
Abstract
Frontotemporal lobar degeneration (FTLD) recognises high familial incidence, with up to 50% of patients reported to have a family history of similar dementia. It has been reported that mutations within progranulin (PGRN) gene are a major cause of FTLD in the USA and worldwide, counting for 5–10% of FTLD and for 20–25% of familiar FTLD cases. The aim of the present study was to define the role of PGRN genetic variations in a large sample of consecutive patients with FTLD in Italy. Two-hundred forty-three FTLD patients were investigated. Each subject performed a clinical and neuropsychological evaluation, a functional and structural brain imaging, and the diagnosis was confirmed by at least 1 year follow-up. PGRN sequencing was performed in all FTLD patients and in 121 healthy age-matched controls drawn from the same geographic area. Only one PGRN pathogenetic mutation was found, consisting of a four-base pair deletion in the coding sequence of exon 8 (delCACT). This mutation was recognised in four patients, being the overall frequency of mutations in our clinical series of 1.64%. Considering only patients with a well-known family history for dementia, the frequency of this mutation was 6%. Moreover, four missense mutations within intron regions (g.100474G>A, g.100674G>A, g.101266G>A, g.102070G>A) were found. The frequency of these genetic variations did not differ in patients compared to controls, and they did not influence on clinical FTLD phenotype. In conclusion, this study supports a lower frequency of PGRN mutations amongst FTLD patients in Italy compared to literature data and further underlies the genetic heterogeneity of FTLD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Frontotemporal lobar degeneration (FTLD) is the overall term for a group of neurodegenerative diseases that accounts for 5–10% of all dementias and between 10–20% of dementia in patients with onset before 65 years of age [1].
The behavioural variant FTD (bvFTD), semantic dementia (SD), and progressive non-fluent aphasia (PNFA) represent the most frequently recognised clinical syndromes [2, 3]. Progressive supranuclear palsy (PSP) and corticobasal degeneration syndrome (CBDS) are also considered under the same label of FTLD because they overlap both clinically and neuropathologically [4].
On the basis of immunohistochemical staining and distribution of intracellular inclusions, FTLD is subgrouped into tau-positive pathology and ubiquitin-positive but tau negative pathology [1].
FTLD is a genetically complex disorder with multiple genetic factors contributing to the disease. A positive family history of dementia is found in 40% of FTLD patients. Genetic linkage studies have revealed FTLD loci and genes on chromosome 3p [5], chromosome 9q [6], chromosome 9p (two loci) [7–9] and chromosome 17q (two loci) [1, 10].
In 1998, the microtubule associated protein tau gene (MAPT) [MIM#157140] mapping on chromosome 17q21 was identified as causative of FTLD in several families [11, 12]. At autopsy, MAPT mutation carriers consistently showed extensive tau pathology [13].
Over the years, however, evidence accumulated for the presence of a second gene at 17q21 involved in FTLD [13].
Only recently, it has been demonstrated that heterogeneity at 17q21 locus was explained by the fact that mutations were identified within progranulin (PGRN) gene [MIM#138945] located close to MAPT. PGRN mutations are associated with ubiquitin-positive tau-negative FTLD cases [14, 15].
PGRN is a secreted factor involved in tissue remodelling, wound repair and inflammation [16]. In the brain, where PGRN is expressed in both neurons and microglia, the functions have not been studied extensively. However, the evidence that reduced concentrations of PGRN can lead to neurodegeneration in FTLD implicates PGRN in neuronal survival [14, 15].
Recent studies have shown that PGRN mutations are a major cause of FTLD in the USA and worldwide, accounting for 5–10% of FTLD and for 20–25% of familial FTLD cases [17, 18]. Up to now, almost 50 pathogenetic PGRN mutations have been described, and all are expected to cause PGRN haploinsufficiency (Alzheimer Disease and Frontotemporal Dementia Database. http://www.molgen.ua.ac.be/FTDmutations/).
The role of PGRN mutations in Italy is still unknown. Only one study is currently available on 78 Italian FTLD, reporting an overall frequency of 1.3%, thus lower than expected [19].
These observations prompted the present study aimed at establishing a PGRN mutation role in determining FTLD in an Italian clinical series. For this purpose, we included a large sample of patients covering all FTLD spectrum.
Materials and methods
Subjects
This work is part of an ongoing study aimed at evaluating the genetic and environmental determinants of FTLD. Patients were recruited from the “Centre for Ageing Brain and Neurodegenerative Disorders”, University of Brescia (n = 237), from the Neurology Unit, University of Milan (n = 6), Italy. These centres are located in Lumbardy, in the Northern Italy.
The Neary and McKhann for FTLD were fulfilled by all subjects [2, 3]. Inclusion and exclusion criteria were previously reported [20].
All subjects underwent a somatic and neurological evaluation and routine laboratory examination, a brain structural magnetic resonance imaging (MRI) study and a brain functional single photon emission tomography (SPECT) study.
Patients considered to have a positive family history were those who had a first-degree relative with dementia, parkinsonism or motor neuron disease. No patients belonging to the same family were included. Patients with family history underwent screening for MAPT mutations, which were excluded (data not shown). Demographic and comorbidities were carefully recorded. Collection of venous blood sample were drawn from each patient for PGRN sequencing.
The diagnostic assessment involved a review of full medical history, a semi-structured neurological examination and a complete mental status evaluation by at least two independent and experienced reviewers (B.B., C.A., E.S., A.P.). Only patients with full consensus agreement by the reviewers were enrolled.
A standardised neuropsychological assessment including global cognitive evaluation and a wide test battery for investigating the main cognitive domains was performed.
Moreover, a control group similar in age and gender composition was recruited in the same Italian area from which the patients were drawn. All controls were found to be cognitively intact, following medical history, presence of comorbidities and neuropsychological examination as well as PGRN genotyping.
Informed consent was obtained for blood collection from venous puncture and genetic analysis from each subject. The work was conformed to the Helsinki Declaration and was approved by local Ethic Committee of our hospital.
Progranulin sequencing
Total genomic DNA was prepared from peripheral blood according to standard procedures. All the 12 exons plus exon 0 of PGRN and at least 30 base pairs (bp) of their flanking introns were evaluated by polymerase chain reaction (PCR). PCR primers were designed to optimise denaturing high-performance liquid chromatography (dHPLC) conditions following previously provided primer pairs (see Appendix 1) [14, 15].
All PCR programs were established using a Touchdown approach, the annealing temperature ranging from 58°C to 66°C.
Preliminary dHPLC analysis was performed on the Wave® nucleic acid fragment analysis system (Transgenomic, Santa Clara, CA, USA), and samples with an altered dHPLC profile were purified with Microcon Centrifugal filter devices (Amicon Bioseparation, Millipore) and sequenced. Sequencing was performed in duplicate, from purified PCR on the 310 DNA sequencer ABI Prism (Applera Biosystems, Italy), according to the manufacturer’s instructions. Sequences were compared with those available at public databases.
The numbering of reported genetic variations within PGRN sequence is relative to the reverse complement of GeneBank accession number AC003043 and starting at nucleotide 1. The numbering of predicted RNA is relative to the largest PGRN transcript (GeneBank Accession Number NM002087.2 and starting at translation initiation codon), and the numbering of predicted protein is relative to the largest PGRN isoform (GeneBank Accession Number NP002078.1, exon numbering starts with non-coding first Exon 0). Automated splice site analyses were conducted according to https://splice.cmh.edu [21].
Haplotype analyses were conducted by using microsatellite markers as reported in Appendix 2.
Statistical analysis
One-way analyses of variance and chi-square tests were performed for socio-demographic and clinical characteristics of patients and control subjects included in the study. Results are expressed as mean ± standard deviation (SD). The significant level was established at P < 0.05. Data analyses were carried out using SPSS 13.0 software (http://www.spss.com).
Haplotype frequency estimation was calculated by using Arlequin 2000 and Phase 2.1.
Results
Subjects
Two-hundred forty-three FTLD patients, i.e. 110 bvFTD, 13 SD, 10 PNFA, 53 CBDS and 57 PSP, entered the study. One-hundred twenty-one healthy controls were recruited as well. The number of control sample was established on the basis of PGRN polymorphism frequencies found in FTLD patients.
Demographic and clinical characteristics of FTLD patients and healthy controls are shown in Table 1.
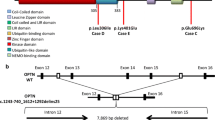
Sequencing of PGRN gene was performed in all FTDL patients. Table 2 shows the PGRN genetic variations in the studied population. Five PGRN genetic variations were detected (see Fig. 1). Four previously reported [17, 22] polymorphisms within intron regions (g.100474G>A, g.100674G>A, g.101266G>A, g.102070G>A) and a four-base pair deletion in the coding sequence of exon 8 (g.102039 delCACT) were found.

PGRN mutations identified in FTLD patients. Schematic representation of the PGRN gene showing the PGRN mutations identified in the Italian population. Mutations are numbered relative to the largest PGRN transcript (GenBank accession number NM_002087.2). IVS intron, Ex exon
Intronic genetic variations within PGRN gene
As shown in Table 2, the prevalence of the four intronic PGRN genetic variations did not differ in FTLD patients and controls.
The IVS2+21 G>A genetic variation showed a low incidence both in FTLD patients (1.64%) and in controls (0.82%). The IVS8+7 G>A mutation prevalence ranged from 3.30% in controls to 4.93% in FTLD, and the IVS3+23 G>A was of 5.8% in both groups. The IVS4+24 G>A genetic variation had a higher prevalence in the overall studied population, being found in 20–25% of subjects.
Despite the comparable prevalence of these genetic variations in both FTLD patients and controls, the automated splice site analysis suggested a decrease in binding site (75.8%) in IVS8+7 G>A and an abolishment of the binding site due to IVS4+24 G>A polymorphism (see Table 2). Thus, further analyses in patients carrying these genetic variations were performed. No significant differences in age at onset was found between FTLD patients carrying no polymorphism (n = 166, 64.0 ± 9.8) compared either to FTLD patients with IVS8+7 G>A polymorphism (65.3 ± 7.2) or to FTLD patients with IVS4+24 G>A polymorphism (66.6 ± 9.0). No differences in gender and family history were found as well.
We can therefore conclude that these polymorphisms do not have any pathogenetic significance in FTLD.
Exon 8 delCACT mutation
Exon 8 delCACT deletion causes a frameshift at codon 272 that introduces a premature termination codon after a read through of ten residues (Tyr272SerfsX10). The resultant mutant protein is predicted to be composed of 282 amino acid in length, instead of the 593 residues of the wild-type PGRN. This deletion was found in four nuclear families and was not present in control subjects. The pathogenetic mutation was previously described in other two families from Italy, in the same county where the present sample was drawn [23].
In our population, only this pathogenetic mutation was found, being the overall prevalence of 1.64 (4/243). Excluding patients with PSP, as PGRN mutations have never been described; the overall prevalence was 2.1% (4/186). Considering only patients with a well-known family history for dementia, the prevalence of this mutation was 6% (4/84).
The pedigrees are reported in Fig. 2. Three of them showed a positive family history; one had no other known affected siblings. The four delCACT index cases have a significant allele sharing (P < 1 × 10−5) demonstrating the same ancestral haplotype (see Table 3).

Pedigree of the four probands carrying PGRN exon 8 delCACT mutation. Three out of four showed a positive family history for dementia
In Table 4, the demographic and clinical characteristics of the four patients are reported. The mean age at onset was 56.2 ± 5.2; two of them were diagnosed as having bvFTD, while the other two patients had a diagnosis of PNFA, demonstrating a clinical variability within the same PGRN mutation. Additionally, in the four delCACT patients, trans alleles are different, and they are not related to the age at onset. The cerebral SPECT perfusion patterns were highly variable as well, but highlighting frontotemporal hypoperfusion (see Fig. 3).

Hypoperfusion pattern in the four FTLD carriers of PGRN Exon 8 delCACT mutation. Each patient was compared to 15 healthy age-matched controls, and the analysis was carried out by statistical parametric mapping (SPM2)
Discussion
The present study aimed at addressing the role of PGRN mutations in a large clinical series of Italian patients affected by FTLD, as represented by different variants, including FTD, PSP, CBDS, and overall 40% with a positive family history.
Recent identification of PGRN as the gene responsible for FTLD with ubiquitin-positive brain pathology linked to chromosome 17 has contributed significantly to our understanding of the genetic etiology of FTLD. Most PGRN mutations reported to date are missense, splice site or frameshift substitutions that lead to loss of mutant transcript and thus functional protein [14, 15].
In our clinical series, we found a previously described four-base pair deletion in exon 8, defining the overall genetic contribution of PGRN mutations of 1.64% (4/243). The haplotype analysis of the four nuclear families carrying the frameshift exon 8 delCACT mutation demonstrated a common founder. No other missense or splice site pathogenic mutations has been identified in our sample.
In comparison to previously published results, the frequency of PGRN pathogenetic mutations is much lower than expected. In fact, previous epidemiologic studies showed PGRN being causative of FTLD in 5–11% of cases in large series worldwide [17, 18, 22]. The frequency in our sample was still low when only familial cases were considered; we observed a frequency of 6% (4/84) compared to 20–25% from literature data [17, 18, 22].
Such a discrepancy could count for a different patient selection; the lack of neuropathological confirmation in our sample might decrease the quote of ubiquitin-positive cases that were highly represented in the already reported clinical series (about 30%), and therefore, we cannot exclude that future brain autopsy studies may contribute to further define the genetic aetiology of the disease. Indeed, in a previous work of pathologically confirmed ubiquitin-positive cases, the frequency of PGRN-related FTLD raised to 60–70% [17].
Our FTLD patients were carefully clinically characterised; they underwent structural and functional brain imaging and had a follow-up evaluation to further confirm the diagnosis. We are confident that our patients are clinically FTLD, but we are aware that tau-positive patients have been included. It also might be considered that dHPLC screening of PGRN does not take into account the possibility to detect a genomic macrodeletion, as it has been recently identified [24]; however, as 40% of our sample shows heterozygosis for PGRN polymorphic variants, we may at least deduce that this portion of patients cannot present a macrodeletion within the region comprising PGRN.
In accordance to current data, a previous study performed in a sample of Southern Italy showed comparable prevalence of PGRN pathogenetic mutation [20]. The authors studied 78 FTLD patients, and they found a novel truncating PGRN mutation (c.1145insA) in a proband from an extended consanguineous kindred, the overall prevalence being about 1%. Indeed, a possible genetic gradient from the North to the South of Europe might be addressed for such a difference, but further studies are warranted. In the same view, it has been observed that also MAPT mutations have a much low prevalence in Italian FTLD patients [25].
The identified four-base pairs frameshift mutation has been already described as pathogenic [23]; functional molecular data are not yet available, but as the mutation causes a premature stop codon and the predicted protein is 282 amino acid in length, it is plausible to consider a deficit in PGRN production and secretion as the possible mechanism responsible for the disease in analogy to other PGRN mutations.
The clinical and neuropsychological features of our mutated patients confirmed the heterogeneity of FTLD associated to the same PGRN mutation. This variability was also supported by imaging data (see Fig. 3). Indeed, although defined by a peculiar ubquitin-positive and tau-negative neuropathological hallmark, PGRN pathogentic variations seem to be responsible of a wide spectrum of clinical phenotypes [26, 27]. The number of reports providing clinical details of patients belonging to the same family or carrying the same mutation is growing rapidly.
The reason of these different clinical presentations due to the same PGRN mutation is still unknown, but it resembles the heterogeneity seen both within families and among families having identical mutations in MAPT as well [28].
Notwithstanding, it is worth to note the gender specificity in phenotype in our patients: the males presented with PNFA, whilst females with bvFTD. PGRN’s role in gender-specific brain development has been previously highlighted [29] and may suggest gender-based differences in presentation, but it warrants further study.
In addition, we firstly included PSP patients, not represented in other PGRN mutation series, to evaluate whether this extrapyramidal syndrome, which belongs to the FTLD spectrum, might share a common genetic mechanism. The absence of PGRN pathogenetic mutations in patients resembling PSP phenotype supports the view of a different genetic background for this disease. Therefore, also excluding patients with PSP, in our clinical series, the overall frequency of pathogenetic PGRN mutations was still low, being 2.1% (4/186).
Finally, we found four intronic genetic variations, the pathogenic significance being evaluated by comparison with healthy controls. The polymorphism analysis in control individuals allowed us to demonstrate that these genetic variations were comparable in frequency in both FTLD and control subjects. This observation indicates that the natural genetic variability of PGRN is high and that the pathogenetic significance strictly depends on the impact on protein structure and stability. In fact, the reported frequencies of polymorphic variations in PGRN gene were similar to previous data, and the analysis of the clinical phenotypes in polymorphism carriers and not-carriers showed comparable results.
In conclusion, the current study further confirms the genetic heterogeneity of FTLD and suggests that PGRN does not represent a major disease cause in these Italian clinical series.
Abbreviations
- bvFTD:
-
behavioural variant frontotemporal dementia
- CBDS:
-
corticobasal degeneration syndrome
- FTLD:
-
frontotemporal lobar degeneration
- SD:
-
semantic dementia
- MAPT:
-
microtubule-associated protein tau
- PGRN:
-
progranulin
- PNFA:
-
progressive non-fluent aphasia
- PSP:
-
progressive supranuclear palsy
References
Spillantini MG, Van Swieten JC, Goedert M (2000) Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Neurogenetics 2(4):193–205
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51(6):1546–1554
McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ, Work Group on Frontotemporal Dementia and Pick’s Disease (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 58(11):1803–1809
Kertesz A (2003) Pick’s complex and FTDP-17. Mov Disord 6:S57–62
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sørensen SA, Gydesen S, Fisher EM, Collinge J (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37(8):806–808
Hosler BA, Siddique T, Sapp PC, Sailor W, Huang MC, Hossain A, Daube JR, Nance M, Fan C, Kaplan J, Hung WY, McKenna-Yasek D, Haines JL, Pericak-Vance MA, Horvitz HR, Brown RH Jr (2000) Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. JAMA 284(13):1664–1669
Morita M, Al-Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, Mitchell JE, Habgood JJ, de Belleroche J, Xi J, Jongjaroenprasert W, Horvitz HR, Gunnarsson LG, Brown RH Jr (2006) A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 66(6):839–844
Vance C, Al-Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, Baas F, de Jong V, Shaw CE (2006) Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain 129(Pt 4):868–876
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36(4):377–381
van der Zee J, Rademakers R, Engelborghs S, Gijselinck I, Bogaerts V, Vandenberghe R, Santens P, Caekebeke J, De Pooter T, Peeters K, Lübke U, Van den Broeck M, Martin JJ, Cruts M, De Deyn PP, Van Broeckhoven C, Dermaut B (2006) A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21-linked tau-negative FTLD. Brain 129(Pt 4):841–852
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A 95(13):7737–7741
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393(6686):702–705
Rademakers R, Cruts M, van Broeckhoven C (2004) The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat 24(4):277–295
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442(7105):916–919
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442(7105):920–924
Eriksen JL, Mackenzie IR (2008) Progranulin: normal function and role in neurodegeneration. J Neurochem 104:287–297
van der Zee J, Le Ber I, Maurer-Stroh S, Engelborghs S, Gijselinck I, Camuzat A, Brouwers N, Vandenberghe R, Sleegers K, Hannequin D, Dermaut B, Schymkowitz J, Campion D, Santens P, Martin JJ, Lacomblez L, De Pooter T, Peeters K, Mattheijssens M, Vercelletto M, Van den Broeck M, Cruts M, De Deyn PP, Rousseau F, Brice A, Van Broeckhoven C (2007) Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat 28(4):416
Le Ber I, van der Zee J, Hannequin D, Gijselinck I, Campion D, Puel M, Laquerrière A, De Pooter T, Camuzat A, Van den Broeck M, Dubois B, Sellal F, Lacomblez L, Vercelletto M, Thomas-Antérion C, Michel BF, Golfier V, Didic M, Salachas F, Duyckaerts C, Cruts M, Verpillat P, Van Broeckhoven C, Brice A, French Research Network on FTD/FTD-MND (2007) Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat 28(9):846–855
Bruni AC, Momeni P, Bernardi L, Tomaino C, Frangipane F, Elder J, Kawarai T, Sato C, Pradella S, Wakutani Y, Anfossi M, Gallo M, Geracitano S, Costanzo A, Smirne N, Curcio SA, Mirabelli M, Puccio G, Colao R, Maletta RG, Kertesz A, St George-Hyslop P, Hardy J, Rogaeva E (2007) Heterogeneity within a large kindred with frontotemporal dementia: a novel progranulin mutation. Neurology 69(2):140–147
Borroni B, Grassi M, Agosti C, Paghera B, Alberici A, Di Luca M, Perani D, Padovani A (2007) Latent profile analysis in frontotemporal lobar degeneration and related disorders: clinical presentation and SPECT functional correlates. BMC Neurol 7:9
Nalla VK, Rogan PK (2005) Automated splicing mutation analysis by information theory. Hum Mutat 25(4):334–342
Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, Josephs K, Pickering-Brown SM, Graff-Radford N, Uitti R, Dickson D, Wszolek Z, Gonzalez J, Beach TG, Bigio E, Johnson N, Weintraub S, Mesulam M, White CL 3rd, Woodruff B, Caselli R, Hsiung GY, Feldman H, Knopman D, Hutton M, Rademakers R (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 15(20):2988–3001
Benussi L, Binetti G, Sina E, Gigola L, Bettecken T, Meitinger T, Ghidoni R (2008) A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol Aging 29(3):427–435
Gijselinck I, van der Zee J, Engelborghs S, Goossens D, Peeters K, Mattheijssens M, Corsmit E, Del-Favero J, De Deyn PP, van Broeckhoven C, Cruts M (2008) Progranulin locus deletion in frontotemporal dementia. Hum Mut 29:53–58
Binetti G, Nicosia F, Benussi L, Ghidoni R, Feudatari E, Barbiero L, Signorini S, Villa A, Mattioli F, Zanetti O, Alberici A (2003) Prevalence of TAU mutations in an Italian clinical series of familial frontotemporal patients. Neurosci Lett 338(1):85–87
Kelley BJ, Haidar W, Boeve BF, Baker M, Graff-Radford NR, Krefft T, Frank AR, Jack CR Jr, Shiung M, Knopman DS, Josephs KA, Parashos SA, Rademakers R, Hutton M, Pickering-Brown S, Adamson J, Kuntz KM, Dickson DW, Parisi JE, Smith GE, Ivnik RJ, Petersen RC (2008) Prominent phenotypic variability associated with mutations in progranulin. Neurobiol Aging (in press)
Rademakers R, Baker M, Gass J, Adamson J, Huey ED, Momeni P, Spina S, Coppola G, Karydas AM, Stewart H, Johnson N, Hsiung GY, Kelley B, Kuntz K, Steinbart E, Wood EM, Yu CE, Josephs K, Sorenson E, Womack KB, Weintraub S, Pickering-Brown SM, Schofield PR, Brooks WS, Van Deerlin VM, Snowden J, Clark CM, Kertesz A, Boylan K, Ghetti B, Neary D, Schellenberg GD, Beach TG, Mesulam M, Mann D, Grafman J, Mackenzie IR, Feldman H, Bird T, Petersen R, Knopman D, Boeve B, Geschwind DH, Miller B, Wszolek Z, Lippa C, Bigio EH, Dickson D, Graff-Radford N, Hutton M (2007) Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C->T (Arg493X) mutation: an international initiative. Lancet Neurol 6(10):857–868
Bird TD, Nochlin D, Poorkaj P, Cherrier M, Kaye J, Payami H, Peskind E, Lampe TH, Nemens E, Boyer PJ, Schellenberg GD (1999) A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain 122(Pt 4):741–756
Chiba S, Suzuki M, Yamanouchi K, Nishihara M (2007) Involvement of granulin in estrogen-induced neurogenesis in the adult rat hippocampus. J Reprod Dev 53(2):297–307
Acknowledgement
The authors are grateful to Dr. Enrico Premi for the clinical assessment and to Mrs. Maria Gervasi for her valuable technical support. The authors are in debt to Dr Francesca Piazza for linguistic editing of the manuscript.
This work was supported by Centre for Behavioural Disturbances and Neurodegenerative Diseases EULO (Ente Universitario Lombardia Orientale) to P.A.
Author information
Authors and Affiliations
Corresponding author
Appendices
Appendix 1. Primers used for the analysis of progranulin sequence
F: forward primer; R: reverse primer; M: middle F/R
Appendix 2. Primers used for microsatellite haplotyping and methods
F: forward primer; R: reverse primer
Microsatellite haplotype and analysis
Polymerase chain reaction (PCR) amplicons were generated using fluorescently end-labelled primers reported in the table above at 500 mM for microsatellite markers D17S1818(HEX) [GenBank Accession: Z52895], D17S1814(TAMRA) [GenBank Accession: Z52854], D17S1787(FAM) [GenBank Accession: Z52130], D17S1793(HEX) [GenBank Accession: Z52280], D17S951(FAM) [GenBank Accession: Z24197], D17S1861(FAM) [GenBank Accession: Z53921] and D17S934(HEX) [GenBank Accession: Z23831]. A loading mix of 1 μl amplicon, 9.75 μl HiDi formamide (ABI) and 0.25 μl 400HD size standard (ABI) was prepared, and DNA products were electrophoresed on an ABI 3130xl automated sequencer. Data were analysed using ABI GeneMapper software v4.0.
The haplotype frequency estimation calculated from the observed genotypes were tested using Arlequin 2000 and Phase 2.1 softwares
Rights and permissions
About this article
Cite this article
Borroni, B., Archetti, S., Alberici, A. et al. Progranulin genetic variations in frontotemporal lobar degeneration: evidence for low mutation frequency in an Italian clinical series. Neurogenetics 9, 197–205 (2008). https://doi.org/10.1007/s10048-008-0127-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-008-0127-3