
Abstract
Granulin (GRN) mutations were first found in frontotemporal dementia (FTD) patients with ubiquitin-positive, tau-negative inclusions in 2006. These inclusions were also found to contain a TAR-DNA binding protein of 43 kDa (TDP-43). PGRN protein itself is not a component of ubiquitin-positive inclusion bodies. Since then, more than 190 GRN mutations have been reported, including substitutions, insertions, and deletions. The common pathological mechanism observed with these mutations was proposed to arise from haploinsufficiency; the amount of functional PGRN was reduced to half of the normal level. In fact, GRN mutation carriers showed significantly reduced expression levels of PGRN in plasma and serum. Immunohistochemical analyses of phosphorylated TDP-43 revealed that cases of FTLD-TDP with a GRN mutation invariably display type A pathology, which is characterized by numerous short dystrophic neurites (DNs) and crescentic or oval shaped neuronal cytoplasmic inclusions (NCIs). GRN mutations were initially found in FTD patients with tau-negative, TDP-43-positive inclusions, however, recent findings suggested that different clinical phenotypes may be seen in the same GRN mutation carriers and additional tau or α-synuclein accumulation may be observed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Frontotemporal dementia (FTD)
- Frontotemporal lobar degeneration (FTLD)
- GRN mutation
- Nonsense-mediated dacay
Introduction
Frontotemporal lobar degeneration (FTLD) is a collective term for a disease group characterized by progressive neurodegeneration limited to frontal and temporal lobes. FTLD is clinically divided into three types: frontotemporal dementia (FTD), semantic dementia (SD) and progressive nonfluent aphasia (PNFA) (Neary et al. 1998). This classification is based on clinical manifestations that reflect differences in the degenerative brain region. They do not reflect specific neuropathological characteristics. FTLD can be subdivided into three neuropathological groups, depending on the presence of inclusion bodies or a certain protein component (McKhann et al. 2001). The first group, exhibiting “tauopathy”, has tau-positive inclusion bodies. This group includes Pick’s disease, corticobasal degeneration (CBD), progressive supranuclear palsy (PSP) and frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17). The second group might be called FTLD-U since it is similar to FTLD but has ubiquitin-positive tau-negative neurocytoplasmic inclusions (Mackenzie et al. 2006a). FTLD is divided into two types, FTLD with motor neuron disease: (FTLD-MND) and FTLD with MND type inclusions but without MND. The third group consist of FTLD without tau- or ubiquitin-positive inclusions, and this group has been considered a dementia lacking distinctive histology (DLDH). However, most cases of the third group consist of FTLD-U with inclusions which are identified using high-sensitivity ubiquitin immunostaining. Rare cases with tau-negative, cytoplasmic and nuclear ubiquitin-positive inclusions have also been found (Mackenzie et al. 2006c).
Some 35–50% of FTLD patients have a family history of dementia and the causative gene loci have been identified on chromosomes 3, 9 and 17. Microtubule-associated protein tau (tau, MAPT), valosin-containing protein (VCP) and charged multivesicular body protein 2b (CHMP2B) have been identified as causative of FTLD. The identification of the tau gene mutation on chromosome 17q21 reminds us of the importance of tau in neurodegenerative disease research (Hutton et al. 1998). However, a considerable number of familial FTLD-U cases linked on chromosome 17q21 with tau-negative, cytoplasmic and nuclear ubiquitin-positive inclusions have been found.
In 2006, Cruts and Baker identified a granulin (GRN) mutation in FTLD-U patients (Baker et al. 2006; Cruts et al. 2006). Since then, more than 190 GRN mutations have been reported including substitutions, insertions and deletions (Tables 1, 2 and 3 and Alzheimer Disease & Frontotemporal Dementia Mutation Database, http://www.molgen.ua.ac.be/FTDMutations/) (Cruts et al. 2012). The common pathological mechanism in these mutations was proposed to arise from haploinsufficiency. Symptoms of haploinsufficiency appear after inactivation of one allele of the causative gene in a dominantly-inherited disease (Wilkie 1994). With GRN mutation, a mutated form of mRNA is degraded by nonsense-mediated decay (NMD) which is likely to create a null (no expression) allele. It is thought that the functional form of the PGRN protein decreases with disease onset.
A TAR-DNA binding protein of 43 kDa (TDP-43), the main component of ubiquitin-positive inclusions, was observed in FTLD and ALS patients and was identified in 2006 by Arai et al. and Neumann et al. (Arai et al. 2006; Neumann et al. 2006). The tau-negative, ubiquitin-positive inclusions that were seen in GRN mutation brains were also identified as containing TDP-43.
GRN Mutations and Pathological Mechanisms
Baker and colleagues examined more than 80 candidate genes within the 3.53-cM (6.19-Mb) critical region clarified by haplotype analysis of Canadian tau-negative FTD families (Baker et al. 2006). They identified an insertion mutation of four base pairs (CTGC) in exon 1 of the GRN gene (g.90_91insCTGC) [g: genomic DNA, ins: insertion]. The numbering is relative to the reverse complement of GenBank accession number AC003043.1, starting at adenine (A) of Met 1. This mutation causes a frame shift at codon 31 that induces a premature termination codon after a read through of 34 amino acids (p.Cys31LeufsX34) [p: protein, fs: frame shifts, X: termination codon]. The p.Cys31LeufsX34 mutation was absent in 550 North American control individuals. They sequenced the GRN gene in affected families in Canada, the USA, UK, Netherlands and Scandinavia and identified an additional eight GRN mutations in nine families. These mutations were as follows: four nonsense mutations: g.1087C>T (p.Gln125X), g.2609G>A (p.Trp386X), g.2923C>T (p.Arg418X), g.3073C>T (p.Gln468X); two flame shift mutations: g.1102_1105delCAGT (p.Gln130SerfsX124), g.2597delC (pThr382SerfsX29); one splicing site mutation: IVS8-1G (p.Val279GlyfsX4); and mutation in start codon: g.2T>C (p.Met1?). Next, they extracted RNA from the lymphoblasts of cases with mutations g.90_91insCTGC (p.Cys31LeufsX34) and c.2923C>T (p.Arg418X), performed quantitative RT-PCR analysis and found that the expression of GRN mRNA was reduced by approximately 50%. They performed sequencing of GRN mRNAs and found that most of them encoded wild type GRN, whereas the mutated type of GRN was rarely detected. These results suggested that the mutated mRNA was degraded by NMD. NMD degrades mRNA with a premature termination codon (PTC) which arise from a splicing error or mutation, and thereby prevent production of an abnormal protein (Maquat 2004).
When the lymphoblasts from patients were treated with the NMD inhibitor, cycloheximide, the mutated mRNA was increased. Immunoblotting analysis revealed that the amount of wild type PGRN protein had decreased compared with the controls and mutated PGRN protein was barely detected. They also detected a significant reduction in the amount of mutated mRNA in the brains of patients with the g.2T>C mutation. They suggested that translation of the protein did not occur because the Kozak sequence was disrupted by the g.2T>C mutation.
Cruts and colleagues also identified five novel GRN mutations, IVS0+5G>C (now termed IVS1+5G>C), g.3G>A (p.Met1?), g.1094_1095delCT (p.Pro127ArgfsX2), g.1872G>A (p.Ala237TrpfsX4), and g.1087C>T (p.Gln125X). IVS1+5G>C indicates a point mutation in the intron 1 splice donor site causing intron 1 retention, resulting in nuclear mRNA degradation (Cruts et al. 2006). Sequence analysis of GRN in 103 Belgian FTD patients identified this mutation in the eight probands belonging to different branches of the Belgian founder family. An in silico analysis of the IVS1+5G>C mutation predicted an intense decline in the binding efficiency of the U1 snRNP complex.
Next, they analyzed full length GRN cDNA from the brains and lymphoblasts of two probands, abnormal transcripts. According to the polymorphism (rs5848) in the 3′ untranslated region of the GRN gene, probands were judged C/T heterogeneous (the T-allele is the disease haplotype). However, on sequence analysis of cDNA from their lymphoblasts or brain tissue, only the C allele was observed. These results suggested a complete disappearance of mutated GRN mRNA. Immunoblot analysis using an extract from the lymphoblasts of a proband showed PGRN protein reduction. They confirmed loss of mRNA and wild type PGRN protein reduction in the cases of the g.1087C>T (p.Gln125X) mutation. Subsequently, Gass and colleagues performed systematic screening for the GRN gene in 378 FTLD and 48 ALS cases at the Mayo Clinic and identified 23 GRN mutations in 39 FTLD cases.
Twenty of these twenty-three mutations (4 nonsense mutations, 12 frame shift mutations and 4 splicing donor site mutations) predicted production of PTC and mutated mRNA degradation by NMD. They also identified novel mutations in the splicing donor site of exon 1 (IVS1+1G>A) as well as a missense mutation (g.26C>A (p.Ala9Asp)). In this study, no mutation was identified in ALS cases. RT-PCR analysis of a brain with an IVS1+1G>A mutation revealed two bands corresponding to mutated GRN mRNA and wild type GRN mRNA, respectively. These results suggested that the IVS1+1G>A mutation did not cause degradation of mutated mRNA by NMD. Initiation of NMD first required a translation process, so that it has been speculated that any IVS1+G>A mutation would escape NMD because no translation would start without the Kozak sequence. The g.26C>A (p.Ala9Asp) mutation was identified as singular missense mutation in this study, the 9th alanine in exon 1 of GRN being replaced by aspartic acid. The 9th alanine corresponds to the hydrophobic core of the signal peptide. Mutated mRNA was reduced in the g.26C>A (p.Ala9Asp) brain by an unknown mechanism. If a mutated allele was translated in this case, it would produce a mutated PGRN protein lacking binding capability to the signal recognition motif and could not be transported to the endoplasmic reticulum. Since 2006, many novel GRN mutations have been found and are listed in Tables 1, 2 and 3.
PGRN Protein Is Not a Component of Ubiquitin-Positive Inclusion Bodies
Immunohistochemical staining using antibodies for all regions of PGRN protein showed that some of the neurons and activated microglia were positive. Ubiquitin-positive neuronal cytoplasmic inclusions (NCI) and neuronal intranuclear inclusions (NII) were negative with PGRN antibodies (Baker et al. 2006; Cruts et al. 2006). These results indicated that PGRN accumulation did not occur during development of the FTD pathology caused by the GRN mutation. PGRN-positive neuron and activated microglia were also observed in the brains of normal elderly individuals and Alzheimer’s disease (AD) cases.
Clinico-Pathological Characterization of GRN Mutation Carriers
Incidence Rate
In the Belgian study, Cruts and colleagues found GRN mutations in 10.7% (11 out of 103) of the FTD cases overall and in 25.6% (11 out of 43) of familial FTD cases (Cruts et al. 2006). MAPT mutation frequencies were 2.9% (3 out of 103) in the non-familial FTD and 7% (3 out of 43) in the familial FTD cases. These results indicated that GRN mutations are approximately a 3.5 times more frequent cause of FTD in Belgian patients. GRN mutation data of Gass and colleagues showed mutations in 10.5% (39 out of 378) of FTD and 25.6% (32 out of 144) of familial FTD cases. However, they pointed out that there was some bias in their cases because the Mayo Clinic treated many familial FTLD patients or FTLD patients with a definitive pathological diagnosis.
To exclude this kind of clinical bias, 167 non-selective FTLD cases were collected between 1990 and 2006 in five different Alzheimer’s disease research centers and analyzed. The frequency of the GRN mutation was 48%. It was noted that the frequencies of the GRN and MAPT mutations were almost the same; the frequency of the MAPT mutation was 44% in the same series of brains. Further investigation of this similarity will be needed. Of 649 dementia cases collected in Minnesota between 1987 and 2006 as part of a dementia research project, 15 were diagnosed with FTLD. Three patients were identified with the GRN mutation. The frequency of the GRN mutation in the dementia patients overall was calculated to be 0.5%.
Pickering-Brown and colleagues reported that the frequency of the GRN mutation was 7.3% (14 out of 192 FTLD patients) (Pickering-Brown et al. 2008) whereas Le Ber and colleagues reported the frequency to be 6.4% (32 out of 502 FTD patients) (Le Ber et al. 2008). The frequency of GRN mutations in probands was 5.7% (20 of 352) in fvFTD, 4.4% (3 of 68) in primary progressive aphasia (PPA) and 3.3% (1 of 30) in corticobasal syndrome (CBS). The authors also mentioned that no mutations were found in the 52 probands with FTD-MND. Yu et al. found the frequency of the GRN mutation to be 6.9% (30 of 434) (Yu et al. 2010).
Age of Onset
The age of onset of FTLD in Belgian patients with the IVS1+5G>C mutation was 45–70 years (average 63.4 ± 6.8) (Cruts et al. 2006). This mutation was identified in a few asymptomatic individuals; one who had died at 41 years of age, two who had died within the normal age of onset at ages (44 and 54 years) and the one who died at 81. Gass et al. found that the age of onset was 48–83 years (average 59.0 ± 7.0) among GRN mutation carriers over all (Gass et al. 2006). Other studies demonstrated that the average age of FTLD onset was 59.0 ± 5 (Pickering-Brown et al. 2008); 59.4 ± 9.4 in FTD, 62.0 ± 7.9 in FTD-MND, 63.8 ± 8.5 in PPA and 61.8 ± 9.7 in CBS (Le Ber et al. 2008). In another study, the average age of onset was 57.7 years, which was calculated from the onset age of 31 GRN mutation-positive patients from 28 different families (Yu et al. 2010). Leverenz et al. investigated two families with the GRN c.709-2A>G mutation (now termed g.1871A>G (p.Ala237TrpfsX6)) (Leverenz et al. 2007). In family 1, the mean age of onset was 55.6 ± 8.9 years (range = 35–69), the mean age at death was 65.5 ± 6.8 years (range = 56–78) and the mean duration was 9.8 ± 5.5 years (range = 4–22). In family 2, the mean age of onset was 61.0 ± 6.6 years (range = 50–67), the mean age at death was 68.6 ± 6.0 years (range = 57–73) and the mean duration was 6.8 ± 0.4 years (range = 6–7) (Leverenz et al. 2007).
Clinicopathological Images of FTLD
Patients with the GRN IVS1+5G>C mutation show non-fluent aphasia (Cruts et al. 2006). Gass et al. indicated that FTLD patients with the GRN mutation often exhibited dysphasia and this was rarely accompanied by motor neuron dysfunction (Gass et al. 2006). Pathological analysis of GRN IVS1+5G>C patients revealed the presence of neuronal cytoplasmic inclusions (NCIs). Neuronal intranuclear inclusions (NIIs) were also observed in all cases. These observations corresponded with previous reports in which NIIs were commonly detected in familial FTLD patients without motor neuron dysfunction (Mackenzie and Feldman 2003; Woulfe et al. 2001). These results suggested that NIIs would be a pathological marker of PGRN mutation cases. However, NIIs were also found in sporadic FTLD cases or FTLD patients with motor neuron dysfunction, indicating that more investigation will be needed (Mackenzie et al. 2006a). Investigating the clinical response to the GRN mutation, Gass et al. found that the most common diagnosis was FTD followed by PA. Other diagnoses were CBD, AD with convulsions and motor dysfunction (PD, parkinsonism and FTD-MND) (Gass et al. 2006). Snowden and colleagues reported that in a single pedigree of the g.3073C>T (p.Gln468X) mutation, patients showed symptoms of FTD and PA (Snowden et al. 2006). Masellis et al. reported that a patient with the GRN IVS7+1G>A (p.Val200GlyfsX18) mutation exhibited CBD-like symptoms (Masellis et al. 2006). Pickering-Brown et al. reported that in patients with the GRN mutation, 57% were diagnosed as FTD, 36% as PNFA and 7% as apraxia and parkinsonism (Pickering-Brown et al. 2008). Le Ber et al. reported that 63% of patients with the GRN mutation were diagnosed as fvFTD with other clinical patterns being PPA (16%), CBS (6%) and Lewy body disease (LBD) (6%) (Le Ber et al. 2008). They also found that 9% of patients had other diagnoses including AD and parkinsonism (Le Ber et al. 2008). The most common diagnosis was FTD including PPA and CBS. Other clinical phenotypes such as AD, AD+PD and LBD were observed (Yu et al. 2010).
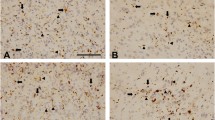
Immunohistochemical analyses for phosphorylated TDP-43 revealed a considerable number of neuronal cytoplasmic inclusions and dystrophic neurites in GRN mutation cases (Fig. 1). In FTLD-TDP, TDP-43 pathology falls within four histological subtypes (types A-D) based on the predominant type of TDP-43-positive structures exhibited (Mackenzie et al. 2011). Type A is characterized by numerous short dystrophic neurites (DNs) and crescentic or oval shaped neuronal cytoplasmic inclusions (NCIs). Cases of FTLD-TDP with a GRN mutation invariably display type A pathology (Cairns et al. 2007; Josephs et al. 2007; Mackenzie et al. 2006b).

Immunohistochemical staining of the temporal lobe of a GRN mutation case with antibody to phosphorylated TDP-43. Numerous neuronal cytoplasmic inclusions (arrows) and dystrophic neurites (arrowheads) were stained with anti-TDP-43-pS409/410 antibody and the section was counterstained with hematoxylin. Scale bar = 100 μm
PGRN Protein Levels in GRN Mutation Carriers
Plasma PGRN protein levels were measured in FTLD patients with the g.1975_1978delCTCA (p.Leu271LeufsX10) mutation or the g.2473 C>T (p.Gln341Arg) mutation and in unaffected individuals with the g.1975_1978delCTCA (p.Leu271LeufsX10) mutation, and in all cases were found to have significantly reduced expression of PGRN (Ghidoni et al. 2008). Plasma PGRN was proposed as a useful biomarker. Sleegers et al. reported that serum PGRN levels were reduced in both affected and unaffected carriers of the PGRN null mutation (IVS1+5G>C) compared with their noncarrier relatives (Sleegers et al. 2009). The authors also measured serum PGRN levels in carriers of the g.1129T>C (p.Cys139Arg) and g.3542C>T (p.Arg564Cys) mutations, and found them to be significantly lower than in controls, but greater than in null mutation carriers. They concluded that the serum PGRN level is a reliable biomarker for diagnosis of FTLD caused by a PGRN null mutation (Sleegers et al. 2009).
Plasma PGRN levels were measured in PGRN loss-of-function mutation carriers, FTLD patients without GRN mutations or symptomatic/asymptomatic GRN mutation carriers (Finch et al. 2009). Pathogenic GRN loss-of-function mutations such as g.26C>A (p.Ala9Asp), g.1098_1101delTAGT (p.Gln130SfsX125), g.2273_2274insTG (p.Trp304LeufsX58fs), g.2450delC (p.Gly333ValfsX28), g.3240C>T (p.Arg493X) and g.3175A>G (p.Ala472_Gln584del) resulted in significantly reduced plasma PGRN levels. Missense mutations (g.2422G>A (p.Ala324Thr)), g.2968C>T (p.Arg433Trp), g.3012C>T (p.His447His) and g.3586G>A (p.Pro578Pro) were associated with plasma PGRN levels equal to those of the controls, but g.55C>T (p.Arg19Trp) and g.1129T>C (p.Cys139Arg) cases showed plasma PGRN levels below the level of detection in controls. These results suggested that g.55C>T (p.Arg19Trp) and g.1129T>C (p.Cys139Arg) mutations might induce a partial loss of PGRN function (Finch et al. 2009). Plasma PGRN levels were also lower than those in carriers of the PGRN g.1A>G (p.Met1), g.1129T>C (p.Cys139Arg), p.Ala89ValfsX41 and p.Ala303AlafsX57 mutations (Gomez-Tortosa et al. 2013).
Mean plasma PGRN levels within the FTLD group were significantly lower in patients with GRN mutations than in those with C9ORF72 expansions, or those without mutation (Gibbons et al. 2015). Meeter and colleagues recently reported that PGRN levels in the plasma and CSF of patients with a loss-of-function GRN mutation (g.366delC (p.Ser82ValfsX174), g.1087C>T (p.Gln125X), g.1102_1105delCAGT (p.Gln130SerfsX125) and g.2902_2903delGT (p.Val411SerfsX2)) and presymptomatic loss-of-function GRN mutation carriers were lower than those of healthy controls (Meeter et al. 2016).
It has been reported that the homozygous carriers of the T-allele of rs5848 have an elevated risk of developing FTD. TT genotype carriers had lower serum PGRN levels than CT or CC carriers (Hsiung et al. 2011). The rs5848 T-allele is known to be a miRNA-659 binding site and rs5848 may enhance translational inhibition of GRN and alter the risk of FTD and other dementias (Hsiung et al. 2011).
The Effect of GRN Mutation and Its Influence on PGRN
The effects of GRN mutation and its influence on PGRN function are as follows.
-
1.
Mutations that introduce a premature termination codon (PTC) induce nonsense-mediated mRNA decay machinery.
-
2.
Mutations in the intron 1 splice-donor site such as IVS1+3A>T and IVS1+5G>C may generate intron 1 read-through mRNA. Such aberrant mRNAs may not be capable of normal transport through the nuclear pore complex, so that they may remain in the nuclear area where they are then liable to be degraded by the nuclear mRNA degradation system.
-
3.
Complete gene deletion such as found in delGRN (Gijselinck et al. 2008) or g.-95_3490del in French patients (Rovelet-Lecrux et al. 2008) may lead to no PGRN at all.
-
4.
Missense mutations in the signal peptide may induce mislocalization of PGRN and insufficient translocation to the endoplasmic reticulum (ER).
-
5.
Missense mutations in other areas may also cause problems. If the mutations exist in the consensus sequence of PGRN, they may be pathological because aberrant protein folding may occur in the ER and reduce PGRN secretion to the extracellular lumen. However, the pathological nature of almost all of them is unknown. The other missense mutations are considered to be benign.
GRN Mutation: Multiple Proteinopathy?
GRN mutations were initially found in tau-negative patients (Baker et al. 2006) (Cruts et al. 2006), but recent findings indicate that these mutations are associated with other neurodegenerative disorders with tau pathology, including AD and CBD. Leverenz et al. found that families with the GRN g.1871A>G (p.Ala237TrpfsX6) mutation had variable clinical presentations such as PD, AD, HD, depression and schizophrenia (Leverenz et al. 2007). Immunohistochemical analyses revealed that six of seven cases had evidence of distinctive tau pathology and two of the seven cases also had α-synuclein pathology (Leverenz et al. 2007).
A reduction in progranulin in tau transgenic mice was associated with an increasing tau accumulation (Hosokawa et al. 2015). A reduction in progranulin in APP transgenic mice was associated with a decrease in Aβ accumulation (Takahashi et al. 2017; Hosokawa et al. 2018).
Human GRN mutation cases were investigated histochemically and biochemically by Hosokawa and colleagues. Results showed a neuronal and glial tau accumulation in 12 of 13 GRN mutation cases (Hosokawa et al. 2017). Tau staining revealed neuronal pretangle forms and glial tau in both astrocytes and oligodendrocytes. Furthermore, phosphorylated α-synuclein-positive structures were also found in oligodendrocytes as well as in the neuropil. Immunoblot analysis of fresh frozen brain tissues revealed that tau and α-synuclein were present in the sarkosyl-insoluble fraction and were composed of three- and four-repeat tau isoforms, resembling those found in AD. These data suggested that PGRN reduction might be the cause of multiple proteinopathies due to the accelerating accumulation of abnormal proteins. These might include TDP-43 proteinopathy, tauopathy and α-synucleinopathy (Hosokawa et al. 2017).
Very recently, Sieben and their colleagues reported that a family with a GRN loss-of-function mutation (IVS1+5G>C) had tau and α-synuclein pathology (Sieben et al. 2018). Of nine members of this family, all were tau-positive and one case had extensive Lewy body pathology. No Aβ pathology or mild accumulation was observed (Sieben et al. 2018).
Recent findings have suggested that different clinical phenotypes may occur in carries of the same GRN mutation and additional tau or α-synuclein accumulation may be observed. It has been also reported that PGRN deficiency causes lysosomal dysfunction (Tanaka et al. 2017). Lysosomal dysfunction may reduce protein degradation in brain cells, allowing aggregation-prone neurodegenerative disease-related proteins to deposit more easily (Hosokawa et al. 2017).
References
Almeida MR et al (2014) Progranulin peripheral levels as a screening tool for the identification of subjects with progranulin mutations in a Portuguese cohort. Neurodegener Dis 13:214–223. https://doi.org/10.1159/000352022
Arai T et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. https://doi.org/10.1016/j.bbrc.2006.10.093
Arosio B et al (2013) GRN Thr272fs clinical heterogeneity: a case with atypical late onset presenting with a dementia with Lewy bodies phenotype. J Alzheimers Dis 35:669–674. https://doi.org/10.3233/JAD-130053
Aswathy PM, Jairani PS, Raghavan SK, Verghese J, Gopala S, Srinivas P, Mathuranath PS (2016) Progranulin mutation analysis: identification of one novel mutation in exon 12 associated with frontotemporal dementia. Neurobiol Aging 39:218 e211–218 e213. https://doi.org/10.1016/j.neurobiolaging.2015.11.026
Baker M et al (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919. https://doi.org/10.1038/nature05016
Barandiaran M et al (2012) Neuropsychological features of asymptomatic c.709-1G>A progranulin mutation carriers. J Int Neuropsychol Soc 18:1086–1090. https://doi.org/10.1017/S1355617712000823
Beck J et al (2008) A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain J Neurol 131:706–720. https://doi.org/10.1093/brain/awm320
Behrens MI et al (2007) Neuropathologic heterogeneity in HDDD1: a familial frontotemporal lobar degeneration with ubiquitin-positive inclusions and progranulin mutation. Alzheimer Dis Assoc Disord 21:1–7. https://doi.org/10.1097/WAD.0b013e31803083f2
Benussi L, Binetti G, Sina E, Gigola L, Bettecken T, Meitinger T, Ghidoni R (2008) A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol Aging 29:427–435. https://doi.org/10.1016/j.neurobiolaging.2006.10.028
Bernardi L et al (2009) Novel PSEN1 and PGRN mutations in early-onset familial frontotemporal dementia. Neurobiol Aging 30:1825–1833. https://doi.org/10.1016/j.neurobiolaging.2008.01.005
Bit-Ivan EN et al (2014) A novel GRN mutation (GRN c.708+6_+9delTGAG) in frontotemporal lobar degeneration with TDP-43-positive inclusions: clinicopathologic report of 6 cases. J Neuropathol Exp Neurol 73:467–473. https://doi.org/10.1097/NEN.0000000000000070
Boeve BF et al (2006) Frontotemporal dementia and parkinsonism associated with the IVS1+1G->A mutation in progranulin: a clinicopathologic study. Brain J Neurol 129:3103–3114. https://doi.org/10.1093/brain/awl268
Borroni B et al (2008) Progranulin genetic variations in frontotemporal lobar degeneration: evidence for low mutation frequency in an Italian clinical series. Neurogenetics 9:197–205. https://doi.org/10.1007/s10048-008-0127-3
Bronner IF et al (2007) Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur J Hum Genet 15:369–374. https://doi.org/10.1038/sj.ejhg.5201772
Brouwers N et al (2007) Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol 64:1436–1446. https://doi.org/10.1001/archneur.64.10.1436
Brouwers N et al (2008) Genetic variability in progranulin contributes to risk for clinically diagnosed. Alzheimer disease. Neurology 71:656–664. https://doi.org/10.1212/01.wnl.0000319688.89790.7a
Bruni AC et al (2007) Heterogeneity within a large kindred with frontotemporal dementia: a novel progranulin mutation. Neurology 69:140–147. https://doi.org/10.1212/01.wnl.0000265220.64396.b4
Cairns NJ et al (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240. https://doi.org/10.2353/ajpath.2007.070182
Calvi A et al (2015) The novel GRN g.1159_1160delTG mutation is associated with behavioral variant frontotemporal dementia. J Alzheimers Dis 44:277–282
Carecchio M et al (2009) Progranulin plasma levels as potential biomarker for the identification of GRN deletion carriers. A case with atypical onset as clinical amnestic Mild Cognitive Impairment converted to Alzheimer’s disease. J Neurol Sci 287:291–293. https://doi.org/10.1016/j.jns.2009.07.011
Carecchio M et al (2014) Evidence of pre-synaptic dopaminergic deficit in a patient with a novel progranulin mutation presenting with atypical parkinsonism. J Alzheimers Dis 38:747–752. https://doi.org/10.3233/JAD-131151
Cerami C, Marcone A, Galimberti D, Villa C, Fenoglio C, Scarpini E, Cappa SF (2013) Novel missense progranulin gene mutation associated with the semantic variant of primary progressive aphasia. J Alzheimers Dis 36:415–420. https://doi.org/10.3233/JAD-130317
Chang KH et al (2018) Genetic and functional characters of GRN p.T487I mutation in Taiwanese patients with atypical parkinsonian disorders. Parkinsonism Relat Disord 51:61–66. https://doi.org/10.1016/j.parkreldis.2018.02.045
Chiang HH et al (2008) Progranulin mutation causes frontotemporal dementia in the Swedish Karolinska family. Alzheimers Dement 4:414–420. https://doi.org/10.1016/j.jalz.2008.09.001
Cioffi SM et al (2016) Non fluent variant of primary progressive aphasia due to the novel GRN g.9543delA(IVS3-2delA) mutation. J Alzheimers Dis 54:717–721. https://doi.org/10.3233/JAD-160185
Clot F et al (2014) Partial deletions of the GRN gene are a cause of frontotemporal lobar degeneration. Neurogenetics 15:95–100. https://doi.org/10.1007/s10048-014-0389-x
Coppola G et al (2008) Gene expression study on peripheral blood identifies progranulin mutations. Ann Neurol 64:92–96. https://doi.org/10.1002/ana.21397
Coppola C et al (2012) A progranulin mutation associated with cortico-basal syndrome in an Italian family expressing different phenotypes of fronto-temporal lobar degeneration. Neurol Sci 33:93–97. https://doi.org/10.1007/s10072-011-0655-8
Cortini F et al (2008) Novel exon 1 progranulin gene variant in Alzheimer’s disease. Eur J Neurol 15:1111–1117. https://doi.org/10.1111/j.1468-1331.2008.02266.x
Cruchaga C et al (2009) Cortical atrophy and language network reorganization associated with a novel progranulin mutation. Cereb Cortex 19:1751–1760. https://doi.org/10.1093/cercor/bhn202
Cruts M et al (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924. https://doi.org/10.1038/nature05017
Cruts M, Theuns J, Van Broeckhoven C (2012) Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33:1340–1344. https://doi.org/10.1002/humu.22117
Cupidi C, Manna I, Navarra V, Vena L, Realmuto S, Cerami C, Quattrone A, Gambardella A, Piccoli F, Piccoli T (2009) Identification of three novel progranulin mutations in a series of patients affected by sporadic and familial frontotemporal lobar degeneration. Alzheimers Demen 5:P406
Davion S et al (2007) Clinicopathologic correlation in PGRN mutations. Neurology 69:1113–1121. https://doi.org/10.1212/01.wnl.0000267701.58488.69
Del Bo R et al (2011) No major progranulin genetic variability contribution to disease etiopathogenesis in an ALS Italian cohort. Neurobiol Aging 32:1157–1158. https://doi.org/10.1016/j.neurobiolaging.2009.06.006
Dopper EG et al (2011) Symmetrical corticobasal syndrome caused by a novel C.314dup progranulin mutation. J Mol Neurosci 45:354–358. https://doi.org/10.1007/s12031-011-9626-z
Finch N et al (2009) Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain J Neurol 132:583–591. https://doi.org/10.1093/brain/awn352
Frangipane FCR, Mirabelli M, Puccia G, Bernardi L, Tomaino C, Anfossi M, Gallo M, Geracitano S, Maletta R, Smirne N, Elder J, Kawarai T, Sato C, Pradella S, Wakutani Y, Dertesz A, St George Hyslop P, Hardy J, Rogaeva E, Momeni P, Bruni AC (2008) A novel progranulin mutation in a large frontotemporal dementia Calabrian kindred. Alzheimers Dement 4:T604
Galimberti DFC, Cortini F, Venturelli E, Guidi I, Scalabrini D, Villa C, Marcone A, Mandelli A, Perini L, Pomati S, Clerici F, Cappa S, Mariani C, Bresolin N, Scarpini E (2008) Progranulin gene mutation scanning in Alzheimer’s disease and frontotemporal lobar degeneration: functional and phenotypic correlations. Alzheimers Dement 4:T585
Gass J et al (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 15:2988–3001. https://doi.org/10.1093/hmg/ddl241
Gazzina S et al (2017) Frontotemporal dementia due to the novel GRN Arg161GlyfsX36 mutation. J Alzheimers Dis 57:1185–1189. https://doi.org/10.3233/JAD-170066
Ghetti B et al (2008) In vivo and postmortem clinicoanatomical correlations in frontotemporal dementia and parkinsonism linked to chromosome 17. Neurodegener Dis 5:215–217. https://doi.org/10.1159/000113706
Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G (2008) Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71:1235–1239. https://doi.org/10.1212/01.wnl.0000325058.10218.fc
Gibbons L et al (2015) Plasma levels of progranulin and interleukin-6 in frontotemporal lobar degeneration. Neurobiol Aging 36:1603 e1601–1604. https://doi.org/10.1016/j.neurobiolaging.2014.10.023
Gijselinck I et al (2008) Progranulin locus deletion in frontotemporal dementia. Hum Mutat 29:53–58. https://doi.org/10.1002/humu.20651
Gomez-Tortosa E et al (2013) Plasma progranulin levels in cortical dementia phenotypes with asymmetric perisylvian atrophy. Eur J Neurol 20:1319–1324. https://doi.org/10.1111/ene.12211
Guerreiro RJ et al (2008) Novel progranulin mutation: screening for PGRN mutations in a Portuguese series of FTD/CBS cases. Move Disord 23:1269–1273. https://doi.org/10.1002/mds.22078
Guerreiro RJ, Washecka N, Hardy J, Singleton A (2010) A thorough assessment of benign genetic variability in GRN and MAPT. Hum Mutat 31:E1126–E1140. https://doi.org/10.1002/humu.21152
Hosaka T, Ishii K, Miura T, Mezaki N, Kasuga K, Ikeuchi T, Tamaoka A (2017) A novel frameshift GRN mutation results in frontotemporal lobar degeneration with a distinct clinical phenotype in two siblings: case report and literature review. BMC Neurol 17:182. https://doi.org/10.1186/s12883-017-0959-2
Hosokawa M et al (2015) Progranulin reduction is associated with increased tau phosphorylation in P301L tau transgenic mice. J Neuropathol Exp Neurol 74:158–165. https://doi.org/10.1097/NEN.0000000000000158
Hosokawa M et al (2017) Accumulation of multiple neurodegenerative disease-related proteins in familial frontotemporal lobar degeneration associated with granulin mutation. Sci Rep 7:1513. https://doi.org/10.1038/s41598-017-01587-6
Hosokawa M, Tanaka Y, Arai T, Kondo H, Akiyama H, Hasegawa M (2018) Progranulin haploinsufficiency reduces amyloid beta deposition in Alzheimer’s disease model mice. Exp Anim 67:63–70. https://doi.org/10.1538/expanim.17-0060
Hsiung GY, Fok A, Feldman HH, Rademakers R, Mackenzie IR (2011) rs5848 polymorphism and serum progranulin level. J Neurol Sci 300:28–32. https://doi.org/10.1016/j.jns.2010.10.009
Huey ED et al (2006) Characteristics of frontotemporal dementia patients with a Progranulin mutation. Ann Neurol 60:374–380. https://doi.org/10.1002/ana.20969
Hutton M et al (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. https://doi.org/10.1038/31508
Jin SC et al (2012) Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease. Ibero-American cohort. Alzheimers Res Ther 4:34. https://doi.org/10.1186/alzrt137
Josephs KA et al (2007) Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol 66:142–151. https://doi.org/10.1097/nen.0b013e31803020cf
Karch CM et al (2016) Missense mutations in progranulin gene associated with frontotemporal lobar degeneration: study of pathogenetic features. Neurobiol Aging 38:215 e211–215 e212. https://doi.org/10.1016/j.neurobiolaging.2015.10.029
Kelley BJ et al (2009) Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol Aging 30:739–751. https://doi.org/10.1016/j.neurobiolaging.2007.08.022
Kelley BJ et al (2010) Alzheimer disease-like phenotype associated with the c.154delA mutation in progranulin. Arch Neurol 67:171–177. https://doi.org/10.1001/archneurol.2010.113
Kim G et al (2016) Asymmetric pathology in primary progressive aphasia with progranulin mutations and TDP inclusions. Neurology 86:627–636. https://doi.org/10.1212/WNL.0000000000002375
Kuuluvainen L et al (2017) A novel loss-of-function GRN mutation p.(Tyr229*): clinical and neuropathological features. J Alzheimers Dis 55:1167–1174. https://doi.org/10.3233/JAD-160647
Le Ber I et al (2007) Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat 28:846–855. https://doi.org/10.1002/humu.20520
Le Ber I et al (2008) Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain J Neurol 131:732–746. https://doi.org/10.1093/brain/awn012
Lee JH et al (2014) Disease-related mutations among Caribbean Hispanics with familial dementia. Mol Genet Genomic Med 2:430–437. https://doi.org/10.1002/mgg3.85
Leverenz JB et al (2007) A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain J Neurol 130:1360–1374. https://doi.org/10.1093/brain/awm069
Lindquist SG, Schwartz M, Batbayli M, Waldemar G, Nielsen JE (2009) Genetic testing in familial AD and FTD: mutation and phenotype spectrum in a Danish cohort. Clin Genet 76:205–209. https://doi.org/10.1111/j.1399-0004.2009.01191.x
Lladó A et al (2007) Late-onset frontotemporal dementia associated with a novel PGRN mutation. J Neural Transm 114:1051–1054. https://doi.org/10.1007/s00702-007-0716-6
López de Munain A et al (2008) Mutations in progranulin gene: clinical, pathological, and ribonucleic acid expression findings. Biol Psychiatry 63:946–952. https://doi.org/10.1016/j.biopsych.2007.08.015
Luzzi S et al (2017) Missense mutation in GRN gene affecting RNA splicing and plasma progranulin level in a family affected by frontotemporal lobar degeneration. Neurobiol Aging 54:214 e211–214 e216. https://doi.org/10.1016/j.neurobiolaging.2017.02.008
Mackenzie IR, Feldman H (2003) Neuronal intranuclear inclusions distinguish familial FTD-MND type from sporadic cases. Acta Neuropathol 105:543–548. https://doi.org/10.1007/s00401-003-0678-1
Mackenzie IR et al (2006a) Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol 112:539–549. https://doi.org/10.1007/s00401-006-0138-9
Mackenzie IR et al (2006b) The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain J Neurol 129:3081–3090. https://doi.org/10.1093/brain/awl271
Mackenzie IR, Shi J, Shaw CL, Duplessis D, Neary D, Snowden JS, Mann DM (2006c) Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol 112:551–559. https://doi.org/10.1007/s00401-006-0123-3
Mackenzie IR et al (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122:111–113. https://doi.org/10.1007/s00401-011-0845-8
Mao Q et al (2017) Disease and region specificity of granulin immunopositivities in Alzheimer disease and frontotemporal lobar degeneration. J Neuropathol Exp Neurol 76:957–968. https://doi.org/10.1093/jnen/nlx085
Maquat LE (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5:89–99
Marcon G et al (2011) Variability of the clinical phenotype in an Italian family with dementia associated with an intronic deletion in the GRN gene. J Alzheimers Dis 26:583–590. https://doi.org/10.3233/JAD-2011-110332
Masellis M et al (2006) Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain J Neurol 129:3115–3123. https://doi.org/10.1093/brain/awl276
McKhann GM et al (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 58:1803–1809
Meeter LH, Patzke H, Loewen G, Dopper EG, Pijnenburg YA, van Minkelen R, van Swieten JC (2016) Progranulin levels in plasma and cerebrospinal fluid in Granulin mutation carriers. Dement Geriatr Cogn Disord Extra 6:330–340. https://doi.org/10.1159/000447738
Mendez MF (2018) Manic behavior and asymmetric right frontotemporal dementia from a novel progranulin mutation. Neuropsychiatr Dis Treat 14:657–662. https://doi.org/10.2147/NDT.S156084
Mesulam M et al (2007) Progranulin mutations in primary progressive aphasia: the PPA1 and PPA3 families. Arch Neurol 64:43–47. https://doi.org/10.1001/archneur.64.1.43
Moreno F et al (2009) “Frontotemporoparietal” dementia: clinical phenotype associated with the c.709-1G>A PGRN mutation. Neurology 73:1367–1374. https://doi.org/10.1212/WNL.0b013e3181bd82a7
Mukherjee O et al (2006) HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol 60:314–322. https://doi.org/10.1002/ana.20963
Mukherjee O et al (2008) Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum Mutat 29:512–521. https://doi.org/10.1002/humu.20681
Neary D et al (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Neumann M et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. https://doi.org/10.1126/science.1134108
Nuytemans K et al (2008) Progranulin variability has no major role in Parkinson disease genetic etiology. Neurology 71:1147–1151. https://doi.org/10.1212/01.wnl.0000327563.10320.2b
Piaceri I et al (2018) Novel GRN mutations in Alzheimer’s disease and frontotemporal lobar degeneration. J Alzheimers Dis 62:1683–1689. https://doi.org/10.3233/JAD-170989
Pickering-Brown SM et al (2006) Mutations in progranulin explain atypical phenotypes with variants in MAPT. Brain J Neurol 129:3124–3126. https://doi.org/10.1093/brain/awl289
Pickering-Brown SM et al (2008) Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 131:721–731. https://doi.org/10.1093/brain/awm331
Pietroboni AM et al (2011) Phenotypic heterogeneity of the GRN Asp22fs mutation in a large Italian kindred. J Alzheimers Dis 24:253–259. https://doi.org/10.3233/JAD-2011-101704
Pires C et al (2013) Phenotypic variability of familial and sporadic Progranulin p.Gln257Profs*27 mutation. J Alzheimers Dis 37:335–342
Rademakers R et al (2007) Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C-->T (Arg493X) mutation: an international initiative. Lancet Neurol 6:857–868. https://doi.org/10.1016/S1474-4422(07)70221-1
Redaelli V et al (2018) Alzheimer neuropathology without frontotemporal lobar degeneration hallmarks (TAR DNA-binding protein 43 inclusions) in missense progranulin mutation Cys139Arg. Brain Pathol 28:72–76. https://doi.org/10.1111/bpa.12480
Rohrer JD et al (2008) Parietal lobe deficits in frontotemporal lobar degeneration caused by a mutation in the progranulin gene. Arch Neurol 65:506–513. https://doi.org/10.1001/archneur.65.4.506
Rohrer JD et al (2009) Corticobasal syndrome associated with a novel 1048_1049insG progranulin mutation. J Neurol Neurosurg Psychiatry 80:1297–1298. https://doi.org/10.1136/jnnp.2008.169383
Rohrer JD, Crutch SJ, Warrington EK, Warren JD (2010a) Progranulin-associated primary progressive aphasia: a distinct phenotype? Neuropsychologia 48:288–297. https://doi.org/10.1016/j.neuropsychologia.2009.09.017
Rohrer JD et al (2010b) Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. NeuroImage 53:1070–1076. https://doi.org/10.1016/j.neuroimage.2009.12.088
Rossi G et al (2011) A novel progranulin mutation causing frontotemporal lobar degeneration with heterogeneous phenotypic expression. J Alzheimers Dis 23:7–12. https://doi.org/10.3233/JAD-2010-101461
Rovelet-Lecrux A et al (2008) Deletion of the progranulin gene in patients with frontotemporal lobar degeneration or Parkinson disease. Neurobiol Dis 31:41–45. https://doi.org/10.1016/j.nbd.2008.03.004
Sassi C et al (2014) Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer’s disease. Neurobiol Aging 35:2881 e2881–2881 e2886. https://doi.org/10.1016/j.neurobiolaging.2014.06.002
Sassi C et al (2016) A novel splice-acceptor site mutation in GRN (c.709-2 A>T) causes frontotemporal dementia Spectrum in a large family from southern Italy. J Alzheimers Dis 53:475–485. https://doi.org/10.3233/JAD-151170
Schlachetzki JC, Schmidtke K, Beckervordersandforth J, Borozdin W, Wilhelm C, Hull M, Kohlhase J (2009) Frequency of progranulin mutations in a German cohort of 79 frontotemporal dementia patients. J Neurol 256:2043–2051. https://doi.org/10.1007/s00415-009-5248-6
Schymick JC et al (2007) Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry 78:754–756. https://doi.org/10.1136/jnnp.2006.109553
Sieben A et al (2018) Extended FTLD pedigree segregating a Belgian GRN-null mutation: neuropathological heterogeneity in one family. Alzheimers Res Ther 10:7. https://doi.org/10.1186/s13195-017-0334-y
Skoglund L, Englund E, Ingelsson M, Lannfelt L, Passant U, Glaser A (2007) Mutation analysis of the progranulin gene in a Scandinavian frontotemporal dementia population. Neurodegener Dis 4:38
Skoglund L et al (2009) Frontotemporal dementia in a large Swedish family is caused by a progranulin null mutation. Neurogenetics 10:27–34. https://doi.org/10.1007/s10048-008-0155-z
Sleegers K et al (2008) Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 71:253–259. https://doi.org/10.1212/01.wnl.0000289191.54852.75
Sleegers K et al (2009) Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol 65:603–609. https://doi.org/10.1002/ana.21621
Snowden JS, Pickering-Brown SM, Mackenzie IR, Richardson AM, Varma A, Neary D, Mann DM (2006) Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain J Neurol 129:3091–3102. https://doi.org/10.1093/brain/awl267
Spina S, Murrell JR, Huey ED, Wassermann EM, Pietrini P, Grafman J, Ghetti B (2007) Corticobasal syndrome associated with the A9D Progranulin mutation. J Neuropathol Exp Neurol 66:892–900. https://doi.org/10.1097/nen.0b013e3181567873
Spina SMJ, Vidal R, Ghetti B (2008) Neuropathologic and genetic characterization of frontotemporal lobar degeneration with Ubiquitin- and/or Tdp-43-positive inclusions: a large series. Alzheimers Dement 4:T431
Takahashi H et al (2017) Opposing effects of progranulin deficiency on amyloid and tau pathologies via microglial TYROBP network. Acta Neuropathol 133:785–807. https://doi.org/10.1007/s00401-017-1668-z
Tanaka Y et al (2017) Progranulin regulates lysosomal function and biogenesis through acidification of lysosomes. Hum Mol Genet 26:969–988. https://doi.org/10.1093/hmg/ddx011
Tremolizzo L et al (2009) Higher than expected progranulin mutation rate in a case series of Italian FTLD patients. Alzheimer Dis Assoc Disord 23:301. https://doi.org/10.1097/WAD.0b013e31819e0cc5
Van Deerlin VM et al (2007) Clinical, genetic, and pathologic characteristics of patients with frontotemporal dementia and progranulin mutations. Arch Neurol 64:1148–1153. https://doi.org/10.1001/archneur.64.8.1148
van der Zee J et al (2007) Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat 28:416. https://doi.org/10.1002/humu.9484
Wilkie AO (1994) The molecular basis of genetic dominance. J Med Genet 31:89–98
Wong SH, Lecky BR, Steiger MJ (2009) Parkinsonism and impulse control disorder: presentation of a new progranulin gene mutation. Move Dis 24:618–619. https://doi.org/10.1002/mds.22429
Woulfe J, Kertesz A, Munoz DG (2001) Frontotemporal dementia with ubiquitinated cytoplasmic and intranuclear inclusions. Acta Neuropathol 102:94–102
Yu CE et al (2010) The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol 67:161–170. https://doi.org/10.1001/archneurol.2009.328
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Hosokawa, M., Arai, T. (2019). Progranulin and Frontotemporal Lobar Degeneration. In: Hara, H., Hosokawa, M., Nakamura, S., Shimohata, T., Nishihara, M. (eds) Progranulin and Central Nervous System Disorders. Springer, Singapore. https://doi.org/10.1007/978-981-13-6186-9_3
Download citation
DOI: https://doi.org/10.1007/978-981-13-6186-9_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-6185-2
Online ISBN: 978-981-13-6186-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)