
Abstract
Pesticides are one of the mandatory defense weapons in this modern world to win over the vast populations of plant pests attacking crops during and after cultivation. But injudicious application of these chemicals creates nuisance to the environment leading to residues, resistance, and pest resurgence problem. These residues bind to the environment and revolve in the food chain resulting in bioaccumulation and biomagnification. As the presence of trace amounts of both pesticide residues and their degradation products could be potential health hazards, the International organizations like FAO, WHO have already raised concerns regarding presence of these toxic chemicals in soil, food, and feed samples. Codex Alimentarius Commission after years of trial determined a value called maximum residue limit (MRL) with the aim to establish restrictive measures to protect the environment against pollution. Due to intensive use of pesticides, their residues have become an intrinsic part of the environment including soil, and they are often detected in various samples and therefore their monitoring has been frequently performed throughout the world. Considering low concentration levels of pesticide residues in soil matrices and the determination of these residues often requires extensive sample extraction and clean-up prior to the analysis. This article describes the different sample preparation techniques including their extraction and clean-up that are widely applied for soil sample analysis for pesticide residues.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


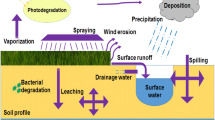
Keywords
10.1 Introduction
Since time immemorial, we have an inseparable relationship with the soil. Soil, being a natural resource, has considerably affected our ability to cultivate crops and influenced the development of civilizations. This relationship between humans, the earth, and food sources affirms soil as the foundation and one of the critical parts of successful agriculture. To enhance production and productivity, the application of pesticide is compulsory and unavoidable. Pesticides are basically a heterogeneous group of compounds including insecticides, herbicides, fungicides, nematicides, etc. having different physicochemical and biological properties. After introduction into the soil, pesticides undergo various movements and transformation processes which ultimately produce their derivatives or metabolites, degradation products, reaction products, and other impurities having toxicological significance, those are collectively called as pesticide residues (Đurović 2011). Owing to the misuse and overuse of these pesticides, their residues are continuously increasing and they have become an unavoidable portion of the pedosphere. Considering the persistence of residues and their deleterious effects, it seems that soil contamination over a long period of time is the biggest threat in terms of food safety as these compounds are mobile and capable of bioaccumulation (Damalas 2009). Exposure to contaminated soil samples may be detrimental to the health of not only humans but also of all other living organisms (Odukkathil and Vasudevan 2013). Therefore, the concentration levels of pesticides and their derivatives in the soil must be frequently monitored. Maximal residue limits (MRLs) for pesticides have been established by the United Nation’s Food and Agriculture Organization (FAO) and the World Health Organization (WHO) for this purpose (Codex 2019) and any quantity above this MRL value is a concern to human health. The MRL is the maximum level of a pesticide residue (expressed in mg kg−1) which is legally permitted in or on food or animal feed (EU, MRL 2019). In recent years, some MRLs have been significantly lowered from ppm to ppb levels to meet the expectations for securing human health at the top level. Therefore, to detect these pesticides meticulously, reliable methods that can analyze dilute mixtures of parent substances and their metabolites are required. So, there is also increasing interest in industrial and government sectors to develop more precise, sophisticated, and cost-effective methods to generate large amounts of residue data on new and existing products.
The current trend in pesticide residues analysis is developing multi-residual methods that not only provide a simultaneous determination of a large number of pesticides but also can be applied to large numbers of samples of different origin. The entire chemical analysis involves several important stages like sample preparation, analyte separation (i.e., quantification and data analysis) of which sample preparation step is considered as the most critical one. Conventional sample preparation techniques (solvent extraction, sonication assisted extraction, etc.) are laborious, expensive, time consuming, and require large amounts of organic solvents and usually involve many steps, leading to loss of some analyte quantity. Additionally, consequences of use of hydrocarbon solvents, such as depletion of ozone layer and generation of considerable carcinogenic waste, lead to a reduction of not only their use but their manufacture also. As a result, modern sample preparation procedures, such as solid phase extraction (SPE), solid phase micro-extraction (SPME), supercritical fluid extraction (SFE), microwave assisted extraction (MAE), microwave-assisted micellar extraction (MAME), accelerated solvent extraction (ASE), matrix solid phase dispersion (MSPD) extraction, and QuEChERS (quick, easy, cheap, effective, rugged and safe) method, have been developed to overcome the limitations of the conventional approaches. SFE, ASE, and MAE are instrumental techniques, and often use SPE (for purification of obtained extracts) and SPME (for purification and concentration of obtained extracts) for desired results.
Most residue analysis procedures fall within the scheme shown in Fig. 10.1. Design of experiment deals with strategic planning for evaluation of several factors such as selection of site, plot size, replications of sample, time element, maximum residue limit, formulation, type or variety of crops, etc. Sampling is the process to obtain a representative quantity from the large consignment, so that the selected representative quantity can be handled conveniently. Sample preparation for laboratory analysis is considered the most crucial step as the success of entire experiment depends on the proficiency at this level. It is done by selecting the components of interest, thereafter mixing, subdividing, and systematically reducing the sample size. Once a valid, representative sub-sample has been selected for residue analysis, it is processed for isolation of pesticide or its metabolites having toxicological significance from the surrounding biological environment. Extraction must be adequate to remove the toxicant in sufficient quantities from sample into a suitable solvent. The method of extraction and the type of solvent or solvent combinations will be dependent on the physical and chemical properties of the pesticide to be extracted, the type of substrate from which it will be quantitatively removed, and the final method of analysis. While extracting the pesticide with solvents from the plant materials, proteins, tannins, lipids, fat, waxes, chlorophyll, and terpenoids, they are co-extracted from matrix of substances (Erwin et al. 1955). These co-extractives can prevent the reaction of pesticides with chromogenic reagents, colored extracts directly interferes in the colorimetric analysis and can also contaminate the columns and detectors in the analysis. To achieve necessary sensitivity, the interfering substances have to be removed from pesticide, and this step is known as clean-up. It usually begins with some form of extraction technique and the degree of clean-up required is dependent on the scope of analysis, the complexity of sample, and the sensitivity and selectivity of detection methods available for the contaminant sought (Handa et al. 1999; Singh 2000). Estimation step, covering both detection and quantification of target compound, wind up the story of residue analysis. It is always desirable that chosen analytical procedure allows the simultaneous determination of large number of pesticides.

Steps of pesticide residue analysis
This article describes the basic principles of sample preparation techniques, especially soil sampling, extraction cum clean-up techniques, both conventional and modern approaches, comparing their advantages and disadvantages, and their ability and applicability for pesticide residues determination, with special emphasis on soil samples.
10.2 Soil Sampling Methods
The sampling of soil is typically done to detect pesticide residues or to routinely monitor environmental samples (Sharma, 2007). Soil samples should be taken from growing fields in the grid pattern uniformly distributed so that each area of the field is sampled. A 3 × 3 grid with nine total sample proportions is suggested for smaller fields, with 4 × 4 (16 sample portions) for the medium-sized fields, and 5 × 5 and even larger grids are used for very large fields. Each sample site represents one portion of the total sample, and at each site, two soil plugs about 15 cm deep and 3–5 cm in diameter are to be taken. The two plugs, when combined, become sample portion of that sample site. Another common soil sampling method for a field or other area is to take “5” portions in a “Z” pattern. An example of a 3 × 3 sampling grid is designed by X-pattern sampling (Fig. 10.2).

Soil sampling patterns: Z-pattern and X-pattern
Sampling tools include soil augers. Place each portion of the soil sample into a separate glass jar covered with aluminum foil. It is recommended to chill soil samples to 4 °C for transport to the laboratory. The glass jars for collecting soil samples should be rinsed thoroughly with acetone or methanol and dried.
10.3 Conventional to Modern Approaches for Extraction and Clean-up: A Paradigm Shift
Traditional sample preparation methods (liquid–liquid extraction, Soxhlet extraction, sonication assisted extraction, etc.) are laborious, time consuming, expensive, require large amounts of organic solvents and usually involve many steps, leading to loss of some analyte quantity. Additionally, consequences of hydrocarbon solvents use, such as ozone depletion and generation of considerable cancer waste, lead to reduction of not only their use but also their manufacture. As a result, modern sample preparation procedures, such as accelerated solvent extraction (ASE), supercritical fluid extraction (SFE), microwave assisted extraction (MAE), solid phase extraction (SPE), solid phase microextraction (SPME), matrix solid phase dispersion (MSPD) extraction and QuEChERS (quick, easy, cheap, effective, rugged and safe), have been developed to overcome the drawbacks of the traditional approaches. It should be noted that some (SFE, ASE and MAE) are instrumental techniques, and often use SPE (for purification of obtained extracts) and SPME (for both purification and concentration of obtained extracts) for desired purpose.
10.3.1 Solvent Extraction
For extraction of toxicants, either any suitable solvent or mixture of solvents is used. Soil samples were extracted by shaking with suitable solvent or solvent mixture in a mechanical shaker for definite period. The mixture was filtered, washed, and stored for further action. Liquid–liquid extraction (LLE) is one of the earliest and most commonly used extraction techniques employed for pesticide residue analysis in complex media (Dean 1998). The principle of LLE is that the sample is distributed or partitioned between two immiscible solvents in which the analyte and matrix have different solubility or it is based on the low value of the partition coefficient for most organic compounds between different solvents. The main advantage of this technique is the wide availability of pure solvents and use of low cost apparatus. Khan et al. (2011) employed ethyl acetate and hexane for the LLE of pentachloronitrobenzene and hexachlorobenzene and its metabolites prior to their HPLC determination. Another method that can be applied for dry materials like soil is Soxhlet extraction. Although the method is very efficient, sometimes formation of fine capillary tubes in the sample mass obstructs complete extraction.
10.3.2 Sonication Assisted Extraction (SAE)
Sonication provides a more efficient contact between the solid and solvent than Soxhlet method, usually resulting in a greater recovery of analyte (Poole et al. 1990). The extraction procedure should be optimized with regard to the solvent amount, the duration of sonication, and the number of extraction steps. The ultrasonic solvent extraction is more rapid than conventional shake-flask or Soxhlet extraction methods, and the solvent consumption is significantly lower. Additionally, the extracts from sonication can be chromatographed without subsequent clean-up step, and the analysis time is considerably reduced. Sonication assisted extraction has been used by Sánchez-Brunete et al. (2003) for carbamate pesticides.
10.3.3 Liquid Solid Extraction (LSE)/Solid Phase Extraction (SPE)
Solid phase extraction is one of the sorbent techniques which is used very often for pesticide residue analysis. It, being less laborious, produces low background interferences and also significantly reduces the use of organic contaminants. This method is based on the exclusion of extracts containing target analytes through a column (cartridge) filled with the appropriate solid phase called sorbent (which was previously conditioned by an appropriate solvent or solvent mixture), or passing of an appropriate solvent through the SPE column to which a suitable amount of sample was previously added (Moors et al. 1994). So, SPE basically separates compounds of interest from impurities in three distinct ways: selective extraction (the compounds of interest retained by the packing material and the impurities are eluted out), selective washing (the column is washed with strong solutions to remove impurities but the solution should not be so strong that it carries away the compound), and selective elution (the compound of interest is eluted in a solvent but the impurities are retained in the column). Method of operation can be divided into five steps: wetting the sorbent, conditioning of the sorbent, loading of the sample, rinsing or washing the sorbent to elute extraneous material, and elution of the analyte of interest. Each step is characterized by the nature and type of solvent used which in turn is dependent upon the characteristics of the sorbent and the sample (Dean 1998). Using selective solvents, first the co-extractants from the SPE column can be successfully eluted, and then the target analytes (Fig. 10.3, A), or the elution of analytes can be direct, where undesirable co-extractants derived from the sample matrix remain in the SPE column (Fig. 10.3, B).

Steps of solid phase extraction technique
The SPE sorbents used frequently in pesticide residues determination include reverse phase octadecyl (C18), normal-phase aminopropyl (–NH2) and primary-secondary amine (PSA), anion-exchanger three-methyl ammonium (SAX) and adsorbents such as graphitized carbon black (GCB). Normal-phase sorbents such as florisil (MgSiO3), aluminum oxide (Al2O3), and silica (SiO2) are usually used in combination with the previously mentioned sorbents. The SPE cartridge should be chosen depending on the physicochemical properties of pesticides that are searched for in a particular sample, and the nature of the sample matrix (Ðurović and Ðorđević 2011). C18 cartridges have been found a good choice for determination of carbamates in soil (Santalad et al. 2010) and silica gel has proven effective in determination of OCPs in soil samples (Lehnik-Habrink et al. 2010).
10.3.4 Solid Phase Micro-Extraction (SPME)
Solid phase micro-extraction, one of the latest extraction techniques, is widely used in the pesticide residues analysis because of the fact that purification and concentration of the sample extract (analytes of interest) run simultaneously here. SPME syringe is the main part of the SPME system that visually resembles on the chromatographic system; however, it also contains a 1 cm long fiber located within a syringe needle, which is made of an appropriate polymer deposited on the holder of fused silica. Micro-extraction process is based on the redistribution of analytes between micro-extraction fiber and sample matrix, i.e., on the selective sorption of target analytes in the active layer of the fiber and direct desorption in the chromatograph injector (thermal in the case of the gas chromatography or, i.e., by solvent elution in the case of liquid chromatography). The basic micro-extraction procedure of analytes from the solution is shown in Fig. 10.4 (Ðurović and Ðorđević 2011).

Procedure for micro-extraction of analytes from solution
Before the analysis, the fiber is drawn into a metal tube of the SPME syringe. After breaking through the vial septum in which a certain sample amount was previously inserted, the fiber is pulled out from the syringe, i.e., it is exposed to the sample by lowering the syringe plunger. After specific time, the fiber with the sorbed analytes is drawn into the needle, which is then pulled out from the vial. Analytes desorption from the fiber is performed by introducing a SPME syringe needle into the injector of the chromatographic system.
SPME is an equilibrium technique, where analytes are distributed between the three phases: sample, gas phase, and fiber. The fiber does not extract all analytes present in the sample, but by the proper calibration, this technique can be used for successful quantification (Đurović et al. 2007a; Pawliszyn 1997). The amount of analytes that would be adsorbed on the fiber will depend on the thickness and polarity of the active fiber layer, sampling mode (direct sampling—micro-extraction from solution, “DM-SPME” and headspace sampling—micro-extraction from gas phase, “HS-SPME”), the nature of the sample and the analyte (analyte polarity, its molecular weight, pH value, nature of matrix), the mode and speed of the sample mixing, the SPME duration, the temperature at which it is performed, and so on.
Today, about 30 different fiber types are in use (different types of polymers and their thickness), so when selecting the fiber it is necessary to take into consideration several factors: molecular weight, structure and polarity of the analyte molecules, the polarity of fibers, the mechanism of extraction (used sampling mode), the detection limit and range of linearity that is desired to be achieved. In order for a fiber to extract specific compounds from a given matrix, it must have a much higher affinity for the given analytes than a matrix, where the general rule applies: non-polar analytes are more efficiently extracted by non-polar active fiber layer, i.e., polar by polar. The research in the field of pesticide residues has indicated that, in the most of the cases, fibers with extremely non-polar polydimethylsiloxane (PDMS) and highly polar polyacrylate (PA) active layers are most effective in the analysis of samples of different origin (Doong and Liao 2001; Sakamoto and Tsutsumi 2004; Đurović et al. 2007b, c, 2010b; Fernandez-Alvarez et al. 2008). After fiber selection, it is necessary to determine optimal conditions for analytes transfer in the chromatographic system. Adsorbed analytes are desorbed from the fiber by introducing the SPME syringe needle into the injector. Defining the parameters of desorption involves determination of the optimal injector temperature, flow of the carrier gas, and desorption time in the case of GC, i.e., proper choice of elution solvent, its flow rate and desorption time, in the case of HPLC.
Although the maximum of SPME sensitivity is achieved at equilibrium times, for practical reasons, extraction time can be shortened (Đurović et al. 2007a, 2010a, b; Pawliszyn 1997). The most effective ways to overcome the kinetics restrictions are heating and efficient sample mixing. The temperature has two opposite effects. On the one side, its increase increases the analytes transfer from the sample to the fiber, while on the other side, due to the simultaneous heating of the fiber during extraction, there is enhanced desorption of analytes from it. Therefore, the necessary step in method development is optimization of the extraction temperature. The speed of extraction is also determined by the sample stirring efficiency. Intensive stirring increases the analytes mobility, and therefore reduces the equilibrium time and increases the analytes amount adsorbed on the fiber. However, in method developing it should be noted that the sample stirring leads to its warming, which may also have non-preferred effects, especially in the case of direct mode.
The matrix nature greatly influences the SPME efficiency, too. Since the analytes distribution coefficients are partially determined by analytes/matrix interaction, appropriate matrix modification can increase the analytes partition coefficients. Thus, for example, the presence of chloride and sulfate ion increases the ionic strength of the solution, which makes a large number of compounds less soluble. In this way, by weakening the matrix/analyte interaction, distribution coefficients can be significantly increased (Arthur et al. 1992). Considering the fact that SPME is a single-stage method that does not require additional purification and concentration of the sample, the problems related to the matrix occur in the analysis of samples with complex matrices. The researches have shown that the negative effect of the matrix could be significantly reduced by adequate dilution of the sample with the distilled water (Simplício and Boas 1999; Đurović and Marković 2005; Đurović et al. 2007c, 2008).
The research results indicate that the most often used SPME fibers in the pesticide residues analysis (PDMS and PA) are a good choice for determination of: OCPs in soils (Zhao et al. 2006; Herbert et al. 2006); pesticides belonging to different chemical groups in soil (Đurović et al. 2010a, b), i.e., in samples of potato, tomato, onion, cabbage, and pepper (Marković et al. 2010). Considering that in the SPME analysis only 1 cm of fiber is exposed to the sample, not only the nature, but also the size of the active surface layer will significantly affect the micro-extraction efficiency. Thus, by adding an additional material into the active layer of the fiber, its outer surface may increase, and therefore often the SPME efficiency, too. On the other side, the added material can significantly change the polarity of the fiber (similar to the GC stationary phase). Thus, for example, by using mixed PDMS/DVB (polydimethylsiloxane/divinylbenzene) fiber, Vega Moreno et al. (2006) provided satisfactory analytical parameters for SPME determination of OCPs in soil.
10.3.5 Supercritical Fluid Extraction (SFE)
Supercritical fluid extraction is the process of separating organic compounds (extractants) from solid matrices using supercritical fluids (CO2, NO2, SO2, NH3, etc.). A substance exists as a supercritical fluid (SCF) when system temperature and pressure are above a critical point (Fig. 10.5). The principle of SFE is based on the solvent power of SCF which is highly dependent on the density of SCF, which in turn depends on the pressure and temperature. Modification of little temperature and pressure changes the property of SCF which is very useful for extraction purpose. Because of low viscosity and higher diffusivity as compared to liquids, SCFs diffuse more rapidly and even penetrate solid samples.

Phase diagram of supercritical fluid
CO2 is the most commonly used SCF for this purpose, as it has relatively low critical temperature (31 °C) and low critical pressure (73 kPa) (Atkins and de Paula 2002). It is non-reactive and non-toxic also, available in a high degree of purity at low cost and shows absence of contamination of final products as CO2 volatilizes off. Changes in temperature and pressure at which the supercritical CO2 is held will increase or decrease the strength of solvent that ensures selective extraction of the target compound. At constant temperature beyond critical temperature, the supercritical CO2 will be able to extract analytes of high polarity at high pressure, and low polarity analytes at low pressure. SFE with CO2 is usually performed at pressures that are not high enough to achieve efficient extraction of polar compounds. In such conditions, the supercritical CO2 is a good extraction medium for non-polar compounds and moderately polar ones, such as polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs), organochlorine (OC), and organophosphorus (OP) pesticides, etc. Supercritical CO2, being non-polar, sometimes requires small amounts of polar co-solvents as modifiers, whose major role is to interact with the sample matrix to promote desorption into the fluid (Langenfeld et al. 1994). Some of the common solvents such as acetone (Kaihara et al. 2002; Ono et al. 2006) and methanol (Rissato et al. 2005a, b) are now mostly used as modifiers.
In general, extraction procedure is completed within 2 h, and further analysis can be accomplished in various ways. According to one, supercritical fluid with analytes is passed through a capillary that is immersed in an appropriate solvent. While in the capillary, it remains as supercritical fluid, but after leaving the capillary it becomes a gas (the pressure falls below the critical pressure). The largest part of this gas passes through the solvent, while the extracted analytes are retained in the solvent (the degree of retention depends on the solvent, i.e., the solubility of the analyte in it). The flow of SF can be directed to a solid sorbent, which will then bind analytes, and its elution by an appropriate solvent, analysts translate into a solution suitable for further analysis (Fig. 10.6). Also, the flow of SF could be directed directly to the capillary column of the gas chromatograph (GC), thus obtaining the on-line SFE. This approach enables the analytical scheme with the highest sensitivity for a limited amount of sample available for analysis. The recent studies have shown that SFE methods, followed by additional purification of the obtained extracts, meet the strict criteria of the pesticide residues analysis. The same sorbent was shown to be the best choice for determination of 32 pesticides in soil using SFE sample preparation (Rissato et al. 2005b).

Supercritical fluid extraction technique
10.3.6 Microwave-Assisted Extraction (MAE)
Microwave-assisted extraction (MAE) is a technique utilizing the microwave energy, and where target compounds can be extracted more selectively and rapidly, with similar or better recovery compared to traditional extraction processes. Microwave energy is a non-ionizing radiation (frequency 300–300,000 MHz), which can penetrate into certain materials and interact with the polar components to generate heat. The MAE causes a direct migration of the desired components out of the matrix, as a result of selective energy application into the matrix. Greater efficiency of extraction method effects in the matrix macrostructure destruction (Lambropoulou and Albanis 2007). During the MAE of solid material, microwave rays travel freely through the solvent and interact selectively with the free matrix water causing localized heating resulting in non-uniform temperature rise with more pronounced effects where the free water is in larger proportions which ultimately leads to a volume expansion within the systems. The walls of these systems cannot countenance the high internal pressures and rupture spontaneously, allowing the organic contents to flow freely toward the relatively cool surrounding solvent that solubilizes them rapidly (Ranz et al. 2008). For method optimization, several variables, such as solvent composition and amount, extraction temperature and time, are usually studied. In order to heat a solvent, part of it must be polar with high dielectric constant to absorb microwave energy efficiently. Non-polar solvents with low dielectric constants can be also used, by adding certain amount of polar solvent that absorbs the microwave radiation and passes it on to other molecules (Caddick 1995). For example, hexane and toluene can be modulated by the addition of small amounts of acetone or methanol (Ericsson and Colmsjö 2000).
Generally, MAE devices comprise a closed extraction vessel under controlled pressure and temperature or a focused microwave oven at atmospheric pressure. These two technologies are commonly named pressurized MAE (PMAE) or focused MAE (FMAE), respectively. The PMAE system consists of a magnetron tube, an oven where the extraction vessels are set upon a turntable and monitoring devices for controlling temperature and pressure. In PMAE, the extractions are performed in some sealed extraction cells with microwave radiation, in static and batch mode. The increase in temperature and pressure accelerates extraction due to the ability of extraction solvent to absorb microwave energy. The closed system offers fast, efficient extraction with less solvent consumption, but it is susceptible to losses of volatile compounds and generally expensive due to its resistance to high pressure and its air-tightness (Zhang et al. 2011). FMAE involves an open MAE system developed to counter the shortcomings of the closed system, such as safety issues. The extractor design is based on the principles of a conventional Soxhlet extractor modified to facilitate accommodation of the sample cartridge compartment in the irradiation zone of a microwave oven. Solvent distillation in the FMAE extractor could be achieved by electrical heating, which is independent of extractant polarity (Luque-García and Luque de Castro 2003, 2004). It is considered more suitable for extracting thermo-labile compounds due to only part of the extraction cell being directly exposed to microwave radiation. Since the upper part of the extraction cell is connected to a reflux unit to condense vaporized solvent, sample throughput is limited (Fig. 10.7).

Microwave-assisted micellar extraction (MAME) procedure
From economical and practical aspects, MAE is a strong competitor to other recent sample preparation techniques. The main MAE advantages are the complete automation, low temperature requirement, high extraction efficiency, and the possibility of extracting different samples at the same time without interference. The main disadvantage of MAE seems to be the lack of selectivity resulting in the co-extraction of significant amounts of interfering compounds. Additional clean-up is therefore needed before chromatographic analysis. Apart from that, the poor efficiency of microwaves when either the target compounds or the solvents are non-polar, or when they are volatile, can be regarded as another disadvantage. Besides, it is important to notice that the application of microwave energy to flammable organic compounds, such as solvents, can pose serious hazards in inexperienced hands, thus an extraordinary level of safety and attention to details when planning and performing experiments must be used by all personnel dealing with microwaves. The first use of MAE technique for pesticide residues determination (parathion and bromophos in soil) was reported by Ganzler et al. (1986). In 1994, 20 OCPs were extracted from six marine sediments and soils (Lopez-Avila et al. 1994). Investigations on MAE extractions of OCPs and OPPs from soil, optimization and comparison of method, was performed by numerous authors (Fuentes et al. 2007; Wang et al. 2007). MAE determination of triazines in soils was reported by Hoogerbrugge et al. (1997) work. MAE determination of imidazolinone herbicides have been reported by Stout et al. (1997). The investigated fungicides were hexaconazole (Frost et al. 1997), and dimethomorph (Stout et al. 1998), both extracted from soils. De Andréa et al. (2001) applied MAE for determination of methyl parathion, p,p′-DDE, HCB, simazine, and paraquat dichloride in soil, Sun and Lee (2003) for carbamates in soil.
10.3.7 Microwave-Assisted Micellar Extraction (MAME)
Microwave assisted micellar extraction, which uses a micellar (surfactant-rich) system to substitute organic solvent as extractant in MAE, has been applied lately to the extraction of different compounds from solid samples including soil (Wang et al. 2016). In order to escalate both extraction rate and efficiency, microwave energy is used while maintaining the sample at a suitable temperature. At this point, micelles of surfactant are formed, with analytes isolated and enriched in them. Figure 10.8 shows the three key steps for the operations of MAME:
-
1.
Introduction of surfactant to the sample,
-
2.
Microwave-assisted micellar extraction for definite time period, and
-
3.
Suitable treatment of the extract.

Steps of microwave-assisted micellar extraction
Before injecting the extract into the analytical instrument, the MAME extract obtained should be suitably prepared (Step III, Fig. 10.8). Separation of two phases requires appropriate experimental conditions depending on the nature of the surfactant. Sometimes, analytes in the MAME extract were concentrated with the help of centrifugation after equilibrium at high temperature and adding salt reagent (Step III a, Fig. 10.8) (Chen et al. 2010). The analytes in the micelle-rich phase could be directly injected into HPLC for subsequent separation and detection. As the micelle-rich phase is viscous and cannot be injected directly into some analysis apparatus (e.g., LC-MS/MS), then additional clean-up and concentration of the MAME extract should be employed, such as solid phase extraction (SPE) (Cueva-Mestanza et al. 2008) or solid-phase micro-extraction (SPME) (Pino et al. 2007) (Step III b and c, Fig. 10.8). For SPE, MAME extracts went through the SPE cartridge, and the retained analytes were eluted and analyzed. For SPME, SPME fibers were directly immersed into the MAME extract under optimized conditions, and thereafter the analytes were desorbed from the fiber by a suitable solvent.
10.3.8 Accelerated Solvent Extraction (ASE)/Pressurized Fluid Extraction (PFE)
Accelerated solvent extraction (ASE™, a Dionex trademark), also known as pressurized liquid extraction (PLE) or enhanced solvent extraction (ESE), is a relatively new extraction technique which is partly derived from SFE (Camel 2001; Richter et al. 1996). It is a solid–liquid extraction process using organic solvents at an elevated temperature (usually between 50 and 200 °C) and applying higher pressure (between 10 and 15 MPa) for short periods (12–18 min) to extract samples in an extraction cell. Extractions are carried out under pressure in order to maintain the solvent in its liquid state, even at temperatures above boiling point. Moreover, pressure allows the extraction cell to be filled more quickly and helps to force the solvent into the matrix pores. Thus, the efficiency of the extraction process is improved. Extraction at elevated temperatures increases solubility, diffusion rate, and mass transfer, along with the ability of the solvent to disrupt the analyte-matrix interactions. PLE thus allows fast extraction due to increased solubility, better desorption, and enhanced diffusion, and rapid extraction.
In practice, the extraction cell is filled with the sample to be examined and placed in a furnace controllable. After the addition of a suitable solvent, the cell is brought to an elevated temperature and pressure (Fig. 10.9). Later, the extract is transferred to a collection vial for clean-up and analysis. At high temperatures, viscosity and the surface tension of the solvent decrease, resulting in a substantial increase in extraction rate (Anastassiades et al. 2003). The solvent is kept below its boiling point by applying high pressure that forces its penetration into the sample pores. The combination of high temperature and pressure results in better extraction efficiency, thus minimizing solvent use. The extraction efficiency is almost independent of sample mass, i.e., is mainly dependent on temperature (Richter et al. 1996; Smith 2002). Often a sample undergoes several extraction cycles. Finally, the extraction cell is flushed with solvent, open the purge valve and the cell, as well as all the lines purged with nitrogen and the apparatus prepared for further extraction. Besides the type of the solvent used, the main parameters which influencing ASE efficiency are extraction temperature and time (Luo et al. 2010). Although high temperatures increase the efficiency, it may lead to degradation of thermo-labile compounds, and to the co-extraction of interfering species. Hence, a compromise between the extraction efficiency and minimization of interfering compounds must be performed carefully, and in addition, usually a further clean-up step involves.

Accelerated solvent extraction technique
ASE is advantageous over conventional techniques as it requires much lesser solvent and shorter extraction times. Using elevated pressure and temperatures with organic solvents, an enhanced analytes extraction can be achieved. Moreover, ASE can reduce waste levels and analysts exposure to harmful solvents. However, samples with high moisture contents are subjected to desiccation before the extraction step (Cervera et al. 2010). ASE was carried out for determination of DDT and its metabolites (Tao et al. 2004), i.e., abamectin in soil samples (Brewer et al. 2004). ASE methods for soil samples were reported for OCPs (Wang et al. 2007), for bromacil and diuron (Pinto and Lanças 2009), and dichlorvos, dimethoate, parathion, malathion, and chlorpyrifos determination (Zhang et al. 2010).
10.3.9 QuEChERS Method
“QuEChERS” is a portmanteau word derived from “Quick, Easy, Cheap, Effective, Rugged, and Safe.” It is a novel multi-residue method for determining pesticide residues in different matrices and appeared to overcome the loopholes of conventional solvent extraction methods (Anastassiades et al. 2003). It is undoubtedly one of the most streamlined sample preparation approaches with excellent results for a wide range of pesticides in different soil samples. The original procedure involves initial single phase extraction of the sample by hand-shaking or vortex mixing with acetonitrile (CH3CN) and simultaneous liquid–liquid partitioning between the aqueous residue and the solvent caused by the introduction of anhydrous magnesium sulfate (MgSO4) and sodium chloride (NaCl) in a suitable ratio (4:1). After vortex mixing and centrifugation, clean-up and exclusion of residual water is performed via a simple step known as dispersive solid phase extraction (d-SPE) that is less time consuming than the traditional SPE. This procedure involves addition of anhydrous MgSO4 with aliquot to remove residual moisture and primary-secondary amine (PSA) adsorbent to get rid of many polar matrix components, such as organic acids, some polar pigments, and sugars (Fig. 10.10).

Steps of QuEChERS method for pesticide residue analysis
Acetonitrile is selected as the QuEChERS solvent because of its high polarity, well miscibility with water, and sufficient dispersive (hydrophobic) properties to extract effectively both polar and non-polar pesticides. The original QuEChERS method was subjected to certain necessary modifications to ensure efficient extraction of pH-dependent compounds (e.g., phenoxyalkanoic acids) and to minimize degradation of susceptible compounds (e.g., base and acid labile pesticides). Anastassiades et al. (2007) realized that buffering at pH 5.0 during extraction could give the optimum balance to achieve acceptably recoveries (>70%) for pH-dependent pesticides, independent of the matrix. On the other hand, Lehotay (2007) modified the method to apply for stronger acetate-buffering conditions. Both of these versions of methods went through extensive laboratory trials and successfully met statistical criteria for acceptability by independent scientific standards organizations. So the acetate-buffering version was labeled as AOAC Official Method 2007.01 (Lehotay 2007) and the citrate-buffering version being entitled as Standard EN 15662 method (www.cen.eu). The QuEChERS advantages are high recovery (>85%), very accurate results (an internal standard is used), low solvent and glassware usage, high sample throughput (10–20 samples analysis in about 30–40 min), less skill, labor and bench space, lower reagent costs, and ruggedness. The main drawback of this method is that the final extract must be concentrated to furnish the necessary sensitivity, i.e., to achieve the desired limits of quantification (LOQ). QuEChERS has been successfully used for determination of metaflumizone (Dong et al. 2009), oxadiargyl (Shi et al. 2010), and 38 pesticides (Yang et al. 2010) in soil samples. As a modified version, it was applied for OCPs (Rashid et al. 2010) and OPPs determination in soil samples (Asensio-Ramos et al. 2010).
10.3.10 Matrix Solid Phase Dispersion (MSPD)
Matrix solid phase dispersion (MSPD) is a new SPE-based extraction and clean-up technique developed for pesticide multi-residue analysis (Kristenson et al. 2006). The MSPD method is based on the homogenization of a viscous, solid, or semi-solid sample with an abrasive solid support material in a glass mortar, in order to perform the complete disruption and dispersal of the sample. After blending, the sample is transferred into a column and analytes are eluted with appropriate solvent. Complete disruption of the sample and its dispersion over the support surface greatly enhance surface area for the sample extraction. Furthermore, interferences are retained on the adsorbent and in that way, extraction and clean-up are performed simultaneously, reducing the analysis time and the amount of solvent used (Barker 2000; Kristenson et al. 2006).
Reversed-phase materials such as C8 and C18-bonded silica are the most commonly used adsorbents, because their lipophilic properties enable good disruption, dispersion, and retention of lipophilic species (Lambropoulou and Albanis 2007). Basically, the adsorbent choice depends on analyte polarity and interferences which could be co-extracted from sample matrix (Fig. 10.11). Also, the nature of the elution solvent is crucial for efficient pesticides elution from the adsorbent (Albero et al. 2003; Blasco et al. 2002a, b; Bogialli et al. 2004). The original MSPD can be modified or combined with other extraction methodologies to improve the extraction yields or simplify the MSPD procedures. The schematic procedure of the original and representative modification of MSPD is shown in Fig. 10.11 (Tu and Chen, 2018).

Schematic procedure of original matrix solid phase dispersion (MSPD), ultrasonic-assisted MSPD (UA-MSPD), vortex-assisted MSPD (VA-MSPD), and magnetically-assisted MSPD (MA-MSPD)
In comparison to traditional extraction methods, MSPD approach has several advantages, including simplified and faster sample-treatment, reduced use of toxic solvents, eliminated emulsion formation, and increased selectivity and sensitivity. In MSPD, the sample extraction and clean-up are performed in the same step by use of small amounts of adsorbent and solvent, thus reducing the cost and analysis time. As a drawback, a number of applications still use large volumes of solvents for extraction and clean-up, which requires solvent evaporation. There is a very reason to believe that solving of this problem will make MSPD more useful in the near future. It has been successfully applied for phenthoate (Li et al. 2002), OCPs (Shen et al. 2005, 2006), and five pesticides in soil (Shen et al. 2007).
10.4 Conclusion
The sample extraction step, the most time determining step, is still the weakest link in the whole analytical procedure and also the prime cause of experimental errors and disparity between laboratories. However, in the recent times, upgradation in the existing techniques and also development of new techniques have unfolded new horizons in the sample preparation techniques in terms of saving time and reducing use of chemicals and thus undoubtedly improved the overall performance of analytical process. As a result of advancement of modern science and technology, several rapid, low cost, environmentally friendly, and readily automated methods of extraction are now available. Besides, because of the complexity of the matrices, extraction is usually followed by very specific clean-up procedures to achieve accurate sample quantification, so the new methods are modified in order to achieve a compromise between cost, selectivity, and sensitivity. Reduced solvent methods, including supercritical fluid extraction (SFE), solid phase extraction (SPE), solid phase micro-extraction (SPME), microwave assisted extraction (MAE), accelerated solvent extraction (ASE), QuEChERS and matrix solid phase dispersion (MSPD) have grown in their maturity, which increased application of these techniques in pesticide analysis of soil matrices. Although the composition of soil matrix varies from place to place, which requires application of different approaches and strategies, the development of a uniform procedure is highly encouraged. Future developments in all areas of analytical sample preparation are expected to continue to be application-driven in a quest for improved recovery, higher sample throughput, and reduced consumption of organic solvent with capability to provide accurate results.
References
Albero B, Sánchez-Brunete C, Tadeo JL (2003) Determination of endosulfan isomers and endosulfan sulfate in tomato juice by matrix solid-phase dispersion and gas chromatography. J Chromatogr A 1007(1–2):137–143
Anastassiades M, Lehotay SJ, Štajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86(2):412–431
Anastassiades M, Scherbaum E, Tasdelen B, Štajnbaher D (2007) Recent developments in QuEChERS methodology for pesticide multiresidue analysis. In: Ohkawa H, Miyagawa H, Lee PW (eds) Pesticide chemistry, crop protection, public health. Wiley-VCH, Weinheim, pp 439–458
Arthur CL, Killam LM, Buchholz KD, Pawliszyn J, Berg JR (1992) Automation and optimization of solid-phase microextraction. Anal Chem 64(17):1960–1966
Asensio-Ramos M, Hernández-Borges J, Ravelo-Pérez LM, Rodríguez-Delgado MA (2010) Evaluation of a modified QuEChERS method for the extraction of pesticides from agricultural, ornamental and Forestal soils. Anal Bioanal Chem 396(6):2307–2319
Atkins P, de Paula J (2002) Atkins` physical chemistry, 7th edn. Oxford University Press, New York, p 18
Barker SA (2000) Applications of matrix solid-phase dispersion in food analysis. J Chromatogr A 880(1–2):63–68
Blasco C, Picó Y, Mañes J, Font G (2002a) Determination of fungicide residues in fruits and vegetables by liquid chromatography–atmospheric pressure chemical ionization mass spectrometry. J Chromatogr A 947(1–2):227–235
Blasco C, Font G, Picó Y (2002b) Comparison of microextraction procedures to determine pesticides in oranges by liquid chromatography–mass spectrometry. J Chromatogr A 970(1–2):201–212
Bogialli S, Curini R, Di Corcia A, Nazzari M, Tamburro D (2004) A simple and rapid assay for analyzing residues of carbamate insecticides in vegetables and fruits: hot water extraction followed by liquid chromatography-mass spectrometry. J Agric Food Chem 52(4):665–671
Brewer BN, Armbrust KL, Mead KT, Holmes WE (2004) Determination of abamectin in soil samples using high-performance liquid chromatography with tandem mass spectrometry. Rapid Commun Mass Spectrom 18(15):1693–1696
Caddick S (1995) Microwave assisted organic reactions. Tetrahedron 51(38):10403–10432
Camel V (2001) Recent extraction techniques for solid matrices-supercritical fluid extraction, pressurized fluid extraction and microwave-assisted extraction: their potential and pitfalls. Analyst 126:1182–1193
Cervera MI, Medina C, Portolés T, Pitarch E, Beltrán J, Serrahima E, Pineda L, Muñoz G, Centrich F, Hernández F (2010) Multi-residue determination of 130 multiclass pesticides in fruits and vegetables by gas chromatography coupled to triple quadrupole tandem mass spectrometry. Anal Bioanal Chem 397(7):2873–2891
Chen L, Zhao Q, Xu Y, Sun L, Zeng Q, Xu H et al (2010) A green method using micellar system for determination of sulfonamides in soil. Talanta 82:1186–1192
Codex (2019) Pesticide residues in food and feeds. Codex pesticides residues in food online database. Available online: http://www.fao.org/fao-who-codexalimentarius/codex-texts/list-standards/en
Cueva-Mestanza R, Sosa-Ferrera Z, Torres-Padrón ME, Santana-Rodríguez JJ (2008) Preconcentration of pharmaceuticals residues in sediment samples using microwave assisted micellar extraction coupled with solid phase extraction and their determination by HPLC-UV. J Chromatogr B 863:150–157
Damalas CA (2009) Understanding benefits and risks of pesticide use. Sci Res Essays 4:945–949
De Andréa MM, Papini S, Nakagawa LE (2001) Optimizing microwave-assisted solvent extraction (MASE) of pesticides from soil. J Environ Sci Health B 36(1):87–93
Dean JR (1998) Extraction methods for environmental analysis. John Wiley & Sons, West Sussex
Dong F, Liu X, Cheng L, Chen W, Li L, Qin D, Zheng Y (2009) Determination of Metaflumizone residues in cabbage and soil using ultra-performance liquid chromatography/ESI-MS/MS. J Sep Sci 32(21):3692–3697
Doong RA, Liao PL (2001) Determination of organochlorine pesticides and their metabolites in soil samples using headspace solid-phase microextraction. J Chromatogr A 918(1):177–188
Đurović R (2011) Procesi koji određuju sudbinu pesticida u zemljištu. Pesticidi i fitomedicina 26(1):9–22
Ðurović R, Ðorđević T (2011) Modern extraction techniques for pesticide residues determination in plant and soil samples. In: Pesticides in the modern world-trends in pesticides analysis, Margarita Stoytcheva. Intech Open. https://doi.org/10.5772/17312
Đurović R, Marković M (2005) Solid phase microextraction in the analysis of Vinclozolin and Procymidone residues in Strawberries. Pesticidi i fitomedicina 20(3):163–169
Đurović R, Marković M, Marković D (2007a) Headspace solid phase microextraction in the analysis of pesticide residues—kinetics and quantification prior to the attainment of partition equilibrium. J Serb Chem Soc 72(8–9):879–887
Đurović R, Milinović J, Cupać S, Marković M (2007b). The book of abstracts, Euroanalysis XIV, p. 75, Antwerp, Belgium, September 9–14, 2007
Đurović R, Milinović J, Marković M, Marković D (2007c) Headspace solid phase Microextraction in pesticide residues analysis: 2. Apple samples. Pesticidi i fitomedicina 22(2):173–176
Đurović R, Gajić Umiljendić J, Đorđević T (2008) Determination of atrazine, acetochlor, clomazone, pendimethalin and oxyfluorfen in soil by a solid phase microextraction method. Pesticidi i fitomedicina 23(4):265–271
Đurović RD, Ðorđević TM, Šantrić LR, Gašić SM, Ignjatović LM (2010a) Headspace solid phase microextraction method for determination of triazine and organophosphorus pesticides in soil. J Environ Sci Health B 45(7):626–632
Đurović RD, Gajić Umiljendić JS, Cupać SB, Ignjatović LM (2010b) Solid phase microextraction as an efficient method for characterization of the interaction of pesticides with different soil types. J Brazil Chem Soc 21(6):985–994
Ericsson M, Colmsjö A (2000) Dynamic microwave-assisted extraction. J Chromatogr A 877(1–2):141–151
Erwin WR, Schiller D, Hoskins WM (1955) Pesticide residue analysis, preassay purification of tissue extracts by wax column. J Agric Food Chem 3(8):676–679
EU, MRL (2019) European Commission (EU) legislation on maximum residue levels. Available online: https://ec.europa.eu/food/plant/pesticides/max_residue_levels/eu_rules/mrls_2019_en
Fernandez-Alvarez M, Llompart M, Pablo Lamas J, Lores M, Garsia-Jares C, Cela R, Dagnac T (2008) Simultaneous determination of traces of pyrethroids, organochlorines and other main plant protection agents in agricultural soils by headspace solid-phase microextraction–gas chromatography. J Chromatogr A 1188(2):154–163
Frost SP, Dean JR, Evans KP, Harradine K, Cary C, Comber MHI (1997) Extraction of hexaconazole from weathered soils: a comparison between soxhlet extraction, microwave-assisted extraction, supercritical fluid extraction and accelerated solvent extraction. Analyst 122(9):895–898
Fuentes E, Báez ME, Labra R (2007) Parameters affecting microwave-assisted extraction of organophosphorus pesticides from agricultural soil. J Chromatogr A 1169(1–2):40–46
Ganzler K, Salgó A, Valkó K (1986) Microwave extraction: a novel sample preparation method for chromatography. J Chromatogr 371:299–306
Handa SK, Agnihotri NP, Kulshreshtha G (1999) Multiresidue methods for estimation of pesticide residues pesticide residues: significance, management and analysis. Research Periodicals and Book Publishing House, New Delhi, pp 136–145
Herbert P, Morais S, Paíga P, Alves A, Santos L (2006) Development and validation of a novel method for the analysis of chlorinated pesticides in soils using microwave-assisted extraction–headspace solid phase microextraction and gas chromatography-tandem mass spectrometry. Anal Bioanal Chem 384(3):810–816
Hoogerbrugge R, Molins C, Baumann RA (1997) Effects of parameters on microwave assisted extraction of Triazines from soil: evaluation of an optimisation trajectory. Anal Chim Acta 348(1–3):247–253
Kaihara A, Yoshii K, Tsumura Y, Ishimitsu S, Tonogai Y (2002) Multi-residue analysis of 18 pesticides in fresh fruits, vegetables and rice by supercritical fluid extraction and liquid chromatography-electrospray ionization mass spectrometry. J Health Sci 48(2):173–178
Khan F, Prakash D, Jain RK (2011) Development of an HPLC method for determination of pentachloronitrobenzene, hexachlorobenzene and their possible metabolites. BMC Chem Biol 11(1):2
Kristenson EM, Brinkman UAT, Ramos L (2006) Recent advances in matrix solid-phase dispersion. Trends Anal Chem 25(2):96–111
Lambropoulou DA, Albanis TA (2007) Methods of sample preparation for determination of pesticide residues in food matrices by chromatography-mass spectrometry-based techniques: a review. Anal Bioanal Chem 389(6):1663–1683
Langenfeld JJ, Hawthorne SB, Miller DJ, Pawliszyn J (1994) Role of modifiers for analytical-scale supercritical fluid extraction of environmental samples. Anal Chem 66(4):909–916
Lehnik-Habrink P, Hein S, Win T, Bremser W, Nehls I (2010) Multi-residue analysis of PAH, PCB, and OCP optimized for organic matter of forest soil. J Soils Sediments 10(8):487–1498
Lehotay SJ (2007) Determination of pesticide residues in foods by acetonitrile extraction and partitioning with magnesium sulfate: collaborative study. J AOAC Int 90(2):485–520
Li ZY, Zhang ZC, Zhou QL, Gao RY, Wang QS (2002) Fast and precise determination of phenthoate and its enantiomeric ratio in soil by the matrix solid-phase dispersion method and liquid chromatography. J Chromatogr A 977(1):17–25
Lopez-Avila V, Young R, Beckert WF (1994) Microwave-assisted extraction of organic compounds from standard reference soils and sediments. Anal Chem 66(7):1097–1106
Luo L, Shao B, Zhang J (2010) Pressurized liquid extraction and cleanup procedure for the determination of Pyrethroids in soils using gas chromatography/tandem mass spectrometry. Anal Sci 26(4):461–465
Luque-García JL, Luque de Castro MD (2003) Where is microwave-based analytical equipment for solid sample pre-treatment going. Trends Anal Chem 22:90–98
Luque-García JL, Luque de Castro MD (2004) Focused microwave-assisted Soxhlet extraction: devices and applications. Talanta 64:571–577
Marković M, Cupać S, Ðurović R, Milinović J, Kljajić P (2010) Assessment of heavy metal and pesticide levels in soil and plant products from agricultural area of Belgrade, Serbia. Arch Environ Contam Toxicol 58(2):341–351
Moors M, Massart DL, Mcdowall RD (1994) Analyte isolation by solid phase extraction (SPE) on silica-bonded phases: classification and recommended practices. Pure Appl Chem 66:277
Odukkathil G, Vasudevan N (2013) Toxicity and bioremediation of pesticides in agricultural soil. Rev Environ Sci Biotechnol 12:421–444
Ono Y, Yamagami T, Nishina T, Tobino T (2006) Pesticide multiresidue analysis of 303 compounds using supercritical fluid extraction. Anal Sci 22(11):1473–1476
Pawliszyn J (1997) Solid phase microextraction – theory and practice. Wiley, New York
Pino V, Ayala JH, González V, Afonso AM (2007) Focused microwave-assisted micellar extraction combined with solid-phase microextraction gas chromatography/mass spectrometry to determine chlorophenols in wood samples. Anal Chim Acta 582:10–18
Pinto JSS, Lanças FM (2009) Design, construction and evaluation of a simple pressurized solvent extraction system. J Brazil Chem Soc 20(5):913–917
Poole SK, Dean TA, Oudsema JW, Poole CF (1990) Sample preparation for chromatographic separations: an overview. Anal Chim Acta 236:3–42
Ranz A, Maier E, Motter H, Lankmayr E (2008) Extraction and derivatization of polar herbicides for GC-MS analyses. J Sep Sci 31(16–17):3021–3029
Rashid A, Nawaz S, Barker H, Ahmad I, Ashraf M (2010) Development of a simple extraction and clean-up procedure for determination of organochlorine pesticides in soil using gas chromatography-tandem mass spectrometry. J Chromatogr A 1217(17):2933–2939
Richter BE, Jones BA, Ezzell JL, Porter NL, Avdalovic N, Pohl C (1996) Accelerated solvent extraction: a technique for sample preparation. Anal Chem 68(6):1033–1039
Rissato SR, Galhiane MS, Apon B, Arruda M (2005a) Multiresidue analysis of pesticides in soil by supercritical fluid extraction/gas chromatography with electron-capture detection and confirmation by gas chromatography−mass spectrometry. J Agric Food Chem 53(1):62–69
Rissato SR, Galhiane MS, De Souza AG, Apon BM (2005b) Development of a supercritical fluid extraction method for simultaneous determination of organophosphorus, organohalogen, organonitrogen and pyrethroids pesticides in fruit and vegetables and its comparison with a conventional method by GC-ECD and GC-MS. J Brazil Chem Soc 16(5):1038–1047
Sakamoto M, Tsutsumi T (2004) Applicability of headspace solid-phase microextraction to the determination of multi-class pesticides in waters. J ChromatogrA 1028(1):63–74
Sánchez-Brunete C, Rodriguez A, Tadeo JL (2003) Multiresidue analysis of carbamate pesticides in soil by sonication-assisted extraction in small columns and liquid chromatography. J Chromatogr A 1007:85
Santalad A, Zhou L, Shang F, Fitzpatrick D, Burakham R, Srijaranai S, Glennon JD, Luong JHT (2010) Micellar electrokinetic chromatography with amperometric detection and off-line solid-phase extraction for analysis of carbamate insecticides. J Chromatogr A 1217(32):5288–5297
Sharma KK (2007) Pesticide residue analysis. In: Pesticide residue analysis manual, 1st edn, pp 8–10
Shen Z, Cai J, Gao Y, Zhu X, Su Q (2005) A new matrix solid phase dispersion-accelerate solvent extraction-gas chromatographic method for determination of organochlorine pesticides residues in soil. Chin J Anal Chem 33(9):1318–1320
Shen X, Cai J, Gao Y, Su Q (2006) Determination of organophosphorus pesticides in soil by MMSPD-GC-NPD and confirmation by GC-MS. Chromatographia 64(1–2):71–77
Shen X, Su Q, Zhu X, Gao Y (2007) Determination of pesticide residues in soil by modified matrix solid-phase dispersion and gas chromatography. Ann Chim 97(8):647–654
Shi C, Gui W, Chen J, Zhu G (2010) Determination of oxadiargyl residues in environmental samples and rice samples. Bull Environ Contam Toxicol 84(2):236–239
Simplício A, Boas L (1999) Validation of a solid-phase microextraction method for the determination of organophosphorus pesticides in fruits and fruit juice. J Chromatogr A 833(1):35–42
Singh B (2000) Significance of clean-up in residue analysis. In: Yadav PR, Kathpal TS, Rohilla HR (eds) Pesticide residue analysis. Department of Entomology, CCSHAU, Hisar, pp 22–33
Smith RM (2002) Extractions with superheated water. J Chromatogr A 975(1):31–46
Stout SJ, Da Cunha AR, Safarpour MM (1997) Simplified determination of Imidazolinone herbicides in soil at parts-per billion level by liquid chromatography electrospray ionization tandem mass spectrometry. J AOAC Int 80(2):426–432
Stout SJ, Babbitt BW, Da Cunha AR, Safarpour MM (1998) Microwave-assisted extraction coupled with gas chromatography with nitrogen-phosphorus detection or electron capture negative chemical ionization mass spectrometry for determination of dimethomorph residues in soil. J AOAC Int 81(5):1054–1059
Sun L, Lee HK (2003) Optimization of microwave-assisted extraction and supercritical fluid extraction of carbamate pesticides in soil by experimental design methodology. J Chromatogr A 1014(1–2):165–177
Tao S, Guo LQ, Wang XJ, Liu WX, Ju TZ, Dawson R, Cao J, Xu FL, Li BG (2004) Use of sequential ASE extraction to evaluate the bioavailability of DDT and its metabolites to wheat roots in soils with various organic carbon contents. Sci Total Environ 320(1):1–9
Tu X, Chen W (2018) A review on the recent progress in matrix solid phase dispersion. Molecules 23(11):2767
Vega Moreno D, Sosa Ferrera Z, Santana Rodriguez JJ (2006) Sample extraction method combining micellar extraction−SPME and HPLC for the determination of organochlorine pesticides in agricultural soils. J Agric Food Chem 54(20):7747–7752
Wang W, Meng B, Lu X, Liu Y, Tao S (2007) Extraction of polycyclic aromatic hydrocarbons and organochlorine pesticides from soils: a comparison between Soxhlet extraction, microwave-assisted extraction and accelerated solvent extraction techniques. Anal Chim Acta 602(2):211–222
Wang H, Ding J, Ren N (2016) Recent advances in microwave-assisted extraction of trace organic pollutants from food and environmental samples. Trends Anal Chem 75:197–208
Yang XB, Ying GG, Kookana RS (2010) Rapid multiresidue determination for currently used pesticides in agricultural drainage waters and soils using gas chromatography-mass spectrometry. J Environ Sci Health B 45(2):152–161
Zhang JS, Pan FD, Cheng H (2010) Determination of organophosphorus pesticide in soil by accelerated solvent extraction-gas chromagraphy/mass spectrometry 2010 4th International conference on bioinformatics and biomedical engineering (iCBBE 2010), art. no. 5517391
Zhang HF, Yang XH, Wang Y (2011) Microwave assisted extraction of secondary metabolites from plants: current status and future directions. Trends Food Sci Tech 22:672–688
Zhao R, Wang X, Fu S, Yuan J, Jiang T, Xu X (2006) A novel headspace solid-phase microextraction method for the exact determination of Organochlorine pesticides in environmental soil samples. Anal Bioanal Chem 384(7–8):1584–1589
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Mondal, P. (2020). Modern Sample Preparation Techniques for Pesticide Residues Analysis in Soil. In: Rakshit, A., Ghosh, S., Chakraborty, S., Philip, V., Datta, A. (eds) Soil Analysis: Recent Trends and Applications. Springer, Singapore. https://doi.org/10.1007/978-981-15-2039-6_10
Download citation
DOI: https://doi.org/10.1007/978-981-15-2039-6_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-2038-9
Online ISBN: 978-981-15-2039-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)