
Abstract
Network pharmacology is a new research strategy to understand the molecular and drug mechanisms of complex diseases based on the structure and function of the “molecular biological network.” The network-based method is a powerful tool for studying complex diseases and the nonlinear modes of drug-disease interactions. The WHO drafted guidelines for traditional medicine from 2014 to 2023. Traditional Chinese Medicine (TCM), as important inheritance of the Chinese nation and the critical constituent of China’s existing medical and health care system, play great role in the field of healthcare of Chinese people. Currently, researches on modernization of traditional medicine, including TCM, have attracted increasing attention both at home and abroad. Based on network visions and methods, it can facilitate the systematically understanding of the mechanism of disease pathology from biological network equilibrium view, promoting study of the relationships among different diseases and also the study of drug repositioning. What’s more, network visions and methods could also promote the systematic deciphering of complex interactions between TCM and disease biomolecular network, and enhance the R&D on combinatorial drugs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

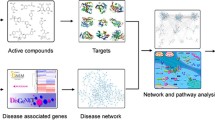
Network pharmacology is a new research strategy [1] to understand the molecular and drug mechanisms of complex diseases based on the structure and function of the “molecular biological network.” The network-based method is a powerful tool for studying complex diseases and the nonlinear modes of drug-disease interactions [2]. The WHO drafted guidelines for traditional medicine from 2014 to 2023. Traditional Chinese Medicine (TCM), as important inheritance of the Chinese nation and the critical constituent of China’s existing medical and health care system, play great role in the field of healthcare of Chinese people [3]. Currently, researches on modernization of traditional medicine, including TCM, have attracted increasing attention both at home and abroad. Based on network visions and methods, it can facilitate the systematically understanding of the mechanism of disease pathology from biological network equilibrium view, promoting study of the relationships among different diseases and also the study of drug repositioning. What’s more, network visions and methods could also promote the systematic deciphering of complex interactions between TCM and disease biomolecular network, and enhance the R&D on combinatorial drugs.
Fan’s research group from Zhejiang University focus on key scientific issues such as network regulation of TCM Formulae’s components compatibility. With application of omics technologies including transcriptomics, several research strategies for TCM network pharmacology with disease network as core have been established, which contributed to the formation of the concept of network formulaology [4, 5]. A series of significant progresses have made, including establishment of several basic databases, such as TCM-PTD, which is a database of potential TCM targets. Based on the characteristics of integrated regulation of TCM, Fan’s research group proposed a TCM Formulae’s components compatibility optimization strategy according to their network balance construction algorithm, after which a series of experimental studies were conducted to establish a comprehensive evaluation method for TCM efficacy analysis. They also created a multi-component/multi-target/multi-pathway network construction method for TCM, and the method was successfully applied for the study of integrated regulation mechanism of TCM, such as Shengmai and Qishen Yiqi Formulae [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. This chapter took ischemic heart disease as an example, together with several research examples [12, 13, 15, 18, 19] to introduce disease-based network pharmacology research, which can provide references for the development and application of network pharmacology associated research works.
8.1 Disease Network Construction and Analysis of Coronary Heart Disease
Coronary heart disease (CHD), as a chronic and complex disease, its occurrence and development involves multi-genes, multi-signaling pathways, and multi-links. The construction of disease network could fully integrate the disease-related gene–gene interactions, and the interactions were abstractly presented as network in the way of network visualization. Meanwhile, network analysis technology helps study of disease-associated gene groups and biological pathways at a holistic level. Therefore, disease networks provide a platform for systematically study interactions among molecules, which shows important scientific research and application value for understanding the pathogenesis of CHD and drug development.
8.1.1 Data Acquisition and Processing
8.1.1.1 Text Mining of CHD-Related Genes
The keywords “Coronary heart disease” were applied to searched in the PubMed database with year limited from 2000/1/1 to 2013/1/23. The search retrieved about 110,000 literatures. In the PubMed retrieved panel, select “Send to” → “Choose Destination” → “File” → “Format” → “Abstract (text)” → “Create File” from pull-down menu to save the .txt file containing abstracts. The abstracts information can be extracted using text mining technology.
Words were extracted from abstracts and the ArrayTrack (V 3.5.0) gene database was queried to get the potentially related genes, and these genes were retrieved in the obtained abstracts to attain genes with literatures support. Three researchers then independently carried out manual confirmations work. Genes associated with CHD were determined by reading relevant abstracts, full text, and other related references to acquire CHD-related genes. The researchers retained 660 CHD-related genes for further research.
8.1.1.2 Extraction of CHD-Related Genes by Manually Reading of Literatures
Compared to text mining technology, manual literature mining is time-consuming and labor-consuming, but it has a high accuracy rate. According to JCR Science Edition report published by Thomson Reuters, Circulation is the most authoritative professional journal in the field of cardiac & cardiovascular systems. Therefore, Circulation was selected as the literature source, with the year set from 2006 to 2011. By reading literature abstracts, gene and protein information were extracted from 151 abstracts and inconsistent formats were standardized. Official names of genes (Official Symbol) and gene numbers Entrez ID in NCBI were applied to normalization of gene names. Standard protein names adopted were “UniProt ID” and “UniProt Consortium Protein Name.” Finally, 252 related genes were obtained.
8.1.1.3 Mining CHD-Related Genes from Public Databases
Public databases, containing information on cardiovascular disease-related genes and proteins, are important resources to study the pathogenesis of cardiovascular diseases and the mechanism of drug action. Cardiovascular disease-related genes were selected from the rat genome research database (RGD) [28] by February 11th, 2014. 161 genes related to the cardiovascular disease portal and myocardial ischemia were selected. These genes were extracted and removed duplications for the construction of CHD@ZJU research platform.
8.1.1.4 Integration of CHD-Related Genes from Different Sources
CHD-related genes were normalized through the three methods by application of their official names (official symbol) and gene Entrez ID in NCBI, after integration and removal of duplicated genes, 1073 genes were finally identified. Compared with the CHD@ZJU version 1.0, 413 new genes were added. Further gene-related information was annotated, including gene name, gene Entrez Gene ID, and gene description, literature evidence of the genes, other literature information related to CHD, PPI relationship information related with the genes, and FDA drug information of the genes, etc., new genes in version 2.0 were especially highlighted in red.
8.1.2 Network Construction and Visualization
CHD@ZJU V2.0 integrates PPI-related information from databases HPRD and BioGRID. We optimized the internal data structure of the CHD@ZJU database and the computation and access speed of the website was improved.
Pairwise relationships of 1073 genes were extracted from the integrated PPI relationships to generate a gene–gene interaction relationship table, including 4030 pairs. Cytoscape Web [29] was used to construct a CHD disease network. The network model includes 1073 nodes and 4030 edges, with the largest sub-network containing 819 nodes and 3988 edges.
In disease network, gene groups with similar biological functions (corresponding to the sub-network or sub-cluster in the network), also known as biological function modules, can concurrently involve in certain phase of disease biogenesis. Thus, further analysis of biological functional modules is expected to reveal the pathogenesis of complex diseases and to provide support for the design and R&D of therapeutic drugs.
8.1.3 Network Analysis and Prediction
Network analysis was conducted using network topological attribute analysis, cluster analysis of sub-network/sub-cluster, and biological function gene ontology (GO) analysis.
8.1.3.1 Network Topological Attribute Analysis
The topological properties of the disease network were evaluated, and results showed that the distribution of node connectivity of the constructed disease network conformed to the power-law distribution (R2 = 0.890), that is, the CHD network indicated scale-free properties and obtain the general characteristics of biological network.
The network analyzer plug-in in Cytoscape software was applied to calculate the node degree (Degree) and betweenness (Betweenness Centrality) of disease network. In biological networks, nodes with large degrees are usually considered as “Hubs” of the network, while nodes with high betweenness are called “bottleneck” (Bottleneck Node). These topological properties represent the importance of nodes in the network [30]. In addition, some studies showed that important genes related to diseases usually tend to form hub nodes in biological networks [31,32,33,34].
The degree and betweenness of nodes in disease network can be calculated and two strategies were adopted for node ranking analysis:
8.1.3.1.1 Betweenness as the Screening Criteria
23 network nodes were obtained with betweenness greater than 0.02 and sorted from large to small according to betweenness values from the CHD-related gene network topology parameters (betweenness > 0.02).
23 genes were imported into ArrayTrack 3.5, “Pathway Enrichment Analysis” was performed to retrieve 32 signaling pathways. Pathways were divided into six categories according to their biological functions: cell adhesion and connection, apoptosis, myocardial and smooth muscle contraction, energy metabolism, immune inflammation, and cell signal transduction-related pathways.
If the threshold value of betweenness is set at 0.01, 65 nodes can be obtained. The signaling pathway enrichment analysis revealed 32 pathways in 7 types. These pathways were basically the same as those obtained when the betweenness threshold was set at 0.02. Three new signaling pathways were different, i.e., Cytosolic DNA-sensing pathway (p = 0.025) related to immune inflammation and RIG-I-like receptor signaling pathway (p = 0.046); Signal molecular related pathways of extracellular matrix: ECM-receptor interaction (p = 0.002).
8.1.3.1.2 Degree as the Screening Standard
Nodes with degrees greater than 10 were sorted in descending order of degree value. Simultaneously, the betweenness was greater than 0.02, and 22 nodes were obtained for the topological parameters of CHD-related gene network (degree > 10, betweenness > 0.02).
The signaling pathway enrichment analysis of these 22 genes indicated that there were mainly 32 signaling pathways in 6 categories.
If the degree is set as greater than 10, it returned 221 nodes meeting the criteria. Signaling pathway enrichment analysis of these genes can be performed to get 35 signaling pathways in 7 categories with 5 newly-added signaling pathways, i.e., PPAR signaling pathway related to energy metabolism (P = 0.019); Immune inflammation-related signaling pathway RIG-I-like receptor signaling pathway (p = 0.000), Cytosolic DNA-sensing pathway (p = 0.006); Extracellular signal molecule related signaling pathway ECM-receptor interaction (p = 0.002), Cytokine-cytokine receptor interaction (p = 0.002).
The pathways enriched using the key genes can reflect the biological functions of the gene group and can provide support for explanation of CHD pathogenesis. Our network analysis results indicated that the abnormal function of one or several signaling pathways in the biological pathways, such as cell adhesion and connection, apoptosis, contraction of myocardium and smooth muscle, energy metabolism, immune inflammation, cell signal transduction, and so on, may be related to the pathogenesis of CHD.
8.1.3.2 Cluster Analysis of Sub-Network/Sub-Cluster
Network medicine investigates human diseases in the view of network. Its theories and methods are based on the associations between human diseases occurrence and the disturbance of the disease module. The tacit hypothesis is that “Topological Modules,” “Functional Modules,” and “Disease Modules” could overlap in the network. Therefore, functional modules are equivalent to topological modules, and disease can be regarded as disturbance and disruption of functional modules [35].
The topological module can be obtained by applying a network clustering algorithm. MCODE plug-in in Cytoscape was adopted to analyze the network topological properties to discover highly interactive areas, namely clusters [36]. The MCODE sub-cluster analysis was carried out for the entire CHD disease network, and a total of 38 sub-clusters were returned. These sub-clusters were further analyzed for their biological functions (GO_Biological_Process, GO_BP).
8.1.3.3 GO Analysis of Biological Functions
BiNGO, another plug-in in Cytoscape, can integrate the molecular interaction networks, visualize and analyze the GO categories of genes in biological networks to discover functional modules of the network [37].
GO_BP analysis was performed on the top 15 sub-clusters obtained from the extraction and analysis of the MCODE sub-clusters. Homo sapiens was selected as the species with other parameters in BiNGO settings dialog box setting to the default, i.e., the statistical significance level is set as 0.05, the whole annotation is taken as the reference set for analysis. Finally, a total of 13 sub-cluster GO analysis results were returned, however, results for sub-cluster 9 were not returned, and sub-cluster 15 had only 2 nodes, hence GO analysis was not performed.
This chapter set sub-clusters 1, 2, and 3 as examples to conduct GO_BP analysis. The analysis results and discussion are listed as follows:
-
1.
Sub-cluster 1, cluster genes included E2F1, IRS2, RELA, SOCS1, ESR1, RB1, SIRT1, IRS1, STAT3, BRCA1, STAT6, HIF1A, HDAC1, JAK1, PARP1, PIK3R1, and GHR. GO_BP analysis found that the functions of these genes were involved in three aspects: intracellular and extracellular stimulation signals, synthesis and metabolism of biological molecules in the body, regulation of cell proliferation and apoptosis.
-
(a)
The responses to intracellular and extracellular stimulation signals may be mainly due to the organisms response to various biomolecules (hormones, cytokines, etc.) during the occurrence and developmental stages of CHD.
-
(b)
The synthesis and metabolic regulation of biomolecules, including glycolipid metabolism and other processes related to body energy metabolism. For example, insulin-related signaling pathways in sub-cluster 1 were involved in regulating the transport and metabolism of glucose, β-oxidation of fatty acids, etc., which show important role in the pathogenesis of CHD. The related GO_BP categories included: GO-ID 10907, positive regulation of glucose metabolic process; GO-ID 45913, positive regulation of carbohydrate metabolic process; GO-ID 10828, positive regulation of glucose transport; GO-ID 19216, regulation of lipid metabolic process; GO-ID 32000, positive regulation of fatty acid beta-oxidation; GO-ID 43550, regulation of lipid kinase activity; GO-ID46321, positive regulation of fatty acid oxidation, etc.
-
(c)
Cell proliferation and apoptosis are involved in the pathological process of myocardial infarction. The main related GO_BP categories included: GO-ID 42127, regulation of cell proliferation; GO-ID 8284, positive regulation of cell proliferation; GO-ID 42981, regulation of apoptosis; GO-ID 43067, regulation of programmed cell death; GO-ID 10,941, regulation of cell death, etc.
-
(a)
-
2.
Sub-cluster 2, cluster genes included PPARA, CAV1, TNF, IL6ST, GRB2, PPARG, NFKBIA, FOXO1, NR3C1, CTNNB1, RPA1, FOS, GATA2, CD44, RAC1, RUNX1, MYC, CCNA2, CHUK, HSPA8, AKT2, NFATC1, IRAK1, MAP 2K1, RXRA, SMAD5, TP53, SMAD3, SMAD1, CDK4, PRKCD, KDR, HDAC4, HDAC3, CDKN1A, ETS1, MAPK3, MAPK8, and MDM4. GO_BP analysis found that the functions of these genes were mainly involved in the responses to intracellular and extracellular stimulation signals, regulation of cell proliferation, differentiation, and apoptosis, and the regulation of immune inflammatory related processes:
-
(a)
The responses to intracellular and extracellular stimulation signals may be produced by the corresponding response of the body caused by myocardial infarction, for example: GO-ID 9611 response to wounding.
-
(b)
Cell proliferation, differentiation, and apoptosis are involved in the pathological process of myocardial ischemia and infarction. The corresponding GO_BP categories are: GO-ID 42127, regulation of cell proliferation; GO-ID 8285, negative regulation of cell proliferation; GO-ID 45595, regulation of cell differentiation; GO-ID 43069, negative regulation of programmed cell death; GO-ID 42981, regulation of apoptosis; GO-ID 43067, regulation of programmed cell death; GO-ID 10941, regulation of cell death; GO-ID 45767, regulation of anti-apoptosis; GO-ID 51726, regulation of cell cycle, etc.
-
(c)
Immune inflammatory reaction occurs throughout the development of CHD, the main GO_BP categories include: GO-ID 2376, immune system process; GO-ID 2682, regulation of immune system process; GO-ID 2520, immune system development; GO-ID 6954, inflammatory response; GO-ID 2673, regulation of acute inflammatory response; GO-ID 50727, regulation of inflammatory response, etc.
-
(a)
-
3.
Sub-cluster 3, cluster genes included: BID, TRAF1, THRA, ERBB2, NFKB1, BCL2L1, FOXO3, SRC, ATF2, IGF1R, VDR, CSNK2A1, CXCR4, RHOA, FAS, APEX1, AR, SOCS3, SMAD2, PTPN11, CCND1, TNFRSF10B, EP300, HDAC2, JUN, MDM2, JAK2, PTPN1, and TNFAIP3. GO_BP analysis found that the main functions of these genes are regulation of cell proliferation, differentiation and apoptosis, and regulation of hypoxic and oxidative stress.
-
(a)
In myocardial infarction, cell proliferation, differentiation, and apoptosis involve myocardial cells, endothelial cells, smooth muscle cells, immune inflammatory cells, etc. The main GO_BP categories include: GO-ID 42981, regulation of apoptosis; GO-ID 43067, regulation of programmed cell death; GO-ID 10941, regulation of cell death; GO-ID 45595, regulation of cell differentiation; GO-ID 8219, cell death; GO-ID 6915, apoptosis; GO-ID 12501, programmed cell death; GO-ID 6916, anti-apoptosis; GO-ID 42127, regulation of cell proliferation; GO-ID 8633, activation of pro-apoptotic gene products; GO-ID 51726, regulation of cell cycle; GO-ID 6917, induction of apoptosis; GO-ID 12502, induction of programmed cell death, etc.
-
(b)
Hypoxia and oxidative stress play an important role in the occurrence and development of systolic heart disease. The GO_BP categories include: GO-ID 51341, regulation of oxidoreductase activity; GO-ID 1666, response to hypoxia; GO-ID 70482, response to oxygen levels; GO-ID 42542, response to hydrogen peroxide; GO-ID 6979, response to oxidative stress; GO-ID 302, response to reactive oxygen species, etc.
-
(a)
Similarly, this chapter also conducted GO_BP analysis of the other 10 sub-clusters, results mainly involving angiogenesis, leukocyte chemotactic migration, and cell apoptosis; oxidative stress injury, damage repair; cell proliferation, migration and apoptosis, fatty acid oxidative metabolism; immune inflammation-related biological processes; calcium ion channel regulation; Cell-extracellular matrix interaction regulation; coagulation function cascade reaction, and other biological pathways.
8.1.4 Validation and Summary
8.1.4.1 Enrichment Results Discussion
Functions of signaling pathways enriched through disease network analysis and their association relationships with diseases are discussed and analyzed as follows:
-
1.
The role of apoptosis and necrosis in CHD: In acute myocardial infarction (AMI), cardiomyocyte apoptosis and necrosis occur rapidly, and the related signaling pathways like cell cycle, apoptosis, and p53 signaling pathway play a key role in the pathological process of cardiomyocyte apoptosis and necrosis [38,39,40].
-
2.
The immune inflammatory response involves throughout the entire occurrence and development process of atherosclerosis (AS), and is the key biological process of CHD [41,42,43,44]. Inflammatory response involves throughout the entire process of AMI. Hypoxia is one of the causes of inflammation. Inflammation involves all stages of ischemic injury [45]. Of all the 37 enriched signaling pathways, 11 are associated with immune inflammation, including: T cell receptor signaling pathway, Chemokine signaling pathway, B cell receptor signaling pathway, Fc epsilon RI signaling pathway, Fc gamma R-mediated phagocytosis, natural killer cell mediated cytotoxicity, toll-like receptor signaling pathway, leukocyte transendothelial migration, NOD-like receptor signaling pathway, cytosolic DNA-sensing pathway, RIG-I-like receptor signaling pathway. Studies indicate that MAPK signaling pathway, with regulatory effects on cell proliferation, differentiation, and apoptosis, plays an important role in inflammatory response, and is a potential target of anti-inflammatory therapy [46].
-
3.
Energy metabolism plays an essential role in the occurrence and development of CHD. Myocardial energy metabolism includes the metabolism and utilization of fatty acids and glucose. Cardiac insulin promotes cardiomyocytes to utilize fatty acids and glucose for ATP synthesis, while myocardial ischemia inhibits normal conduction of insulin signaling pathway [47], with subsequent inducing of the occurrence of adverse left ventricular remodeling after myocardial infarction. This process is related to the decreased function of mitochondrial fatty acid oxidation in cardiomyocytes [48]. For PPAR signaling pathway, it can inhibit myocardial ischemia by promoting glucose utilization and anti-inflammatory effects [49], in which adiponectin is a key adipocytokine that can promote fatty acids β-oxidation and increase the utilization of glucose [50].
-
4.
The role of angiogenesis in CHD: VEGF signaling pathway regulates the proliferation, migration or cell viability of vascular endothelial cells, promotes vasodilation, and improves blood supply post-myocardial infarction [51,52,53]. The mTOR signaling pathway has a regulatory effect on hypoxia induced angiogenesis, and can promote the recovery of blood flow supply to ischemic myocardium post-myocardial infarction [54, 55].
-
5.
The role of cell adhesion and junction in CHD: Focal adhesion is the medium for cells to connect with the ECM through the cytoskeleton, while AS is the internal pathological basis of CHD. The disturbance of blood “Fluid Shear Stress” at the bifurcation point of arteries is transmitted through focal adhesion and causes morphological and functional changes of endothelial cells, which promotes the occurrence and development of AS [56]. At the same time, focal adhesion also mediates endothelial injury due to lipoproteins and promotes the progress of AS [57]. The integrity of the structure and function of endothelial cells is an vital basis for maintaining the normal functions of the cardiovascular system. Adherens junction, Gap junction, and Tight junction are three critical connections between endothelial cells. Abnormal connection between these cells changes the morphology and function of the endothelium, increases the permeability of the endothelium, and promotes the occurrence and development of AS [58]. In addition, injury to endothelial cells causes pro-inflammatory cells to adhere to the surface of damaged endothelial cells, and to migrate and invade farther into the blood vessel wall in the endothelium, participating in the formation of atherosclerotic plaques together with the involving of actin cytoskeleton [59]. Endothelial tissue is a continuous monolayer structure of endothelial cells in the inner wall of blood vessels, which plays the role of a functional barrier between blood and vascular smooth muscle. The changes in endothelial structure and function directly affect the contraction function of blood vessels. On one hand, the functional barrier of endothelial tissue is changed with increased permeability, which induces some vasoactive substances in blood circulation to invade into the vascular wall. On the other hand, the injured endothelium synthesizes and secretes vasoactive substances (such as endothelin), or metabolizes the precursor substances in the blood circulation into vasoactive substances (such as AngI transforming to AngII), which cause vasoconstriction and affect the blood and oxygen supply to the myocardium [60].
-
6.
The role of ventricular remodeling and myocardial injury repair in CHD: The TGF-beta signaling pathway is activated in the repair process post-myocardial infarction, which has a pleiotropic and multifunctional regulatory effect on different types of cells involved in the repair [61]. Myocardial infarction triggers inflammation, which eventually forms scar tissue. In the early stage of infarction repair, TGF-β inhibits the activity of macrophages with subsequent inhibition of synthesis of chemokines and cytokines by endothelial cells to show anti-inflammatory effects. In the followed process, TGF-β causes ECM deposition, activates the fibrosis signaling pathway, and induces fibrosis and hypertrophy of non-infarcted myocardium, which all contribute to left ventricular remodeling [62]. The notch signaling pathway plays a key role in mammalian heart development [63] as well as in the myocardial repair [64] and regeneration process after infarction. The hedgehog signaling pathway, another signaling pathway regulating the development and formation of tissues and organs, plays an important role in cardiac repair post-myocardial infarction. Erythropoietin (EPO) can promote angiogenesis through the hedgehog signaling pathway and plays a protective role [65] in the heart after infarction. Another mechanism of the hedgehog signaling pathway on cardiac function recovery post-myocardial infarction is to upregulate the expression of angiogenic genes and to enhance the migration of bone marrow progenitor cells to the infarcted myocardium [66].
-
7.
The role of other biological processes in CHD: ErbB and Wnt signaling pathways have multiple roles in regulating biological processes, including cardiac development, cardiomyocyte proliferation, myocardial cell viability, etc. ErbB signaling pathway is especially vital for cardiac development and can regulate the balance of both sympathetic vagus nerve and hemodynamics. The activation of ErbB signaling pathway can reverse the declining cardiac function post-myocardial infarction [67]. Wnt signaling pathway plays an significant role in stem cell biological activity, cardiac development and differentiation, angiogenesis, etc. [68,69,70] What’s more, the Jak-STAT signaling pathway has a versatile biological functions, involving cell proliferation, differentiation, migration, and apoptosis, and is an essential signaling cascade for organisms [71]. Studies have found that under pathological conditions, the renin-angiotensin system (RAS) is activated, and Ang II binds to its receptor to further activate Jak-STAT signaling pathway, participating in cardiac dysfunction caused by myocardial ischemia-reperfusion [72]; In addition, the Jak-STAT signaling pathway plays an important role in the occurrence of myocardial infarction and post-myocardial infarction ventricular remodeling, which may be related to the activation of the pro-inflammatory signaling pathway of Jak-STAT signaling pathway [73].
The abovementioned analysis indicates that the pathogenesis of CHD involves immune inflammatory reaction, apoptosis and necrosis, energy metabolism of cells, cell adhesion and connection, angiogenesis, myocardial injury repair, ventricular remodeling, and other related biological processes.
8.1.4.2 Conclusion
-
1.
The disease network of CHD was improved and the CHD@ZJU2.0 disease network research platform was established through application of the integrated text knowledge mining, manually literature validation and public database knowledge methods. With the guidance of network biology, network pharmacology and network medicine concepts and methods, network analysis was applied to identify key genes and biological pathways of the CHD disease network. CHD@ZJU, the disease network research platform, has currently been updated to version 3.0.
-
2.
The results of network analysis preliminarily show that CHD is a polygenic, multi-biological pathways associated disease. Pathogenesis related biological processes mainly include: Immune inflammatory response, cell proliferation, differentiation and apoptosis-related processes, angiogenesis, hypoxia and oxidative stress response, glucose and fatty acid related energy metabolism, myocardial injury repair, and ventricular remodeling. The construction and improvement of the CHD disease network research model can facilitate the subsequent experimental research works.
8.2 Research Practice of the Application of Disease Network in TCM Prescriptions
8.2.1 Integrated Mechanism Study of TCM Prescriptions Modes of Action
The Qishen Yiqi formulae mainly contain water-soluble components from Radix Astragali, Salvia Miltiorrhiza, and Panax Notoginseng, together with volatile oil components from Dalbergia Odorifera. Our previous research results [74, 75] show that the pharmacodynamic material basis for Qishen Yiqi formulae are mainly saponins from Radix Astragali, phenolic acids from Salvia Miltiorrhiza, saponins from Panax Notoginseng, and volatile oil components from Dalbergia Odorifera. Shi Pei-ying [75] studied the non-volatile and volatile chemical constituents of Qishen Yiqi formulae by application of liquid chromatography mass spectrometry (LC/TOF/MS), liquid chromatography ion trap mass spectrometry (LC/IT/MS), and gas chromatography mass spectrometry (GC/MS). A total of 35 non-volatile components were identified, and the compounds with highest contents were of danshensu, salvianolic acid B, salvianolic acid A, isosalvianolic acid C, ginsenoside Rb1, and ginsenoside Rd. 24 volatile components were identified in the Qishen Yiqi extract. By comparing the total ion flow diagram of Qishen Yiqi and Dalbergia Odorifera extracts, 5 volatile components with high content amount were obtained, notably cis-α-santalol, nerolidol, E-nerolidol (ENL), (3S,6R,7R)-3,7,11-trimethyl-3,6-epoxy-1,10-dodecadien-7-ol (RDL), and (3S,6S,7R)-3,7,11-trimethyl-3,6-epoxy-1,10-dodecadien-7-ol (SDL), respectively. Analysis of plasma samples of SD rats after intragastric administration of Qishen Yiqi extract (6 g/kg) revealed that the constituents absorbed into the blood were four phenolic acid compounds and seven saponins compounds.
In addition, our research group’s Li et al. [76, 77] used HPLC-UV-ELSD and HPLC-DAD-ESI-MSn methods to study the content of related components in Qishen Yiqi dropping pills. Combined with results of this study, Shi Pei-ying suggested that astragaloside IV (Ast), Danshensu (DSS), Ginsenoside Rg1 (Rg1), and ginsenoside Rb1 (Rb1) in the Qishen Yiqi formulae may be its representative effective components in blood corresponding to drugs in Qishen Yiqi. Other domestic research groups have also conducted research on the pharmacodynamic material basis of Qishen Yiqi formulae [78].
Based on the research results of the chemical composition of TCM prescription and in vivo pharmacokinetics, effective components of the Qishen Yiqi formulae are the saponins of Radix Astragali, phenolic acids of Salvia Miltiorrhiza, saponins of Panax Notoginseng, and volatile oil of Dalbergia Odorifera. Representative compounds were selected from corresponding TCM fractions, i.e., Ast, DSS, Rg1, and ENL were identified as the four main active compounds. Study was designed to investigate the effects of single compound administration and combined compounds administration on rat AMI models. Gene chip technology and network pharmacology methods were used, together with CHD@ZJU V2.0 ischemic heart disease network, and the synergistic mechanism of four main active components of the Qishen Yiqi formulae against the rat AMI at the transcriptional level.
8.2.1.1 Data Acquisition and Processing
Left Anterior Descending Coronary Artery (LAD) ligation was done under anesthesia in rats. Myocardial infarction model rats were randomly divided into groups with intragastric administration (i.g) on every morning for 7 consecutive days (i.g volume 10 ml/kg). The sham operation group (1% sodium carboxymethyl cellulose solution, 1% CMCNa), model group (1% CMCNa), Ast group (80 mg/kg), DSS group (44 mg/kg), Rg1 group (8 mg/kg), ENL (53.3 mg/kg), 4H group, Qishen Yiqi decoction group (QSYQ). Five rats in each group were used for gene chip research, i.e., sham, model, Ast, DSS, Rg1, ENL, and 4H.
Extraction of rat myocardial tissue for gene chip detection required surgical instruments with DEPC RNase-free water treatment, autoclaved, and dried. Ophthalmic scissors and ophthalmic tweezers were used to obtain about 300 mg of myocardial tissue at the junction between the infarcted and normal myocardium below the site of ligation. The Affymetrix Rat Genome 230 2.0 chip was selected and entrusted to the chip company for testing. The company used Affymetrix GeneChip Command Console software (version 4.0, Affymetrix) to process and extracts the original data from the scanned original images.
The original data document was downloaded from the Arraytrack server, and Excel was used to standardize with median set as 1000 for all data. The standardized calculation is as follows:
The average value of probe expression was calculated. The expression value obtained after normalization was further calculated through a logarithm of base 2 (Log2expression value) to attain the average log2 value of each probe. 1073 ischemic heart disease-related genes were downloaded from the CHD disease network platform CHD@ZJUV2.0, constructed in Sect. 8.1, and 902 ischemic heart disease-related genes were extracted and returned from the 26,430 genes on the chip. Reverse Rate (RR) of each gene was calculated, and finally the signaling pathway enrichment analysis was done in Arraytrack.
The formula for calculating the gene callback rate is as follows:
where RR stands for the callback rate, that is, the ability of drug to regulate gene expression in the opposite direction of the model’s change against the sham operation group, so it can return to the sham operation level. D stands for component or component combination, M stands for model, and S stands for sham operation.
Where a gene is up-regulated relative to the sham operation after LAD ligation, i.e., M > S, the effective callback condition is M > D ≥ S, then the callback rate is 0 < RR ≤ 1. If the gene is down-regulated relative to the sham operation after LAD ligation, i.e., S > M, the effective callback condition is S ≥ D > M, then the callback rate is 0 < RR ≤ 1, that is, the closer the RR value getting to 1, the stronger the callback effect shows.
However, callback rate RR > 1 indicates the over-callback effect, that is, the gene continues to be regulated in the direction of sham surgery after the gene was recalled to the level of sham surgery due to drug intervention, and the callback rate RR ≤ 0 indicates no callback effect.
8.2.1.2 Network Analysis and Prediction
ArrayTrack software was used to conduct signaling pathway enrichment analysis of those effective callback genes. There were 466 effective callback genes for Ast, 365 effective callback genes for DSS, 495 effective callback genes for Rg1, 425 effective callback genes for ENL, and 444 effective callback genes for the 4H group. The respective groups of regulated signaling pathways and signaling pathway network were constructed and referred to as the drug-signaling pathway network. The network attributes included 49 nodes, 179 edges, and 44 signaling pathways. There were 26 signaling pathways with significant regulation of single administration of the four components and combined administration of the four components, accounting for 59.1% of the total enriched signaling pathways. These signaling pathways mainly involved biological processes such as immune inflammation, cell adhesion and connection, angiogenesis, ventricular remodeling and myocardial repair, and energy metabolism. There were 34 signaling pathways that are significantly regulated by Ast (p < 0.05), 35 by Rg1 (p < 0.05), 39 by DSS (p < 0.05), 36 by ENL (p < 0.05), and 35 by 4H (p < 0.05).
The results of network analysis show that each component can regulate multiple ischemic heart disease-related signaling pathways. Some signaling pathways are regulated by only one component, while others are regulated by two to four components simultaneously.
Glycerolipid metabolism signaling pathway is only regulated by Ast, while the three signaling pathways of nicotinate and nicotinamide metabolism, steroid biosynthesis, and cell cycle are only modulated by DSS. And methane metabolism signaling pathway is only regulated by ENL.
Ast and Rg1 can significantly regulate PPAR signaling pathway and pyruvate metabolism. DSS and ENL can both significantly regulate p53 signaling pathway, tight junction, and RIG-I-like receptor signaling pathway.
The three components of Ast, DSS, and Rg1 can significantly regulate cardiac muscle contraction and Fc gamma R-mediated phagocytosis. Ast, DSS, and ENL can significantly regulate ABC transporters; Ast, Rg1, and ENL can significantly regulate ECM-receptor interaction; DSS, Rg1, and ENL can significantly regulate Leukocyte transendothelial migration, Hematopoietic cell lineage, and natural killer cell mediated cytotoxicity.
The four components of Ast, DSS, Rg1, and ENL can significantly regulate arachidonic acid metabolism.
There are 26 signaling pathways with significant regulation of single and combined administration of the four components, including: adherens junction, adipocytokine signaling pathway, apoptosis, B cell receptor signaling pathway, calcium signaling pathway, chemokine signaling pathway, complement and coagulation cascades, cytokine-cytokine receptor interaction, ErbB signaling pathway, Fc epsilon RI signaling pathway, focal adhesion, gap junction, GnRH signaling pathway, insulin signaling pathway, Jak-STAT signaling pathway, MAPK signaling pathway, mTOR signaling pathway, NOD-like receptor signaling pathway, regulation of actin cytoskeleton, renin-angiotensin system, T cell receptor signaling pathway, TGF-beta signaling pathway, toll-like receptor signaling pathway, vascular smooth muscle contraction, VEGF signaling pathway, and Wnt signaling pathway.
8.2.1.3 Validation and Summary
8.2.1.3.1 Experimental Validation
Lipopolysaccharide (LPS)-induced inflammatory cellular model in RAW264.7 mouse mononuclear macrophages was applied to study the anti-inflammatory effect and mechanism of ENL (the main component of the volatile oil of Dalbergia Odorifera) and the two components SDL and RDL (isolated from the oil of Dalbergia Odorifera for the first time). Meanwhile, the synergistic anti-inflammatory effects of the four main components of QSYQ, i.e., Ast, DSS, Rg1, and ENL, were studied.
The effects of ENL, SDL, and RDL were investigated in order to identify their safety concentration ranges which would have no affections to the viability of RAW264.7 cells, and the inhibitory effect of these compounds against the production of NO in RAW264.7 cells induced by LPS was evaluated. The synergistic inhibitory effects of sodium danshensu and ginsenoside Rg1 against LPS-induced secretion of NO in RAW264.7 were also investigated. The inhibitory effects of the combined treatment of the four components against LPS induced NO secretion in RAW264.7 were detected by the Griess method. Further, the effects of small molecule (Tool Compound), i.e., U0126, T0070907, Pioglitazone hydrochloride (Pio), ZnPPIX on the NO secretion in LPS stimulated RAW264.7 were investigated. Among them, U0126 is a selective ERK1/2 phosphorylation inhibitor, T0070907 is an inhibitor of PPARγ phosphorylation, Pio is a PPARγ agonist, and ZnPPIX is a specific inhibitor of HO-1. Western blot method was selected to detect the protein expression levels of ERK1/2, phospho-ERK1/2, PPARγ, and HO-1.
8.2.1.3.2 Main Conclusions
-
1.
Gene chip technology and network pharmacology method were applied, the synergetic mechanisms of the four main components of the QSYQ against AMI in rats were studied at the transcriptome level, and the four main components have regulatory effects on multiple genes related to ischemic heart disease. Among the genes with effective callbacks from the combination of the four components, more than 50% of genes are involved in effective callbacks by at least two components, reflecting the synergistic effects of all four active components on the regulation of related genes. In the component combination administration group, there were 40 genes that are up-regulated and the four components had synergistic effects against LAD ligation. 36 genes that were down-regulated indicated synergistic effects by the four components after modeling. Biological function and signaling pathway enrichment analysis of these genes revealed several pathways, including immune inflammation, angiogenesis, ventricular remodeling and myocardial repair, energy metabolism, etc., which may be the key biological pathways for the four components of the QSYQ to play a multi-target and multi-pathway integrated synergy against rat AMI.
-
2.
Experimental verification studies revealed that ENL, SDL, and RDL inhibit the secretion of NO in LPS-induced RAW264.7 monocytes macrophages. The mechanism of action of the three components (especially ENL and SDL) may be partly through inhibition of ERK1/2 and PPARγ phosphorylation, and increase of PPARγ and HO-1 expression involving NF-κB, ROS, iNOS, and so on, which all contributed to inhibition of LPS-induced NO production in RAW264.7 macrophages. The combined co-culture of DSS and Rg1 with RAW264.7 could synergistically enhance the inhibition effects of LPS induced NO secretion in a dose-effect relationship. This effect of DSS and Rg1 could be attenuated or even abolished by ZnPPIX, an HO-1-specific inhibitor. Thus, DSS and Rg1 may play an anti-inflammatory role by partially promoting the expression of HO-1. Meanwhile, compared to the single component administration group, the combined administration of Ast, DSS, Rg1, and ENL of the four components could synergistically enhance the inhibition effects on the LPS-induced NO secretion and exhibited a good dose-response relationship.
-
3.
The component-target-pathway network of the main pharmacodynamic substance of the QSYQ was constructed, and experimental data revealed that the regulation network of Qishen Yiqi dropping pills consist of 12 effective components and 55 targets acting on 17 pathways, involving both AMI stage (Vasodilation, anti-myocardial apoptosis, anti-inflammatory protection of the endothelium, and improved energy metabolism) and ventricular remodeling stage (promoting angiogenesis, improving cardiac function, anti-myocardial fibrosis, and anti-platelet aggregation). This indicated the integrative regulation effects of QSYQ as multi-components, multi-targets, and multi-pathways modes of action.
8.2.2 Compatibility Law: “Sovereign-Minister-Assistant-Courier” in TCM Formulae
Network pharmacology technology provides a new perspective for the systematic exploration of integrated regulation modes of TCM formulae. Qishen Yiqi formulae were set as an example to explain the scientific connotation of its compatibility law in combating AMI. This section proposes a new research model of integrated regulation of TCM based on biological network equilibrium analysis. Firstly, the Organism Disturbed Network (ODN) model was constructed by integrating disease-related gene information, transcriptome expression, and interaction related information. We also analyze the ability of the Qishen Yiqi formulae and its components of Radix Astragali, Salvia Miltiorrhiza, Panax Notoginseng, and Dalbergia Odorifera on recovering and regulating of the ODN. The network recovery ability algorithm was improved and applied to quantitatively evaluate the efficacy of Qishen Yiqi formulae. Results indicated that the Qishen Yiqi formulae was significantly better than the four TCM drugs of QSYQ administered alone against AMI, and had a synergistic mode of effect. As for the ability of network recovery, the sovereign drug and minister drug (Radix Astragali and Salvia Miltiorrhiza) is significantly better than that of the assistant drug and courier drug (Panax Notoginseng and Dalbergia Odorifera), which complies with the compatibility law of “sovereign-minister-assistant-courier or Jun-Chen-Zuo-Shi rules.” Pathway enrichment analysis sheds light on the compatibility rules of the Qishen Yiqi formulae and its components at the pathway level.
8.2.2.1 Data Acquisition and Processing
8.2.2.1.1 Preparation of Myocardial Tissue Samples from AMI Rats
The AMI rat myocardial tissue samples were provided by the Institute of Pharmacoinformatics, Zhejiang University. A total of seven groups of samples were involved in this study, including normal group (Ctrl), model group (MI), Qishen Yiqi Decoction group (QSYQ), Salvia Miltiorrhiza group (DS), Radix Astragali group (HQ), Panax Notoginseng group (SQ), and Dalbergia Odorifera group (JX). Three biological replicate samples were included in each group.
8.2.2.1.2 Chip Experiment and Data Preprocessing
RNA extraction, purification, sample quality inspection, chip experiment, and data acquisition were completed by chip company. The Affymetrix Rat 230 2.0 chip was used, and the final chip data was saved in the .CEL format files. ArrayTrack software was applied to read the .CEL files and the chip data was exported to Excel in .txt format files for normalization processing. Data from 21 chips was processed for data standardization. The median expression value for each chip was set to 1.000 (Median = 1000) by multiplying with the weight coefficient.
8.2.2.2 Network Construction and Visualization
8.2.2.2.1 Construction of Organism Disturbed Network (ODN)
The whole process can be divided into four parts: data collection, data arrangement, network construction and visualization, and network analysis.
In the data collection process, the AMI-related gene information, correlation, and expression profile information were collected through knowledge mining techniques and transcriptomics experiments. In the data management process, genes related to CHD and myocardial ischemia in CHD@ZJU and RGD databases were integrated. Protein–protein interaction (PPI) relationship information in both HPRD and BioGRID databases was also integrated. Gene expression information was attained through chip expression data processing using data normalization. AMI disease biological network was constructed by using integrated genetic information and PPI relationship as nodes and edges, respectively. Each node in the network represents a gene, and each edge represents the interaction relationship between the corresponding proteins of two genes. Then, the gene expression profile information of AMI was obtained by transcriptomics technology, and it was combined with the disease biological network in the form of network annotation (including node annotation and edge annotation) to form ODN. Cytoscape software was applied to conduct visualization research of the ODN.
8.2.2.2.2 Calculation of Network Recovery Index (NRI) of ODN Model
NRI-ODN, which is oriented to the Organism Disturbed Network model, enables to integrate node topology attributes and callback efficiency, to comprehensively evaluate the ability of network recovery regulation of drugs. In this study, node topology attributes are defined by the degree of nodes. The node callback efficiency is evaluated by “Efficiency of Recovery regulation” (EoR).
EoR is an index describing the callback efficiency of a node based on a quantitative callback state (RL′). RL′ is a continuous variable form of the callback state index RL, and the calculation is shown in Formula 8.1. The EoR calculation is shown in Formula 8.2, and its maximum value is 100%, that is, the drug can eliminate the expression change caused by the AMI modeling. For example, if the mean Log2 value of expression intensity in the normal control group is 5, and that in the model group is 10. EoR = 50% indicates that the expression intensity of this gene was regulated in recovery by 50% (value = 7.5) or over-regulated by 50% (value = 12.5) after drug administration. While EoR = 100% indicates that the gene expression intensity returned to the normal control state after administration (value = 10). If EoR < 0, it means that the drug does not have a callback effect (value < 5) or produces an excessive over-callback effect (>15) on the gene, and this type of gene does not contribute to the efficacy of the drug.
NRIODN was calculated by Formula 8.4. The callback level (Recovery Regulation-ODN, RRODN) comprehensively considers the influence of node topology and callback efficiency on the callback level, as shown in Formula 8.3. Where Wtopo represents the degree of the node and EoRpositive indicates that only those nodes with positive EoR (value > 0) are used in the RRODN calculation. Finally, NRIODN was obtained by calculating the overall network, the sum of the RRODN of significantly up-regulated genes and significantly down-regulated genes.
8.2.2.3 Network Analysis and Prediction
The study applied the NRI-ODN to evaluate the efficacy of the Qishen Yiqi formulae against AMI, and analyzed the impact of the node’s Fold Change (FC) on the callback level RRODN and NRIODN. The callback efficiency of Qishen Yiqi and its single drugs to the nodes of the ODN were calculated through EoR. The genes with effective callback ability were defined with EoR > 50% as the threshold, and the effective callback genes lists of Qishen Yiqi and its four drugs were obtained.
Pathway enrichment analysis methods were used to analyze the biological signaling pathways involved in each gene list. ArrayTrack (version 3.5.0) software was cited to analyze the pathway of the effective callback gene list. The KEGG pathway database was selected as the pathway information source, and fisher p value < 0.05 was set as the standard to identify the significant pathways. The KEGG pathway database contains cellular processes, environmental information processes, metabolism, human diseases, etc. Considering the relevance with PPI, only cellular and environmental information processes were studied in this research.
8.2.2.4 Validation and Summary
8.2.2.4.1 Network Construction and EoR Calculation Results
The AMI-related disease biological network based on the Qishen Yiqi data involves a total of 324 genes and 623 pairs of interaction relationships. The largest sub-network is the ODN, involving 281 genes and 616 interaction relationships. Each node represents a gene, and each edge represents an interaction relationship between the corresponding proteins of the genes. The color of the nodes represents the changed state of expression in the modeling group, where red represents the up-regulated expression of the modeling group as compared to the normal group; and green represents the down-regulated expression of the modeling group versus the normal group (as shown in Fig. 8.1a).

Graph of the AMI-related Organism Disturbed Network (ODN) (Qishen Yiqi network) based on the Qishen Yiqi gene chip expression data
The EoR index was used to annotate the callback ability of the Qishen Yiqi decoction after administration (as shown in Fig. 8.1b). A blue node indicated that the Qishen Yiqi formulae could callback the expression imbalance caused by AMI after its administration, while a gray node indicated that there was no callback effect. The four drugs of Qishen Yiqi formulae—Salvia Miltiorrhiza (Salvia), Radix Astragali (Astragalus), Panax Notoginseng (Notoginseng), and Dalbergia Odorifera (Dalbergia), are shown in Fig. 8.1c.
8.2.2.4.2 Calculation Results of RRODN and NRIODN of Qishen Yiqi Formulae
Among the four drugs, the NRIODN index of Salvia Miltiorrhiza and Radix Astragali was higher than that of Panax Notoginseng and Dalbergia Odorifera. The Qishen Yiqi formulae, Radix Astragali, and Salvia Miltiorrhiza had a very significant callback effect on the ODN (p < 0.01). Dalbergia Odorifera had a significant callback effect (p < 0.05), and the Panax Notoginseng alone showed an insignificant callback effect on the ODN (p > 0.1).
8.2.2.4.3 Results of Network Recovery Regulation Ability
In this study, the absolute value of FC > 0.5 was used as the screening criterion for significantly up-regulated and down-regulated genes, and the influence of this screening criterion on the calculation results of RRODN was investigated. The RR value of Qishen Yiqi formulae is always higher than those of its four drugs alone. When the FC threshold is greater than 1, the RRODN levels of Salvia Miltiorrhiza and Radix Astragali are higher than those of Panax Notoginseng and Dalbergia Odorifera. When the FC is greater than 1, the significant RRODN value of Qishen Yiqi formulae shows little difference with that of Salvia Miltiorrhiza, while the significant RRODN values corresponding to Radix Astragali, Panax Notoginseng, and Dalbergia Odorifera are similar.
8.2.2.4.4 Compatibility Rules of Qishen Yiqi at the Pathway Level
EoR > 50% was used as the threshold value to define the effective callback genes of the Qishen Yiqi formulae and its four drugs against ODN, and the biological pathway enrichment of the genes with EoR > 50% were counted and analyzed. The results indicated that Radix Astragali could affect more pathways, while the other three drugs had similar number of signaling pathways. The 14 pathways regulated by the Qishen Yiqi formulae can be also regulated by at least one of its drugs. Radix Astragali and Salvia Miltiorrhiza could both regulate 10 pathways of the 14 pathways, Panax Notoginseng could regulate 8 pathways, while Dalbergia Odorifera could only regulate 5 pathways, which all indicates that Radix Astragali and Salvia Miltiorrhiza may play a major role in the regulation effects of Qishen Yiqi formulae at the pathway level.
8.2.3 Study of “Tonifying Qi and Activating Blood” Efficacy in TCM Formulae
The Qishen Yiqi formulae is composed of Salvia Miltiorrhiza, Radix Astragali, Panax Notoginseng, and Dalbergia Odorifera, and it is prescribed for treatment of ischemic heart disease [79]. The Qishen Yiqi dripping pills manufactured according to Qishen Yiqi formulae [80] are clinically used for the treatment of CHD due to Qi deficiency and blood stasis with definite curative effects. The four herbs are formulated in accordance with the prescription of “sovereign-minister-assistant-courier,” however, their mechanism of the treatment of ischemic heart disease and related complications needs to be further studied [81]. The preparation was originally called Huangqi Danshen Dropping Pills [82,83,84], and then renamed as Qishen Yiqi dropping pills. TCM theory indicates that Qi deficiency and blood stasis lead to the pathogenesis of ischemic heart disease. Tonifying Qi and activating blood circulation is the basic treatment regimen for ischemic heart disease. Therefore, research on the efficacy of Qi tonifying and blood activating could conducted from the view of the Qi tonifying and blood activating of Radix Astragali and Salvia Miltiorrhiza. Based on the ischemic heart disease network, with the regulation of Salvia Miltiorrhiza and Radix Astragali and network regulation analysis method, it is feasible to carry out research of efficacy of Qi tonifying and blood activating on the disease molecular network view. What’s more, further cellular and molecular biology experiments are warranted to verify related molecular mechanisms.
8.2.3.1 Data Acquisition and Processing
8.2.3.1.1 Construction of Ischemic Heart Disease Network
The disease network was constructed with application of CHD@ZJU cardiovascular disease network pharmacology research platform, which was used to investigate the efficacy of the Qishen Yiqi formulae of its Qi tonifying and blood circulation promoting.
8.2.3.1.2 Construction of Molecular Network of Radix Astragali and Salvia Miltiorrhiza for Regulating of Ischemic Heart Disease
The myocardial tissue samples of AMI rats were provided by the Institute of Pharmacoinformatics, Zhejiang University. A total of seven groups of samples were involved in this study, including normal group (Ctrl), model group (MI), Qishen Yiqi Decoction group (QSYQ), Salvia Miltiorrhiza group (DS), Radix Astragali group (HQ), Panax Notoginseng group (SQ), and Dalbergia Odorifera group (JX). Three biological replicate samples were included in each group.
RNA extraction, purification, sample quality inspection, chip experiment, and data acquisition were completed by chip company. The Affymetrix Rat 230 2.0 chip was used, and the final chip data was saved in the. CEL format files. ArrayTrack software was applied to read the. CEL files and the chip data was exported to Excel in .txt format files for normalization processing. Data from 21 chips was processed for data standardization. The median expression value for each chip was set to 1.000 (Median = 1000) by multiplying with the weight coefficient.
8.2.3.2 Network Construction and Visualization
In the network modeling process, the PPI relationships were obtained with integration of HPRD and BioGRID databases. As for disease-associated genes collection, CHD@ZJU was applied, together with CHD and myocardial ischemia related genes from RGD database were also used. The chip expression data was processed with normalization to attain gene expression information. Gene expression profile information of AMI was obtained through transcriptomics technology, then Cytoscape software (version 3.0.1) was used to conduct the research on visualization of the ODN.
8.2.3.3 Network Analysis and Prediction
Efficacy exploration of Radix Astragali and Salvia Miltiorrhiza in “tonifying Qi and promoting blood circulation” in ischemic heart disease at molecular network level.
In Sect. 8.2.2, the NRIODN was used to study the compatibility rules of the Qishen Yiqi formulae with “sovereign-minister-assistant-courier.” At the molecular network level, the network recovery regulation ability of the sovereign and minister drugs (Radix Astragali and Salvia Miltiorrhiza) is significantly better than that of assistant and courier drugs (Panax Notoginseng and Dalbergia Odorifera), which is accord with the compatibility rules “sovereign-minister-assistant-courier” of these drugs. In our previous pharmacodynamic and proteomics research, results showed that Radix Astragali and Salvia Miltiorrhiza, the sovereign and minister drugs in Qishen Yiqi formulae, targeted on the energy metabolism and blood circulation pathways of the imbalance network, respectively, which was consistent with the traditional efficacy of “replenishing Qi and promoting blood circulation.”
In this section, NRI-ODN algorithm was cited to calculate the callback ability of the drug to the organisms network balance, and the drug efficacy was evaluated systematically. The NRI-ODN algorithm further considered the influence of network topology and node callback efficiency on the overall recovery regulation ability, so it could reflect the influence of TCM drugs on the network more reasonably.
Further pathway enrichment analysis was carried out. Effective callback genes with a threshold value (EoR > 50%) were defined and obtained for Qishen Yiqi formulae and its constituent drugs. ArrayTrack (version 3.5.0) software was cited to analyze the pathway of the effective callback gene list. The KEGG pathway database was selected as the pathway information source, and fisher p value < 0.05 was set as the standard to identify the significant pathways. The KEGG pathway database contains cellular processes, environmental information processes, metabolism, human diseases, etc. Considering the relevance with PPI, only cellular and environmental information processes were studied in this research.
8.2.3.4 Validation and Summary
8.2.3.4.1 “Tonifying Qi” Effects of Radix Astragali: Detecting Energy Metabolism-Related Indicators
The rats LAD ligation model was used on under anesthesia. Myocardial infarction Rats were divided randomly into different groups with intragastric administration of TCM every morning for 7 consecutive days. The rats were administered 10 ml/kg in the Sham operation group (1% CMCNa), model group model (1% CMCNa), high-dose Radix Astragali group (200 mg/kg/day, ARE-H), and Radix Astragali low-dose group (100 mg/kg/day, ARE-L). Radix Astragali extract was provided by Tianjin Tasly Pharmaceutical Co., Ltd.
After 7 days of administration, the rats were anesthetized by intraperitoneal injection of 360 mg/kg chloral hydrate. Blood was collected from the abdominal aorta and allowed to stand for 30 min at room temperature, then was centrifuged at 4000 rpm for 15 min. The supernatant was collected and stored in the refrigerator at −80 °C. Then the heart was perfused with 20 ml of precooled normal saline, and was cut off and washed in precooled normal saline. The connective tissue and right ventricle were removed. Myocardium below the ligation site were placed in a precooled 2 ml cryopreservation tube and stored at −80 °C.
The test kit was used to measure biochemical indicators in the serum and myocardial tissue, HPLC chromatography method was used to detect the content of high-energy phosphate compounds in myocardial tissue, and Western Blot was used to detect the expression of proteins related to energy metabolism in the myocardial tissue.
Serum and myocardial tissue homogenate: detecting of content or activity of lactate dehydrogenase (LDH), pyruvate (PA), lactic acid (LD), creatine kinase (CK), and free fatty acid (FFA).
8.2.3.4.2 Effects of Representative Ingredients of Radix Astragali on Energy Metabolism of Hypoxic Cardiomyocytes
The effects of the three representative compounds, i.e. Astragaloside IV, calycosin, and formononetin, against ATPase activity in H9c2 cardiomyocytes were detected respectively. The hypoxic device consists of a hypoxic culture chamber (Modular Incubator Chamber, Billups-Rothenberg). Hypoxic conditions were created by using mixed air to replace the air in the hypoxic culture chamber (ventilate mixed air for about 15 min). The culture chamber was placed in a 37 °C, 5% CO2 incubator for 12 h. The ATP content of cardiomyocytes was detected using the CellTiter-Glo™ reagent, and the luminescence intensity was measured with a fluorescence chemiluminescence analyzer (Tecan F200, USA).
At the same time, the effects of the three representative compounds against ATPase activity in cardiomyocytes were detected, using a reference ultra-trace ATPase test kit according to kit instructions.
Finally, Western Blot was conducted to detect the effects of the three active compounds on the expression of the PGC-1 and PPAR-α, two proteins which are associated with energy metabolism in hypoxic cardiomyocytes.
8.2.3.4.3 Main Conclusions
-
1.
Based on the ischemic heart disease network and transcriptomics information, NRI-ODN algorithm application can reflect the recovery regulation ability of Radix Astragali and Salvia Miltiorrhiza on the organisms network balance and is able to evaluate TCM drug efficacy systematically. Pathway level studies indicated that the “Qi tonifying and blood circulation activating” of Radix Astragali and Salvia Miltiorrhiza may be associated with the regulation of myocardial energy metabolism and blood circulation.
-
2.
Radix Astragali extract showed effects on LDH activity and CK, FFA, PA, and LA levels in the serum and myocardial tissue of AMI rats. Compared to the sham operation group, the LDH activity and CK level in the serum and ischemic myocardial tissue were significantly increased. After administration of Radix Astragali extract (200 mg/kg/day, 100 mg/kg/day), the LDH activity and CK level in the serum and ischemic myocardium significantly decreased compared to those of the model group. The serum LDH activity and CK levels of ischemic myocardial tissue in the high-dose group were significantly different from those in the model group (P < 0.05). These results show that the Radix Astragali extract could effectively alleviate ischemic injury of AMI rats. Compared with the sham operation group, the levels of FFA, PA, and LA in serum and ischemic myocardial tissue were significantly increased. After intragastric administration of the Radix Astragali extract (200 mg/kg/day, 100 mg/kg/day), the levels of FFA, PA, and LA in serum and ischemic myocardial tissue decreased compared to those in the model group. The levels of FFA, PA, and LA in serum and ischemic myocardium of the high-dose group (200 mg/kg/day) significantly decreased (P < 0.01, P < 0.05), while the FFA level in ischemic myocardium with the low-dose administration (100 mg/kg/day) showed significantly decreased (P < 0.05). The results showed that the Radix Astragali extract could effectively regulate the energy metabolism-related metabolites in serum and myocardial tissue of AMI rats.
-
3.
Effects of Radix Astragali extract on high-energy phosphate compounds in ischemic myocardial tissues of rats with myocardial ischemia. The HPLC detection showed that after LAD ligation, the levels of ATP, ADP, AMP, and TAN in myocardial tissue decreased. Compared to the model group, the concentrations of ATP, ADP, AMP, and TAN increased with treatment of Radix Astragali extract of high and low dosages.
-
4.
Effects of active compounds of Radix Astragali on ATP level of hypoxic cardiomyocytes. Ast can promote the production of ATP in H9c2 cardiomyocytes under hypoxic conditions in a dose-dependent manner. Compounds Cal and For can also increase the production of ATP, showing a good dose-effect relationship.
-
5.
Effects of Radix Astragali active compounds on ATPase activity of hypoxic cardiomyocytes. Compounds Ast, Cal, and For can enhance the activities of Na+-K+-ATPase and Ca2+-Mg2+-ATPase of H9c2 under hypoxic conditions with a good dose-effect relationship.
-
6.
Effects of active compounds of Radix Astragali on PGC-1 and PPAR-α in hypoxic cardiomyocytes. Compared with the normal group, the protein expression of PPAR-α decreased significantly while protein expression of PGC-1 decreased a little. Ast, Cal, and For can significantly promote the expression of PGC-1, but the expression of PPAR-α increases slightly.
In summary, Radix Astragali can improve the energy metabolism of ischemic myocardium, and PPAR pathway activation may be the involved mode of action.
8.3 Practice of Disease Network Application for Holistic Efficacy Evaluation of TCM Drug
The rational application of network pharmacology techniques in the field of TCM research and the development of TCM network pharmacology are currently hotspots of TCM drug research. Currently, the applications of network pharmacology in TCM research mainly involve the construction of TCM information database, construction of TCM network model, study of the TCM components-target relationships, analysis of TCM biological network, and so on. Establishing a scientific and systematic evaluation system to conduct activity evaluation of TCM is a key technical link [85] in the creation and optimal design of modern new TCM drugs, which is also a difficult point in the current research. With the discovery of drug multi-target effects, drug optimization design strategy based on multiple targets is able to more reasonably reflect drug efficacy, side effects, in vivo processes, and other characteristics [86]. Network pharmacology research has further developed the concept of multi-objective optimization. The effects of a drug on the overall network as a comprehensive evaluation index to replace the traditional single-indicator or multi-objective evaluation system, in which pharmacodynamic evaluation based on system biology data has made remarkable progresses [87, 88] in the field of the drug repositioning. By constructing the system network model, drug actions were represented as the characteristics of the holistic network, which makes breakthrough against the bottleneck that single target could not fully represent the overall effects of drugs against the biological system, while it also solves the faced problem of multi-objective optimization when using multi-efficacy indicators for evaluation. What’s more, the network model also reflects changes in the biological system responding to diseases and drugs at the molecular level. This allows for further research on the mode of action and reveals the biological pathways and molecular mechanisms involved during the drug treatment process. This type of research does not rely on the drug structural characteristics, nor does it aim to a specific disease target, which reduces the research limitations. Therefore, compared to the structure-dependent traditional chemical medicine research, it is more suitable for the development of holistic Chinese medicine research.
Holistic biological network analysis is different from the study of drug-target network, which adopts homogeneous network or a hybrid network model integrating both homogeneous and heterogeneous networks to replace the heterogeneous drug-target network. More emphasis is placed on the relationship within the biological system together with the impact of drugs on the whole biological network, rather than only focusing on the direct effect and impact of drugs on the target.
Considering Shenmai injection as an example, the Network Recovery Index (NRI) was used to quantitatively to evaluate the callback effect of Shenmai Injection on acute myocardial ischemia imbalance network.
8.3.1 Data Acquisition and Processing
8.3.1.1 Research on Acute Myocardial Ischemia in Rats
Male SD rats were subjected to LAD ligation, and divided into three groups, sham, model, and drug. The dosage of Shenmai injection was 10 ml/kg, and the rats were intraperitoneally injected for 7 consecutive days (n = 8 in each group). After 7 days of administration, rat cardiac function was evaluated by echocardiography. The ischemic penumbra area between the necrotic area and normal tissue in the myocardium were sampled and used for mRNA extraction and chip analysis research. The animal experiment was conducted by the department of pharmacology, the second military medical university, and the echocardiography measurement results were processed by Zhongshan Hospital affiliated to Fudan University.
8.3.1.2 Chip Experiments and Data Preprocessing
The chip used in this study was the Affymetrix Rat 230 2.0 chip based on rat genome-wide chip, including 31,099 probes. There were 24 samples, n = 4 for red ginseng group, and n = 5 for the Ophiopogon japonicus group, Shenmai injection group, normal group, and model group. Affymetrix expression profiling chip matching kit—GeneChip 3′IVT Express Kit (Cat#901229, Affymetrix, Santa Clara, CA, US) and standard operating procedures (SOPs) were applied to amplify, label, and purify mRNA in the total RNA of the samples to obtain Biotin-labeled cRNAs.
According to the hybrid standard procedure and matching kit provided by the Affymetrix expression profile chip—GeneChip® Hybridization, Wash and Stain Kit (Cat#900720, Affymetrix, Santa Clara, CA, US), rolling hybridization was carried out for 16 h in a 45 °C Hybridization Oven 645 (Cat#00-0331-220V, Affymetrix, Santa Clara, CA, US). After the hybridization was completed, the chip was then washed in a Fluidics Station 450 (Cat#00-0079, Affymetrix, Santa Clara, CA, US) according to the SOPs provided by Affymetrix.
The chip results were scanned by GeneChip® Scanner 3000 (Cat#00-00212, Affymetrix, Santa Clara, CA, US). The original data were read by Command Console Software 3.1 (Affymetrix, Santa Clara, CA, US). Qualified data was normalized using Gene Spring Software 11.0 (Agilent technologies, Santa Clara, CA, US) with application of MAS 5.0 as the algorithm.
The chip experiments were completed by the Shanghai Biotechnology Corporation.
8.3.2 Network Construction and Visualization
The network model, used to evaluate the effects of Shenmai injection against AMI, is referred to the enriched pathway network model, and was mainly based on the significantly enriched signaling pathways and related gene-association information in AMI. The specific construction process is as follows: (1) P value < 0.01 and Fold change > 1.5 were selected as the threshold, the genes were extracted and pathway enrichment analysis was conducted to obtain the corresponding information. (2) KEGG database was used to retrieve all of the genes contained in these pathways. (3) The HPRD database was used to acquire the correlation relationships between these genes, and network model was constructed.
With p value < 0.01 and fold change > 1.5 as the threshold, a total of 1957 probes, with differential expressions produced with comparing of the model and control groups, were obtained and involved a total of 1376 related genes. Pathway enrichment analysis was performed on these genes, and 27 enriched KEGG signaling pathways (Fisher P value < 0.05) were obtained, including 10 metabolism pathways and 10 cellular process pathways, 2 environmental signal transduction pathways, 4 disease pathways, and DNA replication pathway. These pathways involve a total of 1478 related genes. By searching the HPRD database, 905 genes were found to have at least one PPI association with other genes (or themselves), involving a total of 2618 PPIs. 700 genes were found to form the largest sub-network. The enriched pathway network diagram was generated, in which nodes of different colors represented that they belong to different signaling pathways. Network parameter analysis showed that the connectivity distribution is in accordance with the characteristics of scale-free network (R2 = 0.868), indicating that it has the properties of general biological network.
8.3.3 Network Analysis and Prediction
Analysis of the alleviation effects of Shenmai injection against network imbalance induced by AMI.
Changes of gene expression levels were used to create networks to reflect the status of the network before and post-modeling and Shenmai injection administration, as well as the recovery regulation trend post-Shenmai injection administration. In the drug regulation network, the red node represents the up-regulated expression level, while the green node represents the down-regulated node. By conducting network comparing, i.e., Model vs sham, Shenmai vs sham, Shenmai vs Model, holistic network expression changes were analyzed both for post-AMI and post-Shenmai injection administration treatments. Information on regulation of the overall network expression on MI was obtained, and efficacy associated mechanism post-Shenmai administration was found.
Shenmai injection shows the ability to make recovery regulation effects against the network imbalance caused by ischemic modeling. What’s more, NRI index was applied to evaluate the network recovery capability. For NRI calculation, refer to Sect. 8.2.2.
8.3.4 Main Conclusions
-
1.
Echocardiography evaluation of left ventricular function in the rats was done 7 days after AMI as the apparent pharmacodynamic index of Shenmai injection, to calculate the evaluation indexes of left ventricular function, including Ejection Fraction (EF) and Fractional Shortening (FS). The EF and FS values of rats in the modeling group decreased significantly after 7 days of AMI and increased significantly after Shenmai injection administration (P < 0.05), showing myocardial repair effects. However, the changes of EF and FS values after administration of red ginseng and Ophiopogon japonicus alone, respectively, were not statistically significant (p > 0.05).
-
2.
The administration of Shenmai injection alleviates network disorders caused by AMI. It was found that the holistic network expression change-related trend is similar to that of after the administration of the Shenmai injection, that is, the injection does not produce excessively strong regulatory effects that affect the balance of the organisms. Compared to the regulatory trend of MI modeling, the change in expression levels of the Shenmai injection is almost opposite, that is, the Shenmai injection could alleviate the imbalance of the enrichment pathway network caused by AMI and regulate it back to a normal status.
-
3.
By analyzing the expression levels of the top 10 genes with the most significant expression imbalance after MI modeling, it was found that the expression levels of all nodes that were significantly regulated were recalled after the administration of the Shenmai injection, which further confirms the conclusion that the Shenmai injection has a callback to the network imbalance caused by ischemic modeling.
-
4.
Shenmai injection has over 90% recovery regulation ability for genes that are significantly dysregulated after AMI, and the overall network callback ability was 77.9%. In contrast, the network callback ability of red ginseng and Ophiopogon japonicus is relatively weak. Red ginseng can only callback about 50% of the ODN, while the effect of Ophiopogon japonicus is even lower, with only about 16.4–35.4%. Finally, the NRI score of Shenmai injection was 0.876, much higher than the NRI scores of red ginseng and Ophiopogon japonicus (0.498 and 0.269, respectively), indicating that red ginseng and Ophiopogon japonicus have a significant synergistic enhancing effect when administered concurrently in Shenmai injection. They also produce a stronger callback effect on the expression imbalance that occurs in the enriched pathway network.
Human disease spectrum has changed from communicable diseases to non-communicable diseases (NCDs). The mortality and disability rate of NCDs has brought severe challenges to the human medical and health system. The occurrence and development of diseases are often not limited to a single gene, but a manifestation of the interactions among internally associated multi-molecular processes. Humans have made new progress in understanding complex diseases. From the view of structure and function of the “molecular biological network” to systematically reveal the molecular mechanisms of complex diseases, the internal relationships between disease and disease, and the modes of action of drugs is a new research strategy.
Humanity’s understanding of the role of drugs has changed from the traditional “one drug, one target” model to the “network target, multi-component treatment” pattern. As a historical heritage and treasure of China, TCM plays an important role in China’s medical and health care system, and thus makes positive contributions to the world’s medical and health system. TCM is the most important way for preventing and treating diseases in China. As a complex system with multiple ingredients, TCM has a nonlinear interaction relationship with complex diseases. Therefore, the understanding and analysis of this mode of action need to break through the traditional pharmacological research methods. With the rapid development of bioinformatics, systems biology, and polypharmacology, web-based methods have become a powerful tool for the study of complex diseases and nonlinear drug-disease complex modes of action.
In summary, this chapter studies cardiovascular disease, which is rank first in the diseases to cause human deaths, and analyzes the occurrence and development of ischemic heart disease through disease network construction and analysis. Based on the disease network, we conducted the research on the integrated mechanisms of TCM, the law of compatibility, the efficacy of “supplementing Qi and promoting blood circulation,” and the regulation of ODN. In the research process, pharmacodynamics, pharmacokinetics, transcriptomics, proteomics, metabolomics, bioinformatics, network pharmacology, and other methods and technologies were integrated to design experimental studies at the organism, cellular, molecular, and other levels. Examples of several famous TCM products mentioned in this chapter, namely Qishen Yiqi dropping pills, Xuesaitong injection, and Shenmai injection, have obvious clinical efficacy and social benefits. They play integrated regulation roles, including multi-components, multi-targets, and multi-pathways in the prevention and treatment of ischemic heart disease. Therefore, the research in this chapter was also designed and carried out based on a systematic and integrated perspective. The research concepts and methods in this chapter can provide references for the study of other diseases and the effects of other drugs.
Certainly, for disease-based network pharmacology study there will be more extended applications, including deciphering of disease–disease associations (DDAs), drug repositioning research, drug-target prediction, and research on network toxicology of TCM. Future research also needs to integrate multi-dimensional “-omics” information, including genes, RNA, proteins, endogenous metabolites, etc., which could facilitate the network-based research to play a key role in the modernization and internationalization of TCM.
References
Li S. Exploring traditional Chinese medicine by a novel therapeutic concept of network target. Chinese J Integr Med. 2016;22(9):647–52.
Zhang C, Zhou W, Guan DG, et al. Network intervention, a method to address complex therapeutic strategies. Front Pharmacol. 2018;9:754.
World Health Organization. Who traditional medicine strategy 2014–2023. 2013. https://www.who.int/publications/list/traditional_medicine_strategy/zh/ (in Chinese) [世界卫生组织. 世卫组织2014-2023年传统医学战略. 2013. https://www.who.int/publications/list/traditional_medicine_strategy/zh/].
Fan X, Xiao S, Ai Ni, et al. Study on the effective substance group of Xiaoqinglong Decoction based on network prescription science. Chinese J Traditional Chinese Med. 2015;40(13):2634–2638 (in Chinese) [范骁辉, 肖舜, 艾妮, 等. 基于网络方剂学的小青龙汤类方功效物质组研究. 中国中药杂志, 2015, 40(13):2634–2638].
Fan X, Cheng Y, Zhang B. Network pharmacology: a new strategy for modern research of prescriptions. Chinese J Traditional Chinese Med. 2015;40(1):1–6 (in Chinese) [范骁辉, 程翼宇, 张伯礼. 网络方剂学: 方剂现代研究的新策略. 中国中药杂志, 2015, 40(1):1–6].
Fan X, Zhao X, Jin Y, et al. On the establishment of network toxicology and traditional Chinese medicine network toxicology research ideas. Chinese J Traditional Chinese Med. 2011;36(21):2920–2 (in Chinese) [范骁辉, 赵筱萍, 金烨成, 等. 论建立网络毒理学及中药网络毒理学研究思路. 中国中药杂志, 2011, 36(21):2920–2922].
Wu L, Wang Y, Fan X. Internet pharmacology technology tools: network visualization and network analysis. Chinese J Traditional Chinese Med. 2011;36(21):2923–5 (in Chinese) [吴磊宏, 王毅, 范骁辉. 网络药理学技术工具: 网络可视化及网络分析. 中国中药杂志, 2011, 36(21):2923–2925].
Wu L, Gao X, Cheng Y, et al. Research on network pharmacology of decoction pieces of traditional Chinese medicine based on correlation between indications and indications of traditional Chinese medicine. Chinese J Traditional Chinese Med. 2011;36(21):2907–10 (in Chinese) [吴磊宏, 高秀梅, 程翼宇, 等. 基于中医主治关联的中药饮片网络药理学研究. 中国中药杂志, 2011, 36(21):2907–2910].
Wu L, Gao X, Wang L, et al. Prediction of multi-component action target and network pharmacology of Aconitum carmichaeli. Chinese Herbal Med. 2011;36(21):2916–9 (in Chinese) [吴磊宏, 高秀梅, 王林丽, 等. 附子多成分作用靶点预测及网络药理学研究. 中国中药杂, 2011, 36(21):2916–2919].
Li X, Wu L, Fan X, et al. Study on network pharmacology of main active components of Compound Danshen Prescription. Chinese J Traditional Chinese Med. 2011;36(21):2911–5 (in Chinese) [李翔, 吴磊宏, 范骁辉, 等. 复方丹参方主要活性成分网络药理学研究. 中国中药杂志, 2011, 36(21):2911–2915].
Wang Y, Fan XH, Qu HB, et al. Strategies and techniques for multi-component drug design from medicinal herbs and traditional Chinese medicine. Curr Top Med Chem. 2012;12(12):1356–62.
Wu LH, Wang Y, Nie J, et al. A network pharmacology approach to evaluating the efficacy of Chinese medicine using genome-wide transcriptional expression data. Evid Based Complement Alternat Med. 2013;2013:915343.
Wu LH, Li X, Yang JH, et al. CHD@ZJU: a knowledgebase providing network-based research platform on coronary heart disease. Database. 2013;2013:bat047.
Yang ZZ, Yang JH, Liu W, et al. T2D@ZJU: a knowledgebase integrating heterogeneous connections associated with type 2 diabetes mellitus. Database. 2013;2013:bat052.
Wang LL, Li Z, Zhao XP, et al. A network study of Chinese medicine Xuesaitong injection to elucidate a complex mode-of-action with multi-compound, multi-target and multi-pathway. Evid Based Complement Alternat Med. 2013;2013:652373.
Yang JH, Li Z, Fan XH, et al. A three step network based approach (TSNBA) to finding disease molecular signature and key regulators: a case study of IL-1 and TNF-alpha stimulated inflammation. PLoS One. 2014;9(4):e94360.
Wang LL, Li Z, Shao Q, et al. Dissecting active ingredients of Chinese medicine by content-weighted ingredient-target network. Mol BioSyst. 2014;10:1905–11.
Li X, Wu LH, Liu W, et al. A network pharmacology study of Chinese medicine QiShenYiQi to reveal its underlying multi-compound, multi-target, multi-pathway mode of action. PLoS One. 2014;9(5):e95004.
Wu LH, Wang Y, Li Z, et al. Identifying roles of “Jun-Chen-Zuo-Shi” component herbs of QiShenYiQi formula in treating acute myocardial ischemia by network pharmacology. Chinese Med. 2014;9:24.
Yang JH, Li Z, Fan XH, et al. Drug-disease association and drug-repositioning predictions in complex diseases using causal inference-probabilistic matrix factorization. J Chem Inf Model. 2014;54(9):2562–9.
Liu YF, Ai N, Keys A, et al. Network pharmacology for traditional Chinese medicine research: methodologies and applications. Chinese Herbal Med. 2015;7(1):18–26.
Ai N, Fan XH, Ekins S. In silico methods for predicting drug-drug interactions with cytochrome P-450s, transporters and beyond. Adv Drug Deliv Rev. 2015;86:46–60.
Liu YF, Ai N, Liao J, et al. Transcriptomics: a sword to cut the Gordian knot of traditional Chinese medicine. Biomark Med. 2015;9(11):1201–13.
Chen Q, Zheng J, Shao Y, et al. Network-based assessment on chemical-induced cholestatic liver injury. Curr Top Med Chem. 2016;16(30):3668–77.
Chen Q, Ai N, Liao J, et al. Revealing topics and their evolution in biomedical literature using bio-DTM: a case study of ginseng. Chinese Med. 2017;12:27.
Wang Y, Zhao Y, Jiang W, et al. iTRAQ-based proteomic analysis reveals recovery of impaired mitochondrial function in ischemic myocardium by Shenmai formula. J Proteome Res. 2018;17(2):794–803.
Liao J, Hao C, Huang WH, et al. Network pharmacology study reveals energy metabolism and apoptosis pathways-mediated cardioprotective effects of SHENQI FUZHENG. J Ethnopharmacol. 2018;227:155–65.
Dwinell MR, Worthey EA, Shimoyama M, et al. The rat genome database 2009: variation, ontologies and pathways. Nucleic Acids Res. 2009;37(suppl 1):D744–9.
Lopes CT, Franz M, Kazi F, et al. Cytoscape web: an interactive web-based network browser. Bioinformatics. 2010;26(18):2347–8.
Yang M, Chen J-L, Xu L-W, et al. Navigating traditional chinese medicine network pharmacology and computational tools. Evid Based Complement Alternat Med. 2013;2013:731969.
Gandhi T, Zhong J, Mathivanan S, et al. Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet. 2006;38(3):285–93.
Xu J, Li Y. Discovering disease-genes by topological features in human protein–protein interaction network. Bioinformatics. 2006;22(22):2800–5.
Goh K-I, Cusick ME, Valle D, et al. The human disease network. Proc Natl Acad Sci. 2007;104(21):8685–90.
Feldman I, Rzhetsky A, Vitkup D. Network properties of genes harboring inherited disease mutations. Proc Natl Acad Sci. 2008;105(11):4323–8.
Barabási A-L, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12(1):56–68.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4(1):2.
Maere S, Heymans K, Kuiper M. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–9.
Yaoita H, Ogawa K, Maehara K, et al. Apoptosis in relevant clinical situations contribution of apoptosis in myocardial infarction. Cardiovasc Res. 2000;45(3):630–41.
Palojoki E, Saraste A, Eriksson A, et al. Cardiomyocyte apoptosis and ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2001;280(6):H2726–31.
Crow MT, Mani K, Nam Y-J, et al. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95(10):957–70.
Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17(11):1410–22.
Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–12.
Hansson GK, Robertson A-KL, Söderberg-Nauclér C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297–329.
Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685–95.
Schwartz RS, Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–65.
Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta. 2005;1754(1):253–62.
Bertrand L, Horman S, Beauloye C, et al. Insulin signalling in the heart. Cardiovasc Res. 2008;79(2):238–48.
Sena S, Hu P, Zhang D, et al. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J Mol Cell Cardiol. 2009;46(6):910–8.
Finck BN. The PPAR regulatory system in cardiac physiology and disease. Cardiovasc Res. 2007;73(2):269–77.
Yamauchi T, Kamon J, Minokoshi YA, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–95.
Robinson ES, Khankin EV, Karumanchi SA, et al. Hypertension induced by vascular endothelial growth factor signaling pathway inhibition: mechanisms and potential use as a biomarker. Proceedings of the seminars in nephrology. Elsevier; 2010:591–601.
Aramoto H, Breslin JW, Pappas PJ, et al. Vascular endothelial growth factor stimulates differential signaling pathways in in vivo microcirculation. Am J Physiol Heart Circ Physiol. 2004;287(4):H1590–8.
Olsson A-K, Dimberg A, Kreuger J, et al. VEGF receptor signalling? In control of vascular function. Nat Rev Mol Cell Biol. 2006;7(5):359–71.
Kiefer FN, Berns H, Resink TJ, et al. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002;16(8):771–80.
Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(20):3589–94.
Katoh K, Kano Y, Ookawara S. Role of stress fibers and focal adhesions as a mediator for mechano-signal transduction in endothelial cells in situ. Vasc Health Risk Manag. 2008;4(6):1273.
Pellegrino M, Furmaniak-Kazmierczak E, Leblanc JC, et al. The apolipoprotein (a) component of lipoprotein (a) stimulates actin stress fiber formation and loss of cell-cell contact in cultured endothelial cells. J Biol Chem. 2004;279(8):6526–33.
De Caterina R, Libby P. Endothelial dysfunctions and vascular disease. Wiley; 2008.
Thomas T, Advani A. Inflammation in cardiovascular disease and regulation of the actin cytoskeleton in inflammatory cells: the actin cytoskeleton as a target. Cardiovasc Hematol Agents Med Chem. 2006;4(2):165–82.
Vanhoutte PM, Rubanyi GM, Miller VM, et al. Modulation of vascular smooth muscle contraction by the endothelium. Annu Rev Physiol. 1986;48(1):307–20.
Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51(4):600–6.
Bujak M, Frangogiannis NG. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74(2):184–95.
Niessen K, Karsan A. Notch signaling in cardiac development. Circ Res. 2008;102(10):1169–81.
Li Y, Hiroi Y, Liao JK. Notch signaling as an important mediator of cardiac repair and regeneration after myocardial infarction. Trends Cardiovasc Med. 2010;20(7):228–31.
Ueda K, Takano H, Niitsuma Y, et al. Sonic hedgehog is a critical mediator of erythropoietin-induced cardiac protection in mice. J Clin Invest. 2010;120(6):2016.
Roncalli J, Renault M-A, Tongers J, et al. Sonic hedgehog-induced functional recovery after myocardial infarction is enhanced by AMD3100-mediated progenitor-cell mobilization. J Am Coll Cardiol. 2011;57(24):2444–52.
Lemmens K, Doggen K, De Keulenaer GW. Role of neuregulin-1/ErbB signaling in cardiovascular physiology and disease implications for therapy of heart failure. Circulation. 2007;116(8):954–60.
Rao TP, Kühl M. An updated overview on Wnt signaling pathways a prelude for more. Circ Res. 2010;106(12):1798–806.
Goodwin A, D’Amore P. Wnt signaling in the vasculature. Angiogenesis. 2002;5(1–2):1–9.
Cohen ED, Tian Y, Morrisey EE. Wnt signaling: an essential regulator of cardiovascular differentiation, morphogenesis and progenitor self-renewal. Development. 2008;135(5):789–98.
Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117(8):1281–3.
Mascareno E, El-Shafei M, Maulik N, et al. JAK/STAT signaling is associated with cardiac dysfunction during ischemia and reperfusion. Circulation. 2001;104(3):325–9.
Zhang S, Liu X, Goldstein S, et al. Role of the JAK/STAT signaling pathway in the pathogenesis of acute myocardial infarction in rats and its effect on NF-κB expression. Mol Med Rep. 2013;7(1):93–8.
Hong C. Comparative proteomics study on compatibility mechanism of effective components of Qishen Yiqi formula. Hangzhou: Zhejiang University; 2010. (in Chinese) [洪诚韬. 比较蛋白质组学研究芪参益气方有效组分的配伍机制. 杭州:浙江大学, 2010].
Shi P. Fragmentation of four components of traditional Chinese medicine by mass spectrometry and pharmacokinetics of Qishen Yiqi formula. Hangzhou: Zhejiang University; 2010. (in Chinese) [史培颖. 中药四类成分质谱裂解规律及芪参益气方药代动力学研究. 杭州:浙江大学, 2010].
Li YF, Qu HB, Cheng YY. Identification of major constituents in the traditional Chinese medicine “QI-SHEN-YI-QI” dropping pill by high-performance liquid chromatography coupled with diode array detection-electrospray ionization tandem mass spectrometry. J Pharm Biomed Anal. 2008;47(2):407–12.
Li Y, Wang Y, Qu H, et al. Simultaneous determination of seven bioactive compounds in Chinese medicine “QI-SHEN-YI-QI” dropping pill by LC-UV and LC-ELSD. Chromatographia. 2008;67(3–4):293–7.
Wang J. Chemical basis and mechanism of Qishen Yiqi dropping pills. Tianjin: Nankai University; 2011. (in Chinese) [王静. 复方中药芪参益气滴丸化学物质基础与作用机理研究. 天津:南开大学, 2011].
Hong C, Wang Y, Lou J, et al. The effect of Qi Shen Yiqi prescription on myocardial proteome in rats with acute myocardial infarction. Chinese J Traditional Chinese Med. 2009;34(8):1018–21 (in Chinese) [洪诚韬, 王毅, 楼剑洲, 等. 芪参益气方对急性心肌梗死大鼠心肌蛋白质组的影响. 中国中药杂志, 2009, 34(8): 1018–1021].
Yan X, Wu N, Guo Z, et al. Traditional Chinese medicine preparation for treating angina pectoris of coronary heart disease and its preparation method. China. 2005 (in Chinese) [闫希军, 吴迺峰, 郭治昕, 等. 治疗冠心病心绞痛的中药制剂及其制备方法. 中国. 2005].
Wei C, Zhu M, Du W, et al. Research progress on clinical application of Qishen Yiqi dropping pills. Western Chinese Med. 2011;24(10):102–6 (in Chinese) [魏聪聪, 朱明丹, 杜武勋, 等. 芪参益气滴丸临床应用研究进展. 西部中医药, 2011, 24(10): 102–106].
Zhu X. Clinical observation of Huangqi Danshen dripping pills in treating angina pectoris of coronary heart disease. Beijing: Beijing University of Traditional Chinese Medicine; 2002. (in Chinese) [朱晓泉. 黄芪丹参滴丸治疗冠心病心绞痛临床观察. 北京:北京中医药大学, 2002].
Li Y. Clinical study on Huangqi Danshen dripping pill in treating angina pectoris of coronary heart disease with qi deficiency and blood stasis syndrome. Beijing: Beijing University of Traditional Chinese Medicine; 2003. (in Chinese) [李玉峰. 黄芪丹参滴丸治疗冠心病心绞痛气虚血瘀证的临床研究. 北京:北京中医药大学, 2003].
Wang Y, Gao X. Experimental study on the anti-acute myocardial ischemia induced by pituitrin with Huangqi Danshen drop pills. New Chinese Med Clin Pharmacol. 2003;14(2): 91–3 (in Chinese) [王怡, 高秀梅. 黄芪丹参滴丸抗垂体后叶素致大鼠急性心肌缺血的实验研究. 中药新药与临床药理, 2003, 14(2): 91–93].
Zhang B, Wang Y. Basic research on key scientific problems of prescriptions--development of modern Chinese medicine based on component compatibility. Chinese Nat Med. 2005;3(5): 258–61 (in Chinese) [张伯礼,王永炎. 方剂关键科学问题的基础研究--以组分配伍研制现代中药. 中国天然药物, 2005, 3(5):258–261].
Nicolaou CA, Brown N. Multi-objective optimization methods in drug design. Drug Discov Today Technol. 2013;10(3):e315–450.
Sirota M, Dudley JT, Kim J, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med. 2011;3(96):96ra77.
Dudley JT, Sirota M, Shenoy M, et al. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med. 2011;3(96):96ra76.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Fan, X., Li, X. (2021). Disease-Based Network Pharmacology Practice Process. In: Li, S. (eds) Network Pharmacology. Springer, Singapore. https://doi.org/10.1007/978-981-16-0753-0_8
Download citation
DOI: https://doi.org/10.1007/978-981-16-0753-0_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-0752-3
Online ISBN: 978-981-16-0753-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)