
Abstract
The formation of the head and face is a complex process which involves many different signaling cues regulating the migration, differentiation, and proliferation of the neural crest. This highly complex process is very error-prone, resulting in craniofacial defects in nearly 10,000 births in the United States annually. Due to the highly conserved mechanisms of craniofacial development, animal models are widely used to understand the pathogenesis of various human diseases and assist in the diagnosis and generation of preventative therapies and treatments. Here, we provide a brief background of craniofacial development and discuss several rare diseases affecting craniofacial bone development. We focus on rare congenital diseases of the cranial bone, facial jaw bones, and two classes of diseases, ciliopathies and RASopathies. Studying the animal models of these rare diseases sheds light not only on the etiology and pathology of each disease, but also provides meaningful insights towards the mechanisms which regulate normal development of the head and face.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


6.1 Overview
The formation of the head and face is a complex process which involves many different signaling cues regulating the migration, differentiation, and proliferation of the neural crest. This highly complex process is very error-prone, resulting in craniofacial defects in nearly 10,000 births in the United States annually. Due to the highly conserved mechanisms of craniofacial development, animal models are widely used to understand the pathogenesis of various human diseases and assist in the diagnosis and generation of preventative therapies and treatments. Here, we provide a brief background of craniofacial development and discuss several rare diseases affecting craniofacial bone development. We focus on rare congenital diseases of the cranial bone, facial jaw bones, and two classes of diseases, ciliopathies and RASopathies. Studying the animal models of these rare diseases sheds light not only on the etiology and pathology of each disease, but also provides meaningful insights towards the mechanisms which regulate normal development of the head and face.
6.2 Significance
Craniofacial defects are complex and often impact multiple structures of the head and face. One of the most severe craniofacial defects is craniosynostosis, or premature fusion of the skull bones, that occurs in 1/2500 births with detrimental consequences for brain and sensory organ development [1, 2]. The only current treatment for craniosynostosis is surgical separation of the sutures, and in some cases, the sutures become fused again over time requiring additional surgeries [1]. Similar survival treatments are available for many craniofacial disorders; however, these treatments are often limited to repeated surgical intervention and can result in lifelong economic burden to the patient’s family or community. One example of such a financial burden stems from analysis of the treatment cost for the repair of cleft lip and/or cleft palate, estimated to be nearly $700 million dollars just within the United States each year [3]. Rare genetic diseases affect a smaller proportion of the population occurring in less than 1 in 2000 births annually [4]. For example, only 1 in 500,000 individuals have been diagnosed with Robinow syndrome, described below [5]. Over 700 rare craniofacial disorders have been categorized and thus encompass a large percentage of the patients born each year with craniofacial defects [3, 4, 6]. Mouse models are valuable to understand rare human craniofacial defects because they have comparative anatomy and physiology and a complex assortment of genetic tools to study the function of genes in spatiotemporal manner. Animal models of rare diseases improve our understanding of disease etiology and pathology and oftentimes can result in the identification of novel treatment options, as we discuss below.
6.3 Introduction into Craniofacial Development
6.3.1 Conservation of Craniofacial Development
During embryonic development across many species, the regulatory mechanisms that govern pattern formation and mechanisms of development are highly conserved [7]. An example of this highly conserved phenomenon can be observed when analyzing embryo patterning across highly diverse organisms. The Hox gene cluster, originally identified for its role in Drosophila body patterning, is also required for vertebrate patterning of the axial skeleton [8]. Across vertebrate model organisms, craniofacial development is highly conserved at the molecular and morphological levels [9, 10]. For this reason, animal models are critical for our understanding of many aspects of human biology. Much of our understanding of human craniofacial development came from studying various embryonic models of mouse, frog, and chick [11]. Here we focus on the insights obtained from genetic mouse models of rare craniofacial defects affecting craniofacial bone development.
6.3.2 Craniofacial Development Is Highly Error-Prone
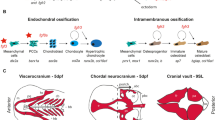
Craniofacial development begins with the migration of cranial neural crest cells (CNCC), a cell population unique to vertebrates [12, 13], from the neural tube to the frontonasal process (FNP), first branchial arch, and supraorbital arch (SOA) [1, 3, 4, 13, 14] (Fig. 6.1, and described in detail below). Many signaling pathways are vital for maintaining cellular and tissue movements and cell fate specification. These processes are dependent on specific signaling cues at specific spatial and temporal intervals to properly form the various structures of the head and face. For this reason, normal craniofacial development culminates from a series of events, beginning with initiation and migration and ending with differentiation of specialized cell types dependent on crucial spatiotemporal signaling [15]. If any of the steps of these events are altered, the result would be detrimental for normal development; thus, the process of craniofacial development is highly error-prone. In fact, one-third of all congenital defects result in craniofacial anomalies [3]. The precise timing of these various cellular mechanisms is not fully understood. However, the mechanisms that regulate the morphogenesis of various facial and cranial structures are still being investigated and our understanding of these pathways is rapidly expanding as new animal models emerge.

Craniofacial bones are derived from cranial neural crest and paraxial mesoderm. (a) Cranial neural crest and paraxial mesoderm migrate from adjacent to the neural tube into the frontonasal process and first branchial arch. (b) Schematic delineating the direction of differentiation and ossification from basal to apical to form the frontal (Fb) and parietal bones (Pb). (c) Schematic comparing the cranial and facial bones in a newborn human and mouse skull (F frontal, P parietal, N nasal, Mx maxilla, Z zygoid, Sp sphenoid, T temporal, O occipital, IP interparietal, Mn mandibular). (Panels (a) and (b) are reprinted and adapted from Ferguson and Atit 2018 with permission)
6.3.3 The Need for Animal Models of Craniofacial Defects
Due to the high prevalence of congenital craniofacial defects, there is a demand for an improved understanding of their etiology to enable proper diagnosis and discovery of therapeutic or preventative treatments [16, 17]. To this end, researchers rely on the use of animal models for a variety of reasons. First, testing various treatments and therapies is not ethically feasible in humans in most cases. Second, we are able to genetically and chemically model human diseases effectively and affordably in an animal model to understand the molecular basis of a disease and to test the aforementioned treatments in vivo. Third, because of the highly conserved nature of craniofacial development, there is a wide variety of vertebrate species that can be utilized to understand not only disease mechanisms, but also the mechanisms and processes that lead to normal craniofacial development.
In this review, we focus on three regions of the head and face and describe findings from recent studies that illuminate the etiology of several craniofacial defects. We first focus on cranial bone development and two rare cranial bone diseases, progressive osseous heteroplasia (POH) and fibrous dysplasia (FD). Second, we review the development of facial structures and etiology of micrognathia within Robinow syndrome and Nager syndrome. Finally, we will discuss two categories of rare diseases, ciliopathies and RASopathies. While there are many useful and relevant animal models, we have limited our discussions to focus on mouse genetic models. This is due to the high conservation of signaling pathways that regulate craniofacial structures between humans and mice and the ability to spatiotemporally test gene function in specific tissue and adequately model therapeutic treatments in mammalian models.
6.4 Defects in Cranial Bone
6.4.1 Brief Introduction to Calvarial Bone Development
6.4.1.1 Origins of Calvarial Bones
The calvaria consists of the skull bones that cover and protect the brain and other sensory organs. Mammalian skull bones are derived from cranial NCC and the paraxial mesoderm (PM) [1, 18, 19]. The CNCC give rise to the frontal bones while the PM gives rise to the parietal, squamous part of the temporal and occipital bones. Both contribute to the formation of the interparietal bones [12, 14, 19, 20] (Fig. 6.1).
6.4.1.2 Calvarial Bone Morphogenesis
In mice, the frontal and parietal bone precursors originate in the CNCC and PM mesenchyme and then migrate into the supraorbital arch (SOA) between embryonic day 9.5 (E9.5) and E12.5 [1, 19,20,21,22]. Morphogenesis of the frontal and parietal bones begins at E12.5 when the SOA mesenchyme begins to condense [23]. This condensation is pivotal to the formation of the progenitors/rudiments of the cranial bones and specification towards an osteogenic lineage [23, 24]. Upon specification, the calvarial bone progenitors express bone lineage-specific markers, such as runt-related transcription factor 2 (RUNX2) and alkaline phosphatase (AP) [25, 26]. From E13.5, the calvarial bone progenitors differentiate into committed calvarial bone osteoblasts and the cells expand apically and laterally until reaching the apex through mechanisms that remain to be defined [18, 21, 27, 28]. Unlike long bones of the body, calvarial bones form by intramembranous ossification. The condensed SOA mesenchyme directly differentiates into committed calvarial bone osteoblasts to form mineralized bone beginning at E12.5. This is in contrast to endochondral ossification of long bones, which requires formation of a cartilage template prior to mineralization and formation of mature bone. The calvarial bone mineralization for the frontal and parietal bone initiates in the SOA at approximately E14.5 and progresses apically following the wave of differentiation laid out by the osteogenic front [14, 29] (Fig. 6.1). Normal development of calvarial bones requires complex cellular signaling for proper cell differentiation and growth [20, 30]. This process is highly regulated and defects in cell signaling or cell migration results in various cranial defects including craniosynostosis [1, 27] and various skeletal dysplasias [31] (see below). Thus, it is important to understand the biological mechanisms that regulate normal morphogenesis of the cranial bones.
6.4.1.3 Bone Transcription Program
Upon migrating from the neural tube at E9.5, a subpopulation of the cranial mesenchyme in the SOA is fated to become calvarial bone progenitors and begin to express osteogenic genes from E11.5 onwards [14]. There are a growing number of genes and signaling pathways that are reported to govern this cell fate decision, and the transcription cascade that is involved in the specification of cranial bone progenitors is well understood [14, 20, 32]. The “skull bone initiation program” consists of a cascade of three major transcription factors, Msh homeobox 1 and 2 (Msx1 and Msx2), Runx2, and Osterix (Osx/Sp7) [14, 20]. Msx1 and Msx2 are expressed in the early migrating cranial mesenchyme at E10.5 and the calvarial bone precursors by E12.5 [28]. Runx2 expression requires expression of MSX proteins and is required for establishment of calvarial bone progenitors. As the calvarial bone precursors differentiate into calvarial bone progenitors, Runx2 expression is decreased and expression of Osx is increased. Osx is required for ossification of intramembranous bone. The function of these transcription factors has been well-studied using conditional mouse models with targeted genetic deletions of each gene [20, 32]. The bone morphogenetic protein (BMP), Wnt, Hedgehog (Hh), and Notch signaling pathways, among others, have been shown to regulate various aspects of cranial bone development and have been reviewed elsewhere [14, 20, 32, 33].
6.4.2 Rare Cranial Bone Defects
6.4.2.1 Rationale
There is a high prevalence of craniofacial defects and disease, in part due to the complexity of the many processes involved in normal skull bone development [11]. Collectively, craniofacial defects comprise one-third of all congenital defects and there are relatively few known preventative treatments or therapies [3]. Understanding the etiology of rare genetic diseases may elucidate new mechanisms of development and help to identify new therapeutic targets [17]. Here, we will explore two rare bone disorders to demonstrate the value of utilizing animal models in our endeavor to understand the cellular mechanisms of cranial bone defects in humans.
Heterotopic ossification refers to the ectopic formation of bone in soft tissues [34, 35]. This can occur as a result of trauma, cerebral injury or surgery, or more rarely an underlying genetic disorder. One type of hereditary heterotopic ossification is progressive osseous heteroplasia (POH). On the other end of this type of abnormal bone growth is fibrous dysplasia (FD). FD is characterized as the growth of fibrous tissue in place of normal bone [36, 37]. FD presents across multiple bones, including cranial bones, in McCune-Albright syndrome (MAS) [36, 37].
Here we will discuss the current understanding of the genetic and molecular mechanisms governing these diseases. We will also review how animal models have helped to advance the clinical understanding of POH and FD and how these models have provided new insights into normal intramembranous bone development.
6.4.2.2 Progressive Osseous Heteroplasia
6.4.2.2.1 Etiology and Molecular Mechanisms of POH
POH (OMIM: #166350) is an autosomal dominant disease linked to loss-of-function mutations in the GNAS gene [34, 35, 38, 39]. The disease presents as ectopic formation of intramembranous bone in the dermis and underlying subcutaneous tissues.
GNAS encodes the alpha subunit of the G-protein complex, Gαs [40,41,42]. G-protein signaling begins when a G-protein ligand binds to G-protein-coupled receptors (GPCRs) at the cell membrane. Different G-protein subunits bind to specific GPCRs to elicit a variety of cellular responses. In the context of osteogenic differentiation, Gαs binding to the receptor results in upregulation of cAMP/PKA signaling which inhibits Hh signaling by preventing nuclear translocation of GLI (Fig. 6.2) [39, 42]. When Gαs is lost, Hh signaling is no longer inhibited and thus there is increased ossification of cranial bone [43]. Outlined below are the animal models which delineated the mechanism and etiology of Gαs in POH.

Overview of signaling pathways linked with rare craniofacial diseases. (a) Noncanonical Wnt signaling (Robinow syndrome), (b) MAPK signaling (RASopathies), (c) Hh signaling in primary cilia (ciliopathies), (d) GαS signaling (progressive osseous heteroplasia and Fibrous dysplasia). (e) Canonical Wnt/β-catenin signaling (FD) increase or decrease in β-catenin leads to dysregulated bone differentiation. Red asterisks indicate disease associated proteins
6.4.2.2.2 Insights from Animal Models of POH
The first animal model of POH was established in heterozygous null Gnas mice [43]; however, this model did not fully recapitulate the unique phenotype observed in human patients with POH. First, while the heterozygous mouse models present heterotopic ossification in the dermis, there is no ossification present in the deeper subcutaneous cell populations, including muscle and connective tissues, and onset of heterotopic ossification occurred late in life, whereas POH in human patients normally presents during infancy [34, 41, 44,45,46]. Second, POH patients tend to exhibit heterotopic ossification biased laterally, in some cases the observed ossification failed to cross the midline—this pattern was not observed in the previously described heterozygous Gnas mouse models [43, 45].
Cairns et al. utilized a chick model to more accurately represent the human disease [45]. By injecting a dominant negative form of GNAS into specific somites, the authors demonstrated a mosaic effect of ectopic ossification similar to that observed in clinical patients of POH. Within this model, the authors were able to glean new mechanistic insight on the etiology of POH in humans. They determined that the likely mechanism of inheritance of GNAS mutations is dependent on a stem cell population, or nearby adjacent niche cells, which results in a mosaic distribution of ectopic bone. However, the source of the cell population responsible for driving the ectopic formation of intramembranous bone within dermal cell population is not known.
Conditional deletion of Gnas in specific murine tissues provided new insights into the mechanisms of osteoblast differentiation during embryonic development. Tissue-specific deletion of Gnas in limb mesenchyme using Prx1-cre resulted in efficient deletion of Gnas by mouse embryonic day 14.5 (E14.5) and heterotopic ossification as early as E16.5 which progressed through postnatal day 20 (P20), by which time most mutant animals died [47]. Similar levels of heterotopic ossification occurred in two different models targeting Gnas in mesenchyme more broadly using Dermo1-cre and Ap2-cre and in adult dermis and subcutaneous tissues following injection of adenovirus-Cre in Gnasfl/fl animals [47]. The introduction of mesenchyme-specific deletion of Gnas provided unique opportunities to elucidate the mechanism that drives the ectopic formation of intramembranous bone. Regard et al. used the Prx1-cre Gnas mutants to demonstrate that heterotopic ossification in the mesenchyme was a result of dysregulated Hh signaling downstream of decreased cAMP/PKA signaling [47]. Their studies established the paradigm that Hh signaling is necessary and sufficient to induce ectopic intramembranous ossification similar to that observed in POH patients. Furthermore, Gαs, encoded by GNAS, is necessary to regulate levels of Hh signaling during development and into adulthood [47]. These findings were pivotal in developing an understanding of the etiology and pathology of POH, which could lead towards the development of new therapeutic targets.
6.4.2.2.3 New Therapeutic Targets
Understanding the mechanisms behind disease states can also lead to new insights towards normal cranial development. Considering that heterotopic ossification forms through intramem-branous ossification of mesenchymal cells, it stands to reason that this signaling pathway may be conserved during normal development of cranial intramembranous bone. Xu et al. hypothesized that the signaling pathway implicated in POH heterotopic ossification is conserved during normal cranial intramembranous bone ossification [40]. By using comprehensive mouse genetics targeting Gnas and Hh signaling effectors Smoothened (Smo) and Gli and small-molecule inhibitors of Hh signaling, Xu et al. were able to partially rescue the increased intramembranous bone formation observed in Gnas loss-of-function mutants [40]. These data indicate that Hh signaling inhibition may serve as a therapeutic target towards treatments of diseases such as POH and possibly similar diseases demonstrating increased intramembranous bone formation, such as craniosynostosis.
6.4.2.3 Fibrous Dysplasia
6.4.2.3.1 Etiology
Similar to POH, FD is caused by a somatic mutation in GNAS during early development [36]. This is an activating missense mutation that occurs in a somatic cell in a post-zygotic embryo [36]. FD can affect a single bone (monostotic) or multiple bones (polyostotic) [37]. Mutations leading to polyostotic FD are thought to occur pre-gastrulation as the affected bones consist of both neural crest and mesoderm-derived populations [36]. McCune-Albright syndrome (MAS) (OMIM: #174800) is a form of polyostotic FD that spans multiple bones and organs. More than half of all MAS cases include craniofacial bone defects [36]. FD lesions consist of mesenchyme-like mutated cells and normal osteoblasts resulting in deficient mineralization.
The missense mutation in GNAS results in a selective inhibition of its GTPase activity, leading to increased activity of PKA signaling pathways. Gαs has been demonstrated to regulate Wnt signaling in many contexts [48]. Wnt/β-catenin signaling is required for normal cranial bone development [21, 49, 50]. Loss of canonical Wnt signaling in early cranial mesenchyme results in diminished skull bone formation [21, 50, 51]. However, in mature osteoblast cells, Wnt/β-catenin signaling prevents bone mineralization and maturation [52]. Loss of Gαs results in increased Wnt/β-catenin signaling in affected tissues, which inhibits osteoblast maturation (Fig. 6.2) [48, 53].
6.4.2.3.2 New Insights from Animal Models of FD
Bianco et al. first used mice to study the pathology of FD by transplanting human bone marrow cells from FD lesions into immunocompromised mice and observed similar lesions as to those observed in human FD/MAS affected bone [54]. Their techniques were pivotal in understanding the role of mosaicism in FD bone lesions [54, 55]. One of the earliest genetic mouse models of FD was characterized in 2014 by Saggio et al. [56]. This mouse model was able to recapitulate some aspects of the human phenotype by overexpressing Gnas using viral vectors to model the most common genetic mutations observed in humans [56]. The authors described a unique pattern of bone lesion formation consisting of three phases beginning with excess bone formation, followed by bone remodeling, and culminating in fibrotic bone lesions. They identified unique histological patterns for each phase of lesion development and postulate that different stages require different treatment regimens. Although humans begin presenting pathological symptoms beginning in childhood [36], Saggio et al. described the formation of bone lesions occurring in mice aged beyond 1 year [56]. This suggests that the mechanisms elucidated in this model are not significant during juvenile stages of bone development. There may be some undescribed human mechanism resulting in the age discrepancies between the onset of FD in humans and the model described above.
More recently, a new mouse model has been developed that provides new insight into FD using a Prx1-Cre-activated tetracycline inducible system to conditionally activate Gnas in skeletal stem cells [57]. Activating this system resulted in FD lesion presentation within 2 weeks of doxycycline exposure, which improved following doxycycline withdrawal. Khan et al. utilized two different Cre lineages, Prx1-Cre and Osx-Cre, and determined that activation of Gnas led to an increase in Wnt signaling and a decrease in Hh signaling [53]. This study more completely defines a mechanism of pathology of FD in a mouse model and highlight potential therapies more relevant to the defects observed in patients with FD.
6.4.2.3.3 New Therapeutic Targets
Currently, there is no cure for FD/MAS, and treatments are limited to surgical intervention for extreme cases [36, 53, 57]. However, the use of animal models has provided valuable insight into the pathophysiology of FD/MAS. The unique mouse model characterized by Zhao et al. demonstrated reversal of FD bone lesions following reduction in ectopic Gnas expression [57]. The authors postulate that inhibition of Gαs may serve as a new therapeutic outlet in the treatment of FD/MAS. Khan et al. demonstrated that inhibition of Wnt signaling in their Gnas mouse models also attenuates FD symptoms, highlighting yet another biological mechanism of FD and possible therapeutic target of FD/MAS [53]. Xu et al. demonstrate that inhibition of Wnt signaling using a chemical inhibitor, LGK-974, and genetic ablation of a single Lrp6 allele can partially rescue the effects of FD in a mouse model of craniofacial FD [40].
6.4.3 Summary
In this section, we have discussed two rare craniofacial diseases that involve abnormal Gαs signaling in the cranial bones. These diseases result from defects in cranial bone ossification that is dependent on a balance of Gαs signaling to differentiate into normal cranial bone. This process seems to be dependent on two downstream signaling pathways that are differentially regulated by Gαs signaling, Hh and Wnt signaling. With various animal models, researchers have been able to identify the mechanisms by which Gαs regulates these two developmentally important signaling pathways. In some instances, putative treatments and therapies have been proposed and tested in a mouse model, demonstrating clear benefits of animal models of disease [40, 53, 57]. Next, we will discuss defects in the facial region focusing on one particular phenotype, micrognathia, across two rare genetic disorders.
6.5 Facial Defects
6.5.1 Facial Outgrowth and Patterning
The facial skeleton is derived from the mesenchymal cells that populate the frontonasal process (FNP) and first branchial arch (Fig. 6.1). The FNP consists of NCC and head mesoderm and is divided into the medial and lateral nasal processes [58]. Alongside the FNP are the maxillary (MXP) and mandibular (MNP) processes. Together the FNP, MXP, and MNP give rise to facial primordia [4, 58, 59]. The facial prominences are observed as facial swellings by mouse embryonic day 9.5 (E9.5). By E11.5, the medial and lateral nasal processes and MXP fuse to form the upper lip and initiate primary palate development [59]. The medial nasal processes meet at the midline to form the nasal septum, which can regulate facial outgrowth (Fig. 6.3) [59]. Secondary palatogenesis is dependent on the mandibular and maxillary prominences. Finally, the jaw is derived from the first branchial arch. The branchial arches are tissue structures with an outer ectodermal layer and inner endodermal layer with a mesodermal core, surrounded by neural-crest-derived mesenchyme. Many complex signaling pathways regulate the formation of these major facial structures. Abnormal signaling, cell growth, and cell differentiation can lead to a variety of facial defects including cleft lip and palate and micrognathia, among others. Here we will describe the processes that regulate facial morphogenesis and two human diseases that result in abnormal craniofacial development.

Facial structures derived from the frontonasal process and first branchial arch. Schematic delineating the frontonasal process and first branchial arch at E9.5 (left) and the structures derived from these two cell populations that make up the facial primordia by E10.5 (right). FNP frontonasal process, MP medial nasal process, LP lateral nasal process, MXP maxillary process, MNP mandibular process. (E9.5 embryo adapted with permission from Ferguson and Atit 2018)
6.5.1.1 Palate Development
The FNP is formed from migrating NCC originating from the caudal forebrain/midbrain by E9.5 [13, 59, 60]. By E10.5 the cells of the FNP have been divided into the medial and lateral nasal processes which begin to fuse with the MXP by E11.5 to form the upper lip and primary palate (Fig. 6.3) [61, 62]. Following formation of the primary palate (palatogenesis) is secondary palatogenesis. The palatal shelves arise from the MXP and grow vertically and laterally adjacent to the tongue. Prior to palatal shelf fusion, the tongue descends and the palatal shelves rotate and expand towards the midline where they fuse by E15.5 [59]. The molecular mechanisms that govern palate development have been reviewed extensively [58,59,60]. The normal formation of lip, primary palate, and secondary palate is heavily dependent on normal cell movements, cell differentiation, programmed cell death, and concerted actions across multiple adjacent tissues and cell populations. Cleft lip and cleft palate can occur when any of these processes is dysregulated during development [3, 6, 58]. The etiology and pathophysiology of cleft lip and cleft palate have been reviewed extensively [62,63,64,65] and thus will not be reviewed here.
6.5.1.2 Transcriptional Control of Jaw Development
The first branchial arch is subdivided into maxilla and mandibular prominences through pattern-specific expression of key Dlx (distal-less) genes [66]. Expression of DLX1/2 corresponds to MXP, while expression of DLX1/2/5/6 correlates with the MNP. The MNP is further patterned into an oral-aboral axis dependent on signaling from Lim-homeobox (Lhx) genes and goosecoid expression. Prior to formation of the mandibular bones, the formation of a specialized cartilage structure, Meckel’s cartilage (MC), is required. The formation of MC begins with condensation of CNC mesenchyme at E12.5, which then differentiates into chondrocytes forming the template for the future jaw [66]. MC expands outwards first ventromedially and then dorsolaterally until they fuse at the most distal tip. The posterior regions of MC ultimately develop into the malleus and incus bones of the middle ear, while the intermediate MC is degraded and differentiated to form connective tissues of the jaw [60, 66]. Intramembranous ossification of the jaw is driven through expression of Dlx5, which is required for the upregulation of the bone marker Runx2 [66]. Ossification of the lower jaw bone begins with the formation of a primary ossification center lateral to the MC at stage E13.5 [60, 66]. The upper jawbones are derived from the MXP and are physically unique from the lower jawbones. Recent work in zebrafish has identified two novel identity markers of the upper and lower jaw. The upper jaw requires expression of the nuclear receptor subfamily 2 group F, Nrf2, nuclear receptors, while the lower jaw requires endothelin 1 (Edn1) to repress expression of Nrf2 and maintain proper lower jaw identity [67].
The size and shape of the lower jaw bones are dependent on the underlying MC, and abnormal development of the MC or ossification of the mandibular bone presents as micrognathia, a shortened/decreased jaw, or macrognathia, an oversized/lengthened jaw. Several side effects can occur as a result from micrognathia including feeding disturbances in infants [68]. Here, we will describe the occurrence of two rare craniofacial disorders which present as syndromes including micrognathia and other facial defects.
6.5.2 Robinow Syndrome
6.5.2.1 Etiology
Robinow syndrome (RS) (OMIM: #180700, #268310, #616894, #616331) is a genetic disease that manifests as skeletal dysplasia with distinct craniofacial defects [5]. RS results from mutations in components of the noncanonical Wnt and planar cell polarity (PCP) signaling pathways. The most common mutations resulting in RS are located within the Wnt family member 5a (WNT5A) or Receptor Tyrosine Kinase-like Orphan Receptor 2 (ROR2) loci [5, 69]. However, mutations in other pathway components, such as Disheveled (DVL) proteins, have also been identified [70, 71]. Apart from skeletal phenotypes such as short stature, the craniofacial defects are described as “fetal facies” which include but are not limited to macrocephaly, micrognathia, and midface hypoplasia [69, 72]. There are a high amount of genetic heterogeneity surrounding this disease owing to the large number of mutations across multiple genes that vary in the level of inheritance across patients. However, despite this RS remains a rare disorder affecting only 1 in 500,000 individuals [5].
WNT5a is a secreted protein that regulates cellular patterning and morphology through noncanonical Wnt signaling. During noncanonical signaling, ROR2 acts as a coreceptor for frizzled receptors of WNT5a [73]. Binding of the ligand to the receptor results in signaling cascades involving the cellular effector DVL, ultimately directing cellular outgrowth, planar cell polarity, and cell migration (Fig. 6.2) [74]. It is hypothesized that heterozygous missense mutations result in loss of function of the affected WNT5A allele [75]. Alternatively, homozygous ROR2 mutations leading to decreased ROR2 activity also result in RS [76, 77]. Finally, mutations in DVL1 or DVL3 also result in autosomal dominant RS [70, 71].
6.5.2.2 New Insights from Animal Models
The first animal model of RS characterized the recessive mutations in Ror2 using a knockout mouse model [69]. They identified several defects in the craniofacial region of the developing mouse embryo. Most notably, they identified a defect in craniofacial outgrowth including smaller mandible and associated defects in Meckel’s cartilage. Person et al. described a similarity in the phenotypes of Ror2 and Wnt5a null mice and identified key missense mutations in WNT5A in human patients of RS presenting autosomal dominant inheritance [75]. They confirmed the role of human WNT5A missense mutations in RS by selectively inactivating wnt5a alleles in zebrafish and Xenopus embryos. Their work linked WNT5A and ROR2 in a functional pathway that is highly regulated and involved in normal craniofacial outgrowth and development [75]. The role of specific missense mutations of human WNT5A in RS was further delineated in another vertebrate model using chick embryos [5]. Here, the authors utilized retroviral injections into the developing chick mandible to highlight the specific effect of a human C83S WNT5a missense mutation on outgrowth of the vertebrate jaw and formation of normal Meckel’s cartilage. The effect of the C83S WNT5a missense mutations resulted in micrognathia in the chick embryos, resembling the Ror2 and Wnt5a null mice described by Person et al. [75]. However, their data did not highlight a definitive loss-of-function effect of C83S WNT5A on the resulting micrognathia as previously hypothesized. Instead, they postulate that the specific C83S WNT5A mutations identified in RS patients have neither a gain nor loss-of-function effect and ultimately result in diversion of WNT5A towards an unknown, alternative signaling pathway, independent and separate from noncanonical Wnt signaling, thus improving upon the current etiology of micrognathia in RS patients.
These experiments in mouse and chick highlighted the importance of balance in noncanonical Wnt signaling pathways during normal craniofacial development. Due to the genetic heterogeneity of noncanonical Wnt signaling, it was hypothesized that dysregulation in other aspects of this key WNT5a-ROR2 signaling pathway may also account for the occurrence of Robinow syndrome in humans that has not been associated with mutations in WNT5A or ROR2 [78]. More recently, frameshift mutations in DVL1 and DVL3 have been identified in patients of dominant RS [70, 71]. DVL proteins are involved in both canonical and noncanonical Wnt signaling, and in the case of RS, it is hypothesized that the mutations in DVL proteins exist within the WNT5a-ROR2-DVL noncanonical Wnt signaling pathway. A recent genome-wide association study (GWAS) identified a variant in Dvl2 in bulldog breeds associated with widened skull shape and decreased snout length, reminiscent of RS defects [79]. This unique animal model can provide insights into the etiology of distinct craniofacial defects associated with RS and highlights the need for further research into the molecular mechanisms of the WNT5a-ROR2-DVL pathways. Interestingly, Mansour et al. hypothesized that the Dvl2 mutations are hypomorphic in the context of the vertebral phenotypes characterized in the dog model, similar to the conclusions of Hosseini-Farahabadi et al. regarding Wnt5a mutations with respect to mandible outgrowth in chick [5, 79]. New biophysical approaches to understand the cellular basis of Robinow syndrome has shed light on the complex etiology. Recent studies with live Wnt5a−/− mouse embryos reveal that cell movement and cytoskeletal organization during branchial arch morphogenesis are disrupted leading to misshapen arches and diminished MXP [80]. Understanding the molecular mechanisms of WNT5a/ROR2/DVL can provide insights into the etiology of RS and normal development of the head and face.
6.5.3 Nager Syndrome
6.5.3.1 Etiology
Nager syndrome (OMIM: #154400) is a rare genetic disease reported in only 100 individuals as of 2017 [81, 82]. First identified in 1948 [83], Nager syndrome presents with facial defects, including midface retrusion and severe micrognathia, and distinct limb phenotypes unique from other types of face and limb bone diseases [81, 84, 85]. Similar to RS, Nager syndrome has been reported to be inherited as either an autosomal recessive or autosomal dominant disease. Mutations of SF3B4, splicing factor 3B subunit 4, resulting in heterozygous loss of function, have been identified in 57–64% patients with Nager syndrome [81, 86]. Patients with Nager syndrome primarily present de novo mutations of SF3B4; however, there have been observed instances of both autosomal dominant and autosomal recessive inheritance [81, 82, 84, 85]. SF3B4 encodes a spliceosomal protein which has been reported to directly interact with BMP proteins to inhibit osteogenic and chondrogenic differentiation in vitro [87]. Current understanding of the etiology of this disease is limited due to the small number of affected individuals and genetic heterogeneity of individuals affected [82].
6.5.3.2 Insights from Animal Models
Thus far there has only been one characterized animal model of Nager syndrome [88]. Devotta et al. utilize morpholinos to inhibit SF3B4 function during early development in Xenopus embryos [88]. The authors first demonstrated a requirement of sf3b4 in normal neural crest development. In addition to regulating the differentiation of neural crest during early development, the authors have also characterized craniofacial defects similar to those observed in Nager syndrome patients, including decreased formation of Meckel’s cartilage, among other key facial cartilage structures [88]. This was accompanied by decreased levels of sox10 mRNA, but researchers saw no evidence of interaction with the BMP signaling pathway. The authors hypothesize a spatiotemporal requirement for spliceosome mechanisms during early neural crest cell specification [88]. There is still a necessity for the development of appropriate animal models of Nager syndrome to fully understand the etiology of this rare congenital disease. Understanding the etiology of this and other rare facial defects can inform our understanding of normal facial outgrowth and morphology during early embryonic development.
6.6 Ciliopathies/RASopathies
6.6.1 What Are Ciliopathies?
Ciliopathies are a group of diseases classified by defects in the function or formation of primary cilia [4]. Primary cilia are complex organelles found in all vertebrate cell types [4, 89, 90] and are required for many basic cellular processes, including transduction of major signaling pathways either by housing receptors or transducing signaling ligands. Cilia comprise three major functional domains, the axoneme, basal body, and ciliary membrane. Each domain is critical for the overall function of the cilium and its role in signal transduction. The axoneme is composed of microtubules and extends the cilium into the extracellular space [4, 90]. The axoneme builds from the basal body, a specialized centrosome structure that connects the axoneme to the cell. Intraflagellar transport (IFT) is required for the assembly of the axoneme as well as the transport of signal transduction proteins in a functioning cilium [4]. A specialized membrane encompasses the fully formed cilium where various signal transduction receptors are located.
Several key developmental signaling pathways are regulated by cilia, including Hh, Wnt, and platelet-derived growth factor-alpha (PDGFRα). Sonic Hedgehog (SHH) has been extensively reported to be regulated by primary cilia during development [4, 16, 90,91,92]. SHH receptors are localized to the ciliary membrane, and upon binding of a Hh ligand, SMO can enter the ciliary axoneme [16, 92]. Additionally, the transcription factor GLI is trafficked through the ciliary axoneme which is believed to process GLI into a transcriptional activator or transcriptional repressor (Fig. 6.2) [16, 92]. PDGFRα signaling is required during normal cell migration and proliferation of mesenchyme during development and is localized to primary cilia [93, 94]. Loss of Pdgfrα in mouse neural crest results in several craniofacial defects, including cleft palate and decreased formation of cranial bone [93, 95, 96]. There are many different ciliopathies that have been described in recent years [4, 16]. Here we will describe one type of ciliopathy, oral-facial-digital syndrome (OFDS) type-1, as the molecular etiology and pathologies are not well understood and there are new insights from animal models.
6.6.2 Oral-Facial-Digital Syndrome (OFDS)
6.6.2.1 Etiology
OFDS-1 (OMIM: #311200) is a ciliopathic disease linked with varying craniofacial defects, including cleft palate and micrognathia, as well as digit defects such as syndactyly and brachydactyly, and occasionally kidney defects [97]. OFDS-1 is an X-linked dominant disorder that is embryonically lethal in males and occurs in 1 of every 50–250,000 births [4, 98]. Ferrante et al. identified a variety of mutations in the OFD1 gene resulting in loss of protein function [97]. OFD1 encodes a protein found in the basal bodies of primary cilia that is required for normal ciliary function [98,99,100]. Interestingly, mutations in OFD1 have been identified in other ciliopathies including Meckel-Gruber syndrome (OMIM: #249000) and Joubert syndrome (OMIM: # 213300) [99]. This suggests that OFD1 is important for normal primary cilia function and that there may be multiple genes involved across these ciliopathic syndromes to account for the variability in presented phenotypes. The range and severity of defect types in individuals with OFDS-1 is variable even across familial inheritance, suggesting a complex etiology of OFDS-1 in humans that has yet to be fully defined.
6.6.2.2 New Insights from Animal Models
Ferrante et al. generated a mouse model of OFDS-1 by targeted deletion of Ofd1 during the four-cell stage of development [100]. Mutant mice presented with severe craniofacial defects including shortened skull and facial regions and cleft palate, polysyndactyly, cystic kidney, and death at birth [100]. Similar to OFDS-1 in humans, deletion of Ofd1 in male mice was embryonically lethal. Analysis of affected male embryos indicated a loss of primary cilia in the node and coinciding loss in left-right asymmetry. Furthermore, Shh and Shh signaling targets, Ptch1 and Gli1, were decreased in the neural tube following loss of Ofd1 correlating with the observed neural tube defects in mutant embryos [100]. These results highlighted the requirement of cilia in normal craniofacial development and specifically linked the observed defects to decreased Shh signaling in murine development.
A later study conducted by Ferrante et al. elucidated a novel function of ofd1 in zebrafish linking primary cilia to noncanonical Wnt/PCP signaling [101]. Using morpholinos to block expression of ofd1 in zebrafish in vivo, the authors observed delayed cellular migration and decreased body axis length. The cell migration phenotypes were exacerbated with simultaneous injection of a wnt11 morpholino. WNT11 is a noncanonical Wnt/PCP ligand that is required for normal cell migration and convergent extension [101]. Van Gogh-like protein 2, VANGL2, is a PCP receptor protein [101]. When the authors co-injected morpholinos for vangl2 alongside ofd1, they observed a significantly decreased body axis relative to single injected mutants and uninjected controls. Delayed cell migration and shortened body lengths are indicative of defects in convergent extension, and Wnt/PCP signaling is required for normal convergent extension in vertebrates during early development [101].
Both of these studies have begun to elucidate the role of Ofd1 on ciliary function and have identified two key developmental signaling pathways, HH and Wnt/PCP, which are regulated by normal cilia function. The results from these studies helped to classify OFDS-1 as a ciliopathy and have begun to fill in the blanks of the etiology of this disease. Furthermore, these animal models can be used to address other questions on other types of ciliopathies. Mutations in Ofd1 have been identified in other ciliopathies including Joubert syndrome, a ciliopathy that presents with craniofacial anomalies and neural disorders. To better understand the etiology of OFDS-1 and other ciliopathies, more studies to elucidate novel binding partners and genetic interaction with major signaling pathways will be useful. At the clinical level, identification of other genes involved in either ciliary morphogenesis, or ciliary signaling that are impacted in patients, will help to further define the complex genetic heterogeneity and pathophysiology of various ciliopathies.
6.6.3 What Are RASopathies?
RASopathies are a group of disorders caused by mutations in genes within the RAS/MAPK (mitogen-activated protein kinase) signaling pathways. RAS proteins are small GTPases that are activated in response to growth factors binding to a receptor protein [6, 103]. Activation of RAS leads to a signaling cascade first by activation of RAF, a MAPK kinase, which phosphorylates and activates MEK (mitogen-activated protein kinase kinase, which then phosphorylates and activates Erk. Activated ERK (extracellular signal-related kinase) proteins can then function on a variety of cellular processes including cell differentiation and cell cycle regulation (Fig. 6.2) [103,104,105]. Each RASopathy presents with its own unique phenotypes; however, they typically present with craniofacial defects, cardiac malformations, and musculoskeletal defects [6]. Here, we will discuss Noonan syndrome and recent advances made with animal models towards the understanding of this specific RASopathy and other RASopathies in general.
6.6.4 Noonan Syndrome
6.6.4.1 Etiology
Noonan syndrome (NS) is an autosomal dominant disorder that presents with short stature and craniofacial dysmorphologies including broad forehead, hypertelorism (increased distance between the eyes), and high arched palate [6, 103]. NS affects 1 in every 1000–2500 individuals [106]. At least 50% of these individuals have missense mutations in the gene PTPN11 leading to increased phosphatase activity [6, 106]. PTPN11 encodes a tyrosine phosphatase protein that is required for normal activation of RAS/ERK cascades [107]. Mutations have been identified in numerous other genes involved in the RAS/MAPK signaling pathway including KRAS, SOS1, RAF1, NRAS, BRAF, and RIT1 among others [6, 106]. Each mutation is characterized into one of ten subtypes of NS currently being defined. KRAS and NRAS are two types of RAS genes, and RAF1 and BRAF encode RAF genes. SOS and RIT1 encode proteins required for normal signal transduction of the RAS/RAF/MAPK cascade.
6.6.4.2 New Insights from Animal Models
Araki et al. generated a mouse model of NS using an activating gain-of-function mutation of Ptpn11 [107]. Homozygous activation of Ptpn11 was embryonically lethal and heterozygous activation resulted in increased Ptpn11 activation in vitro and in vivo. Heterozygous mutants presented facial phenotypes similar to those observed in humans including triangular facial features such as a wider and more blunt snout. Interestingly, the authors report increased ERK activation only in a subset of tissues indicating tissue-specific effects of Ptpn11 activation.
Recently, several new animal models have targeted different genes affected in NS. Chen et al. characterized a gain-of-function mutation in Sos1, a gene required for activation of RAS, in a mouse model [108]. Sos1 homozygous and heterozygous mutants exhibited short stature and triangular facial features, similar to those in patients with NS and in mice with Ptpn11 mutations. Homozygous mice typically died in utero; however, some were reported to be born and survive. Prenatal treatment with PD0325901, a small-molecule inhibitor of MEK, resulted in increased survival of the Sos1 homozygous mutants and improved stature and facial feature of Sos1 homozygous and heterozygous mutants [108, 109].
Wu et al. generated an overexpression Raf1 mouse model which recapitulates many of the phenotypes associated with NS, including short stature and craniofacial dysmorphology [104]. Postnatal inhibition of MEK/ERK signaling using PD0325901 over the course of 6 weeks was sufficient to rescue the growth defects and craniofacial abnormalities observed in Raf1 mutants.
Hernández-Porras et al. observed characteristic NS phenotypes, including short stature and craniofacial dysmorphology in a mouse model of KRAS gain of function [110]. Similarly to Chen et al. and Wu et al. [104, 108, 110], they observed significant improvement of growth and craniofacial defects in KRAS mutants when rescued prenatally with MEK inhibitor PD0325901. In contrast to observations in the Raf1 model, MEK inhibition postnatally in juvenile or adult mice did not ameliorate any physical defects, although it did improve survivability by 40% [110].
These data are among the first to elucidate the pathology of MEK/ERK signaling in NS and to identify a potential treatment mechanism for patients. In addition to determining the pathology of various distinct gene mutations involved in NS, these studies have determined the role of RAF/MEK/ERK signaling in craniofacial defects across multiple experiments and furthermore elucidated potential therapies to rescue the craniofacial and growth defects associated with NS. Future studies should focus on identifying the downstream factors that are functionally important to normal craniofacial development. Specifically, understanding how MAPK signaling changes result in the observed phenotypes associated with NS will help to understand the etiology of the disease and may also inform researchers on new pathways that are relevant during normal craniofacial development.
6.7 Concluding Remarks
In this chapter, we have discussed several animal models of various craniofacial diseases and their associated defects. While we have focused primarily on mouse models, other vertebrate systems are valuable for studying craniofacial development. There are non-model organisms that are being used currently to address many unanswered questions of craniofacial development. For instance, Werren et al. utilize a unique invertebrate system, the Nasonia wasps, to study the genetics of craniofacial development [111]. These authors study two hybrid male species and identify quantitative trait loci that are associated with cranial morphology and identify several cranial anomalies among hybrids including cleft palate. The blind Mexican cavefish, Astyanax mexicanus, is used to study the natural genetic and environmental causes that result in various craniofacial anomalies, including craniosynostosis [112].
Beyond understanding the basis of normal craniofacial development and the etiology of various human diseases, animal models are also pivotal in the search for preventative treatments and cures. Various animal models, especially mice and rats, are often poised to test different therapies and treatments due to the high levels of conservation among disease pathways. Here, we have identified several research strategies for identifying cures or treatments of various craniofacial diseases such as Hh inhibition in progressive osseous heteroplasia [40], Wnt inhibition in fibrous dysplasia [40, 53], and ERK inhibition in Noonan syndrome [104, 108, 110].
Furthermore, the use of animal models has allowed for the development of many biological tools that can be utilized to understand developmental processes and help to define etiologies of various developmental diseases. While there has been considerable progress towards understanding craniofacial diseases with various animal models, there is still more to be learned for therapeutic development. Novel technologies, such as in utero stem cell transplantation, have been utilized to treat genetic disorders [113], but their potential is limited by our understanding of the genetic causes of the many syndromes associated with craniofacial defects. Understanding the biophysical and cellular basis of craniofacial development will reveal new etiologies of craniofacial disorders. As we have discussed, many craniofacial diseases are associated with genetic heterogeneity and the high number of genetic isoforms within the human genome is just one possible explanation for this [5, 78, 82]. Advancements in sequencing technologies have allowed researchers to gather high-throughput molecular information during early development to make conclusions about cell fate and lineage decisions in early development. Hooper et al. have mapped out tissue-specific genetic programs that regulate facial morphogenesis in early mouse embryo [114]. There have been several studies that have highlighted the importance of epigenetic variation in normal craniofacial development and efforts to characterize the epigenome during development [114,115,116,118]. A greater understanding of the etiology of the various craniofacial diseases will emerge as analyses of the different genetic and epigenetic variations are identified within human patients and as animal models are developed to assess the complex signaling pathways associated with craniofacial defects and ultimately will improve our understanding of normal craniofacial development [17].
References
Morriss-Kay GM, Wilkie AOM. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. 2005;207(5):637–53.
Wilkie A. Craniosynostosis: genes and mechanisms. Hum Mol Genet. 1997;6(10):1647–56.
Trainor PA. Craniofacial birth defects: the role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am J Med Genet Part A. 2010;152A(12):2984–94.
Achilleos A, Trainor PA. Mouse models of rare craniofacial disorders. Curr Top Dev Biol. 2015;115:413–58.
Hosseini-Farahabadi S, Gignac SJ, Danescu A, Fu K, Richman JM. Abnormal WNT5A signaling causes mandibular hypoplasia in Robinow syndrome. J Dent Res. 2017;96(11):1265–72.
Goodwin AF, Kim R, Bush JO, Klein OD. From bench to bedside and back: improving diagnosis and treatment of craniofacial malformations utilizing animal models. Curr Top Dev Biol. 2015;115:459–92.
Hu D, Marcucio RS, Helms JA. A zone of frontonasal ectoderm regulates patterning and growth in the face D. Development. 2003;130:1749–58.
Mallo M. Reassessing the role of Hox genes during vertebrate development and evolution. Trends Genet. 2018;34(3):209–17.
Mork L, Crump G. Zebrafish craniofacial development: a window into early patterning. Curr Top Dev Biol. 2015;115:235–69.
Schneider RA, Hu D, Helms JA. From head to toe: conservation of molecular signals regulating limb and craniofacial morphogenesis. Cell Tissue Res. 1999;296:103–9.
Van Otterloo E, Williams T, Artinger KB. The old and new face of craniofacial research: how animal models inform human craniofacial genetic and clinical data. Dev Biol. 2016;415(2):171–87.
Frisdal A, Trainor PA. Development and evolution of the pharyngeal apparatus. Wiley Interdiscip Rev Dev Biol. 2014;3(6):403–18.
Minoux M, Rijli FM, Paleari L, Zerega B, Postiglione MP, Mantero S, et al. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development. 2010;137(16):2605–21.
Ferguson JW, Atit RP. A tale of two cities: the genetic mechanisms governing calvarial bone development. Genesis. 2019;57(1):e23246.
Evans DJR, Francis-West PH. Craniofacial development: making faces. J Anat. 2005;207(5):435–6.
Schock EN, Brugmann SA. Discovery, diagnosis, and etiology of craniofacial ciliopathies. Cold Spring Harb Perspect Biol. 2017;9(9):a028258.
Zelzer E, Olsen BR. The genetic basis for skeletal diseases. Nature. 2003;423:343–8.
Yoshida T, Vivatbutsiri P, Morriss-Kay G, Saga Y, Iseki S. Cell lineage in mammalian craniofacial mesenchyme. Mech Dev. 2008;125(9–10):797–808.
Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241(1):106–16.
Ishii M, Sun J, Ting M-C, Maxson RE. The development of the calvarial bones and sutures and the pathophysiology of craniosynostosis. Curr Top Dev Biol. 2015;115:131–56.
Tran TH, Jarrell A, Zentner GE, Welsh A, Brownell I, Scacheri PC, et al. Role of canonical Wnt signaling/β-catenin via Dermo1 in cranial dermal cell development. Development. 2010;137:3973–84.
Deckelbaum RA, Majithia A, Booker T, Henderson JE, Loomis CA. The homeoprotein engrailed 1 has pleiotropic functions in calvarial intramembranous bone formation and remodeling. Development. 2006;133:63–74.
Goodnough LH, Dinuoscio GJ, Atit RP. Twist1 contributes to cranial bone initiation and dermal condensation by maintaining Wnt signaling responsiveness. Dev Dyn. 2016;245(2):144–56.
Dunlop LL, Hall BK. Relationships between cellular condensation, preosteoblast formation and epithelial-mesenchymal interactions in initiation of osteogenesis. Int J Dev Biol. 1995;39(2):357–71.
Han J, Ishii M, Bringas P Jr, Maas RL, Maxson RE Jr, Chai Y. Concerted action of Msx1 and Msx2 in regulating cranial neural crest cell differentiation during frontal bone development. Mech Dev. 2007;124:729–45.
Ishii M, Merrill AE, Chan Y-S, Gitelman I, Rice DPC, Sucov HM, et al. Msx2 and Twist cooperatively control the development of the neural crest-derived skeletogenic mesenchyme of the murine skull vault. Development. 2003;130(24):6131–42.
Ting M-C, Wu NL, Roybal PG, Sun J, Liu L, Yen Y, et al. EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development. 2009;136(5):855–64.
Roybal PG, Wu NL, Sun J, Ting M, Schafer CA, Maxson RE. Inactivation of Msx1 and Msx2 in neural crest reveals an unexpected role in suppressing heterotopic bone formation in the head. Dev Biol. 2010;343(1–2):28–39.
Cesario JM, Landin Malt A, Chung JU, Khairallah MP, Dasgupta K, Asam K, et al. Anti-osteogenic function of a LIM-homeodomain transcription factor LMX1B is essential to early patterning of the calvaria. Dev Biol. 2018;443(2):103–16.
Twigg SRF, Wilkie AOM. New insights into craniofacial malformations. Hum Mol Genet. 2015;24(R1):R50–9.
Krakow D, Rimoin DL. The skeletal dysplasias. Genet Med. 2010;12(6):327–41.
Fan X, Loebel DAF, Bildsoe H, Wilkie EE, Tam PPL, Qin J, et al. Tissue interactions, cell signaling and transcriptional control in the cranial mesoderm during craniofacial development. AIMS Genet. 2016;3(1):74–98.
Pan A, Chang L, Nguyen A, James AW. A review of hedgehog signaling in cranial bone development. Front Physiol. 2013;4:61.
Kaplan FS, Hahn GV, Zasloff MA. Heterotopic ossification: two rare forms and what they can teach us. J Am Acad Orthop Surg. 1994;2(5):288–96.
Meyers C, Lisiecki J, Miller S, Levin A, Fayad L, Ding C, et al. Heterotopic ossification: a comprehensive review. JBMR Plus. 2019;3(4):e10172.
Feller L, Wood NH, Khammissa RAG, Lemmer J, Raubenheimer EJ. The nature of fibrous dysplasia. Head Face Med. 2009;5:22.
Hakim DN, Pelly T, Kulendran M, Caris JA. Benign tumours of the bone: a review. J Bone Oncol. 2015;4(2):37–41.
Kaplan FS, Pignolo RJ, Al Mukaddam M, Shore EM. Genetic disorders of heterotopic ossification. In: Primer on the metabolic bone diseases and disorders of mineral metabolism. Wiley; 2018. p. 865–70.
Pignolo RJ, Ramaswamy G, Fong JT, Shore EM, Kaplan FS. Progressive osseous heteroplasia: diagnosis, treatment, and prognosis. Appl Clin Genet. 2015;8:37–48.
Xu R, Khan SK, Zhou T, Gao B, Zhou Y, Zhou X, et al. Gαs signaling controls intramembranous ossification during cranial bone development by regulating both Hedgehog and Wnt/β-catenin signaling. Bone Res. 2018;6(1):33.
Bastepe M. GNAS mutations and heterotopic ossification. Bone. 2018;109:80–5.
Cong Q, Xu R, Yang Y. Gαs signaling in skeletal development, homeostasis and diseases. Curr Top Dev Biol. 2019;133:281–307.
Pignolo RJ, Xu M, Russell E, Richardson A, Kaplan J, Billings PC, et al. Heterozygous inactivation of Gnas in adipose-derived mesenchymal progenitor cells enhances osteoblast differentiation and promotes heterotopic ossification. J Bone Miner Res. 2011;26(11):2647–55.
Happle R. Progressive osseous heteroplasia is not a Mendelian trait but a type 2 segmental manifestation of GNAS inactivation disorders: a hypothesis. Eur J Med Genet. 2016;59(5):290–4.
Cairns DM, Pignolo RJ, Uchimura T, Brennan TA, Lindborg CM, Xu M, et al. Somitic disruption of GNAS in chick embryos mimics progressive osseous heteroplasia. J Clin Invest. 2013;123(8):3624–33.
Shore EM, Kaplan FS. Inherited human diseases of heterotopic bone formation. Nat Rev Rheumatol. 2010;6(9):518–27.
Regard JB, Malhotra D, Gvozdenovic-Jeremic J, Josey M, Chen M, Weinstein LS, et al. Activation of Hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat Med. 2013;19(11):1505–12.
Regard JB, Cherman N, Palmer D, Kuznetsov SA, Celi FS, Guettier J-M, et al. Wnt/β-catenin signaling is differentially regulated by Gα proteins and contributes to fibrous dysplasia. Proc Natl Acad Sci U S A. 2011;108(50):20101–6.
Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8(5):739–50.
Hill TP, Später D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8(5):727–38.
Goodnough LH, DiNuoscio GJ, Ferguson JW, Williams T, Lang RAR, Atit RP, et al. Distinct requirements for cranial ectoderm and mesenchyme-derived Wnts in specification and differentiation of osteoblast and dermal progenitors. PLoS Genet. 2014;10(2):e1004152. Barsh GS, editor.
Kahler RA, Galindo M, Lian J, Stein GS, Van Wijnen AJ, Westendorf JJ. Lymphocyte enhancer-binding factor 1 (Lef1) inhibits terminal differentiation of osteoblasts. J Cell Biochem. 2006;97:969–83.
Khan SK, Yadav PS, Elliott G, Hu DZ, Xu R, Yang Y. Induced Gnas R201H expression from the endogenous Gnas locus causes fibrous dysplasia by up-regulating Wnt/β-catenin signaling. Proc Natl Acad Sci U S A. 2018;115(3):E418–27.
Bianco P, Kuznetsov SA, Riminucci M, Fisher LW, Spiegel AM, Robey PG. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gsalpha-mutated skeletal progenitor cells. J Clin Invest. 1998;101(8):1737–44.
Robinson C, Collins MT, Boyce AM. Fibrous dysplasia/McCune-Albright syndrome: clinical and translational perspectives. Curr Osteoporos Rep. 2016;14(5):178–86.
Saggio I, Remoli C, Spica E, Cersosimo S, Sacchetti B, Robey PG, et al. Constitutive expression of Gsα(R201C) in mice produces a heritable, direct replica of human fibrous dysplasia bone pathology and demonstrates its natural history. J Bone Miner Res. 2014;29(11):2357–68.
Zhao X, Deng P, Iglesias-Bartolom R, Amornphimoltham P, Steffen DJ, Jin Y, et al. Expression of an active Gαs mutant in skeletal stem cells is sufficient and necessary for fibrous dysplasia initiation and maintenance. Proc Natl Acad Sci U S A. 2018;115(3):E428–37.
Lan Y, Xu J, Jiang R. Cellular and molecular mechanisms of palatogenesis. Curr Top Dev Biol. 2015;115:59–84.
Szabo-Rogers HL, Smithers LE, Yakob W, Liu KJ. New directions in craniofacial morphogenesis. Dev Biol. 2010;341(1):84–94.
Bumann EE, Kaartinen V. Craniofacial morphogenesis. In: Primer on the metabolic bone diseases and disorders of mineral metabolism. Wiley; 2018. p. 891–900.
Losa M, Risolino M, Li B, Hart J, Quintana L, Grishina I, et al. Face morphogenesis is promoted by Pbx-dependent EMT via regulation of Snail1 during frontonasal prominence fusion. Development. 2018;145(5):dev157628.
Jiang R, Bush JO, Lidral AC. Development of the upper lip: morphogenetic and molecular mechanisms. Dev Dyn. 2006;235(5):1152–66.
Marcucio R, Hallgrimsson B, Young NM. Facial morphogenesis: physical and molecular interactions between the brain and the face. Curr Top Dev Biol. 2015;115:299–320.
Shkoukani MA, Chen M, Vong A. Cleft lip—a comprehensive review. Front Pediatr. 2013;1:53.
Vanderas AP. Incidence of cleft lip, cleft palate, and cleft lip and palate among races: a review. Cleft Palate J. 1987;24(3):216–25.
Parada C, Chai Y. Mandible and tongue development. Curr Top Dev Biol. 2015;115:31–58.
Barske L, Rataud P, Behizad K, Del Rio L, Cox SG, Crump JG. Essential role of Nr2f nuclear receptors in patterning the vertebrate upper jaw. Dev Cell. 2018;44(3):337–347.e5.
Tan TY, Farlie PG. Rare syndromes of the head and face-Pierre Robin sequence. Wiley Interdiscip Rev Dev Biol. 2013;2(3):369–77.
Schwabe GC, Trepczik B, Süring K, Brieske N, Tucker AS, Sharpe PT, et al. Ror2 knockout mouse as a model for the developmental pathology of autosomal recessive Robinow syndrome. Dev Dyn. 2004;229(2):400–10.
White J, Mazzeu JF, Hoischen A, Jhangiani SN, Gambin T, Alcino MC, et al. DVL1 frameshift mutations clustering in the penultimate exon cause autosomal-dominant Robinow syndrome. Am J Hum Genet. 2015;96(4):612–22.
White JJ, Mazzeu JF, Hoischen A, Bayram Y, Withers M, Gezdirici A, et al. DVL3 alleles resulting in a -1 frameshift of the last exon mediate autosomal-dominant Robinow syndrome. Am J Hum Genet. 2016;98(3):553–61.
Spranger JW, Brill PW, Hall C, Nishimura G, Superti-Furga A, Unger S. Bone dysplasias: an atlas of genetic disorders of skeletal development. 4th ed. New York: Oxford Univeristy Press; 2018. p. 412–8.
Oishi I, Suzuki H, Onishi N, Takada R, Kani S, Ohkawara B, et al. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes to Cells. 2003;8(7):645–54.
Yamaguchi TP, Bradley A, McMahon AP, Jones SA. Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–23.
Person AD, Beiraghi S, Sieben CM, Hermanson S, Neumann AN, Robu ME, et al. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev Dyn. 2009;239(1):327–37.
Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, et al. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat Genet. 2000;25(4):419–22.
Wang B, Sinha T, Jiao K, Serra R, Wang J. Disruption of PCP signaling causes limb morphogenesis and skeletal defects and may underlie Robinow syndrome and brachydactyly type B. Hum Mol Genet. 2011;20(2):271–85.
White JJ, Mazzeu JF, Coban-Akdemir Z, Bayram Y, Bahrambeigi V, Hoischen A, et al. WNT signaling perturbations underlie the genetic heterogeneity of Robinow syndrome. Am J Hum Genet. 2018;102(1):27–43.
Mansour TA, Lucot K, Konopelski SE, Dickinson PJ, Sturges BK, Vernau KL, et al. Whole genome variant association across 100 dogs identifies a frame shift mutation in DISHEVELLED 2 which contributes to Robinow-like syndrome in Bulldogs and related screw tail dog breeds. PLOS Genet. 2018;14(12):e1007850. Leeb T, editor.
Tao H, Zhu M, Lau K, Whitley O, Samani M, Xiao X, et al. Oscillatory cortical forces promote three dimensional cell intercalations that shape the mandibular arch. bioRxiv. 2018;309120.
Bernier FP, Caluseriu O, Ng S, Schwartzentruber J, Buckingham KJ, Innes AM, et al. Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am J Hum Genet. 2012;90(5):925–33.
Terrazas K, Dixon J, Trainor PA, Dixon MJ. Rare syndromes of the head and face: mandibulofacial and acrofacial dysostoses. Wiley Interdiscip Rev Dev Biol. 2017;6(3):e263.
Kennedy SJ, Teebi AS. Newly recognized autosomal recessive acrofacial dysostosis syndrome resembling Nager syndrome. Am J Med Genet. 2004;129A(1):73–6.
Hall BD. Nager acrofacial dysostosis: autosomal dominant inheritance in mild to moderately affected mother and lethally affected phocomelic son. Am J Med Genet. 1989;33(3):394–7.
Chemke J, Mogilner BM, Ben-Itzhak I, Zurkowski L, Ophir D. Autosomal recessive inheritance of Nager acrofacial dysostosis. J Med Genet. 1988;25(4):230–2.
Petit F, Escande F, Jourdain AS, Porchet N, Amiel J, Doray B, et al. Nager syndrome: confirmation of SF3B4 haploinsufficiency as the major cause. Clin Genet. 2014;86(3):246–51.
Watanabe H, Shionyu M, Kimura T, Kimata K, Watanabe H. Splicing factor 3b subunit 4 binds BMPR-IA and inhibits osteochondral cell differentiation. J Biol Chem. 2007;282(28):20728–38.
Devotta A, Juraver-Geslin H, Gonzalez JA, Hong C-S, Saint-Jeannet J-P. Sf3b4-depleted Xenopus embryos: a model to study the pathogenesis of craniofacial defects in Nager syndrome. Dev Biol. 2016;415(2):371–82.
Liu KJ. Craniofacial ciliopathies and the interpretation of Hedgehog signal transduction. PLoS Genet. 2016;12(12):e1006460.
Eggenschwiler JT, Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007;23:345–73.
Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–21.
Lui JC, Garrison P, Nguyen Q, Ad M, Keembiyehetty C, Chen W, et al. EZH1 and EZH2 promote skeletal growth by repressing inhibitors of chondrocyte proliferation and hypertrophy. Nat Commun. 2016;7:13685.
Hoch RV, Soriano P. Roles of PDGF in animal development. Development. 2003;130:4769–84.
Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, et al. PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15(20):1861–6.
Soriano P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124:2691–700.
Tallquist MD, Soriano P. Cell autonomous requirement for PDGFRα in populations of cranial and cardiac neural crest cells. Development. 2003;130:507–18.
Ferrante MI, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, et al. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68(3):569–76.
Ferrante MI, Romio L, Castro S, Collins JE, Goulding DA, Stemple DL, et al. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum Mol Genet. 2009;18(2):289–303.
Bruel A-L, Franco B, Duffourd Y, Thevenon J, Jego L, Lopez E, et al. Fifteen years of research on oral-facial-digital syndromes: from 1 to 16 causal genes. J Med Genet. 2017;54(6):371–80.
Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38(1):112–7.
Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006 Jan 27;38(1):112–7.
Torban E, Kor C, Gros P. Van Gogh-like2 (Strabismus) and its role in planar cell polarity and convergent extension in vertebrates. Trends Genet. 2004;20(11):570–7.
Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19(3):230–6.
Wu X, Simpson J, Hong JH, Kim K-H, Thavarajah NK, Backx PH, et al. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest. 2011;121(3):1009–25.
Zhang Y, Pizzute T, Pei M. A review of crosstalk between MAPK and Wnt signals and its impact on cartilage regeneration. Cell Tissue Res. 2014;358:633–49.
Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–8.
Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, et al. Mouse model of Noonan syndrome reveals cell type– and gene dosage–dependent effects of Ptpn11 mutation. Nat Med. 2004;10(8):849–57.
Chen P-C, Wakimoto H, Conner D, Araki T, Yuan T, Roberts A, et al. Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome–associated Sos1 mutation. J Clin Invest. 2010;120(12):4353–65.
Tatum R, Zhang Y, Salleng K, Lu Z, Lin J-J, Lu Q, et al. Renal salt wasting and chronic dehydration in claudin-7-deficient mice. Am J Physiol Renal Physiol. 2010;298(1):F24–34.
Hernández-Porras I, Fabbiano S, Schuhmacher AJ, Aicher A, Cañamero M, Cámara JA, et al. K-RasV14I recapitulates Noonan syndrome in mice. Proc Natl Acad Sci U S A. 2014;111(46):16395–400.
Werren JH, Cohen LB, Gadau J, Ponce R, Baudry E, Lynch JA. Dissection of the complex genetic basis of craniofacial anomalies using haploid genetics and interspecies hybrids in Nasonia wasps. Dev Biol. 2016;415(2):391–405.
Gross JB, Powers AK. A natural animal model system of craniofacial anomalies: the blind Mexican cavefish. Anat Rec. 2020;303(1):24–9.
Chowdhury N, Asakura A. In utero stem cell transplantation: potential therapeutic application for muscle diseases. Stem Cells Int. 2017;2017:3027520.
Hooper JE, Feng W, Li H, Leach SM, Phang T, Siska C, et al. Systems biology of facial development: contributions of ectoderm and mesenchyme. Dev Biol. 2017;426(1):97–114.
Wilderman A, VanOudenhove J, Kron J, Noonan JP, Cotney J. High-resolution epigenomic atlas of human embryonic craniofacial development. Cell Rep. 2018;23(5):1581–97.
Minoux M, Holwerda S, Vitobello A, Kitazawa T, Kohler H, Stadler MB, et al. Gene bivalency at Polycomb domains regulates cranial neural crest positional identity. Science. 2017;355(6332):eaal2913.
Brinkley JF, Fisher S, Harris MP, Holmes G, Hooper JE, Jabs EW, et al. The FaceBase Consortium: a comprehensive resource for craniofacial researchers. Development. 2016;143(14):2677–88.
Weinberg SM, Cornell R, Leslie EJ. Craniofacial genetics: where have we been and where are we going? PLoS Genet. 2018;14(6):e1007438.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Ibarra, B.A., Atit, R. (2020). What Do Animal Models Teach Us About Congenital Craniofacial Defects?. In: Liu, A. (eds) Animal Models of Human Birth Defects. Advances in Experimental Medicine and Biology, vol 1236. Springer, Singapore. https://doi.org/10.1007/978-981-15-2389-2_6
Download citation
DOI: https://doi.org/10.1007/978-981-15-2389-2_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-2388-5
Online ISBN: 978-981-15-2389-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)