
Abstract
Mutations of the progranulin gene lead to progranulin haploinsufficiency and to frontotemporal lobar degeneration (FTD) with TDP-43 positive inclusions. It is assumed that unknown genetic, epigenetic and environmental factors are responsible for the observed marked degree of phenotypic variability among mutation carriers. This is the first published series of German FTD cases screened for progranulin mutations. Mean age at onset was 62 years, 19 patients (24%) had a positive family history of dementia, and 11 patients (14%) had a positive family history for probable FTD. Data on FTD subtypes are presented. Two mutations were identified (3%), one of which has been described previously. Clinically, both patients showed the frontal-behavioural variant type of FTD. Remarkably, a sibling of one case presented with progressive nonfluent aphasia, clinically distinct from the brother. We also performed quantitative PCR analyses to detect potential whole progranulin gene and exon deletions. Here, results were negative.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Frontotemporal dementia, or frontotemporal lobar dementia (FTLD), is a degenerative brain disease with variable clinical phenotype, pathology and genetic background. FTLD is clinically characterized by behavioural changes and/or language impairment, defined as primary progressive aphasia or semantic dementia. Disease onset occurs mostly at presenile age.
The most frequent histopathological subtype of FTLD shows ubiquinated, tau-negative inclusions in the neurites and cytoplasm of neocortical and hippocampal neurons (FTLD-U). The main constituent of the neuronal inclusions was found to be TAR-DNA binding protein 43 (TDP-43), a highly conserved 43 kDa nuclear transcription factor [10, 29]. Four major subtypes of FTLD-U have been delineated based on the distribution of neuronal cytoplasmic inclusions (NCI), dystrophic neurites and the presence of neuronal intranuclear inclusions (NIIs), [9, 25]. In the same year TDP-43 was identified, mutations of the progranulin (PGRN) gene were described in FTLD-U [2, 11]. Neuropathologically, all cases with PGRN mutations share a common FTLD-U subtype, characterized by NCIs and irregular dystrophic neurites in the neocortex and subcortical nuclei [3, 14, 18, 24, 26, 38, 39]. This subtype is referred to as type I by Mackenzie or type 3 by Sampathu and co-workers [26, 35].
Human PGRN contains 13 exons and is located on chromosome 17q21 in close vicinity to MAPT (Microtubulus-associated protein tau), another gene identified as being involved in FTLD. PGRN encodes for the 88 kDa glycoprotein progranulin. Progranulin is a pluripotent growth factor that has been shown to be involved in cell differentiation, inflammation, tissue repair, tumorigenesis and brain development in mice [13, 16, 43]. The pathophysiological pathway by which progranulin mutations are associated with the deposition of TDP-43 protein and with neuronal degeneration is still unclear.
At present, more than 60 PGRN mutations have been identified in patients with FTLD (http://www.molgen.ua.ac.be/FTDMutations/). Pathogenic mutations include missense and nonsense mutations, or small insertions or deletions in the exons or introns of the gene [14]. Most of the mutations lead to frameshift and premature stop codons. Pathogenic mutations in PGRN invariably lead to mutant mRNA transcripts, which undergo nonsense-mediated decay, thereby resulting in haploinsufficiency [2, 11].
Prevalence of mutations in PGRN is suggested to account for 1–15% of all cases with FTLD [8, 14, 22], but up to 26% of familial cases [7, 12, 30]. In a large series from the USA, mutations were found in 10% of all patients with FTLD and 23% in cases of familial FTD [14]. Several other studies from France, Italy, the Netherlands, the UK, Belgium, Finland, and the USA have reported lower frequencies of an average 5% in unselected FTD groups and 4–10% in groups of cases of familial FTD [6–8, 11, 14, 15, 17, 20–22, 31]. The differences in the reported frequencies may be due to differences in the mode of ascertainment of patients, in ethnic variations as well as to founder effects. Overall, the frequency of PGRN mutations is similar to that of mutations in MAPT [33].
Mean age at onset of FTLD patients with mutations in PGRN is around 60 years. The majority of patients with PGRN mutations show the behavioural-variant phenotype with apathy and social withdrawal as prominent characteristics [42]. PGRN mutations have also been found in patients who present with language impairment early in the course of the disease, diagnosed as primary non-fluent progressive aphasia (PPA) [14, 17, 18, 27, 37]. Patients from different families with the same mutation do not necessarily show the same clinical phenotype or age at onset [17].
The aim of our study was to determine the frequency and type of progranulin mutations in a series of 79 unselected FTLD patients from an ethnically largely homogeneous south-western German population. To date, no mutation screening for PGRN has been published for Germany. In order to ascertain all types of mutations, we performed—in addition to the mutation analysis—quantitative PCR analyses, which additionally allows for detection of whole gene and exon deletions, a technique that has only been applied as an exception in earlier PGRN mutation screening studies.
Materials and methods
Patients
79 consecutive outpatients, who attended our university hospital memory clinic between 2001 and 2007 and received a clinical diagnosis of FTLD, were included. Routine neurological and psychiatric exams, neuropsychological testing and cerebral imaging (CT- or MRI-scan) were conducted in all cases. In addition, HMPAO-SPECT was performed in a subset of cases. The diagnosis of FTLD was made in a case conference, based on established criteria [1]. Screening for other known dementia-related mutations, including tau protein mutations, was not performed. Several of our patients have died, but none have been subjected to autopsy.
PGRN mutation analysis
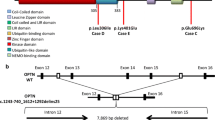
PGRN mutation analysis was performed in all 79 patients. All 13 exons including exon-intron boundaries were sequenced. DNA of EDTA blood samples of patients were extracted with the BACC-2 Nucleon Extraction Kit (Amersham Bioscience, Freiburg, Germany) according to the manufacturer’s instructions. All PGRN exons were first amplified by Fast-PCR using primers listed in Table 1. Amplification products were purified with the QIAquick® Gel Extraction Kit (Qiagene, Hilden, Germany), followed by sequencing using BigDYE®Ready Terminator Sequencing Kit (Applied Biosystems, Foster City, CA, USA). For sequencing, previously-described primers were used [2] (Table 2). Sequencing was performed on the ABI 3130 XL Genetic Analyzer (16 capillary DNA sequencer, Applied Biosystems, Foster City, CA, USA). Sequences were analyzed using SeqPilot software (JSI medical systems, Kippenheim, Germany) (Fig. 1).

PGRN mutation analysis
PGRN deletion analysis
Copy number changes of PGRN were tested by quantitative real-time PCR (qPCR) using SYBR® GREEN l (Qiagene, Hilden, Germany) as described previously [4]. PCR amplicons listed in Table 3 were designed using PRIME program (Genetic Computer Group, Wisconsin, USA) and quantified against two reference amplicons in human subtelomer region 3p26.3 and 4p15.2 [4, 5]. qPCR was performed on ABI Prism 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Data was analyzed with the help of Sequence Detection Software (SDS version 2.2.1, Applied Biosystems, Foster City, CA, USA).
Results
Clinical characterization of the FTD patients
The series was comprised of 43 men and 36 women. Mean age at onset was 62 years, ranging between 41 and 83 years. 19 patients (24%) had a positive family history of dementia. Among these, 11 patients (14%) had a positive family history for FTD with instances of early-onset in at least one first-degree relative (n = 8), or with age at onset >65 years (n = 3). In the other 8 cases with FTD, at least one parent had suffered dementia at a much higher age, presumably Alzheimer’s disease. In three cases, sufficient information on family history was not available.
The clinical FTD subtype was “frontal” with leading behavioural and executive cognitive disorder in 44, primary progressive aphasia (PPA) in 24, semantic dementia (SD) in 8, mixed behavioural variant and PPA in 1, mixed FTD and CBD in 1, and FTD with Parkinsonism in 1. Mean age of disease onset of patients with the “frontal type” was 61.2 years, and 64.9 and 57.0 years in patients with PNA and SD, respectively (Table 4).
DNA sequence analysis of PGRN
Sequence analysis revealed two pathogenic PGRN mutations (3%). Both mutations were nonsense mutations (p.R418X, p.K259X) leading to the generation of a premature stop codon (Fig. 1A).
PGRN deletion Analysis: qPCR analysis revealed no deletions within or including the PGRN gene in our cohort.
Clinical phenotype of the two patients with mutations within the PGRN gene
Case 1
This woman presented with progressive symptoms of language impairment at age 58. One year later, she had become quiet, withdrawn, and inefficient in all her activities. Upon examination, she appeared friendly, cooperative, not depressed. Spontaneous speech was markedly reduced. There were few semantic, and no phonematic paraphasias. She showed difficulties in comprehending instructions, utilisation behaviour, rule breaks, and marked symptoms of perseveration in fluency, clock reading and drawing tasks. The Mini Mental score was 18. Further testing showed dysnomia (9 of 15 in the short Boston naming test), and impaired fluency (five animals, no S-words in one minute). CERAD figure copying was borderline (8). Clock drawing (Shulman score 3–4) and clock reading (score 8) [36], as well as trail making “A” performance (118 s) were pathological. Verbal memory performance was also impaired (15 of 30 in three trials of CERAD word list learning, 4 at delayed recall, 95% at delayed recognition).
An MRI was described as showing external and internal brain atrophy, mostly temporo-parietal. A brain HMPAO perfusion SPECT performed at our clinic showed extensive bilateral frontotemporal hypoperfusion, mostly on the left side, extending to the parietal cortex and also involving the thalami and basal ganglia.
A diagnosis of frontotemporal dementia was made. The presence of early and pronounced general cognitive and behavioural symptoms and the pattern of imaging findings suggest classification as a case of the predominantly “frontal” type FTD (Fig. 1B).
Family history
Both parents of this patient had reached an age >75 years. Her mother was chronically ill and bedridden before she died. More precise information on her medical diagnosis was not available. This case of FTD was therefore considered to be sporadic.
PGRN mutation analysis
Sequence analysis showed a nonsense mutation in exon 10 in a heterozygous state, leading to the generation of a premature stop codon.
Case 2
This man presented with a change of behaviour and with cognitive deficits that became evident at age 58: increased talking, irritability, neglect of former activities, emotional indifference, word finding difficulty, forgetfulness and adherence to irrelevant stimuli. Upon examination, he was cooperative, not depressed, but highly stimulus-bound, making irrelevant remarks and reading aloud whatever was in sight. His sentences tended to be long, complicated, and were sometimes not finished. His overall speech production was reduced. There was no impairment of language comprehension. He showed symptoms of perseveration. The Mini Mental score was 26. Further testing showed normal Token Test, CERAD figure drawing and recall performance. Boston naming (13 of 15) was borderline, and performance at another naming test was pathological (Aachen Aphasia Test, 98/120). Verbal fluency was borderline (16 animals in one minute). Verbal memory performance was pathological (11 of 30, delayed recall 2, delayed recognition 90%).
A brain MRI showed no clear atrophy. A brain HMPAO perfusion SPECT performed at our clinic showed symmetric temporo-polar and fronto-basal hypoperfusion.
As in case 1, the pattern of clinical and imaging findings supports classification of this patient as a case of the predominantly “frontal” type of FTD.
Family history
A brother of this patient suffers a degenerative disease which started at age 51 with word finding difficulties. 21 months later, he showed severe aphasia, but only a minor change of personality was noted. There was atrophy mainly of the left temporal lobe (MRI). When we saw him 4 months later, he was still pursuing his job and said to be unimpaired in all technical and practical matters. His drive, mood and behaviour appeared normal. Speech was reduced to “yes” and “no”. Speech comprehension, reading aloud and writing were markedly impaired. Upon testing, the Mini Mental score was 19, Boston naming score was pathological (5/15), verbal memory borderline (15/30; 5 at delayed recall), figure copying and delayed recall were normal. He was classified as a case of progressive nonfluent aphasia. Genotyping could not be performed since the patient was not able any more to give consent to genetic testing. The parents of case 2 were said to have died of cancer at the age of 70 and 73 years, without known dementia. Further information was not available. This case was classified to be a familial case of FTD.
PGRN mutation analysis
Sequence analysis revealed a nonsense mutation in exon 7 in a heterozygous state not described before, leading to the generation of a premature stop codon (Fig. 1C).
Discussion
This is the first report of a German FTD case series screened for progranulin mutations. Patients live or lived in South Baden, which represents about one-quarter of the territory of the federal state of Baden-Württemberg in the south-west part of Germany.
The overall frequency of positive family history for dementia in our FTD patient cohort was 24%. This proportion is below reported frequencies in several earlier series with a positive family history of up to 40–50% of FTLD cases [28, 40]. The possibility remains that the true proportion of dominantly inherited cases is obscured by instances of early death of mutation carriers in the parental generation, siblings that carry mutations but are yet undiagnosed, or illegitimate descent.
Point mutations were identified in two cases of our cohort of 79 patients. In both cases, a point mutation generated a premature termination codon that causes nonsense-mediated mRNA decay and leads eventually to the absence of mutant PGRN transcript.
The mutation p.R418X as identified in case 1 has been reported before [2, 14, 41]. Mean age at onset in a Canadian family (UBC15) described by Baker and colleagues was 60, comparable to the age at onset of our index patient [2]. Gass and coworkers identified another FTLD family in the USA (01-01) with the p.R418X mutation; here age at onset was 49 [14].
The nonsense mutation of case 2 in exon 7 has not been reported before. The clinical phenotypes of case 2 and his brother, who has not been sequenced for PGRN mutations, differed from each other in regard to age at onset and clinical phenotype (frontal type and PNA type). Pronounced phenotypic variability has previously been described in pedigrees with progranulin mutations with respect to clinical picture [19, 32], age at onset and pattern of histopathological changes. In one family, post-mortem examinations showed atypical tau and alpha-synuclein pathology [23]. Assuming that progranulin mutations are always null mutations and uniformly lead to haploinsufficiency, other as yet unknown genetic, epigenetic and environmental factors must be responsible for this variability within and across families.
Two of our patients had a progranulin gene mutation, corresponding to a low proportion of 3% of the entire sample. In previous FTD series that were unselected for family history, the reported rates of progranulin mutations were 2% [17], 1% [8], 5% [14], and 6% [31]. In apparently familial cases, reported rates were 13% in a French series [22], 22% in a North American series [14], and 4–7% in a Dutch series [7]. In contrast to most other mutation screening studies, we performed quantitative real-time PCR analysis to be sure to identify single exon and larger deletions of PGRN. Previously, two patients with heterozygous genomic deletions in PGRN were reported [15, 34]. Thus, our study provides a reliable frequency for PGRN mutations for south-west Germany.
Age at onset was 58 years in our cases 1 and 2. Information on the status of the mother of case 1 is incomplete. Both parents of case 2 and his affected brother lived to their seventies and were apparently not demented. This situation is, in principle, compatible with illegitimate descent, de novo mutation, incomplete penetrance or with a high variability of age at onset. Previous reports of families with progranulin mutation have shown that age at onset can indeed vary greatly, e.g. from 35 to 75 years within one family [23], from 49 to 88 years across affected families [19], and from 35 to 87 years in one kindred [8]. In some affected families, age at onset is not only variable, but clearly higher than the average presenile age range typical of FTD, e.g. 75.8 ± 5.0 years in one generation of kindred 2 reported by Kelley et al. [19].
We conclude that PGRN mutations in unselected populations may be lower due to a founder effect in selected studies and may explain regional differences in PGRN mutations. In addition, it is important to perform deletion analysis to accurately determine the frequency of pathogenic gene mutations.
References
(1994) Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. Journal of neurology, neurosurgery, and psychiatry 57:416–418
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919
Behrens MI, Mukherjee O, Tu PH, Liscic RM, Grinberg LT, Carter D, Paulsmeyer K, Taylor-Reinwald L, Gitcho M, Norton JB, Chakraverty S, Goate AM, Morris JC, Cairns NJ (2007) Neuropathologic heterogeneity in HDDD1: a familial frontotemporal lobar degeneration with ubiquitin-positive inclusions and progranulin mutation. Alzheimer Dis Assoc Disord 21:1–7
Boehm D, Herold S, Kuechler A, Liehr T, Laccone F (2004) Rapid detection of subtelomeric deletion/duplication by novel real-time quantitative PCR using SYBR-green dye. Hum Mutat 23:368–378
Borozdin W, Boehm D, Leipoldt M, Wilhelm C, Reardon W, Clayton-Smith J, Becker K, Muhlendyck H, Winter R, Giray O, Silan F, Kohlhase J (2004) SALL4 deletions are a common cause of Okihiro and acro-renal-ocular syndromes and confirm haploinsufficiency as the pathogenic mechanism. J Med Genet 41:e113
Borroni B, Archetti S, Alberici A, Agosti C, Gennarelli M, Bigni B, Bonvicini C, Ferrari M, Bellelli G, Galimberti D, Scarpini E, Di Lorenzo D, Caimi L, Caltagirone C, Di Luca M, Padovani A (2008) Progranulin genetic variations in frontotemporal lobar degeneration: evidence for low mutation frequency in an Italian clinical series. Neurogenetics 9:197–205
Bronner IF, Rizzu P, Seelaar H, van Mil SE, Anar B, Azmani A, Kaat LD, Rosso S, Heutink P, van Swieten JC (2007) Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur J Hum Genet 15:369–374
Bruni AC, Momeni P, Bernardi L, Tomaino C, Frangipane F, Elder J, Kawarai T, Sato C, Pradella S, Wakutani Y, Anfossi M, Gallo M, Geracitano S, Costanzo A, Smirne N, Curcio SA, Mirabelli M, Puccio G, Colao R, Maletta RG, Kertesz A, St George-Hyslop P, Hardy J, Rogaeva E (2007) Heterogeneity within a large kindred with frontotemporal dementia: a novel progranulin mutation. Neurology 69:140–147
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114:5–22
Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL 3rd, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM, Mackenzie IR (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924
Cruts M, Kumar-Singh S, Van Broeckhoven C (2006) Progranulin mutations in ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Curr Alzheimer Res 3:485–491
Daniel R, Daniels E, He Z, Bateman A (2003) Progranulin (acrogranin/PC cell-derived growth factor/granulin-epithelin precursor) is expressed in the placenta, epidermis, microvasculature, and brain during murine development. Dev Dyn 227:593–599
Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, Josephs K, Pickering-Brown SM, Graff-Radford N, Uitti R, Dickson D, Wszolek Z, Gonzalez J, Beach TG, Bigio E, Johnson N, Weintraub S, Mesulam M, White CL 3rd, Woodruff B, Caselli R, Hsiung GY, Feldman H, Knopman D, Hutton M, Rademakers R (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 15:2988–3001
Gijselinck I, van der Zee J, Engelborghs S, Goossens D, Peeters K, Mattheijssens M, Corsmit E, Del-Favero J, De Deyn PP, Van Broeckhoven C, Cruts M (2008) Progranulin locus deletion in frontotemporal dementia. Hum Mutat 29:53–58
He Z, Bateman A (1999) Progranulin gene expression regulates epithelial cell growth and promotes tumor growth in vivo. Cancer Res 59:3222–3229
Huey ED, Grafman J, Wassermann EM, Pietrini P, Tierney MC, Ghetti B, Spina S, Baker M, Hutton M, Elder JW, Berger SL, Heflin KA, Hardy J, Momeni P (2006) Characteristics of frontotemporal dementia patients with a progranulin mutation. Ann Neurol 60:374–380
Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR, Uitti RJ, Rademakers R, Adamson J, Baker M, Hutton ML, Dickson DW (2007) Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol 66:142–151
Kelley BJ, Haidar W, Boeve BF, Baker M, Graff-Radford NR, Krefft T, Frank AR, Jack CR Jr, Shiung M, Knopman DS, Josephs KA, Parashos SA, Rademakers R, Hutton M, Pickering-Brown S, Adamson J, Kuntz KM, Dickson DW, Parisi JE, Smith GE, Ivnik RJ, Petersen RC (2009) Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol Aging 30:739–751
Kruger J, Kaivorinne AL, Udd B, Majamaa K, Remes AM (2009) Low prevalence of progranulin mutations in Finnish patients with frontotemporal lobar degeneration. Eur J Neurol 18:27–30
Le Ber I, Camuzat A, Hannequin D, Pasquier F, Guedj E, Rovelet-Lecrux A, Hahn-Barma V, van der Zee J, Clot F, Bakchine S, Puel M, Ghanim M, Lacomblez L, Mikol J, Deramecourt V, Lejeune P, de la Sayette V, Belliard S, Vercelletto M, Meyrignac C, Van Broeckhoven C, Lambert JC, Verpillat P, Campion D, Habert MO, Dubois B, Brice A (2008) Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 131:732–746
Le Ber I, van der Zee J, Hannequin D, Gijselinck I, Campion D, Puel M, Laquerriere A, De Pooter T, Camuzat A, Van den Broeck M, Dubois B, Sellal F, Lacomblez L, Vercelletto M, Thomas-Anterion C, Michel BF, Golfier V, Didic M, Salachas F, Duyckaerts C, Cruts M, Verpillat P, Van Broeckhoven C, Brice A (2007) Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat 28:846–855
Leverenz JB, Yu CE, Montine TJ, Steinbart E, Bekris LM, Zabetian C, Kwong LK, Lee VM, Schellenberg GD, Bird TD (2007) A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain 130:1360–1374
Lopez de Munain A, Alzualde A, Gorostidi A, Otaegui D, Ruiz-Martinez J, Indakoetxea B, Ferrer I, Perez-Tur J, Saenz A, Bergareche A, Barandiaran M, Poza JJ, Zabalza R, Ruiz I, Urtasun M, Fernandez-Manchola I, Olasagasti B, Espinal JB, Olaskoaga J, Ruibal M, Moreno F, Carrera N, Masso JF (2008) Mutations in progranulin gene: clinical, pathological, and ribonucleic acid expression findings. Biol Psychiatry 63:946–952
Mackenzie IR, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM (2006) Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol 112:539–549
Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, Gass J, Cannon A, Rademakers R, Hutton M, Feldman HH (2006) The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129:3081–3090
Mesulam M, Johnson N, Krefft TA, Gass JM, Cannon AD, Adamson JL, Bigio EH, Weintraub S, Dickson DW, Hutton ML, Graff-Radford NR (2007) Progranulin mutations in primary progressive aphasia: the PPA1 and PPA3 families. Arch Neurol 64:43–47
Neary D, Snowden J, Mann D (2005) Frontotemporal dementia. Lancet Neurol 4:771–780
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, NY) 314:130–133
Pickering-Brown SM, Baker M, Gass J, Boeve BF, Loy CT, Brooks WS, Mackenzie IR, Martins RN, Kwok JB, Halliday GM, Kril J, Schofield PR, Mann DM, Hutton M (2006) Mutations in progranulin explain atypical phenotypes with variants in MAPT. Brain 129:3124–3126
Pickering-Brown SM, Rollinson S, Du Plessis D, Morrison KE, Varma A, Richardson AM, Neary D, Snowden JS, Mann DM (2008) Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 131:721–731
Rademakers R, Baker M, Gass J, Adamson J, Huey ED, Momeni P, Spina S, Coppola G, Karydas AM, Stewart H, Johnson N, Hsiung GY, Kelley B, Kuntz K, Steinbart E, Wood EM, Yu CE, Josephs K, Sorenson E, Womack KB, Weintraub S, Pickering-Brown SM, Schofield PR, Brooks WS, Van Deerlin VM, Snowden J, Clark CM, Kertesz A, Boylan K, Ghetti B, Neary D, Schellenberg GD, Beach TG, Mesulam M, Mann D, Grafman J, Mackenzie IR, Feldman H, Bird T, Petersen R, Knopman D, Boeve B, Geschwind DH, Miller B, Wszolek Z, Lippa C, Bigio EH, Dickson D, Graff-Radford N, Hutton M (2007) Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C–>T (Arg493X) mutation: an international initiative. Lancet Neurol 6:857–868
Rosso SM, Donker Kaat L, Baks T, Joosse M, de Koning I, Pijnenburg Y, de Jong D, Dooijes D, Kamphorst W, Ravid R, Niermeijer MF, Verheij F, Kremer HP, Scheltens P, van Duijn CM, Heutink P, van Swieten JC (2003) Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain 126:2016–2022
Rovelet-Lecrux A, Deramecourt V, Legallic S, Maurage CA, Le Ber I, Brice A, Lambert JC, Frebourg T, Hannequin D, Pasquier F, Campion D (2008) Deletion of the progranulin gene in patients with frontotemporal lobar degeneration or Parkinson disease. Neurobiol Dis 31:41–45
Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM (2006) Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol 169:1343–1352
Schmidtke K, Olbrich S (2007) The Clock Reading Test: validation of an instrument for the diagnosis of dementia and disorders of visuo-spatial cognition. Int Psychogeriatr 19:307–321
Snowden J, Neary D, Mann D (2007) Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol 114:31–38
Snowden JS, Pickering-Brown SM, Mackenzie IR, Richardson AM, Varma A, Neary D, Mann DM (2006) Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 129:3091–3102
Spina S, Murrell JR, Huey ED, Wassermann EM, Pietrini P, Grafman J, Ghetti B (2007) Corticobasal syndrome associated with the A9D Progranulin mutation. J Neuropathol Exp Neurol 66:892–900
Stevens M, van Duijn CM, Kamphorst W, de Knijff P, Heutink P, van Gool WA, Scheltens P, Ravid R, Oostra BA, Niermeijer MF, van Swieten JC (1998) Familial aggregation in frontotemporal dementia. Neurology 50:1541–1545
Van Deerlin VM, Wood EM, Moore P, Yuan W, Forman MS, Clark CM, Neumann M, Kwong LK, Trojanowski JQ, Lee VM, Grossman M (2007) Clinical, genetic, and pathologic characteristics of patients with frontotemporal dementia and progranulin mutations. Arch Neurol 64:1148–1153
van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, de Koning I, Kamphorst W, Ravid R, Spillantini MG, Niermeijer HeutinkP (1999) Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol 46:617–626
Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Ding A (2002) Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell 111:867–878
Author information
Authors and Affiliations
Corresponding author
Additional information
J. C. M. Schlachetzki and K. Schmidtke contributed equally to this work.
Rights and permissions
About this article
Cite this article
Schlachetzki, J.C.M., Schmidtke, K., Beckervordersandforth, J. et al. Frequency of progranulin mutations in a German cohort of 79 frontotemporal dementia patients. J Neurol 256, 2043–2051 (2009). https://doi.org/10.1007/s00415-009-5248-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-009-5248-6