings, or soil types (Hayden et al. 2018; Poretsky et al. 2009; Turner et al. 2013).
9.4.2.4 Metaproteomics
Metaproteomics refers to the study of all proteins and peptides present in environmental samples. Mass spectrometry combined with various chromatography techniques is usually used for extraction and analysis of protein samples and latest advancements in this field has resulted in various high efficiency analyzing systems for protein and peptide identification. Metaproteomics is used for the characterization of soil microbial communities, biological samples based not only on taxonomy but also their functional activity and protein expression. For soil ecosystems, it provides an analysis of the functional proteins and helps in understanding various metabolic processes and signal transductions involved in the soil biotic community. Metaproteomic analysis and characterization have been done in different soil systems and crops giving an insight into the metabolic activities inside the soil by the microbes or the plants, molecular (secreted protein) interactions between microbes and plants and the effect of the proteins on rhizosphere community (Bona et al. 2019; Knief et al. 2012; Mattarozzi et al. 2017; Wang et al. 2011). Apart from characterizing bacterial community, metaproteomics analysis can be used to evaluate the role of particular microbes in specific processes such as nitrogen fixation, methane oxidation (Bao et al. 2014), or effect of fertilizers and related crop yield improvement (Chen et al. 2019).
9.4.2.5 Metabolomics
Metabolomics is the whole-community sampling of all the metabolites (i.e., sugars, lipids) represented within a microbial community, representing the functional substrate and products of metabolomic pathways within an ecosystem (White et al. 2017). Metabolomics uses mass spectroscopy-chromatography-based systems for analyzing metabolites to draw conclusions about different cellular, biochemical processes, interactions with environmental factors, and microbial contributions to metabolic varieties thereby determining the functional diversity of given microbial samples based on their biochemical activity. It has been used for studying the effects of specific exogenous metabolites or its hydrolyzed products produced on the rhizospheric microbial community and also evaluating the effect of engineered plants with altered metabolic profiles (Bressan et al. 2009, 2013). Non-sterile soils have been studied for the presence of plant-derived metabolites and their suitable application for crop agriculture (Pétriacq et al. 2017). Using a combined approach of comparative genomics and exometabolomics, it has been demonstrated that root exudate chemistry during developmental stages of a plant and microbial preferential substrate uptake is correlated and aid in predicting microbial response to root growth apart from determining the rhizospheric diversity (Zhalnina et al. 2018). Along with these, the effect of diverse soil microbiome on leaf metabolome, plant growth patterns and herbivore feeding habits has also been successfully investigated (Badri et al. 2013).
9.5 Pros and Cons of the Omics-Based Approach
Omics-based approaches have gained rapid popularity among researchers as they have made it possible to study entire genome sequences, transcripts, proteins, and metabolites from environmental samples providing a more comprehensive view of genetic diversity of the culture-independent microbiome. They provide more in-depth information about composition, the function of a whole microbial community, their interactions with biotic/abiotic factors, metabolic processes occurring inside the soil microbe community (Lagos et al. 2015). However, these approaches have a few drawbacks such as huge data volume and complex data for data analysis, complex extraction methods of analytes from samples, shorter read length of NGS platforms leading to overestimation of taxonomic classifications, high error rate (Pal et al. 2019), and lack of adequate libraries and databases.
9.6 Future Prospects
Future prospects include advancements in existing techniques with the objective of discovering the lesser-known rhizospheric bacterial communities; predict the responses of various species in their native environment, activities, and prosperity of such communities in soil (Lagos et al. 2015). Advancements in software development are needed for qualitative improvement of data generated from recent NGS platforms, developing robust reference libraries, spectral databases for metaproteomic and metabolomic studies, and adequate data analysis tools to make an integrated multi-omics approach possible (White et al. 2017). An integrated multi-omics study will enable us to look at the complete picture of soil microbiome with all its aspects and will help in deciding and implementing strategies for the selection of native bacterial strains capable of beneficial use, to comprehend the significance and function of the rhizosphere microbial activity to support healthy plant growth, improved yield and much more.
References
Abou-Shanab RA, Van Berkum P, Angle JS, Delorme TA, Chaney RL, Ghozlan HA, Ghanem K, Moawad H (2010) Characterization of Ni-resistant bacteria in the rhizosphere of the hyperaccumulator Alyssum murale by 16S rRNA gene sequence analysis. World J Microbiol Biotechnol 26(1):101–108
Ali A, Mustofa M, Asmara W, Rante H, Widada J (2018) Diversity and functional characterization of antifungal-producing Streptomyces-like microbes isolated from the rhizosphere of cajuput plants (Melaleuca leucodendron L.). Malaysian J Microbiol 14(7):663–673
Alzubaidy H, Essack M, Malas TB, Bokhari A, Motwalli O, Kamanu FK, Jamhor SA, Mokhtar NA, Antunes A, Simões MF, Alam I (2016) Rhizosphere microbiome metagenomics of gray mangroves (Avicennia marina) in the Red Sea. Gene 576(2):626–636
Andreote FD, Jiménez DJ, Chaves D, Dias ACF, Luvizotto DM, Dini-Andreote F, Fasanella CC, Lopez MV, Baena S, Taketani RG, de Melo IS (2012) The microbiome of Brazilian mangrove sediments as revealed by metagenomics. PLoS One 7(6):e38600
Badri DV, Zolla G, Bakker MG, Manter DK, Vivanco JM (2013) Potential impact of soil microbiomes on the leaf metabolome and on herbivore feeding behavior. New Phytol 198(1):264–273
Bai Y, Liang J, Liu R, Hu C, Qu J (2014) Metagenomic analysis reveals microbial diversity and function in the rhizosphere soil of a constructed wetland. Environ Technol 35(20):2521–2527
Bai Y, Huo Y, Liao K, Qu J (2017) Influence of microbial community diversity and function on pollutant removal in ecological wastewater treatment. Appl Microbiol Biotechnol 101(19):7293–7302
Bao Z, Okubo T, Kubota K, Kasahara Y, Tsurumaru H, Anda M, Ikeda S, Minamisawa K (2014) Metaproteomic identification of diazotrophic methanotrophs and their localization in root tissues of field-grown rice plants. Appl Environ Microbiol 80(16):5043–5052
Benidire L, Pereira SI, Naylo A, Castro PM, Boularbah A (2020) Do metal contamination and plant species affect microbial abundance and bacterial diversity in the rhizosphere of metallophytes growing in mining areas in a semiarid climate? J Soils Sediments 20(2):1003–1017
Bhattacharyya P, Roy KS, Das M, Ray S, Balachandar D, Karthikeyan S, Nayak AK, Mohapatra T (2016) Elucidation of rice rhizosphere metagenome in relation to methane and nitrogen metabolism under elevated carbon dioxide and temperature using whole genome metagenomic approach. Sci Total Environ 542:886–898
Bona E, Massa N, Novello G, Boatti L, Cesaro P, Todeschini V, Magnelli V, Manfredi M, Marengo E, Mignone F, Berta G (2019) Metaproteomic characterization of the Vitis vinifera rhizosphere. FEMS Microbiol Ecol 95(1):204
Bressan M, Roncato MA, Bellvert F, Comte G, el Zahar HF, Achouak W, Berge O (2009) Exogenous glucosinolate produced by Arabidopsis thaliana has an impact on microbes in the rhizosphere and plant roots. ISME J 3(11):1243–1257
Bressan M, Achouak W, Berge O (2013) Exogenous glucosinolate produced by transgenic Arabidopsis thaliana has an impact on microbes in the rhizosphere and plant roots. Mol Microb Ecol Rhizosphere 1:1173–1179
Cao HX, Schmutzer T, Scholz U, Pecinka A, Schubert I, Vu GT (2015) Metatranscriptome analysis reveals host-microbiome interactions in traps of carnivorous Genlisea species. Front Microbiol 6:526
Chaparro JM, Badri DV, Vivanco JM (2014) Rhizosphere microbiome assemblage is affected by plant development. ISME J 8(4):790–803
Chen J, Arafat Y, Din IU, Yang B, Zhou L, Wang J, Letuma P, Wu H, Qin X, Wu L, Lin S (2019) Nitrogen fertilizer amendment alter the bacterial community structure in the rhizosphere of rice (Oryza sativa L.) and improve crop yield. Front Microbiol 10:2623
Da Silva AC, da Costa Rachid CT, de Jesus HE, Rosado AS, Peixoto RS (2017) Predicting the biotechnological potential of bacteria isolated from Antarctic soils, including the rhizosphere of vascular plants. Polar Biol 40(7):1393–1407
de los Reyes AM, Ocampo ET, Manuel MC, Mendoza BC (2020) Analysis of the bacterial and fungal community profiles in bulk soil and rhizospheres of three mungbean [Vigna radiata (L.) R. Wilczek] genotypes through PCR-DGGE. Int Lett Nat Sci 77:1–26
Ghosh UD, Singh PK, Ganguli S, Saha C, Chandra A, Seal A, Ghosh MM (2019) Rhizospheric soil of Typha angustifolia L. from heavy metal contaminated and free sites: comparative profiling reveals selective abundance of γ-proteobacteria and β-proteobacteria. IJEB 57(10):733–740
Hayden HL, Savin KW, Wadeson J, Gupta VV, Mele PM (2018) Comparative metatranscriptomics of wheat rhizosphere microbiomes in disease suppressive and non-suppressive soils for Rhizoctonia solani AG8. Front Microbiol 9:859
Ibekwe AM, Poss JA, Grattan SR, Grieve CM, Suarez D (2010) Bacterial diversity in cucumber (Cucumis sativus) rhizosphere in response to salinity, soil pH, and boron. Soil Biol Biochem 42(4):567–575
Knief C, Delmotte N, Chaffron S, Stark M, Innerebner G, Wassmann R, Von Mering C, Vorholt JA (2012) Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J 6(7):1378–1390
Kothari V, Kothari C, Rank J, Joshi A, Singh RP, Kothari R (2017) Metatranscriptomic studies of the plant rhizosphere for finding biological agents. In: Understanding host-microbiome interactions-an omics approach. Springer, Singapore, pp 267–275
Kröber M, Wibberg D, Grosch R, Eikmeyer F, Verwaaijen B, Chowdhury SP, Hartmann A, Pühler A, Schlüter A (2014) Effect of the strain Bacillus amyloliquefaciens FZB42 on the microbial community in the rhizosphere of lettuce under field conditions analyzed by whole metagenome sequencing. Front Microbiol 5:252
Kumar V, AlMomin S, Al-Aqeel H, Al-Salameen F, Nair S, Shajan A (2018) Metagenomic analysis of rhizosphere microflora of oil-contaminated soil planted with barley and alfalfa. PLoS One 13(8):e0202127
Lagos L, Maruyama F, Nannipieri P, Mora ML, Ogram A, Jorquera MA (2015) Current overview on the study of bacteria in the rhizosphere by modern molecular techniques: a mini-review. J Soil Sci Plant Nutr 15(2):504–523
Lopez-Fuentes E, Ruiz-Valdiviezo VM, Martinez-Romero E, Gutierrez-Miceli FA, Dendooven L, Rincon-Rosales R (2012) Bacterial community in the roots and rhizosphere of Hypericum silenoides Juss. 1804. Afr J Microbiol Res 6(11):2704–2711
Marquez-Santacruz HA, Hernandez-Leon R, Orozco-Mosqueda MD, Velazquez-Sepulveda I, Santoyo G (2010) Diversity of bacterial endophytes in roots of Mexican husk tomato plants (Physalis ixocarpa) and their detection in the rhizosphere. Genet Mol Res 9(4):2372–2380
Mattarozzi M, Manfredi M, Montanini B, Gosetti F, Sanangelantoni AM, Marengo E, Careri M, Visioli G (2017) A metaproteomic approach dissecting major bacterial functions in the rhizosphere of plants living in serpentine soil. Anal Bioanal Chem 409(9):2327–2339
Mendes LW, Kuramae EE, Navarrete AA, Van Veen JA, Tsai SM (2014) Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J 8(8):1577–1587
Mirete S, Mora-Ruiz MR, Lamprecht-Grandío M, de Figueras CG, Rosselló-Móra R, González-Pastor JE (2015) Salt resistance genes revealed by functional metagenomics from brines and moderate-salinity rhizosphere within a hypersaline environment. Front Microbiol 6:1121
Mittal D, Shukla R, Verma S, Sagar A, Verma KS, Pandey A, Negi YS, Saini RV, Saini AK (2019) Fire in pine grown regions of Himalayas depletes cultivable plant growth promoting beneficial microbes in the soil. Appl Soil Ecol 139:117–124
Nimnoi P, Pongsilp N, Lumyong S (2011) Actinobacterial community and diversity in rhizosphere soils of Aquilaria crassna Pierre ex Lec assessed by RT-PCR and PCR-DGGE. Biochem Syst Ecol 39(4–6):509–519
Pal S, Roy A, Kazy SK (2019) Exploring microbial diversity and function in petroleum hydrocarbon associated environments through omics approaches. In: Microbial diversity in the genomic era. Academic, Cambridge, pp 171–194
Pétriacq P, Williams A, Cotton A, McFarlane AE, Rolfe SA, Ton J (2017) Metabolite profiling of non-sterile rhizosphere soil. Plant J 92(1):147–162
Poretsky RS, Hewson I, Sun S, Allen AE, Zehr JP, Moran MA (2009) Comparative day/night metatranscriptomic analysis of microbial communities in the North Pacific subtropical gyre. Environ Microbiol 11(6):1358–1375
Puranik S, Pal RR, More RP, Purohit HJ (2016) Metagenomic approach to characterize soil microbial diversity of Phumdi at Loktak Lake. Water Sci Technol 74(9):2075–2086
Rampadarath S, Bandhoa K, Puchooa D, Jeewon R, Bal S (2018) Metatranscriptomics analysis of mangroves habitats around Mauritius. World J Microbiol Biotechnol 34(4):59
Reddy GC, Goyal RK, Puranik S, Waghmar V, Vikram KV, Sruthy KS (2020) Biofertilizers toward sustainable agricultural development. In: Plant microbe symbiosis. Springer, Cham, pp 115–128
Ridl J, Kolar M, Strejcek M, Strnad H, Stursa P, Paces J, Macek T, Uhlik O (2016) Plants rather than mineral fertilization shape microbial community structure and functional potential in legacy contaminated soil. Front Microbiol 7:995
Saccà ML, Manici LM, Caputo F, Frisullo S (2019) Changes in rhizosphere bacterial communities associated with tree decline: grapevine esca syndrome case study. Can J Microbiol 65(12):930–943
Sánchez-Pérez BN, Zenteno-Rojas A, Rincón-Molina CI, Ruíz-Valdiviezo VM, Gutiérrez-Miceli FA, Vences-Guzmán MA, Villalobos-Maldonado JJ, Rincón-Rosales R (2020) Rhizosphere and endophytic bacteria associated to Ocimum basilicum L. with decaclorobiphenyl removal potential. Water Air Soil Pollut 231(3):1–15
Shu W, Pablo GP, Jun Y, Danfeng H (2012) Abundance and diversity of nitrogen-fixing bacteria in rhizosphere and bulk paddy soil under different duration of organic management. World J Microbiol Biotechnol 28(2):493–503
Singh A, Kumar M, Verma S, Choudhary P, Chakdar H (2020) Plant microbiome: trends and prospects for sustainable agriculture. In: Plant microbe symbiosis. Springer, Cham, pp 129–151
Srivastava M, Kaushik MS, Mishra AK (2016) Linking the physicochemical properties with the abundance and diversity of rhizospheric bacterial population inhabiting paddy soil based on a concerted multivariate analysis of PCR-DGGE and RISA. Geomicrobiol J 33(10):894–905
Tanim MT, Chowdhury MM, Bari L, Rahaman MM, Rahman SR, Rahman MM (2019) Genetic diversity of Rhizobiums: isolated from soil samples of Bangladesh. Bangladesh J Microbiol 36(1):7–10
Tian L, Shi S, Ma L, Zhou X, Luo S, Zhang J, Lu B, Tian C (2019) The effect of Glomus intraradices on the physiological properties of Panax ginseng and on rhizospheric microbial diversity. J Ginseng Res 43(1):77–85
Turner TR, Ramakrishnan K, Walshaw J, Heavens D, Alston M, Swarbreck D, Osbourn A, Grant A, Poole PS (2013) Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J 7(12):2248–2258
Unno Y, Shinano T (2012) Metagenomic analysis of the rhizosphere soil microbiome with respect to phytic acid utilization. In: Microbes and environments, p ME12181
Uroz S, Buée M, Murat C, Frey-Klett P, Martin F (2010) Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ Microbiol Rep 2(2):281–288
Verma P, Yadav AN, Khannam KS, Mishra S, Kumar S, Saxena AK, Suman A (2019) Appraisal of diversity and functional attributes of thermotolerant wheat associated bacteria from the peninsular zone of India. Saudi J Biol Sci 26(7):1882–1895
Wang HB, Zhang ZX, Li H, He HB, Fang CX, Zhang AJ, Li QS, Chen RS, Guo XK, Lin HF, Wu LK (2011) Characterization of metaproteomics in crop rhizospheric soil. J Proteome Res 10(3):932–940
Wang J, Wu L, Tantai H, Khan MU, Letuma P, Wu H, Zhang S, Chen T, Lin S, Lin W (2019) Properties of bacterial community in the rhizosphere soils of Achyranthes bidentata tolerant to consecutive monoculture. Plant Growth Regul 89(2):167–178
Wei Z, Hu X, Li X, Zhang Y, Jiang L, Li J, Guan Z, Cai Y, Liao X (2017) The rhizospheric microbial community structure and diversity of deciduous and evergreen forests in Taihu Lake area, China. PLoS One 12(4):e0174411
White RA, Borkum MI, Rivas-Ubach A, Bilbao A, Wendler JP, Colby SM, Köberl M, Jansson C (2017) From data to knowledge: the future of multi-omics data analysis for the rhizosphere. Rhizosphere 3:222–229
Willms IM, Kamran A, Aßmann NF, Krone D, Bolz SH, Fiedler F, Nacke H (2019) Discovery of novel antibiotic resistance determinants in forest and grassland soil metagenomes. Front Microbiol 10:460
Yan Y, Kuramae EE, de Hollander M, Klinkhamer PG, van Veen JA (2017) Functional traits dominate the diversity-related selection of bacterial communities in the rhizosphere. ISME J 11(1):56–66
Yergeau E, Tremblay J, Joly S, Labrecque M, Maynard C, Pitre FE, St-Arnaud M, Greer CW (2018) Soil contamination alters the willow root and rhizosphere metatranscriptome and the root–rhizosphere interactome. ISME J 12(3):869–884
Ying YX, Ding WL, Li Y (2012) Characterization of soil bacterial communities in rhizospheric and nonrhizospheric soil of Panax ginseng. Biochem Genet 50(11–12):848–859
Zhalnina K, Louie KB, Hao Z, Mansoori N, da Rocha UN, Shi S, Cho H, Karaoz U, Loqué D, Bowen BP, Firestone MK (2018) Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat Microbiol 3(4):470–480
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Gupta, T., Chakraborty, D., Sarkar, A. (2021). Structural and Functional Rhizospheric Microbial Diversity Analysis by Cutting-Edge Biotechnological Tools. In: Pudake, R.N., Sahu, B.B., Kumari, M., Sharma, A.K. (eds) Omics Science for Rhizosphere Biology. Rhizosphere Biology. Springer, Singapore. https://doi.org/10.1007/978-981-16-0889-6_9
Download citation
DOI: https://doi.org/10.1007/978-981-16-0889-6_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-0888-9
Online ISBN: 978-981-16-0889-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)