
Abstract
Biofilm-based bacterial infections are a significant health threat due to their chronic nature and lack of susceptibility to both the host immune response and to treatment with conventional antibiotics. There are numerous complex and interrelated mechanisms underling this tolerance, and strategies to overcome them are required in order to combat the considerable threat posed by biofilm-based bacterial infections. Several such strategies that have been explored toward the eradication of biofilm-based bacterial infections are discussed in this chapter. One strategy involves developing new antibiotics that are active against biofilm cells, while other approaches center on enhancing the activity of conventional antibiotics against biofilm cells with compounds that interfere with quorum sensing and other bacterial signaling and communication pathways, target biofilm-specific genes, or target the biofilm matrix.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Biofilm formation is often considered to be a significant factor in the failure of bacterial infections to respond to antibiotic treatment, with an estimated 65–80% of all infections thought to be biofilm-related (Van Acker et al. 2014). Biofilm-based infections are recognized as a significant health threat, with an estimated 17 million new biofilm infections arising each year in the U.S., resulting in up to 550,000 fatalities (Quave et al. 2012). This has led the CDC to declare that biofilms are one of the most pressing clinical obstacles of this century (Donlan 2002; Davies 2003). A significant economic burden is placed upon healthcare systems both as a result of protracted hospital stays and increased fatalities, and also due to persistent colonization of hospital facilities by bacteria within a biofilm state (Smith and Hunter 2008).
The discovery and development of therapeutic agents that are active against biofilm-based bacteria is of vital importance. In this chapter, we discuss the various strategies that have been explored toward the development of therapeutics for the eradication of biofilm-based infections, and give examples of compounds that act via these strategies. We do not cover empirical screening for compounds with anti-biofilm activity, which has been extensively reviewed elsewhere (Worthington et al. 2012; Rabin et al. 2015).
1.1 Overview of Bacterial Biofilms
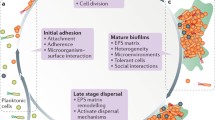
The development of strategies to eradicate biofilm-based infections requires an understanding of many aspects of the biofilm, including structure, the biofilm life cycle, matrix composition, and the signaling and regulatory networks that mediate the biofilm lifestyle. Bacterial biofilms are highly complex surface-associated communities comprising microcolonies of bacterial cells that are embedded in a matrix of extracellular polymeric substances (EPS) (see below) and separated from one another by interstitial voids that act as channels for the transport of water, nutrients, and waste (Donlan 2002). Biofilms are pervasive, occurring across almost all bacterial species and across many environmental settings, including within the human body in both normal and pathogenic processes. Biofilm formation is an adaptive response that is initiated in response to environmental triggers such as nutrient depletion, pH change, and the presence of antibiotics, to enhance survival under these conditions (Webb et al. 2003; Jefferson 2004). Biofilms are heterogeneous and highly structured, and although each biofilm is unique and dependent upon the constituent microorganisms and specific environment, some structural features are common.
The highly structured and cooperative nature of biofilms has led them to be likened to multicellular organisms (Nikolaev Iu and Plakunov 2007). This multicellular existence confers distinct advantages to the constituent cells that include better access to resources, improved ability to colonize new territories, increased survival in intermicrobial conflicts, and a markedly increased tolerance to antimicrobial agents and the host immune response (Lyons and Kolter 2015). Additionally, multicellularity allows for specialization of different cells, analogous to cellular differentiation seen in multicellular organisms (Jefferson 2004). This differentiation allows a division of labor among cells and optimizes population survival. It has even been reported that there exists an altruistic component to biofilm behavior, one theory being that programmed cell death of a subpopulation of the biofilm is activated to benefit surviving cells (Webb et al. 2003), while another theory centers on the adoption of a persister state by a fraction of the biofilm population to enable survival in the occurrence of unfavorable environmental conditions (Lewis 2001).
1.1.1 Biofilm Life Cycle
Biofilms are dynamic entities, both spatially and temporally, and while specific changes occur in response to external and internal processes, the overall lifecycle of a biofilm can be generally described as being made up of five stages that are controlled by a complex developmental cascade of signaling and regulatory molecules. Stage 1 involves the initial attachment of cells to a substratum, mediated by bacterial adhesins (Hancock et al. 2011). During this stage, only a small amount of EPS is present, the cells remain capable of independent movement, and attachment at this stage is thus reversible. A subsequent increase in EPS production marks stage 2, in which cells begin to adhere irreversibly. In stage 3, the biofilm matures and complex architecture such as water channels develops. This architecture continues to develop in stage 4, during which time individual microcolonies may begin to detach to leave hollow remnants that become part of the mature water channels. Finally, stage 5 encompasses dispersion of single cells from the biofilm that become planktonic revertants and are able to colonize additional sites (Stoodley et al. 2002).
1.1.2 Matrix
As mentioned above, bacterial cells within a biofilm are encased in a self-produced matrix. The matrix forms the three-dimensional architecture of the biofilm, provides mechanical stability, mediates adhesion, and allows for transport within the biofilm. The matrix also affords protection against desiccation, ultraviolet radiation, some predators, metallic cations, attack from host immune defenses, and certain biocides and antibiotics (Flemming and Wingender 2010). Water accounts for up to 97% of the biofilm matrix, while the three-dimensional structure of the matrix is afforded by the EPS. Biofilm formation and maintenance is critically dependent upon EPS production, which can account for up to 90% of the dry mass of the biofilm. The composition and therefore the properties of the EPS vary considerably depending on the bacterial species and the environment; however, the major constituent is typically polysaccharides, which are indispensible for biofilm formation in many bacteria. The polysaccharides that comprise the EPS vary in nature and can be anionic, such as the widely studied alginate produced by Pseudomonas aeruginosa, cationic such as the partially deacetylated β-1,6-linked N-acetylglucosamine (PNAG) produced by Staphylococcus aureus, or neutral (Flemming and Wingender 2010). Other macromolecules that constitute the EPS include proteins, extracellular DNA (eDNA), and lipids. Extracellular enzymes in the biofilm matrix have several functions: to act as an external digestive system by degrading biopolymers to low molecular weight compounds that can be taken up by cells and used as energy sources, to effect structural degradation of parts of the EPS to promote detachment of cells from the biofilm, and finally, some extracellular enzymes serve as virulence factors during biofilm-based infections. Non-enzymatic proteins, such as lectins and amyloids, play a role in the formation and stabilization of the matrix, forming a link between bacterial cell surfaces and exopolysaccharides, abiotic surfaces and host cells. eDNA is also an integral part of the biofilm matrix, and has roles as an adhesin and as an intercellular connector. Extracellular surfactants produced by some bacteria play a role in attachment, and also function to render hydrophobic materials bioavailable.
Not surprisingly, nutrient availability affects EPS production, with levels increased in response to the availability of excess carbon and to low levels of other nutrients such as nitrogen, potassium, or phosphate. Slow growth rates also enhance EPS production (Sutherland 2001). The immobilization of cells within the biofilm matrix keeps them proximal to one another and facilitates cell—cell communication (see below) and horizontal gene transfer via conjugation, while DNA from lysed cells present in the matrix can also act as a reservoir of genes for horizontal gene transfer.
1.1.3 Signaling and Communication Within a Biofilm
Bacterial biofilm formation and maintenance is mediated by a complex system of signaling and regulatory networks that include quorum sensing (QS), second messenger signaling, indole signaling, and two-component signal transduction systems (TCS).
Perhaps, the best-studied bacterial communication system with respect to biofilm formation and maintenance is QS. QS allows a bacterial community to coordinate gene expression, and therefore behaviors including biofilm formation, in response to changes in population density (Camilli and Bassler 2006). This is achieved by the production, release, and detection of small diffusible signaling molecules known as autoinducers. QS is an extremely complex process, but at a basic level can be described as comprising two primary proteins: a synthase that produces the autoinducer in response to population changes and environmental stresses, and a receptor protein that binds and responds to the autoinducer. The autoinducer must accumulate above a threshold level to effect an alteration in gene expression, thus ensuring that QS-regulated genes are only expressed when the ensuing phenotypes would be most beneficial. Several classes of signaling molecules exist, some of which are species specific and some of which are universal signaling molecules produced by, and regulating behaviors of, multiple bacterial species.
Gram-negative bacteria utilize a class of signaling molecules known as acyl homoserine lactones (AHLs). In P. aeruginosa, which has become a model organism for QS research, two major QS systems involved in biofilm regulation include the related las and rhl systems, which employ 3-oxo-C12-AHL 1 and N-butyrylhomoserine lactone (C4-AHL) 2 (Fig. 1), respectively, as signaling molecules (Parsek and Greenberg 2000). These systems influence biofilm formation in multiple ways including rhamnolipid production, which is required for maintaining the open spaces between biofilm aggregates, and also affects swarming, which is important in biofilm development. Additionally, these systems regulate production of lectins and siderophores that are important for biofilm formation. Another QS system that plays a role in biofilm regulation in P. aeruginosa is the PQS system, which utilizes 2-alkyl-4-quinolones (AQs), including 3 as signaling molecules, and regulates production of the matrix component eDNA, which is important in generating the initial scaffold of the biofilm (Passos da Silva et al. 2017).

Structures of compounds used by bacteria for signaling and communication
In Gram-positive bacteria, the predominant QS systems utilize autoinducing peptides (AIPs) as signaling molecules. S. aureus produces at least four classes of AIPs, including 4 (Fig. 1) (Baldry et al. 2016), which, upon reaching a critical concentration, activate the Agr QS system (Malone et al. 2007). This system plays a role in biofilm regulation by driving expression of a small non-coding RNA (RNA-III) upon binding of the AIP to the histidine kinase AgrC. RNA-III downregulates the expression of surface adhesins including protein A and the fibronectin-binding protein (Yarwood et al. 2004), and repression of the Agr system is necessary for biofilm formation to occur, while reactivation of the system in established biofilms has been shown to trigger biofilm dispersal (Boles and Horswill 2008).
A third class of autoinducer molecules is the autoinducer-2 (AI-2) class, which is employed by both Gram-negative and Gram-positive bacteria, and as such has been termed the universal QS system. AI-2 molecules are derived from a common precursor; (S)-4,5-dihydroxy-2,3-pentanedione (DPD) 5 (Fig. 1), which is a by-product of the activated methyl cycle and is synthesized by LuxS during the conversion of S-ribosylhomocysteine to homocysteine (Vendeville et al. 2005). AI-2 molecules are cyclic derivatives of DPD that are generated spontaneously and vary between bacterial species, such as the Vibrio harveyi AI-2 6 and the Salmonella typhimurium AI-2 7. In Escherichia coli, AI-2 is phosphorylated by LsrK following uptake into the cell, and the phospho-AI-2 binds the repressor LsrR. Upon sensing AI-2, LsrK and LsrR alter the expression of genes involved in the regulation of aggregation, attachment, and biofilm formation (Jani et al. 2017).
In addition to QS systems, other signaling pathways also play a role in the complex process of biofilm regulation. The ubiquitous second messenger bis-(3′5’)-cyclic di-guanylic acid (c-di-GMP) 8 (Fig. 1) is synthesized and metabolized by diguanylate cyclases (DGCs) and phosphodiesterases (PDEs), respectively, in response to both environmental and intercellular signals (Yan and Chen 2010). c-Di-GMP allosterically regulates downstream proteins, known as effectors, which respond by regulating phenotype changes and cellular functions, one of which is the transition from a planktonic to a biofilm state. For example, in Pseudomonas fluorescens Pf0–1, when cellular levels of c-di-GMP are high, the messenger binds the effector LapD, which is a transmembrane protein involved in the export of the adhesin LapA to the outer membrane. Allosteric changes in LapD upon binding of c-di-GMP result in a conformation that allows export of LapA, and subsequent adoption of a biofilm phenotype, while at low cellular levels of c-di-GMP, during which the messenger is not bound, LapA is not exported and the bacteria remain in a planktonic state (Yan and Chen 2010).
Another proposed class of universal signaling molecules that play a role in numerous bacterial behaviors including biofilm regulation is the indole class (Melander et al. 2014). Indole itself 9 (Fig. 1) is known to be produced by at least 85 bacterial species, and even species that are not themselves indole producers have been shown to respond to the presence of indole (Lee and Lee 2010). Indole has been reported to affect biofilm formation in E. coli by inducing the transcription regulator SdiA, which in turn leads to repression of motility and biofilm formation (Lee et al. 2007b).
Finally, TCS also mediate biofilm formation and regulation across many bacterial species. TCS are regulatory systems found predominantly in prokaryotes that allow the organism to sense and respond to changes in their environment, and regulate many bacterial phenotypes. TCS consist of a sensor histidine kinase and a DNA-binding response regulator. In response to an extracellular stimulus, the histidine kinase undergoes autophosphorylation and in turn transfers the phosphate group to a conserved aspartate residue on the cognate response regulator. This induces a conformational change that results in up- or downregulation of gene expression, leading to changes in biofilm formation (Worthington et al. 2013).
1.1.4 Role of Biofilms in Chronic Infections and Tolerance to Antibiotics and the Host Immune Response
Biofilms are implicated in many chronic infections, and these infections are often characterized by persisting and progressive pathology (Hoiby et al. 2015). Biofilm-based infections include lung infections in cystic fibrosis (CF) patients, chronic otitis media, chronic wound infections, bacterial endocarditis, tooth osteomyelitis, and infections of indwelling medical devices (IMDs).
The chronic nature of biofilm-based infections is in part due to their ability to evade the host immune response. Furthermore, cells residing within a biofilm exhibit high levels of antibiotic tolerance, which renders antibiotics ineffective even against bacterial strains that do not harbor genetic resistance determinants. Tolerance, by definition, involves a reduction in the rate of antibiotic-induced killing of a whole bacterial population by comparison with the behavior of cultures of the strain from which the tolerant strain was derived (Tuomanen et al. 1986). In contrast to acute infections caused by planktonic bacteria, which (outside of drug-resistant strains) can typically be successfully treated with antibiotics, biofilm-based infections are often recalcitrant to antibiotic treatment, exacerbating the development into a chronic state. Importantly, cells exhibiting antibiotic tolerance as a result of the adoption of a biofilm phenotype will exhibit susceptibility to the antibiotic upon dissociation from the biofilm (Bjarnsholt 2013). Both tolerance and genetic resistance mechanisms contribute to the ability of bacteria within a biofilm to withstand antimicrobial challenge.
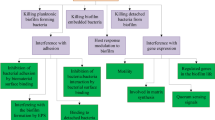
The impervious nature of biofilms can be attributed to numerous complex and interrelated mechanisms (Fig. 2), and an in-depth review of these mechanisms was recently published by Hall and Mah (2017). Understanding the mechanisms that govern antibiotic tolerance in biofilms is necessary for the development of strategies to overcome them, which may ultimately provide a means to eradicate biofilm-based infections.

Mechanisms of antibiotic tolerance within a biofilm
One important factor is the matrix, which contributes to the tolerance of bacterial cells within a biofilm to antibiotics and immune effectors, primarily by limiting access to the bacterial cells. This may be a physical effect whereby the antimicrobial is unable to diffuse through the EPS, or may be a result of degradation or inactivation of the antibacterial agent within the matrix (Van Acker et al. 2014). For example, it has been shown that the penetration of ciprofloxacin and tobramycin is significantly impeded in P. aeruginosa biofilms, while Klebsiella pneumoniae has been shown to secrete a ß-lactamase into the matrix that degrades ampicillin before it is able to reach the cells (Hall and Mah 2017).
As mentioned earlier, biofilms are heterogeneous entities with nutrient and oxygen levels varying in different areas of the biofilm, which leads to altered bacterial growth rates in different areas of the biofilm (Mah and O’Toole 2001). Cells in nutrient and oxygen poor areas exhibit reduced metabolic activity and exist in a stationary phase-like state, leading to decreased susceptibility to most conventional antibiotics that predominantly target one of five biosynthetic processes that occur only in actively growing bacteria: the biosynthesis of proteins, RNA, DNA, peptidoglycan, and folic acid (Hurdle et al. 2011). Most antibiotics that effectively kill or inhibit the growth of dividing bacterial cells tend to be very inefficient at killing non-multiplying bacteria and are therefore inactive against biofilm cells (Coates and Hu 2008).
Biofilm cells exhibit significantly altered gene expression profiles, both compared to planktonic cells and as a function of their location within the biofilm. For example, a comparison of the transcriptome profiles of planktonic and biofilm cells of Acinetobacter baumannii ATCC 17978 showed that 1621 genes were overexpressed in biofilm cells relative to stationary phase planktonic cells, and 55 genes were expressed only in biofilm cells (Rumbo-Feal et al. 2013). Some of these changes directly contribute to antibiotic tolerance in addition to having biofilm-specific functions; for example, general efflux pump expression is often upregulated in biofilm cells, and it has been postulated that functional efflux systems are required for full biofilm formation, possibly due to their role in effecting waste removal under cramped biofilm conditions (Van Acker and Coenye 2016). Indeed, inactivation of any of the efflux systems of Salmonella enterica serovar Typhimurium has been shown to result in the inability of the bacterium to form a biofilm (Baugh et al. 2012). Some antibiotic-specific efflux systems are also upregulated in biofilm cells, for example, in P. aeruginosa, the MexCD-OprJ and MexAB-OprM efflux pumps are thought to be biofilm-specific defense mechanisms against azithromycin and colistin, respectively (Van Acker and Coenye 2016).
Horizontal gene transfer is a major driver in bacterial evolution and rapid adaptation, and the transfer of plasmids by conjugation is one of the best-understood mechanisms for dissemination of genetic information (Davey and O’Toole 2000). Many antibiotic resistance determinants are spread by horizontal gene transfer (Barlow 2009), and the high population density and proximal nature of cells within a biofilm provide a favorable environment for the efficient transfer of genetic material, with rates of 1000–16,000-fold higher than those found in planktonic cells reported(Hausner and Wuertz 1999; Savage et al. 2013), though rates have been shown to be dependent on location within the biofilm and rate of cell growth (Stalder and Top 2016). Thus, facile transfer of antibiotic resistance genes in biofilms is another mechanism contributing to the lack of efficacy of antibiotics against biofilm cells.
Another contributory factor to antibiotic recalcitrance within a biofilm is the phenomenon of hypermutation. Hypermutable strains mutate at higher rates than the general population, and environmental stresses such as antibiotic challenge select for (and in some cases promote the generation of) these strains (Blazquez 2003). The production of reactive oxygen and nitrogen intermediates, formed as a result of the vast oxygen and nutrient gradients found with biofilm microcolonies, and the presence of oxidative stress in these environments have been linked to the occurrence of hypermutation in P. aeruginosa (Conibear et al. 2009). One study that examined 128 P. aeruginosa isolates from 30 cystic fibrosis patients reported that 36% of patients were colonized by a hypermutable strain that persisted for years in most patients, while such strains were not present in non-cystic fibrosis patients presenting with acute P. aeruginosa infections (Oliver et al. 2000).
It has also been suggested that biofilm cells exhibit a greater tolerance to oxidative stress, such as the antibiotic-induced generation of reactive oxygen species (ROS), which have been hypothesized to contribute to the killing of bacterial cells by bactericidal antibiotics. The molecular basis of this increased tolerance is not yet fully understood, but enzymes such as the catalase KatA, which detoxifies hydrogen peroxide, are thought to play a role, and it has been shown that KatA expression is higher under anaerobic conditions (Su et al. 2014) such as those experienced by cells located in oxygen-limited areas of a biofilm. Another way in which biofilm cells combat oxidative stress is via the production of polyamines that reduce intracellular ROS levels, a process controlled by two major transcriptional regulators, OxyR and SoxRS (Slachmuylders et al. 2018).
The stringent response is also thought to contribute to antibiotic tolerance in biofilms. The stringent response is a signaling pathway controlled by the alarmone (p)ppGpp, a second messenger that is activated under nutrient-limiting conditions, as experienced by cells in certain areas of a biofilm, and involves a comprehensive restructuring of the metabolic gene expression profile from one that facilitates growth to one allowing for prolonged survival in the stationary phase (Traxler et al. 2008). It has been shown that nutrient supplementation increases the antibiotic susceptibility of biofilm cells and that biofilms formed by a P. aeruginosa mutant deficient in (p)ppGpp exhibit significantly increased susceptibility to several antibiotics with diverse mechanisms of action compared to wild-type biofilms (Hall and Mah 2017).
Finally, the presence of persister cells has also been posited to contribute to antibiotic tolerance in biofilms and the chronic nature of biofilm-based infections. Persister cells comprise a subpopulation of cells (about 1% in biofilms) that are genetically homogeneous to the rest of the population and appear susceptible to antibiotic exposure, yet are phenotypically heterogeneous and are able to withstand the antibiotic, ultimately ensuring the survival of the population (Poole 2012). The mechanisms of persister cell formation and tolerance are not well understood and have been discussed in depth by Wood et al. (Wood et al. 2013) and Conlon et al. (Conlon et al. 2015). It is generally accepted that persister cells arise as a result of entering a state of dormancy in which they become metabolically inactive and thus impervious to the effects of antibiotics.
1.1.5 Current Clinical Approaches to Treating Biofilm-Based Infections
Current clinical approaches for the treatment of biofilm-based infections are limited, and the nature of the treatment is dependent upon the location of the infection, and the causative agent. In the case of colonization of IMDs or other foreign bodies, treatment involves removal of the foreign body, surgical debridement, and aggressive antibiotic therapy (Stewart and Costerton 2001; Wu et al. 2015). When the infection results in the formation of an abscess, which has been likened to a biofilm state (May et al. 2014), drainage of the abscess is necessary. In the absence of a foreign body or abscess, treatment encompasses long-term high-dose antibiotic regimens, often involving combinations of antibiotics with different mechanisms of action. If treatment is not started early when the biofilm is immature, this may only result in a reduction of the biofilm, and subsequent chronic biofilm suppressive treatment will likely be necessary (Wu et al. 2015). In the case of chronic biofilm-based lung infections in CF patients, treatment involves long-term suppressive therapy with both topical (nebulized) and systemic antibiotics; both routes of administration are necessary in order to achieve adequate concentrations in the respiratory and conductive compartments of the lungs (Hoiby et al. 2015).
2 Strategies to Eradicate Biofilms
Alternative strategies to the traditional antibacterial approach of developing compounds that kill or inhibit the growth of metabolically active bacteria are necessary to effectively treat biofilm-based infections. Such strategies include: developing compounds that exhibit bactericidal activity against biofilm cells and the more widely taken approach of developing compounds that counteract biofilm tolerance mechanisms and enhance the activity of conventional antibiotics against the biofilm.
2.1 The Development of Antimicrobial Agents that Kill Biofilm Cells
As described above, most clinically approved antibiotics have limited efficacy against bacteria within a biofilm, and are effective only against bacteria that are actively growing and dividing. The development of antibiotics that are active against bacteria within a biofilm would have a significant impact on the outcome of treating biofilm-based infections.
One class of antimicrobial agents that has demonstrated microbicidal activity against biofilm cells is antimicrobial peptides (AMPs). AMPs are short cationic amphipathic peptides that comprise part of the innate immune response of many eukaryotic and prokaryotic organisms (Reddy et al. 2004). AMPs exhibit broad-spectrum activity against most bacterial pathogens, and importantly some AMPs exhibit activity against metabolically inactive cells, including biofilm cells (Di Luca et al. 2014). AMPs act upon biofilms via multiple mechanisms of action in addition to simply killing, including preventing biofilm formation by targeting the stringent response (Pletzer et al. 2016), which will be discussed later. One example of an AMP that is active against biofilm cells is human ß-defensin 3 (hBD-3) 10 (Fig. 3), which exhibits potent broad-spectrum antibacterial activity and has been shown to kill biofilm cells within a 3-week-old polymicrobial biofilm composed of Actinomyces naeslundii, Lactobacillus salivarius, Streptococcus mutans, and Enterococcus faecalis (Di Luca et al. 2014).

Compounds that kill bacterial biofilm cells
Carolacton 11 (Fig. 3), a secondary metabolite produced by the myxobacterium Sorangium cellulosum, demonstrates selective killing of biofilm cells of S. mutans, exhibiting only minor effects on planktonic cell growth (Kunze et al. 2010). Carolacton affects expression of several TCS including VicKR, which is required for the response to oxidative stress (Banu et al. 2010) likely mediated through the serine/threonine protein kinase PknB (Reck et al. 2011). Similarly, the thiazolidinone 12, which was identified from a structure-based virtual screen (SBVS) of 85,000 potential drug-like molecules for inhibitors of YycG in Staphylococcus epidermidis, also exhibits bactericidal effects on biofilm cells (though this compound is also bactericidal toward planktonic cells) and was shown to bind to YycG and inhibit autophosphorylation in vitro (Qin et al. 2006). A class of halogenated phenazines based upon a marine phenazine antibiotic has been reported to possess antibacterial activity against methicillin-resistant S. aureus (MRSA) biofilm cells, with lead compounds from this class 13 and 14, exhibiting near equipotent killing of biofilm and planktonic cells. Compounds 13 and 14 also eradicated biofilms of other Gram-positive species including methicillin-resistant S. epidermidis (MRSE), and vancomycin-resistant enterococci (VRE), and interestingly some members of this class were also able to kill non-biofilm persister cells of MRSA (Garrison et al. 2015).
The acyldepsipeptide antibiotic (ADEP 4) 15 (Fig. 3), which targets the ClpP protease, is known to be active against growing S. aureus cells (Brotz-Oesterhelt et al. 2005) and has recently been shown to also kill biofilm and persister cells. ADEP4 works by activating ClpP leading it to become a non-specific protease that degrades over 400 intracellular targets, and also removes the ATP requirement for ClpP explaining the activity observed against dormant biofilm and persister cells. ADEP4 effected considerable killing of biofilm cells following 24 hours of treatment, and although the population rebounded after 72 hours, due to the rapid emergence of clpP mutants as a result of the non-essential nature of ClpP in S. aureus, these clpP mutants displayed increased susceptibility to several conventional antibiotics compared to persister or biofilm non-mutant cells, and remarkably a combination of ADEP4 with rifampicin was able to eradicate living cells in a biofilm below the limit of detection (Conlon et al. 2013).
2.2 The Development of Non-antibiotic Compounds that Reduce Biofilm Tolerance to Antibiotics and Immune Response
While the identification of compounds that are able to directly kill biofilm cells is one avenue for the development of therapeutics to eradicate biofilm-based infections, perhaps the more widely explored approach is that of identifying compounds that circumvent the tolerance mechanisms exhibited by bacteria within a biofilm, rendering them susceptible to conventional antibiotics and the host immune response. Such compounds have the potential to be utilized as adjuvants, and co-administered with conventional antibiotics. Several approaches have been explored to this end, including compounds that interfere with the bacterial signaling and communication systems that regulate the biofilm and compounds that degrade the biofilm matrix. One potential advantage of this approach is the fact that compounds that do not elicit direct microbicidal activity will not exert the same pressure on bacteria to evolve resistance as compared to conventional bactericidal therapeutics.
2.2.1 Compounds that Target Bacterial Signaling and Communication
The numerous signaling and communication pathways described earlier that are used by bacteria to regulate biofilm formation and maintenance represent a myriad of potential targets for the development of anti-biofilm therapeutics.
QS Inhibitors
Early approaches in this field centered on the development of QS inhibitors, particularly inhibitors of AHL QS pathways. The use of native AHLs is impeded by their instability and immunomodulatory activity, making them unsuitable for use as a therapeutic (Yates et al. 2002). Several classes of synthetic analogues that are not beset by these issues have been developed as potential anti-biofilm agents, in particular against P. aeruginosa biofilms (Mattmann and Blackwell 2010). The non-native AHL analogues 16 and 17 (Fig. 4) inhibit biofilm formation by P. aeruginosa PAO1 at low micromolar concentrations, by acting as potent antagonists of LasR (Geske et al. 2005), while analogues 18 and 19 inhibit biofilm formation by A. baumannii via antagonism of the LuxR-type receptor, AbaR (Stacy et al. 2012). Taking a slightly different approach, Amara et al. developed isothiocyanate 20, which acts as a covalent inhibitor of LasR in P. aeruginosa, selectively binding Cys 79 in the AHL-binding pocket and inhibiting QS and biofilm formation (Amara et al. 2009). Replacement of the lactone moiety of native AHLs with a thiolactone isostere has led to the identification of a series of analogues including compound 21, which inhibits biofilm formation by P. aeruginosa in vitro and exhibits in vivo activity in a Caenorhabditis elegans P. aeruginosa infection model. (O’Loughlin et al. 2013).

Compounds that exhibit anti-biofilm activity by targeting QS pathways
Non-lactone compounds that target AHL-based QS pathways and interfere with biofilm formation and regulation include the brominated furanone class of marine algae secondary metabolites. The naturally occurring (5Z)-4-bromo-5-(bromomethylene)-3-butyl-2(5H)-furanone 22 (Fig. 4) inhibits biofilm formation by E. coli and Bacillus subtilis (Ren et al. 2001; 2002), while the synthetic furanone C-30 23 exhibits a host of activities, increasing the susceptibility of P. aeruginosa biofilms to tobramycin, promoting clearance in a mouse pulmonary P. aeruginosa infection model (Hentzer et al. 2003), and inhibiting biofilm formation by the Gram-positive Streptococcus intermedius and S. mutans (Lonn-Stensrud et al. 2007). Transcriptome analysis of furanone C-30-treated P. aeruginosa showed that this compound specifically targeted QS systems, while another synthetic furanone 24 was shown to penetrate microcolonies and inhibit QS in P. aeruginosa biofilm cells; compound 24 did not prevent initial attachment, but rather affected biofilm architecture and enhanced bacterial detachment (Hentzer et al. 2002).
In addition to the development of small molecule QS antagonists, enzymatic degradation of native AHLs as a means of inhibiting QS pathways and interfering with biofilm regulation has also been investigated. This is a natural phenomenon known as quorum quenching and is utilized by numerous organisms, either as a means of clearing self-produced QS signals or as a competitive mechanism to degrade QS signals produced by other organisms (Grandclement et al. 2016). One of the first examples of the potential of exploiting quorum quenching as an anti-biofilm strategy was the recombinant production of the hydrolase BpiB05, which had been reported to inhibit AHL activity in the plant pathogens Erwinia carotovora and Agrobacterium tumefaciens, and its inhibition of biofilm formation, motility, and pyocyanin synthesis in P. aeruginosa (Bijtenhoorn et al. 2011). Similarly, the lactonase SsoPox-I, an engineered variant of the Sulfolobus solfataricus, produced SsoPox, inhibits P. aeruginosa QS and biofilm formation in vitro and reduces mortality and histological lung damage in a rat respiratory P. aeruginosa infection model (Hraiech et al. 2014).
Synthetic analogues of the Gram-positive QS signals, AIPs, have also been developed as potential anti-biofilm agents (Gordon et al. 2013). FS3 25 (Fig. 4), an analogue of the S. aureus RNA-III inhibiting peptide (RIP) that acts to inhibit biofilm formation, has been shown to enhance tigecycline efficacy in a rat model of staphylococcal vascular graft infection (Simonetti et al. 2013). The small molecule hamamelitannin 26, which was identified from a virtual screen of an RIP-based pharmacophore against a database of commercially available compounds, has been shown to prevent S. aureus- and S. epidermidis-mediated device-associated infections in vivo (Kiran et al. 2008). Hamamelitannin 26 effects significantly increased killing of S. aureus biofilm cells by several classes of antibiotics, including glycopeptides, ß-lactams, lipopeptides, oxazolidinones, and aminoglycosides, and is thought to act through the TraP receptor, affecting cell wall synthesis and eDNA release (Brackman et al. 2016).
Inhibitors of AI-2 signaling have also shown promise as anti-biofilm agents, and while DPD itself is unstable at high concentrations, several synthetic AI-2 analogues have been investigated for their ability to interfere with AI-2 signaling and subsequently affect biofilm formation (Guo et al. 2013). One example is the acetylated DPD analogue Ac2-DPD 27 (Fig. 4), which affects AI-2-mediated behaviors in Bacillus cereus, including the inhibition of biofilm formation, most likely a result of the release of DPD by in situ hydrolysis (Frezza et al. 2007). C4-Alkoxy-5-hydroxy-2,3-pentanediones 28 and 29 act as AI-2 system agonists and activate the AI-2 pathway in V. harveyi more potently than DPD (Tsuchikama et al. 2012), while isobutyl-DPD 30 acts as an AI-2 system antagonist and has been shown to both inhibit maturation of E. coli biofilms and to achieve near complete biofilm clearance when administered in combination with gentamicin (Roy et al. 2013).
In addition to developing compounds that directly intercept the AI-2 pathway, signaling can also be disrupted by inhibiting biosynthesis of the native AI-2 molecule. The nucleoside analogue 31 is an inhibitor of 5′-methylthioadenosine nucleosidase (MTAN), which is involved in AI-2 biosynthesis and as such inhibits AI-2 production and subsequently biofilm formation in both E. coli and Vibrio cholerae (Gutierrez et al. 2009).
Compounds that Interfere with TCS
Given the ubiquitous nature of TCS among bacteria, their conserved nature across multiple species, the numerous behaviors they regulate, and the lack of an analogous system in eukaryotes, they represent an appealing drug target and are attracting increasing attention for the development of anti-biofilm compounds (Worthington et al. 2013). As mentioned above, some compounds that display bactericidal activity against biofilm cells have been shown to target TCS; in contrast, other compounds described below do not individually kill biofilm cells, but enhance susceptibility to conventional antibiotics. Early efforts at exploiting TCS, both for anti-biofilm and other applications, centered on compounds that target the histidine kinase; for example, walkmycin C 32 (Fig. 5) is a histidine kinase inhibitor isolated from Streptomyces sp. strain MK632-100F11. Walkmycin C inhibits the autophosphorylation activity of the S. mutans kinases VicK and CiaH, which play a role in sucrose-dependent biofilm formation and cause the formation of abnormal biofilms (Qi et al. 2004; Eguchi et al. 2011).

Compounds that exhibit anti-biofilm activity by targeting TCS
Alternatively, compounds that target the response regulator have been investigated as a means of controlling biofilm formation and maintenance. This approach confers some advantages over targeting the histidine kinase, predominantly the avoidance of issues with cross talk, in which a response regulator can be phosphorylated by non-cognate histidine kinases, and as such inhibition of the histidine kinase may not result in complete inhibition of function of the targeted TCS. Directly targeting the response regulator increases the likelihood of interference with only the TCS of interest. The 2-aminoimidazole (2-AI) class of small molecules, which are derived from the marine sponge alkaloids oroidin and bromoageliferin (Melander et al. 2016), are potent broad-spectrum anti-biofilm compounds, some of which have been shown to target response regulators. 2-AI 33 (Fig. 5), which belongs to the reverse amide class of 2-AI compounds, inhibits and disperse biofilms of both P. aeruginosa and A. baumannii (Ballard et al. 2008) and has been shown to bind the response regulator BfmR, which plays an important role in biofilm formation (Tomaras et al. 2008) in A. baumannii (Thompson et al. 2012). A series of aryl 2-AI compounds including 34 and 35 were also shown to bind both BfmR and the Francisella spp. response regulator QseB and to inhibit biofilm formation by both species, with a correlation between inhibition and binding observed across the series (Milton et al. 2018). Another 2-AI 36 inhibits biofilm formation by S. mutans and inhibits accumulation of Porphyromonas gingivalis on a substratum of Streptococcus gordonii, and downregulates the histidine kinase comD in S. mutans (Liu et al. 2014; Wright et al. 2014). The 2-aminoimidazole triazole compound 37 inhibits and disperses biofilms by a broad spectrum of bacteria including P. aeruginosa, A. baumannii, and S. aureus (Rogers and Melander 2008) and also exhibits synergy with novobiocin against S. aureus and S. epidermidis, with tobramycin against P. aeruginosa and with colistin against A. baumannii biofilms (Rogers et al. 2010). The related triazole-containing compound 38 inhibits biofilm formation by S. mutans and reduces bacterial colonization and the incidence of dental caries incidence in vivo in a rat infection model (Pan et al. 2015).
Compounds that Interfere with Other Signaling and Stress Response Systems
Compounds that interfere with signaling by second messengers such as c-di-GMP have also been explored to control biofilm formation. Examples include sulfathiazole 39 (Fig. 6), which acts as a DGC inhibitor and inhibits E. coli biofilm formation at low micromolar concentrations (Antoniani et al. 2010) and benzimidazole 40, which has broad-spectrum anti-biofilm activity, including against P. aeruginosa, K. pneumoniae, Shigella boydii, S. aureus, and several other species (Sambanthamoorthy et al. 2011). The organoselenium compounds ebselen 41 and ebselen oxide 42 reduce DGC activity by covalently modifying cysteine residues within the protein, and have been shown to inhibit biofilm formation by P. aeruginosa (Lieberman et al. 2014). Benzamide 43 was identified from a high-throughput screen for inhibitors of DGC enzymes and was shown to reduce c-di-GMP levels in V. cholerae and to inhibit biofilm formation by both V. cholerae and P. aeruginosa (Sambanthamoorthy et al. 2012).

Compounds that exhibit anti-biofilm activity by targeting other signaling and stress response systems
Another second messenger, the alarmone (p)ppGpp, which plays a role in the stringent response as described earlier, has also been targeted as an anti-biofilm strategy. The immunomodulatory peptide IDR (innate defense regulator) 1018 44 (Fig. 6), which exhibits broad-spectrum activity, preventing biofilm formation and eradicating pre-formed biofilms of P. aeruginosa, E. coli, A. baumannii, K. pneumoniae, MRSA, and several other species (Mansour et al. 2015), exhibits synergy with ceftazidime against biofilms of A. baumannii, S. enterica, and MRSA, and with tobramycin against biofilms of E. coli, A. baumannii, and K. pneumoniae (Reffuveille et al. 2014), and is thought to bind to (p)ppGpp causing its degradation.
Exogenous indole has been used to perturb various aspects of indole signaling-mediated behavior, and various naturally occurring indole metabolites and derivatives have also been shown to influence bacterial behaviors including biofilm formation. For example, the oxidized metabolite 7-hydroxyindole 45 (Fig. 6) inhibits biofilm formation by enterohemorrhagic E. coli (EHEC) to a greater degree than indole at the same concentration (Lee et al. 2007a) and the plant metabolite indole-3-acetaldehyde, 46, which is produced by the plant pathogen Rhodococcus sp. BFI 332, inhibits biofilm formation by EHEC (Wood et al. 2008). Other indole-derived plant metabolites that affect biofilm formation include 3-indolylacetonitrile (IAN) 47 and indole-3-carboxyaldehyde (I3CA) 48, which inhibit biofilm formation by E. coli O157:H7 to a greater extent than indole and also weakly inhibit biofilm formation by P. aeruginosa, a phenotype that is enhanced by indole (Lee et al. 2011).
Numerous synthetic indole derivatives have been investigated in efforts to augment biological activity; these include simple synthetic derivatives such as 7-fluoroindole (7FI) 49 and 7-formylindole 50, both of which inhibit biofilm formation by P. aeruginosa (Lee et al. 2012). These simple derivatives, along with indole itself and naturally occurring metabolites, however require high concentrations that are too high to be therapeutically useful, often up to 1 mM, in order to exert their effects. This has led to the construction of complex synthetic indole-containing compounds that act at much lower concentrations. The indole-containing compound 51 was reported to inhibit biofilm formation by S. enterica serovar Typhimurium, and a subsequent structure–activity relationship study led to the discovery of the more active 8-fluoro-4-[4-(3-phenyl-2-propen-1-yl)-1-piperazinyl]-5H-pyrimido[5,4-b] indole 52, which inhibits biofilm formation at low micromolar concentrations (Robijns et al. 2012). Synthetic indole-containing compounds derived from the bryozan secondary metabolite desformylflustrabromine (dFBr) 53 (Peters et al. 2003) include compounds 54 and 55, which potently inhibit biofilm formation by E. coli and S. aureus. Their activity in E. coli was shown to be dependent on the same factors as the activity of indole itself, i.e., temperature, SdiA, and tryptophanase (TnaA), suggesting that these compounds modulate bacterial behavior through the indole-signaling pathway (Bunders et al. 2011b; Minvielle et al. 2013b). Related flustramine-derived small molecules that have been reported to inhibit biofilm formation by various bacterial species include compound 56, which inhibits biofilm formation by A. baumannii, compound 57, which inhibits biofilm formation by E. coli, and the Gram-positive acting compounds 58 and 59, which inhibit biofilm formation by S. aureus (Bunders et al. 2011a; Minvielle et al. 2013a).
The flavonoid baicalin hydrate 60 (Fig. 6) is known to increase the susceptibility of Burkholderia cenocepacia biofilms to tobramycin and was initially thought to act via QS inhibition (Brackman et al. 2011). Recently, however, it has been demonstrated that the mechanism of action of this compound involves modulation of the oxidative stress response, whereby baicalin hydrate affects multiple pathways including oxidative phosphorylation, glucarate metabolism and modulates biosynthesis of the polyamine putrescine. This leads to increased ROS production in the presence of tobramycin and a subsequent increase in tobramycin-mediated killing of biofilm cells (Slachmuylders et al. 2018).
2.2.2 Targeting the Biofilm Matrix
As discussed earlier, the matrix is vital to numerous aspects of biofilm integrity, and as such targeting the matrix has received much attention as a means to eradicate biofilms and treat biofilm-based infections. This has been predominantly pursued by the use of enzymes such as glycosidases, proteases, and DNases that degrade the major constituents of the matrix; indeed, the endogenous secretion of such enzymes is an innate phenomenon used by many bacteria to initiate dispersion from the biofilm (Kaplan 2010).
It has been demonstrated that biofilms formed in the presence of DNases exhibit reduced biomass and decreased antibiotic tolerance (Tetz and Tetz 2010), and recombinant human DNase I (rhDNase), also known as dornase alfa and marketed as Pulmozyme by Genentech, inhibits and disperses S. aureus biofilms in vitro, increases the susceptibility of S. aureus biofilm cells to several antimicrobials, and enhances the efficacy of tobramycin in S. aureus-infected C. elegans (Kaplan et al. 2012). Pulmozyme also increases the susceptibility of P. aeruginosa biofilm cells to aminoglycosides (Alipour et al. 2009; Kaplan et al. 2012) and is marketed for the treatment of pulmonary disease in cystic fibrosis (CF) patients, (Parsiegla et al. 2012) in whom it leads to reduced demand for antibiotics and improved lung function (Frederiksen et al. 2006). Other DNases that exhibit anti-biofilm activity include NucB, which is an extracellular DNase produced by Bacillus licheniformis that has been shown to cause rapid biofilm dispersal against a range of bacteria including B. subtilis and E. coli (Bayer et al. 1992).
Dispersin B is a soluble glycoside hydrolase that degrades poly-N-acetylglucosamine (PGA), which is a major matrix component of several bacterial biofilms (Ramasubbu et al. 2005). Dispersin B inhibits and disperses biofilms by several medically relevant bacterial species including S. aureus, S. epidermidis, and E. coli and also sensitizes S. epidermidis biofilm cells to the action of various antimicrobials and lowers the rate of catheter colonization by S. aureus in combination with triclosan in a rabbit model of infection (Kaplan et al. 2004; Itoh et al. 2005; Donelli et al. 2007; Izano et al. 2008; Darouiche et al. 2009). Alginate lyases catalyze the degradation of the matrix component alginate and have been shown to enhance the microbicidal activity of aminoglycosides against P. aeruginosa biofilms in vitro (Lamppa and Griswold 2013) and to augment the effectiveness of amikacin in clearing P. aeruginosa in a rabbit model of endocarditis (Bayer et al. 1992).
Proteases that have been investigated for this approach include the serine protease Esp from S. epidermidis, which inhibits and eradicates preformed biofilms of S. aureus, enhances the susceptibility of S. aureus biofilms to hBD2, and eliminates human nasal colonization by S. aureus in vivo (Iwase et al. 2010). Similarly, the metalloprotease serratopeptidase (SPEP), a widely used anti-inflammatory therapeutic, enhances the activity of ofloxacin against biofilms of P. aeruginosa and S. epidermidis (Selan et al. 1993).
Non-enzymatic approaches to targeting components of the biofilm matrix as a means of eradicating biofilms have also been investigated. A screen of multivalent fucosyl-peptide dendrimers for the inhibition of binding of the P. aeruginosa lectin LecB led to the identification of FD2 (C-Fuc-LysProLeu)4(LysPheLysIle)2LysHisIleNH2, which was subsequently shown to inhibit P. aeruginosa biofilm formation and to induce complete dispersion of established biofilms in several P. aeruginosa clinical isolates (Johansson et al. 2008).
2.2.3 Compounds that Target Biofilm-Upregulated Genes
Efflux Pump Inhibitors
As mentioned earlier, efflux pump genes are often upregulated in biofilm cells, and it is thought that functional efflux systems are required for full biofilm formation, leading to investigation into the effect of efflux pump inhibitors on biofilm cells both in the absence and presence of antibiotics. The efflux inhibitors chlorpromazine 61, cyanide 3-chlorophenylhydrazone (CCCP) 62, and PAßN 63 (Fig. 7), all of which have different mechanisms of efflux inhibition, effectively prevent biofilm formation in E. coli, P. aeruginosa, and S. aureus (Baugh et al. 2014). Similarly, PaβN 63, thioridazine 64, and 1-(1-naphthylmethyl)-piperazine (NMP) 65 significantly repress biofilm formation by E. coli and K. pneumoniae (Van Acker and Coenye 2016). Finally, boeravinone B 66 (Fig. 7), an inhibitor of the S. aureus multidrug efflux pump NorA, was recently shown to act synergistically with ciprofloxacin to inhibit biofilm formation by S. aureus (Singh et al. 2017).

Efflux pump inhibitors as anti-biofilm compounds
3 Conclusions
Biofilm-based bacterial infections are inherently unresponsive to the host immune response, and are highly tolerant to conventional antibiotic regimens, rendering them a significant health threat. Multiple mechanisms govern this tolerance, and compounds that overcome or circumvent these mechanisms and eradicate the biofilm have the potential to lead to the development of sorely needed therapeutics for such infections. In addition to the development of compounds that directly kill biofilm cells, an adjuvant approach whereby conventional antibiotics are paired with compounds that potentiate their activity against biofilm cells to eradicate the biofilm has received much attention. Compounds that interfere with various aspects of biofilm regulation and antibiotic tolerance have the potential to be used in this manner. These include: compounds that interfere with bacterial signaling and communication pathways such as QS and TCS, compounds that target stress response pathways, compounds that target efflux pumps, and agents that act upon the biofilm matrix. Several promising examples are discussed in this chapter; however, much work is still needed to develop new strategies and therapeutics to tackle the considerable problem of biofilm-based bacterial infections.
References
Alipour, M., Suntres, Z. E., & Omri, A. (2009). Importance of DNase and alginate lyase for enhancing free and liposome encapsulated aminoglycoside activity against Pseudomonas aeruginosa. The Journal of Antimicrobial Chemotherapy, 64(2), 317–325.
Amara, N., Mashiach, R., Amar, D., Krief, P., Spieser, S. A., Bottomley, M. J., Aharoni, A., & Meijler, M. M. (2009). Covalent inhibition of bacterial quorum sensing. Journal of the American Chemical Society, 131(30), 10610–10619.
Antoniani, D., Bocci, P., Maciag, A., Raffaelli, N., & Landini, P. (2010). Monitoring of diguanylate cyclase activity and of cyclic-di-GMP biosynthesis by whole-cell assays suitable for high-throughput screening of biofilm inhibitors. Applied Microbiology and Biotechnology, 85(4), 1095–1104.
Baldry, M., Kitir, B., Frokiaer, H., Christensen, S. B., Taverne, N., Meijerink, M., Franzyk, H., Olsen, C. A., Wells, J. M., & Ingmer, H. (2016). The agr inhibitors solonamide B and analogues alter immune responses to Staphylococccus aureus but do not exhibit adverse effects on immune cell functions. PLoS One, 11(1), e0145618.
Ballard, T. E., Richards, J. J., Wolfe, A. L., & Melander, C. (2008). Synthesis and antibiofilm activity of a second-generation reverse-amide Oroidin library: A structure-activity relationship study. Chemistry—a European Journal, 14(34), 10745–10761.
Banu, L. D., Conrads, G., Rehrauer, H., Hussain, H., Allan, E., & van der Ploeg, J. R. (2010). The Streptococcus mutans serine/threonine kinase, PknB, regulates competence development, bacteriocin production, and cell wall metabolism. Infection and Immunity, 78(5), 2209–2220.
Barlow, M. (2009). What antimicrobial resistance has taught us about horizontal gene transfer. Methods in Molecular Biology, 532, 397–411.
Baugh, S., Ekanayaka, A. S., Piddock, L. J., & Webber, M. A. (2012). Loss of or inhibition of all multidrug resistance efflux pumps of Salmonella enterica serovar typhimurium results in impaired ability to form a biofilm. The Journal of Antimicrobial Chemotherapy, 67(10), 2409–2417.
Baugh, S., Phillips, C. R., Ekanayaka, A. S., Piddock, L. J., & Webber, M. A. (2014). Inhibition of multidrug efflux as a strategy to prevent biofilm formation. The Journal of Antimicrobial Chemotherapy, 69(3), 673–681.
Bayer, A. S., Park, S., Ramos, M. C., Nast, C. C., Eftekhar, F., & Schiller, N. L. (1992). Effects of alginase on the natural history and antibiotic therapy of experimental endocarditis caused by mucoid Pseudomonas aeruginosa. Infection and Immunity, 60(10), 3979–3985.
Bijtenhoorn, P., Schipper, C., Hornung, C., Quitschau, M., Grond, S., Weiland, N., & Streit, W. R. (2011). BpiB05, a novel metagenome-derived hydrolase acting on N-acylhomoserine lactones. Journal of Biotechnology, 155(1), 86–94.
Bjarnsholt, T. (2013). The role of bacterial biofilms in chronic infections. APMIS. Supplementum, 136, 1–51.
Blazquez, J. (2003). Hypermutation as a factor contributing to the acquisition of antimicrobial resistance. Clinical Infectious Diseases, 37(9), 1201–1209.
Boles, B. R., & Horswill, A. R. (2008). Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathogens, 4(4), e1000052.
Brackman, G., Breyne, K., De Rycke, R., Vermote, A., Van Nieuwerburgh, F., Meyer, E., Van Calenbergh, S., & Coenye, T. (2016). The quorum sensing inhibitor Hamamelitannin increases antibiotic susceptibility of Staphylococcus aureus biofilms by affecting peptidoglycan biosynthesis and eDNA release. Scientific Reports, 6, 20321.
Brackman, G., Cos, P., Maes, L., Nelis, H. J., & Coenye, T. (2011). Quorum sensing inhibitors increase the susceptibility of bacterial biofilms to antibiotics in vitro and in vivo. Antimicrobial Agents and Chemotherapy, 55(6), 2655–2661.
Brotz-Oesterhelt, H., Beyer, D., Kroll, H. P., Endermann, R., Ladel, C., Schroeder, W., Hinzen, B., Raddatz, S., Paulsen, H., Henninger, K., Bandow, J. E., Sahl, H. G., & Labischinski, H. (2005). Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nature Medicine, 11(10), 1082–1087.
Bunders, C., Cavanagh, J., & Melander, C. (2011a). Flustramine inspired synthesis and biological evaluation of pyrroloindoline triazole amides as novel inhibitors of bacterial biofilms. Organic & Biomolecular Chemistry, 9(15), 5476–5481.
Bunders, C. A., Minvielle, M. J., Worthington, R. J., Ortiz, M., Cavanagh, J., & Melander, C. (2011b). Intercepting bacterial indole signaling with flustramine derivatives. Journal of the American Chemical Society, 133(50), 20160–20163.
Camilli, A., & Bassler, B. L. (2006). Bacterial small-molecule signaling pathways. Science, 311(5764), 1113–1116.
Coates, A. R., & Hu, Y. (2008). Targeting non-multiplying organisms as a way to develop novel antimicrobials. Trends in Pharmacological Sciences, 29(3), 143–150.
Conibear, T. C., Collins, S. L., & Webb, J. S. (2009). Role of mutation in Pseudomonas aeruginosa biofilm development. PLoS One, 4(7), e6289.
Conlon, B. P., Nakayasu, E. S., Fleck, L. E., LaFleur, M. D., Isabella, V. M., Coleman, K., Leonard, S. N., Smith, R. D., Adkins, J. N., & Lewis, K. (2013). Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature, 503(7476), 365–370.
Conlon, B. P., Rowe, S. E., & Lewis, K. (2015). Persister cells in biofilm associated infections. Advances in Experimental Medicine and Biology, 831, 1–9.
Darouiche, R. O., Mansouri, M. D., Gawande, P. V., & Madhyastha, S. (2009). Antimicrobial and antibiofilm efficacy of triclosan and DispersinB combination. The Journal of Antimicrobial Chemotherapy, 64(1), 88–93.
Davey, M. E., & O’Toole, G. A. (2000). Microbial biofilms: From ecology to molecular genetics. Microbiology and Molecular Biology Reviews, 64(4), 847–867.
Davies, D. (2003). Understanding biofilm resistance to antibacterial agents. Nature Reviews Drug Discovery, 2(2), 114–122.
Di Luca, M., Maccari, G., & Nifosi, R. (2014). Treatment of microbial biofilms in the post-antibiotic era: Prophylactic and therapeutic use of antimicrobial peptides and their design by bioinformatics tools. Pathogens and Disease, 70(3), 257–270.
Donelli, G., Francolini, I., Romoli, D., Guaglianone, E., Piozzi, A., Ragunath, C., & Kaplan, J. B. (2007). Synergistic activity of dispersin B and cefamandole nafate in inhibition of staphylococcal biofilm growth on polyurethanes. Antimicrobial Agents and Chemotherapy, 51(8), 2733–2740.
Donlan, R. M. (2002). Biofilms: Microbial life on surfaces. Emerging Infectious Diseases, 8(9), 881–890.
Eguchi, Y., Kubo, N., Matsunaga, H., Igarashi, M., & Utsumi, R. (2011). Development of an antivirulence drug against Streptococcus mutans: Repression of biofilm formation, acid tolerance, and competence by a histidine kinase inhibitor, walkmycin C. Antimicrobial Agents and Chemotherapy, 55(4), 1475–1484.
Flemming, H. C., & Wingender, J. (2010). The biofilm matrix. Nature Reviews. Microbiology, 8(9), 623–633.
Frederiksen, B., Pressler, T., Hansen, A., Koch, C., & Hoiby, N. (2006). Effect of aerosolized rhDNase (Pulmozyme) on pulmonary colonization in patients with cystic fibrosis. Acta Paediatrica, 95(9), 1070–1074.
Frezza, M., Soulere, L., Balestrino, D., Gohar, M., Deshayes, C., Queneau, Y., Forestier, C., & Doutheau, A. (2007). Ac2-DPD, the bis-(O)-acetylated derivative of 4,5-dihydroxy-2,3-pentanedione (DPD) is a convenient stable precursor of bacterial quorum sensing autoinducer AI-2. Bioorganic & Medicinal Chemistry Letters, 17(5), 1428–1431.
Garrison, A. T., Abouelhassan, Y., Kallifidas, D., Bai, F., Ukhanova, M., Mai, V., Jin, S., Luesch, H., & Huigens, R. W., 3rd. (2015). Halogenated phenazines that potently eradicate biofilms, MRSA persister cells in non-biofilm cultures, and Mycobacterium tuberculosis. Angewandte Chemie (International Ed. in English), 54(49), 14819–14823.
Geske, G. D., Wezeman, R. J., Siegel, A. P., & Blackwell, H. E. (2005). Small molecule inhibitors of bacterial quorum sensing and biofilm formation. Journal of the American Chemical Society, 127(37), 12762–12763.
Gordon, C. P., Williams, P., & Chan, W. C. (2013). Attenuating Staphylococcus aureus virulence gene regulation: A medicinal chemistry perspective. Journal of Medicinal Chemistry, 56(4), 1389–1404.
Grandclement, C., Tannieres, M., Morera, S., Dessaux, Y., & Faure, D. (2016). Quorum quenching: Role in nature and applied developments. FEMS Microbiology Reviews, 40(1), 86–116.
Guo, M., Gamby, S., Zheng, Y., & Sintim, H. O. (2013). Small molecule inhibitors of AI-2 signaling in bacteria: State-of-the-art and future perspectives for anti-quorum sensing agents. International Journal of Molecular Sciences, 14(9), 17694–17728.
Gutierrez, J. A., Crowder, T., Rinaldo-Matthis, A., Ho, M. C., Almo, S. C., & Schramm, V. L. (2009). Transition state analogs of 5 ’-methylthioadenosine nucleosidase disrupt quorum sensing. Nature Chemical Biology, 5(4), 251–257.
Hall, C. W., & Mah, T. F. (2017). Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiology Reviews, 41(3), 276–301.
Hancock, V., Witso, I. L., & Klemm, P. (2011). Biofilm formation as a function of adhesin, growth medium, substratum and strain type. International Journal of Medical Microbiology, 301(7), 570–576.
Hausner, M., & Wuertz, S. (1999). High rates of conjugation in bacterial biofilms as determined by quantitative in situ analysis. Applied and Environmental Microbiology, 65(8), 3710–3713.
Hentzer, M., Riedel, K., Rasmussen, T. B., Heydorn, A., Andersen, J. B., Parsek, M. R., Rice, S. A., Eberl, L., Molin, S., Hoiby, N., Kjelleberg, S., & Givskov, M. (2002). Inhibition of quorum sensing in Pseudomonas aeruginosa biofilm bacteria by a halogenated furanone compound. Microbiology-Sgm, 148, 87–102.
Hentzer, M., Wu, H., Andersen, J. B., Riedel, K., Rasmussen, T. B., Bagge, N., Kumar, N., Schembri, M. A., Song, Z. J., Kristoffersen, P., Manefield, M., Costerton, J. W., Molin, S., Eberl, L., Steinberg, P., Kjelleberg, S., Hoiby, N., & Givskov, M. (2003). Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO Journal, 22(15), 3803–3815.
Hoiby, N., Bjarnsholt, T., Moser, C., Bassi, G. L., Coenye, T., Donelli, G., Hall-Stoodley, L., Hola, V., Imbert, C., Kirketerp-Moller, K., Lebeaux, D., Oliver, A., Ullmann, A. J., Williams, C., Biofilms, E. S. G. F., & Z Consulting External Expert Werner. (2015). ESCMID guideline for the diagnosis and treatment of biofilm infections 2014. Clinical Microbiology and Infection, 21(Suppl 1), 1–25.
Hraiech, S., Hiblot, J., Lafleur, J., Lepidi, H., Papazian, L., Rolain, J. M., Raoult, D., Elias, M., Silby, M. W., Bzdrenga, J., Bregeon, F., & Chabriere, E. (2014). Inhaled lactonase reduces Pseudomonas aeruginosa quorum sensing and mortality in rat pneumonia. PLoS One, 9(10), e107125.
Hurdle, J. G., O’Neill, A. J., Chopra, I., & Lee, R. E. (2011). Targeting bacterial membrane function: An underexploited mechanism for treating persistent infections. Nature Reviews. Microbiology, 9(1), 62–75.
Itoh, Y., Wang, X., Hinnebusch, B. J., Preston, J. F., 3rd, & Romeo, T. (2005). Depolymerization of beta-1,6-N-acetyl-D-glucosamine disrupts the integrity of diverse bacterial biofilms. Journal of Bacteriology, 187(1), 382–387.
Iwase, T., Uehara, Y., Shinji, H., Tajima, A., Seo, H., Takada, K., Agata, T., & Mizunoe, Y. (2010). Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature, 465(7296), 346–349.
Izano, E. A., Amarante, M. A., Kher, W. B., & Kaplan, J. B. (2008). Differential roles of poly-N-acetylglucosamine surface polysaccharide and extracellular DNA in Staphylococcus aureus and Staphylococcus epidermidis biofilms. Applied and Environmental Microbiology, 74(2), 470–476.
Jani, S., Seely, A. L., Peabody, V. G., Jayaraman, A., & Manson, M. D. (2017). Chemotaxis to self-generated AI-2 promotes biofilm formation in Escherichia coli. Microbiology. https://doi.org/10.1099/mic.0.000567.
Jefferson, K. K. (2004). What drives bacteria to produce a biofilm? FEMS Microbiology Letters, 236(2), 163–173.
Johansson, E. M., Crusz, S. A., Kolomiets, E., Buts, L., Kadam, R. U., Cacciarini, M., Bartels, K. M., Diggle, S. P., Camara, M., Williams, P., Loris, R., Nativi, C., Rosenau, F., Jaeger, K. E., Darbre, T., & Reymond, J. L. (2008). Inhibition and dispersion of Pseudomonas aeruginosa biofilms by glycopeptide dendrimers targeting the fucose-specific lectin LecB. Chemistry & Biology, 15(12), 1249–1257.
Kaplan, J. B. (2010). Biofilm dispersal: Mechanisms, clinical implications, and potential therapeutic uses. Journal of Dental Research, 89(3), 205–218.
Kaplan, J. B., LoVetri, K., Cardona, S. T., Madhyastha, S., Sadovskaya, I., Jabbouri, S., & Izano, E. A. (2012). Recombinant human DNase I decreases biofilm and increases antimicrobial susceptibility in staphylococci. Journal of Antibiotics (Tokyo), 65(2), 73–77.
Kaplan, J. B., Ragunath, C., Velliyagounder, K., Fine, D. H., & Ramasubbu, N. (2004). Enzymatic detachment of Staphylococcus epidermidis biofilms. Antimicrobial Agents and Chemotherapy, 48(7), 2633–2636.
Kiran, M. D., Adikesavan, N. V., Cirioni, O., Giacometti, A., Silvestri, C., Scalise, G., Ghiselli, R., Saba, V., Orlando, F., Shoham, M., & Balaban, N. (2008). Discovery of a quorum-sensing inhibitor of drug-resistant staphylococcal infections by structure-based virtual screening. Molecular Pharmacology, 73(5), 1578–1586.
Kunze, B., Reck, M., Dotsch, A., Lemme, A., Schummer, D., Irschik, H., Steinmetz, H., & Wagner-Dobler, I. (2010). Damage of Streptococcus mutans biofilms by carolacton, a secondary metabolite from the myxobacterium Sorangium cellulosum. BMC Microbiology, 10, 199.
Lamppa, J. W., & Griswold, K. E. (2013). Alginate lyase exhibits catalysis-independent biofilm dispersion and antibiotic synergy. Antimicrobial Agents and Chemotherapy, 57(1), 137–145.
Lee, J., Bansal, T., Jayaraman, A., Bentley, W. E., & Wood, T. K. (2007a). Enterohemorrhagic Escherichia coli biofilms are inhibited by 7-hydroxyindole and stimulated by isatin. Applied and Environmental Microbiology, 73(13), 4100–4109.
Lee, J., Jayaraman, A., & Wood, T. K. (2007b). Indole is an inter-species biofilm signal mediated by SdiA. BMC Microbiology, 7, 42.
Lee, J., & Lee, J. H. (2010). Indole as an intercellular signal in microbial communities. FEMS Microbiology Reviews, 34(4), 426–444.
Lee, J. H., Cho, M. H., & Lee, J. (2011). 3-indolylacetonitrile decreases Escherichia coli O157:H7 biofilm formation and Pseudomonas aeruginosa virulence. Environmental Microbiology, 13(1), 62–73.
Lee, J. H., Kim, Y. G., Cho, M. H., Kim, J. A., & Lee, J. (2012). 7-fluoroindole as an antivirulence compound against Pseudomonas aeruginosa. FEMS Microbiology Letters, 329(1), 36–44.
Lewis, K. (2001). Riddle of biofilm resistance. Antimicrobial Agents and Chemotherapy, 45(4), 999–1007.
Lieberman, O. J., Orr, M. W., Wang, Y., & Lee, V. T. (2014). High-throughput screening using the differential radial capillary action of ligand assay identifies ebselen as an inhibitor of diguanylate cyclases. ACS Chemical Biology, 9(1), 183–192.
Liu, C., Worthington, R. J., Melander, C., & Wu, H. (2014). A new small molecule specifically inhibits the cariogenic bacterium Streptococcus mutans in multispecies biofilms. Antimicrobial Agents and Chemotherapy, 55(6), 2679–2687.
Lonn-Stensrud, J., Petersen, F. C., Benneche, T., & Scheie, A. A. (2007). Synthetic bromated furanone inhibits autoinducer-2-mediated communication and biofilm formation in oral streptococci. Oral Microbiology and Immunology, 22(5), 340–346.
Lyons, N. A., & Kolter, R. (2015). On the evolution of bacterial multicellularity. Current Opinion in Microbiology, 24, 21–28.
Mah, T. F. C., & O’Toole, G. A. (2001). Mechanisms of biofilm resistance to antimicrobial agents. Trends in Microbiology, 9(1), 34–39.
Malone, C. L., Boles, B. R., & Horswill, A. R. (2007). Biosynthesis of Staphylococcus aureus the Synechocystis autoinducing peptides by using DnaB mini-intein. Applied and Environmental Microbiology, 73(19), 6036–6044.
Mansour, S. C., de la Fuente-Nunez, C., & Hancock, R. E. (2015). Peptide IDR-1018: Modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. Journal of Peptide Science, 21(5), 323–329.
Mattmann, M. E., & Blackwell, H. E. (2010). Small molecules that modulate quorum sensing and control virulence in Pseudomonas aeruginosa. The Journal of Organic Chemistry, 75(20), 6737–6746.
May, J. G., Shah, P., Sachdeva, L., Micale, M., Kruper, G. J., Sheyn, A., & Coticchia, J. M. (2014). Potential role of biofilms in deep cervical abscess. International Journal of Pediatric Otorhinolaryngology, 78(1), 10–13.
Melander, R. J., Liu, H. B., Stephens, M. D., Bewley, C. A., & Melander, C. (2016). Marine sponge alkaloids as a source of anti-bacterial adjuvants. Bioorganic & Medicinal Chemistry Letters, 26(24), 5863–5866.
Melander, R. J., Minvielle, M. J., & Melander, C. (2014). Controlling bacterial behavior with indole-containing natural products and derivatives. Tetrahedron, 70(37), 6363–6372.
Milton, M. E., Minrovic, B. M., Harris, D. L., Kang, B., Jung, D., Lewis, C. P., Thompson, R. J., Melander, R. J., Zeng, D., Melander, C., & Cavanagh, J. (2018). Re-sensitizing multidrug resistant Bacteria to antibiotics by targeting bacterial response regulators: Characterization and comparison of interactions between 2-Aminoimidazoles and the response regulators BfmR from Acinetobacter baumannii and QseB from Francisella spp. Frontiers in Molecular Biosciences, 5, 15.
Minvielle, M. J., Bunders, C. A., & Melander, C. (2013a). Indole/triazole conjugates are selective inhibitors and inducers of bacterial biofilms. MedChemComm, 4(6), 916–919.
Minvielle, M. J., Eguren, K., & Melander, C. (2013b). Highly active modulators of indole signaling alter pathogenic behaviors in gram-negative and gram-positive bacteria. Chemistry, 19(51), 17595–17602.
Nikolaev Iu, A., & Plakunov, V. K. (2007). Biofilm—“City of microbes” or an analogue of multicellular organisms? Mikrobiologiia, 76(2), 149–163.
O’Loughlin, C. T., Miller, L. C., Siryaporn, A., Drescher, K., Semmelhack, M. F., & Bassler, B. L. (2013). A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proceedings of the National Academy of Sciences of the United States of America, 110(44), 17981–17986.
Oliver, A., Canton, R., Campo, P., Baquero, F., & Blazquez, J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science, 288(5469), 1251–1254.
Pan, W., Fan, M., Wu, H., Melander, C., & Liu, C. (2015). A new small molecule inhibits Streptococcus mutans biofilms in vitro and in vivo. Journal of Applied Microbiology, 119(5), 1403–1411.
Parsek, M. R., & Greenberg, E. P. (2000). Acyl-homoserine lactone quorum sensing in gram-negative bacteria: A signaling mechanism involved in associations with higher organisms. Proceedings of the National Academy of Sciences of the United States of America, 97(16), 8789–8793.
Parsiegla, G., Noguere, C., Santell, L., Lazarus, R. A., & Bourne, Y. (2012). The structure of human DNase I bound to magnesium and phosphate ions points to a catalytic mechanism common to members of the DNase I-like superfamily. Biochemistry, 51(51), 10250–10258.
Passos da Silva, D., Schofield, M. C., Parsek, M. R., & Tseng, B. S. (2017). An update on the sociomicrobiology of quorum sensing in gram-negative biofilm development. Pathogens, 6(4), E51.
Peters, L., Konig, G. M., Wright, A. D., Pukall, R., Stackebrandt, E., Eberl, L., & Riedel, K. (2003). Secondary metabolites of Flustra foliacea and their influence on bacteria. Applied and Environmental Microbiology, 69(6), 3469–3475.
Pletzer, D., Coleman, S. R., & Hancock, R. E. (2016). Anti-biofilm peptides as a new weapon in antimicrobial warfare. Current Opinion in Microbiology, 33, 35–40.
Poole, K. (2012). Bacterial stress responses as determinants of antimicrobial resistance. The Journal of Antimicrobial Chemotherapy, 67(9), 2069–2089.
Qi, F., Merritt, J., Lux, R., & Shi, W. (2004). Inactivation of the ciaH gene in Streptococcus mutans diminishes mutacin production and competence development, alters sucrose-dependent biofilm formation, and reduces stress tolerance. Infection and Immunity, 72(8), 4895–4899.
Qin, Z., Zhang, J., Xu, B., Chen, L., Wu, Y., Yang, X., Shen, X., Molin, S., Danchin, A., Jiang, H., & Qu, D. (2006). Structure-based discovery of inhibitors of the YycG histidine kinase: New chemical leads to combat Staphylococcus epidermidis infections. BMC Microbiology, 6, 96.
Quave, C. L., Estevez-Carmona, M., Compadre, C. M., Hobby, G., Hendrickson, H., Beenken, K. E., & Smeltzer, M. S. (2012). Ellagic acid derivatives from Rubus ulmifolius inhibit Staphylococcus aureus biofilm formation and improve response to antibiotics. PLoS One, 7(1), e28737.
Rabin, N., Zheng, Y., Opoku-Temeng, C., Du, Y., Bonsu, E., & Sintim, H. O. (2015). Agents that inhibit bacterial biofilm formation. Future Medicinal Chemistry, 7(5), 647–671.
Ramasubbu, N., Thomas, L. M., Ragunath, C., & Kaplan, J. B. (2005). Structural analysis of dispersin B, a biofilm-releasing glycoside hydrolase from the periodontopathogen Actinobacillus actinomycetemcomitans. Journal of Molecular Biology, 349(3), 475–486.
Reck, M., Rutz, K., Kunze, B., Tomasch, J., Surapaneni, S. K., Schulz, S., & Wagner-Dobler, I. (2011). The biofilm inhibitor carolacton disturbs membrane integrity and cell division of Streptococcus mutans through the serine/threonine protein kinase PknB. Journal of Bacteriology, 193(20), 5692–5706.
Reddy, K. V., Yedery, R. D., & Aranha, C. (2004). Antimicrobial peptides: premises and promises. International Journal of Antimicrobial Agents, 24(6), 536–547.
Reffuveille, F., de la Fuente-Nunez, C., Mansour, S., & Hancock, R. E. (2014). A broad-spectrum antibiofilm peptide enhances antibiotic action against bacterial biofilms. Antimicrobial Agents and Chemotherapy, 58(9), 5363–5371.
Ren, D., Sims, J. J., & Wood, T. K. (2001). Inhibition of biofilm formation and swarming of Escherichia coli by (5Z)-4-bromo-5-(bromomethylene)-3-butyl-2(5H)-furanone. Environmental Microbiology, 3(11), 731–736.
Ren, D., Sims, J. J., & Wood, T. K. (2002). Inhibition of biofilm formation and swarming of Bacillus subtilis by (5Z)-4-bromo-5-(bromomethylene)-3-butyl-2(5H)-furanone. Letters in Applied Microbiology, 34(4), 293–299.
Robijns, S. C., De Pauw, B., Loosen, B., Marchand, A., Chaltin, P., De Keersmaecker, S. C., Vanderleyden, J., & Steenackers, H. P. (2012). Identification and characterization of 4-[4-(3-phenyl-2-propen-1-yl)-1-piperazinyl]-5H-pyrimido[5,4-b]indole derivatives as Salmonella biofilm inhibitors. FEMS Immunology and Medical Microbiology, 65(2), 390–394.
Rogers, S. A., Huigens, R. W., 3rd, Cavanagh, J., & Melander, C. (2010). Synergistic effects between conventional antibiotics and 2-aminoimidazole-derived antibiofilm agents. Antimicrobial Agents and Chemotherapy, 54(5), 2112–2118.
Rogers, S. A., & Melander, C. (2008). Construction and screening of a 2-aminoimidazole library identifies a small molecule capable of inhibiting and dispersing biofilms across bacterial order, class, and phylum. Angewandte Chemie International Edition, 47(28), 5229–5231.
Roy, V., Meyer, M. T., Smith, J. A., Gamby, S., Sintim, H. O., Ghodssi, R., & Bentley, W. E. (2013). AI-2 analogs and antibiotics: A synergistic approach to reduce bacterial biofilms. Applied Microbiology and Biotechnology, 97(6), 2627–2638.
Rumbo-Feal, S., Gomez, M. J., Gayoso, C., Alvarez-Fraga, L., Cabral, M. P., Aransay, A. M., Rodriguez-Ezpeleta, N., Fullaondo, A., Valle, J., Tomas, M., Bou, G., & Poza, M. (2013). Whole transcriptome analysis of Acinetobacter baumannii assessed by RNA-sequencing reveals different mRNA expression profiles in biofilm compared to planktonic cells. PLoS One, 8(8), e72968.
Sambanthamoorthy, K., Gokhale, A. A., Lao, W., Parashar, V., Neiditch, M. B., Semmelhack, M. F., Lee, I., & Waters, C. M. (2011). Identification of a novel benzimidazole that inhibits bacterial biofilm formation in a broad-spectrum manner. Antimicrobial Agents and Chemotherapy, 55(9), 4369–4378.
Sambanthamoorthy, K., Sloup, R. E., Parashar, V., Smith, J. M., Kim, E. E., Semmelhack, M. F., Neiditch, M. B., & Waters, C. M. (2012). Identification of small molecules that antagonize diguanylate cyclase enzymes to inhibit biofilm formation. Antimicrobial Agents and Chemotherapy, 56(10), 5202–5211.
Savage, V. J., Chopra, I., & O’Neill, A. J. (2013). Staphylococcus aureus biofilms promote horizontal transfer of antibiotic resistance. Antimicrobial Agents and Chemotherapy, 57(4), 1968–1970.
Selan, L., Berlutti, F., Passariello, C., Comodi-Ballanti, M. R., & Thaller, M. C. (1993). Proteolytic enzymes: A new treatment strategy for prosthetic infections? Antimicrobial Agents and Chemotherapy, 37(12), 2618–2621.
Simonetti, O., Cirioni, O., Mocchegiani, F., Cacciatore, I., Silvestri, C., Baldassarre, L., Orlando, F., Castelli, P., Provinciali, M., Vivarelli, M., Fornasari, E., Giacometti, A., & Offidani, A. (2013). The efficacy of the quorum sensing inhibitor FS8 and tigecycline in preventing prosthesis biofilm in an animal model of staphylococcal infection. International Journal of Molecular Sciences, 14(8), 16321–16332.
Singh, S., Kalia, N. P., Joshi, P., Kumar, A., Sharma, P. R., Kumar, A., Bharate, S. B., & Khan, I. A. (2017). Boeravinone B, a novel dual inhibitor of NorA bacterial efflux pump of Staphylococcus aureus and human P-glycoprotein, reduces the biofilm formation and intracellular invasion of Bacteria. Frontiers in Microbiology, 8, 1868.
Slachmuylders, L., Van Acker, H., Brackman, G., Sass, A., Van Nieuwerburgh, F., & Coenye, T. (2018). Elucidation of the mechanism behind the potentiating activity of baicalin against Burkholderia cenocepacia biofilms. PLoS One, 13(1), e0190533.
Smith, K., & Hunter, I. S. (2008). Efficacy of common hospital biocides with biofilms of multi-drug resistant clinical isolates. Journal of Medical Microbiology, 57(Pt 8), 966–973.
Stacy, D. M., Welsh, M. A., Rather, P. N., & Blackwell, H. E. (2012). Attenuation of quorum sensing in the pathogen Acinetobacter baumannii using non-native N-acyl homoserine lactones. ACS Chemical Biology, 7(10), 1719–1728.
Stalder, T., & Top, E. (2016). Plasmid transfer in biofilms: A perspective on limitations and opportunities. NPJ Biofilms Microbiomes, 2, 16022.
Stewart, P. S., & Costerton, J. W. (2001). Antibiotic resistance of bacteria in biofilms. Lancet, 358(9276), 135–138.
Stoodley, P., Sauer, K., Davies, D. G., & Costerton, J. W. (2002). Biofilms as complex differentiated communities. Annual Review of Microbiology, 56, 187–209.
Su, S., Panmanee, W., Wilson, J. J., Mahtani, H. K., Li, Q., Vanderwielen, B. D., Makris, T. M., Rogers, M., McDaniel, C., Lipscomb, J. D., Irvin, R. T., Schurr, M. J., Lancaster, J. R., Jr., Kovall, R. A., & Hassett, D. J. (2014). Catalase (KatA) plays a role in protection against anaerobic nitric oxide in Pseudomonas aeruginosa. PLoS One, 9(3), e91813.
Sutherland, I. (2001). Biofilm exopolysaccharides: A strong and sticky framework. Microbiology, 147(Pt 1), 3–9.
Tetz, V. V., & Tetz, G. V. (2010). Effect of extracellular DNA destruction by DNase I on characteristics of forming biofilms. DNA and Cell Biology, 29(8), 399–405.
Thompson, R. J., Bobay, B. G., Stowe, S. D., Olson, A. L., Peng, L., Su, Z., Actis, L. A., Melander, C., & Cavanagh, J. (2012). Identification of BfmR, a response regulator involved in biofilm development, as a target for a 2-Aminoimidazole-based antibiofilm agent. Biochemistry, 51(49), 9776–9778.
Tomaras, A. P., Flagler, M. J., Dorsey, C. W., Gaddy, J. A., & Actis, L. A. (2008). Characterization of a two-component regulatory system from Acinetobacter baumannii that controls biofilm formation and cellular morphology. Microbiology-Sgm, 154, 3398–3409.
Traxler, M. F., Summers, S. M., Nguyen, H. T., Zacharia, V. M., Hightower, G. A., Smith, J. T., & Conway, T. (2008). The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Molecular Microbiology, 68(5), 1128–1148.
Tsuchikama, K., Zhu, J., Lowery, C. A., Kaufmann, G. F., & Janda, K. D. (2012). C4-alkoxy-HPD: A potent class of synthetic modulators surpassing nature in AI-2 quorum sensing. Journal of the American Chemical Society, 134(33), 13562–13564.
Tuomanen, E., Durack, D. T., & Tomasz, A. (1986). Antibiotic tolerance among clinical isolates of bacteria. Antimicrobial Agents and Chemotherapy, 30(4), 521–527.
Van Acker, H., & Coenye, T. (2016). The role of efflux and physiological adaptation in biofilm tolerance and resistance. The Journal of Biological Chemistry, 291(24), 12565–12572.
Van Acker, H., Van Dijck, P., & Coenye, T. (2014). Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends in Microbiology, 22(6), 326–333.
Vendeville, A., Winzer, K., Heurlier, K., Tang, C. M., & Hardie, K. R. (2005). Making ‘sense’ of metabolism: Autoinducer-2, LuxS and pathogenic bacteria. Nature Reviews. Microbiology, 3(5), 383–396.
Webb, J. S., Givskov, M., & Kjelleberg, S. (2003). Bacterial biofilms: Prokaryotic adventures in multicellularity. Current Opinion in Microbiology, 6(6), 578–585.
Wood, T. K., Knabel, S. J., & Kwan, B. W. (2013). Bacterial persister cell formation and dormancy. Applied and Environmental Microbiology, 79(23), 7116–7121.
Wood, T. K., Lee, J., Zhang, X. S., Hegde, M., Bentley, W. E., & Jayaraman, A. (2008). Indole cell signaling occurs primarily at low temperatures in Escherichia coli. ISME Journal, 2(10), 1007–1023.
Worthington, R. J., Blackledge, M. S., & Melander, C. (2013). Small-molecule inhibition of bacterial two-component systems to combat antibiotic resistance and virulence. Future Medicinal Chemistry, 5(11), 1265–1284.
Worthington, R. J., Richards, J. J., & Melander, C. (2012). Small molecule control of bacterial biofilms. Organic & Biomolecular Chemistry, 10(37), 7457–7474.
Wright, C. J., Wu, H., Melander, R. J., Melander, C., & Lamont, J. (2014). Disruption of heterotypic community development by Porphyromonas gingivalis with small molecule inhibitors. Molecular Oral Microbiology, 29(5), 185–193.
Wu, H., Moser, C., Wang, H. Z., Hoiby, N., & Song, Z. J. (2015). Strategies for combating bacterial biofilm infections. International Journal of Oral Science, 7(1), 1–7.
Yan, H., & Chen, W. (2010). 3′,5′-cyclic diguanylic acid: A small nucleotide that makes big impacts. Chemical Society Reviews, 39(8), 2914–2924.
Yarwood, J. M., Bartels, D. J., Volper, E. M., & Greenberg, E. P. (2004). Quorum sensing in Staphylococcus aureus biofilms. Journal of Bacteriology, 186(6), 1838–1850.
Yates, E. A., Philipp, B., Buckley, C., Atkinson, S., Chhabra, S. R., Sockett, R. E., Goldner, M., Dessaux, Y., Camara, M., Smith, H., & Williams, P. (2002). N-acylhomoserine lactones undergo lactonolysis in a pH-, temperature-, and acyl chain length-dependent manner during growth of Yersinia pseudotuberculosis and Pseudomonas aeruginosa. Infection and Immunity, 70(10), 5635–5646.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Melander, R.J., Melander, C. (2019). Strategies for the Eradication of Biofilm-Based Bacterial Infections. In: Ahmad, I., Ahmad, S., Rumbaugh, K. (eds) Antibacterial Drug Discovery to Combat MDR. Springer, Singapore. https://doi.org/10.1007/978-981-13-9871-1_22
Download citation
DOI: https://doi.org/10.1007/978-981-13-9871-1_22
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-9870-4
Online ISBN: 978-981-13-9871-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)