
Abstract
Aging signals a gradual deregulation of physiological homeostasis. Steroid hormone actions are an important contributor to this deregulation because of their key involvements in the growth, metabolism, survival, and functional vigor of cells, tissues, and organs. Accumulated evidences show that aging is associated with reduced circulating levels of male and female sex hormones, i.e., androgens and estrogens, respectively, which in turn alter physiological milieu and lead to specific deficits in the organismal vitality. While a number of articles in the literature have provided a generalized description of age-related decline of physiological control mechanisms, in the current chapter, we have focused specifically on the role of sex hormones and sex steroid receptors in age-related bodily dysfunctions. Various segments of our article delved into the current understanding on influences of sex steroids and steroid receptors. Specifically, the roles of androgens, estrogens, and cognate sex steroid receptors in age-accompanied physiological and pathophysiological changes in gene expression and organ functions are discussed. In addition to citing our own studies, information from diverse fields of biology and medicine is taken into consideration in order to present a comprehensive view of sex steroid action with advancing age.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Aging is a multidimensional process marked by progressive loss of cell and tissue functions, making organisms less fit for reproduction and survival [1]. A steady decline in bodily functions during aging is initially associated with a failure to establish homeostasis in response to stress. Thereafter, functional deficits set in even under rested, non-stressed conditions, thereby compromising workings of all vital organs at old age. Many countries in the twenty-first century will experience a large demographic shift toward the age 65+ population as a result of significantly increased longevity afforded by the progress in medical sciences and healthcare management. This shift in demography requires new measures that ensure less stressful aging with a goal to achieve reduced disease burden and improved cognitive and physical fitness along with active engagement in everyday life. Steroid hormones, which are important regulators of human metabolism, are likely to play a key role in these improvements [2]. The interplay between steroid hormones and aging is complex – the aging process impacting steroid hormone biosynthesis, while steroid hormones influencing aging at the molecular level. Elucidation of this interplay may uncover new therapeutics and novel approaches that afford prolonged health span in conjunction with extended life span.
2 Steroid Hormone Biosynthesis
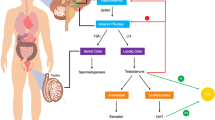
Steroid hormones are lipid-soluble, low-molecular-weight compounds that are synthesized from cholesterol ([3], Fig. 13.1).

Steroid hormones are usually made by steroidogenic glands including the ovary, testis, and adrenals and by the placenta during pregnancy and then released into the bloodstream [3]. They are classified into three categories based on their physiological functions: the sex steroids, glucocorticoids, and mineralocorticoids. The sex steroids include androgens and estrogens. The Leydig cells of testis are the primary sites of synthesis of the principal male hormone, testosterone [5]. Three major sex steroids are also secreted by the ovaries, viz., the estrogens, androgens, and progestin [6]. The adrenal cortex produces three different classes of steroid hormones, namely, glucocorticoids (cortisol and corticosterone), mineralocorticoids (aldosterone and deoxycorticosterone), and dehydroepiandrosterone (DHEA) and androstenedione, the precursors for the sex steroids androgens and estrogens. The zona glomerulosa zone produces the mineralocorticoids, whereas the zona fasciculata in the adrenal cortex produces the glucocorticoids. The zona reticularis of the adrenal cortex primarily produces the adrenal androgens [7]. The principal estrogen, 17β-estradiol, is secreted by the theca cells in ovary, which also secretes androgens such as androstenedione, DHEA, testosterone, and dihydrotestosterone. Androstenedione, the ovarian androgen, is synthesized by the theca cells and then transported to granulosa cells for estrogen synthesis. These cells also produce major progestins such as pregnenolone, progesterone, and 17-hydroxyprogesterone. Of these, pregnenolone is used as a precursor for the synthesis of all the steroid hormones (Fig. 13.1). The corpus luteum secretes progesterone and allows the fertilized ovum to be implanted for the maintenance of pregnancy, in the first 6–8 weeks of gestation. During pregnancy, placenta is the primary producer of progesterone [6].
Testosterone, a major androgen in circulation, is produced by Leydig cells of the testes [3]. The 5α-reduced testosterone, i.e., 5α-dihydrotestosterone (5α-DHT), is the active androgen in many androgen-targeted tissues including the prostate and liver. Androgens are also produced by ovaries in limited quantities. The androgens synthesized by the ovaries include DHEA, androstenedione, testosterone, and dihydrotestosterone. Estrogen, the steroid hormone mostly associated with female-specific phenotype, is secreted primarily by the ovaries [8] and the placenta. It is also produced in lesser quantities by steroidogenic conversion, in the testes of men [8]. The amount of estrogen in women is approximately four times that of men [9]. Indeed, estrogens cause the development of female genital organs, endometrium growth, and inhibition of follicle-stimulating hormone secretion by the pituitary gland.
The hypothalamic gonadotropin-releasing hormone (GnRH) stimulates both synthesis and pulsatile release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) by the anterior pituitary [10]. FSH is required for sperm production; LH is needed for testosterone secretion. Major functions of steroid hormones are tabulated in Table 13.1.
3 Sex Steroid Receptors: Their Nuclear Localization
The sex hormones androgen and estrogen, being lipophilic, enter target cells by passive diffusion through the lipid bilayer membrane of cell. The full-length receptor to androgen, i.e., androgen receptor (AR), remains in the cytoplasmic compartment as an inactive protein in the absence of the hormone. The androgen-bound AR transitions to an active state, which enables the receptor to translocate to the nucleus and function as the mediator of androgen-responsive nuclear signaling [11, 12]. Using cells with the green fluorescent protein-labeled AR (GFP-AR), our study provided the initial evidence for the predominantly cytoplasmic residency of AR in the absence of androgen and its nuclear import to distinct foci when the cells were exposed to the active male sex steroid 5α-DHT (Fig. 13.2; [13]). Conversion of AR from an inactive to active state and its role as a ligand-inducible transcription factor will be further elaborated in Sect. 13.4.

Influence of sex steroids on receptor localization. (a) Androgen-dependent nuclear translocation and formation of nuclear foci of GFP-AR in transiently transfected cells. (b) 17β-estradiol (17βE)-dependent formation of nuclear foci of GFP-ERα. COS-1 cells, transfected with either 500ng of GFP-AR or GFP-ERα, were cultured in steroid-free medium for studying protein expression and localization. After 24 h of expression period, the cells were treated with DMSO:ethanol(1:1), or 10-8M DHT or 10-8M 17βE. After 2 h of DHT or 17βE treatment, cells were observed under a fluorescence microscope. Nuclear dye Hoechst was used to visualize the nuclei
In contrast to AR, ERα is predominantly a nuclear protein in the absence of estrogen (17β-estradiol), and upon treatment of cells with estrogen, ERα localizes to distinct nuclear foci, which likely indicates hormone-dependent transition of the estrogen receptor to a transcriptionally competent active form (Fig. 13.2; [14]).
4 Androgen Receptor (AR), Androgen Action, and Aging
Diverse physiology, encompassing reproductive and nonreproductive processes, is regulated by the androgen receptor (AR), which is a ligand-inducible transcription factor and the initial conduit for transmitting androgen signaling to the transcriptional apparatus in the nucleus. Similar to other members of the nuclear receptor superfamily, AR has a modular primary structure, with each module representing a distinct functional unit [15,16,17]. In the absence of androgen (or other AR agonists/antagonists), the cytoplasmic AR remains sequestered as a multiprotein complex with protein partners that include molecular chaperones like HSP70, HSP90, HSP40, and co-chaperones like immunophilins (forskolin-binding proteins) and p23 [18, 19]. The ligand-bound AR, upon release from the multiprotein complex, undergoes a conformational rearrangement, which exposes its nuclear localization signal (NLS) for binding to importins, allowing nuclear translocation of the receptor (Fig. 13.2). Androgen-induced nuclear import of AR in living cells and its subsequent multiple rounds of nucleocytoplasmic shuttling was first reported in 2000 by us, using green fluorescent protein-labeled AR [13].
The transcriptionally active AR accumulates within microscopically visualized nuclear foci, which are thought to be the sites where AR interacts with target genes via androgen-responsive DNA elements. These foci are also the convergence nodes for coregulators and other components of the transcriptional apparatus [20]. The pioneer transcription factor FOXA1, bound at AR-driven regulatory regions, facilitates AR binding to cognate elements. However, examples are also known where AR, rather than directly binding to an androgen-responsive element, is tethered to a second DNA-bound transcription factor. Coregulators, which associate with AR, relay signals to RNA polymerase II and the basal transcriptional machinery via a multiprotein mediator complex. The p160 coactivators (SRC-1/SRC-2/SRC-3), which physically associate with AR, generate a surface for assembling various classes of coregulators such as histone-modifying enzymes (e.g., acetyltransferases/deacetylases, methylases/demethylases, kinases/phosphatases, ubiquitinases/deubiquitinases), chromatin-remodeling proteins (such as SWI/SNF, INO80, ISWI, and CHD complexes, chromodomain proteins, bromodomain, and extra-terminal (BET) family proteins). Long noncoding RNAs (lncRNAs), such as enhancer RNAs (eRNAs) and other types of lncRNAs (e.g., HOTAIR), are essential components of a coregulator complex. These entities coordinate gene induction or repression by AR [21,22,23].
Androgen/AR signaling has also transcription-independent roles in cellular functions. Non-genomic AR signaling can initiate at cell surface via membrane androgen receptor (mAR) and/or in the cytoplasm. The response occurs rapidly (within seconds to minutes) in the presence of inhibitors of transcription and translation, and it regulates several pathways including MAPK/ERK, PKA, FAK, p38, p53, calcium, and zinc [24, 25]. The membrane-located AR (mAR) is distinct from the cytoplasmic/nuclear AR described above. Cross coupling of genomic and non-genomic androgen signaling may influence cellular proliferation, survival, and apoptosis [26]. In another example, AR associates with telomeres and plays a role in telomere stability independent of its transcription function [27]. Telomeres, which cap each end of a chromosome as repeats of a core nucleotide sequence, are essential for genomic stability. Without telomere repeats, each round of DNA replication would shorten the functional nucleotide sequences at chromosomal ends. Cells will eventually recognize shortened chromosomal ends as DNA damages and trigger chromosomal degradation.
4.1 AR Activity in the Liver and Its Age-Dependent Regulation
Normal liver functions and several liver diseases are influenced by AR activity [28]. For example, hepatic glucose and lipid metabolism are deregulated by liver-specific ablation of AR in mice [29]. AR regulates hepatic steroid, drug, and nutrient metabolism, as evident from the AR-mediated transcriptional regulation of certain cytochrome P450 phase I enzymes and the phase II sulfotransferase SULT2A1; additionally, the liver abundance of these enzymes shows gender differences [30,31,32]. AR confers a protective influence against high-fat diet-induced NAFLD (nonalcoholic fatty liver disease), since liver-specific loss of AR in a mouse model led to insulin insensitivity and type 2 diabetes [28]. Testicular feminized (Tfm) mice, which lack functional AR, were resistant to liver cancer upon carcinogen exposure, which revealed a role for AR in liver cancer [33]. Notably, in a yin-yang relationship, AR promoted initiation of hepatocellular carcinoma (HCC) in mice but suppressed HCC metastasis [34]. Sorafenib, a multiple kinase inhibitor against HCC, was more effective in inhibiting HCC progression in the presence of functional AR in a preclinical HCC metastasis model [34]. Finally, in the context of normal aging, AR-ablated, not AR-intact, male livers developed microvesicle steatosis at advanced age in mice [29].
During physiological aging, male rodents show a gradual reduction in hepatic AR expression, and AR protein levels are undetectable beyond 20–24 months of age (equivalent to an ~80-year-old human) (Fig. 13.3, [35]). Loss of hepatic AR mRNAs parallels the loss of AR protein in old rats [35, 36]. Dietary calorie restriction, a proven means for extending invertebrate and vertebrate life spans and retarding age-related diseases, prevented AR loss and restored androgen sensitivity of the aging rat liver [36,37,38]. Age-associated steady increase in the liver expression of SULT2A1 sulfotransferase (a phase II sulfate-conjugating enzyme for bile acid) and DHEA (dehydroepiandrosterone) is a consequence of the relief of the SULT2A1 gene from AR-mediated repression [30]. Consistent with this repression, calorie restriction prevented age-associated rise of SULT2A1 levels in the aging male rat liver (Fig. 13.4, [37]).

Loss of AR in the livers of male rats during aging. Arrows in the Western blot indicate positions of AR and phosphorylated AR [35]

Loss of SULT2A1 in livers of old male rats by calorie restriction (CR). Liver lysates from ad libitum (AL)-fed rats (21-, 24-, 27-month-old) had high SULT2A1 levels [37]
At the molecular level, positive and negative changes in specific transcription factor activities are linked to the loss of hepatic AR during aging. Roy, Chatterjee, and colleagues conducted detailed investigations of the underlying mechanisms for these changes [35, 39, 40]. The NF-κB transcription factor is a negative regulator of human and rat AR gene transcription [40, 41]. This negative regulation is consistent with the findings that the NF-κB activity in hepatic and extrahepatic tissues is elevated during aging in response to chronic inflammation and oxidative stress, which are hallmarks of physiological aging [40, 42]. In contrast, an age-dependent factor (ADF) gradually declines in activity during aging [39]. ADF can stimulate AR promoter activity by inducing a 20-base pair DNA element (ADF element) located in an upstream region of the rat AR promoter [35, 39]. The nuclear ADF activity was detected in hepatic and non-hepatic cells. AR gene repression is partly a consequence of the replacement of a PARP-1-associated, p/CAF-containing coactivator assembly at the ADF element by a corepressor complex, which associates with the p53 tumor suppressor protein. Beyond its classic role in DNA repair, PARP-1, i.e., poly (ADP-ribose) polymerase-1, can coactivate transcription factors [43], and p/CAF, a histone acetyltransferase, is a core component of coactivator complexes in many contexts. The corepressors mSin3A and Groucho/TLE-1 are part of the corepressor complex. They help stabilizing the corepressor complex due to association with p53. Age-associated switch from a coactivator to corepressor assembly at the ADF element is coordinated by the B-Myb and c-Myb transcription factors, which directly bind to the 20-base pair ADF DNA element. As core components of the ADF activity, B-Myb and c-Myb associate with PARP-1 (in the context of coactivator assembly), or p53 (in the context of corepressor assembly). The heterogeneous ribonucleoprotein K (hnRNPK), which associates with PARP-1, serves as a platform for the convergence of coregulators. The B-Myb level in the rat liver is markedly reduced during aging, and B-Myb is undetectable at the ADF-regulated chromatin region. In contrast, irrespective of age, c-Myb continues to occupy this region. Figure 13.5 depicts a model showing coregulator dynamics at the ADF-regulated AR promoter in young vs. old liver.

Schema for AR gene regulation by ADF during aging. ADF-bound B-Myb/c-Myb and a coactivator complex, which contains PARP-1, hnRNPK, and p/CAF, mediate AR gene induction at young age. AR gene repression at old age is coordinated at the ADF site by c-Myb bound to a p53-associated corepressor complex, which contains Groucho/TLE1 and mSin3A corepressors. (Modified from [35])
Similar to aging, AR gene repression due to oxidative stress involves recruitment of p53, Groucho/TLE-1, and mSin3A to the ADF-regulated chromatin in exchange for the departure of PARP-1, hnRNPK, and p/CAF factors that occupy this region under normoxia. Unlike aging, however, the cellular abundance for B-Myb is not altered by oxidative stress, so that p53 is anchored to the regulated region by association with both B-Myb and c-Myb [35].
We speculate that AR gene suppression in old animals is triggered by the age-associated decline in PARP-1 activity. The intracellular level of NAD+ (oxidized nicotinamide-adenine dinucleotide), a cofactor for PARP-1, declines steadily during aging [44,45,46], which in turn reduces PARP-1 activity [46]. The reduced NAD+ level is detrimental to mitochondrial homeostasis and to the overall metabolic vitality of an organism. Restoration of NAD+ levels can reverse this decline and delay aging, leading to health span and life span extension [44, 46].
4.2 Androgen Dependence of Normal Prostate and Prostatic Diseases of Old Age
AR levels in the rat prostate were found to decline during aging [47]. Androgen action plays an obligatory role in the development and function of the prostate, which is an exocrine gland in adult males. Congenital defects for functional AR or 5-alpha-reductase in genetic XY males cause incomplete development or complete lack of this secondary reproductive organ. The alveolar-ductal structures of the prostate are embedded in a fibromuscular stroma, with the basement membrane providing the stroma-epithelia barrier. Androgen action in the stromal tissue is essential for prostate development [48]. AR-expressing luminal epithelial cells produce a multitude of proteins, including the prostate-specific antigen (PSA), which secrete into the ductal lumen. On the other hand, the basal epithelium contains AR-negative epithelial cells, neuroendocrine-type cells, as well as macrophages and lymphocytes. Embryonic development of prostate begins with the androgen-stimulated synthesis and secretion of various growth factors from AR-expressing stromal fibroblasts and fibromyoblasts. Glandular development subsequent to epithelial cell growth is in turn induced by the paracrine action of stroma-derived secreted factors. In adult prostate, direct androgen action on AR-positive luminal epithelial cells preserves glandular integrity. In castrated rodents, ~ 90% of luminal epithelial cells are lost due to apoptosis. Androgen replenishment restores prostate structure, when stem cell-like basal cells differentiate into AR-negative basal epithelial cells, which progress to mature, AR-positive luminal cells [49].
During aging, accumulated pathologic assaults from insults including chronic inflammation, oxidative stress, and genetic changes (oncogene activation, tumor suppressor inactivation) disrupt the homeostasis between proliferation and apoptosis and lead to prostate cell hyperstimulation that culminates in either prostate hypertrophy, which clinically manifests as lower urinary tract symptoms (LUTS) and benign prostate hyperplasia (BPH), or hyperplasia and dysplasia of prostate acini that progress to adenocarcinoma, viz., prostate cancer. Mechanisms that distinguish the path to noncancer prostate enlargement from malignant progression remain unknown.
The most prevalent prostate disease for aging men is BPH/LUTS, which usually begins from the fifth decade of life. BPH, manifesting as new glandular or stromal growth at the transition zone surrounding the upper portion of the prostatic urethra, causes disruptive urinary symptoms [50]. The disease is initially a quality of life problem; however, left untreated, the symptoms can grow into serious complications such as bladder and kidney damage, inability to urinate and urinary tract infections. Signs of BPH/LUTS begin in nearly half of all 50-year-old men, and by age 80, ~80% of all men have BPH. AR/androgen signaling is closely linked to BPH etiology. For example, eunuchs (castrated men) do not develop BPH, and 5-α reductase inhibitors (such as finasteride, dutasteride), which block testosterone conversion to the prostate-active potent androgen, viz., 5α-dihydrotestosterone (DHT), are effective in alleviating clinical symptoms of BPH in many (but not all) patients. Cellular senescence, which irreversibly blocks senescent cells at the G1/S cell cycle checkpoint, is thought to contribute to BPH etiology [51]. Normally, cellular senescence is a tumor suppression mechanism that prevents proliferation of the cells that are damaged by various insults such as oxidative stress arising from cell’s normal metabolic activities, chromosomal instability due to shortened telomeres in replicating cells, and DNA damage from exposure to chemicals or ionizing radiation [52]. In a contrarian response, a senescence-associated secretory phenotype (SASP) of senescent cells, characterized by the secretion of pro-inflammatory cytokines, chemokines, growth factors, and certain proteases, generates a tissue microenvironment which promotes enhanced proliferation of nearby cells and recruitment of inflammatory cells, both leading to hyperplastic prostate growth. Prostate-originated epithelial cells proliferated faster in the presence of conditioned media from the prostatic fibroblasts of elderly donors (ages 63–81 years) compared to younger donors (ages 40–51 years) [53]. The molecular basis for BPH development is under active investigation.
Precancerous prostatic intraepithelial neoplasia (PIN), which over time can emerge as adenocarcinoma, is histologically distinct from BPH. The risk for prostate cancer increases with age. The median age of men at diagnosis of the cancer is >75 years. Prostate cancer in its early stage is androgen-dependent. Androgen deprivation therapy (ADT) involving medical or surgical castration is the standard of care for managing the disseminated disease when remission occurs due to cancer cell apoptosis. However, post-ADT relapse is almost a total certainty, and for about 70% cases, the re-emerged disease progresses to a therapy-resistant terminal stage within an average of 20 months [54]. Recurrent cancer, which arises in a castrated background, still depends on AR/androgen signaling for tumor growth. Sustained AR levels and elevated intratumoral de novo androgen biosynthesis in post-ADT patients have been extensively documented [55]. Second-generation AR antagonists (e.g., enzalutamide) and androgen biosynthesis blockers (e.g., abiraterone) are used to control post-ADT and post- (or pre-)chemotherapy tumor growth, although responses are non-durable (lasting 4–5 months on average) [56, 57]. An example of AR expression in nontumor and tumor tissue in a clinical specimen of primary prostate cancer is shown in Fig. 13.6.

AR-expressing prostate tumor and adjacent normal prostate acini of a primary human prostate cancer specimen. AR was detected in a paraffin section cut from a formalin-fixed prostatectomy sample by immunohistochemistry using a polyclonal rabbit antibody against human AR. Horseradish peroxidase conjugated anti-rabbit IgG and diaminobenzidine was used for staining. Magnification: 4× (B. Chatterjee, unpublished)
In summary, using the prostate and liver as examples, this section highlights a role for androgen/AR signaling in the physiology and pathophysiology of reproductive and nonreproductive tissues and a role of aging in AR signaling.
5 Androgen, Estrogen, and Aging
During aging, circulating androgen and estrogen levels decline in males and females, respectively (Fig. 13.7). Androgen deficiency in aging males (ADAM) is a common observation. Declining estrogen levels in females are apparent in the perimenopausal phase (by 36–45 years), and thereafter, due to cessation of ovarian function, estrogen levels fall precipitously in menopausal women (46–55 years of age) [4]. Physiological parameters such as muscle mass, bone strength, cardiovascular health and cognitive competency decline concurrently with reduced sex hormone levels [10]. Since women tend to live longer, they are more vulnerable to age-related health problems.

5.1 Testosterone in Aging Males
Androgens promote diverse male physiology including spermatogenesis, muscle mass, bone health, hair growth, nitrogen retention, and development of secondary sexual characteristics [58]. Testosterone levels, 95% of which are produced by testicular Leydig cells, gradually decline in aging males [59], whereas peripheral estrogen (as estrone) steadily rises [60]. The net effect is a reduced testosterone to estrogen ratio that leads to diminished musculature in men [61]. Studies with brown Norway rats, whose aging of the reproductive tract is similar to that of humans, showed that testosterone biosynthesis is lower in 23-week-old than 13-week-old rats. The age-related decline in testosterone production is thought to be due to accumulation of redox particles or other toxic materials that are byproducts of steroidogenesis [62, 63].
Reduction of testosterone in aging men has been documented in multiple ways: (i) taking samples directly from the spermatic vein in older men, (ii) meta-analysis of cross-sectional samples, and (iii) longitudinal investigations performed in healthy groups or cohorts [64]. Plasma testosterone in males decreases significantly after the age of 50 years. Males in the 80- to 90-year age group have about 40 % less circulating testosterone compared to males under the 50-year age group. The mean plasma testosterone concentration for young males ranges from 5.0 to 8.5 ng/ml; for older males circulating testosterone falls in the range of 1.5–5.3 ng/ml [65]. It is estimated that total testosterone (free testosterone + steroid hormone-binding globulin (SHBG)-bound testosterone) levels decline approximately 1.6% per year, while free testosterone levels fall by 2%–3% per year [64].
A 15-year-long study conducted in New Mexico (USA) showed that total testosterone levels decline by ~110 ng/dL/decade in men older than 60 years of age [61]. A study in Massachusetts (USA) predicted a 0.8%–1.3% annual decrease in bioavailable testosterone concentrations; and a longitudinal study of aging in Baltimore (USA) predicted a yearly decline of 4.9 pmol testosterone/nmol SHBG (sex hormone-binding globulin) [69,70,71]. Medications, illness and comorbidity accentuate androgen deficiency [72]. The Baltimore study showed significant age-variant longitudinal effects of age in testosterone (T) and T/SHBG, after compensating for variables [61]. In humans, the urinary excretion of androgenic and non-androgenic neutral 17-ketosteroids is considered a fair measure of androgen production in the body. A gradual but definite decrease in the urinary steroid excretion in aging men was observed when the 24-h urinary androgen excretion was analyzed in 50 men in the age group of 30 and 80 years, using the capon’s comb method [73]. Twenty percent of men aged over 60 years and 50% of men over 80 years had net testosterone below normal levels.
In women also, the urinary androgenic steroid excretion, which is about one-third of male excretion levels, declines with advancing years. This is likely due to reduced androgenic steroid production by the adrenal gland, which is thought to occur from age-associated reduction in testicular responses to gonadotrophin stimuli, along with hypothalamo-pituitary compensation for reduced testosterone levels [74].
5.2 Estrogen and Female Aging
The ovaries and placenta are the primary source for estrogens, which induce female features such as genital organ and breast development, endometrium growth, and inhibition of FSH secretion by the pituitary. Estrogen receptors (ERα, ERβ) mediate estrogen action in the nucleus. ERα is expressed significantly in the breast, ovary, uterus, prostate (stroma), bone, white adipose tissue, testes, muscle, liver, and neuronal tissue, whereas ERβ is expressed most substantially in the colon, bone marrow, salivary gland, testes, prostate, and vascular endothelium [75]. Estrogen is the primary mediator of normal sexual and reproductive behavior in women; additionally, it impacts many nonreproductive functions in men and women [76, 77]. Similar to non-genomic androgen action (discussed in Sect. 13.4), estrogens mediate rapid signaling by activating membrane estrogen receptor [78].
The female reproductive system offers the best example of aging in mammals. “Menopause” results due to a transition from complete ovarian function to ovarian non-function due to lack of estrogen synthesis in women close to 50 years of age. Besides its role in female reproductive tissues, estrogens influence functions of both sexes in diverse nonreproductive tissues such as the brain, adipose tissue, bones, skeletal muscle, vascular system, colon, and skin. Loss of estrogen action leads to increased incidence of osteoporosis, nervous system disorders, and cardiovascular diseases [79]. Within the first year of menopause, estrogen loss is approximately 80% in women [80]. This is accompanied by a speedy decline in muscle mass and strength [81, 82], caused by loss of estrogens [81]. Estrogens are also produced in men through aromatase-catalyzed conversion of testosterone to estradiol in the limbic system and brain [83]. Leydig cell development and function may also be estrogen-dependent [84].
How age-related changes in sex hormone levels affect a subset of age-related health issues is discussed below (Sect. 13.6).
6 Influence of Sex Steroids on Diseases Prevalent in Aging
6.1 Osteoporosis
Osteoporosis is the pathological loss in bone density and strength usually associated with aging. It leads to bone fractures, majorly in the hip region, vertebra, and forearm. Both estrogen and testosterone are required for normal growth and maintenance of bones. Lowering of sex hormone levels or impaired hormone function causes osteoporosis and minimal trauma fractures. The higher bone mass in men, as compared to women, implies that they are less prone to the development of osteoporosis [85]. The difference in age-related bone loss in men and women is depicted in Fig. 13.8.

Estrogen deficiency causes postmenopausal osteoporosis due to termination of ovarian function [88]. Bone cells are conspicuous estrogen targets since estrogen administration prevented bone loss induced by oophorectomy in perimenopausal women [89, 90]. At menopause, women undergo an accelerated phase of bone loss, which becomes more pronounced during the subsequent decade when 20–30% of cancellous bone and 5–10% of cortical bone are lost [91]. This accelerated phase of bone loss in women continues into a phase of slow bone loss, which lasts indefinitely.
Bone loss in menopausal women can be prevented by estrogen therapy. In osteoblasts [92, 93] and osteoclasts [94], estrogen action takes place through high-affinity ERs to regulate bone turnover. During menopause, the control is lost and bone turnover increases. Also, estrogen deficiency makes the bones more sensitive toward parathyroid hormone (PTH) [95] causing further enhancement in resorption. Increase in urinary calcium excretion [96, 97] and decrease in intestinal calcium absorption [96, 98] prevent the outflow of calcium from the skeleton to the extracellular fluids. Although it is clear that estrogen deficiency leads to an acceleration of bone loss in early postmenopausal years in women, the role that the deficiency plays in causing secondary hyperparathyroidism and increase in bone turnover in late postmenopausal women is not clear. A comparative study by Mckane et al. [97], in three separate groups of women, pre-menopausal, untreated elderly, and treated elderly, showed no effects on serum PTH and bone resorption, due to aging after correcting for estrogen deficiency [97]. Also, it was demonstrated by Heshmati et al. [99] in postmenopausal women (with an average age of 69 years) that the reduction of the serum estrogen levels by an aromatase inhibitor, letrozole, to almost undetectable levels, blocks androgen to estrogen conversion in target tissues [97], resulting in a 15% increase in bone resorption markers [99]. This study proved that the minimal levels of sex steroids present in late postmenopausal women are significant in influencing bone turnover. In fact, estrogen affects bone turnover both directly by influencing bone cells and indirectly by affecting bone turnover, by its effects on calcium homeostasis. Age-related bone loss is caused by both increase in bone resorption and impairment in bone formation; however, at menopause, bone resorption exceeds bone formation [100, 101]. Estrogen deficiency during menopause increases both bone resorption and impaired bone formation. Decreased bone formation during late post-menopause is usually attributed to age-related factors, such as growth factors [91, 102] or lower growth hormone and IGF-1 levels [103, 104]. However, estrogen itself might influence the production of IGF-I [105] and other hormones in osteoblastic cells in vitro [94].
A gradual and steady bone loss is also observed in aging men [91]. Bone mineral density in men depends on androgen levels [106, 107]. In young men, testosterone levels correlate with bone size [108], and hypogonadism or lowered testosterone levels are risk factors for hip fracture [109]. Testosterone treatment can improve bone density. Since, estrogen is produced by conversion of testosterone by aromatase, during androgen therapy for males, it is important to use an androgen which can be acted upon by aromatase for maximal beneficial effect on bones. Estrogen action is an additional determinant of bone density in males [110]. Men with deficiency of aromatase enzyme or mutant, inactivated estrogen receptors have below normal bone density, despite normal testosterone levels [111, 112].
6.2 Sex Steroids and Sarcopenia (Muscle Loss)
Aging is accompanied by a progressive decrease in muscle mass [113]. Decrease in the skeletal muscle is particularly significant as it is essential for locomotion. Sarcopenia, or loss of muscle mass, is different from muscle wasting and has its root in a Greek word meaning “loss of flesh” [114]. Age-related muscle loss can lead to disability, hospitalization, and death in older adults and creates a huge financial burden. Typically, in young adults, 50% of total bodyweight is lean muscle mass which decreases to approximately 25% of total bodyweight by the age of 75–80 years. The lowering of muscle mass in the lower limbs with aging is most important with regard to mobility status. The muscle cross-sectional area in quadriceps decreases up to 40% between 20 and 80 years [115]. The decrease in muscle mass is usually correlated with decrease in muscle function. However, studies demonstrate that decrease in muscle strength is more than what it should be on the basis of loss of muscle mass during aging [116], especially beyond 60–70 years [117]. This mismatch between muscle mass and strength results probably due to the deterioration of muscle quality. The reasons for deterioration of muscle quality are not known and may occur due to oxidative stress, mitochondrial dysfunction, a pro-inflammatory state, or metabolic problems. Also, non-muscle-related factors such as loss of motor neurons, changes in nerve-muscle communication, and hormonal changes may be other causes [118].
Androgen and Muscle Mass
Androgen-induced changes on muscle mass have been documented [119]. For example, androgen deprivation therapy against prostate cancer induces a decrease in the skeletal muscle and lean tissue and an increase in body fat [120]. Randomized trials have demonstrated that higher than normal doses of testosterone in men lead to an enhancement of fat-free muscle mass, size, and strength [121]. Androgen treatment in older men with restricted mobility and low testosterone caused improvements in muscle strength and ability to climb stairs. Testosterone induces muscle protein synthesis probably by increasing the use of cellular amino acids in skeletal muscles. Higher amounts of testosterone also increased expression of androgen receptor. Augmentation of the growth hormone (GH) axis may be beneficial as well, since dual therapy with GH and testosterone in older men enhanced lean body mass to a greater extent than the no-treatment control group or the group receiving either growth hormone or testosterone [122]. However, increased lean mass after treatment with testosterone and GH may not translate into improved muscle performance.
Loss of Muscle in Menopausal Women
In women, muscle mass starts declining gradually after 30 years, and the decline is accelerated after 50 years of age, and after menopause, muscle mass is lost at 0.6% per year [123, 124]. In postmenopausal women, important factors that may contribute to the loss of muscle mass include physical inactivity, protein intake, and oxidative stress [125, 126]. Older individuals who had participated in resistance training exercises had higher levels of muscle mass when compared with their sedentary counterparts. Protein intake is important for maintaining muscle mass in postmenopausal women. In younger men and women, the recommended protein intake is 0.8 g/kilogram of body weight daily [127]; however, 1.2 g per kg body weight works better for older men and women. Interestingly, consuming essential amino acids containing proteins from animal sources leads to increase in muscle mass in elderly women [128]. This finding demonstrates the importance of protein intake in delaying sarcopenia. During menopause, oxidative stress increases [125] which in turn may provoke cell apoptosis [129]. The mechanism of apoptosis is not completely understood but may results from reduced energy production by mitochondria, provoking muscle fiber atrophy and muscle loss [80]. Another factor that plays an important role is vitamin D3 along with calcium [130]. Vitamin D3 production by the skin is affected by age, season, latitude, and skin pigmentation [131]. Vitamin D3 receptor is present on muscle cells, and association between vitamin D3 and muscle function has been demonstrated [132, 133]. Loss of muscle strength can be associated with estrogen loss that occurs at menopause [82, 134]. Estrogen can promote muscle buildup by stimulating IGF-1 receptors [135]. In addition, ERα and ERβ are present in muscle fibers, and their number is greater in children, men, and women as compared to postmenopausal women [136]. The transcriptional activity of estrogen receptors is triggered by both circulating estrogen and IGF-1 [137, 138]. Hence, estrogen receptor action may be mediated by both estrogen and IGF-I, and a drop in their levels at menopause can possibly affect muscle mass and strength.
6.3 Sex Steroids, Cardiovascular Diseases, and Aging
Cardiovascular disease and atherosclerosis are one of the major causes of morbidity and death worldwide, and there is a clear age dependence of the disease. Also, the disease is more common in males as compared to females [139], indicating a potential role of sex hormones in the occurrence of atherosclerosis.
Testosterone and Heart Disease
Various epidemiological studies have found a correlation between coronary heart disease and low testosterone levels [140, 141]. Radiological studies suggest an inverse correlation between total and free testosterone and aortic atherosclerosis in men [142]. Epidemiological data showed association between low testosterone levels and atherogenic lipid parameters, such as lower HDL cholesterol and higher total as well as LDL cholesterol and triglyceride levels [143,144,145]. A negative correlation between testosterone and blood pressure also exists [146, 147]. Studies demonstrated reduced levels of testosterone in men with heart failure [148, 149], and administration of mechanical circulatory support in cardiac failure patients controls testosterone levels and other changes [150]. In summary, low testosterone levels are associated with cardiovascular problems, and testosterone treatment may alleviate some of the problems, depending on the patient.
Among women, cardiovascular disease (CVD) is rampant accounting for nearly 50% of female deaths [151]. Women develop CVD, around 10–15 years later in life as compared to men, and the risk increases after menopause [152]. This has led to much investigation regarding the role of estrogen as a potential cardioprotective agent. With increase in age, the difference in the number of cardiac events among pre-menopausal women versus age-matched postmenopausal women dramatically decreases. Also, there is no difference between the events occurring among postmenopausal and pre-menopausal women aged between 50 and 54 years [153]. After adjusting for age and smoking status, menopause does not seem to be associated with a higher risk for CVD [154]. Nevertheless, estrogen/progestin replacement to nearly 3000 postmenopausal women below 80 years of age, who were suffering from coronary artery disease, did not show significant differences in cardiovascular death, myocardial infarction, or any secondary outcome (including peripheral arterial disease, stroke, and congestive heart failure), between the treated and control groups after a follow-up of 4 years [155]. In fact, more women in the hormone-treated group suffered coronary events and thrombosis in the first year of treatment than the placebo-treated group. Therefore, to understand the role of estrogen in cardiac health, further studies are warranted.
6.4 Sex Hormones and Neuroprotection
Sex steroids play pivotal roles in the development and function of the central nervous system (CNS), and androgen, estrogen, and progestin are known to influence brain and CNS functions after binding to cognate receptors. The presence of sex steroid receptors outside of the CNS region and in the pituitary and hypothalamus indicates that sex hormones exert a wide range of influences on brain functions [156]. Sex steroids are also thought to play a role on the survival and programmed cell death of neurons. Neuronal diseases including dementia and Alzheimer’s increase significantly with aging and causes disability among the elderly people worldwide [157, 158]. Gonadal and adrenal steroids regulate not only reproduction but also various neuronal and glial functions [159]. Studies predict that over 33% of women and 20% of men aged 65 years and older may develop dementia [160]. Multiple factors such as genetic makeup and environment play a role in the development of these diseases [161]. Subtle changes occur with age in the nervous system, and treatment with pharmacological agents may reverse these effects. Steroid treatment may be useful in this regard since these hormones are neuroprotective and their levels decrease during aging [162]. Levels of different steroids and neurosteroids in the brain and nervous system have been measured by immunocytochemistry and GC-MS.
Estrogen and cognition
Estrogen is thought to protect against the reduction of cognitive functions that occurs during aging. A low estrogen level negatively impacts learning and memory in postmenopausal females, and it increases susceptibility to neurodegenerative diseases. ERs are distributed in several parts in the hippocampus and in the brain frontal lobes that control verbal and working memory. Many neuronal functions are mediated by the gonadal steroid hormones [159]. Steroids called neurosteroids are synthesized in the nervous system, and sex steroids regulate their synthesis [163]. Differences in brain areas in females and males are largely dependent upon the action of sex steroids, and there is a difference in expression of steroid receptors in both sexes [164]. These differences in organization occur during development, and they remain permanent. Sex differences in brain development are well documented [165].
Within the CNS, specific receptors for estrogen are seen in different brain regions, and glial cells demonstrating the role of estrogen in controlling memory and cognitive functions in females [166]. The estrogen-induced genomic mechanisms have long-term effects on neurons including the synthesis and release of many neuropeptides and neurotransmitters [167]. For example, in the rat brain, there is differential expression of ERα and ERβ, providing evidence for distinct roles of each receptor subtype [168, 169]. In addition, rapid effects of steroid hormones in the brain via non-genomic mechanisms are observed [170] that are exerted through receptors present on the plasma membrane [171, 172]. Estrogen effects on the different nervous systems, such as, serotonergic, dopaminergic, cholinergic, and noradrenergic, may contribute to various aspects of brain function, including movement and cognitive function [173]. Estrogens also modulate brain opioid peptide mRNA levels and signal transduction [174, 175]. A biologically active opioid, called β-endorphin (β-EP), modulates behavioral, analgesic, temperature-related, and neuroendocrine properties. In older female rats, brain levels of β-EP are reduced with respect to fertile female rats [176]. The reduction in β-EP levels in plasma is proposed to play a role in the mechanism of hot flushes and sweating in postmenopausal women [177]. Lowered β-EP levels are also correlated with mood, behavior, and sleep disturbances that occur in older women [178]. Indeed, treatment with estrogens, as hormone replacement therapy, seems to alleviate some of these problems [179]. Oral estrogen replacement therapy, subsequent to menopause, increases circulating β-EP levels significantly [180].
The pineal gland secretes the hormone melatonin. A circadian rhythm is observed in melatonin synthesis and secretion, since production and release is low during the day time and high at the night time [181]. Melatonin is associated with antioxidant property [182]. The administration of melatonin to female mice undergoing senescence prevents the oxidative DNA damage caused by aging in the brain [183]. In humans, melatonin administration induces a tendency to sleep, reduces body temperature, and increases secretion of luteinizing hormone and prolactin [184, 185]. Melatonin reduces blood pressure in hypertensive individuals [186]. Aging influences production and biological responses of melatonin. Due to aging, melatonin levels and melatonin receptors are reduced in animals [177]. Studies in rat showed the presence of gonadal steroid receptors in the pineal gland, suggesting an influence of gonadal steroids on melatonin synthesis [187]. In female rats, estrogens regulate melatonin synthesis [188]; however, the effect of gonadal steroids on melatonin secretion in humans is still controversial.
6.5 Testosterone and Erectile Dysfunction
Aging males require adequate testosterone levels in order to maintain normal sexual behavior. Erectile dysfunction is a common problem in aging males. Over 70% of men, older than 70 years suffer from erectile dysfunction [189]. Erectile dysfunction can be treated by phosphodiesterase-5 (PDE-5) inhibitors in most but not all men [190]. Erectile dysfunction is prevalent in men with low testosterone levels [191]. In a study of healthy men, 60–75 years of age, spontaneous erections and libido are observed after testosterone treatment that produces normal testosterone levels [192]. For patients with low testosterone levels, who do not respond to treatment with PDE-5 inhibitors, testosterone treatment along with PDE-5 inhibitors improved erectile function [193, 194].
6.6 Estrogen and Skin Aging
Menopause in women results in a speedy deterioration of skin quality. Post-menopause, the skin becomes thinner. It has reduced collagen content and elasticity and increased dryness and wrinkling. Estrogen therapy increases skin hydration, elasticity, and skin thickness in aging women. Estrogen replacement can reduce skin wrinkle, improve collagen quality, and enhance vascularization. ERα and ERβ are expressed to similar extents in the skin of males and females. ERβ expression in the epidermis is significantly reduced in individuals over 70 years of age [195]. In postmenopausal women with estrogen deficiency, skin thickness reduces approximately 1.13% and collagen by 2% per year [196]. Type I and III skin collagen decrease by 30% in the first 5 years post-menopause [197, 198] paralleling the reduction in bone mass [196]. Skin thickness and collagen content in elderly females match with the duration of estrogen deficiency rather than age [196,197,198]. The collagen subtypes are also different in pre- and postmenopausal women [198]. In response to estrogen therapy, the collagen content and composition improve. Skin wrinkling can be caused due to aging, but may also get enhanced due to hormones and environmental factors. Wrinkling occurs due to reduction in connective tissue and skin elasticity [199]. Topical estrogen administration thickens the elastic fibers and skin in postmenopausal women.
Another hallmark of aging is reduced wound healing. Estrogen might play a major role in skin physiology and wound healing. Studies indicated that estrogens influence wound healing by modifying the inflammatory responses, speeding up reepithelialization, and regulating proteolysis [200]. The healing of wounds in postmenopausal, estrogen-deficient women is slower as compared to young women, while in women taking estrogen replacement, it was comparable to women in younger age group [201].
7 Hormone Replacement Therapy (HRT) and Aging
Several studies showed that supplementation with male and female sex hormones such as testosterone, estrogen, progesterone, or growth and thyroid hormones (popularly referred as “fountain of youth”) has the potential to improve the quality of life and prevent many age-related symptoms such as fatigue, depression, loss of libido, osteoporosis, and cardiovascular diseases. Recent studies showed various symptoms of menopause, and the risk of osteoporotic fractures decreased significantly by administration of HRT [202]. Though there are many studies supportive of hormone replacement therapy, the side effects in long-term use cannot be denied which may also result in the development of cancer [203, 204]. In consideration to the risk-benefit ratio, the most suitable method for the younger women is the lifestyle management in comparison to the HRT due to lack of proper strategy in use. HRT can be successfully used as a prevention strategy to decrease the severity of many age-related disorders like coronary heart disease, etc. [202].
8 Discussion and Conclusion
The primary sex hormones, androgens and estrogens, are vital for a number of physiological functions. Extensive evidences indicate that aging causes a general decline in both of these hormonal levels. Aging of the reproductive glands (ovaries or testes), which are source for the release of sex steroids, results in undesirable clinical manifestation. With aging both males and females suffer from the reduction of sex hormones, though the effects are much more evident in females with the onset of menopause. Age-related effects of hormonal changes on numerous biological processes are summarized in Fig. 13.9.

Effect of aging on various biological processes and linkage with sex steroids and vice versa. Aging causes a decline in both male and female sex hormonal levels influencing the body and its various biological processes
All steroid hormones are synthesized from cholesterol, the main source of which is LDL cholesterol [3]. The type of steroid hormone produced by a specific steroidogenic cell type depends on its response to tropic hormones and steroidogenic enzymes. Studies in aging rats have shown that the reason for lowering of steroid hormonal levels is that the cholesterol is not transported in optimal amounts to adrenal and testes for the initiation of steroid biosynthesis, namely, cholesterol conversion to pregnenolone that occurs in the inner mitochondrial membrane. The reduction in levels of two important proteins StAR (steroidogenic acute regulatory protein) and PBR/TSPO (peripheral benzodiazepine receptor/translocator protein) which enable cholesterol transport to the inner mitochondrial membrane appear to be the cause for decline in cholesterol availability. Further studies have demonstrated that age-induced increase in production of ROS (reactive oxygen species) is responsible for the changes in expression of these proteins. “ROS” generation and consequent oxidative damage to macromolecules seem to be the primary determinants of longevity in mammals.
Biological macromolecules, including lipids, nucleic acids, and proteins, are susceptible to oxidative damage induced by free radicals [205]. As cellular membranes produce these free radicals, lipid peroxidation in membranes is the major cause of ROS damage during aging [206, 207]. This hypothesis is supported by the observation that lipid peroxidation is enhanced with age [208, 209] and leads to formation of lipofuscin, a fluorescent pigment that accumulates in tissues with age [208]. In steroidogenic cells, the risk of lipid peroxidation is especially high as they use molecular oxygen for steroid biosynthesis [210, 211] and other cellular functions [206, 207, 212, 213]. The cytochrome P450 enzymes required for steroid hormone synthesis use molecular oxygen to hydroxylate substrates and may lead to production of ROS such as hydroxyradicals, superoxide anions, and other oxygen free radicals [210, 211, 214, 215]. These oxyradicals cause oxidative changes leading to cell damage and death. Since lipid peroxidation in membranes could affect the structure, function, and composition of membranes, almost all the steps associated with cholesterol utilization and steroidogenesis which are entirely dependent on membrane integrity will be adversely affected [5]. As a result, steroidogenic cells are well equipped with various antioxidants. For example, in young rat adrenals, minimal lipid peroxidation and highest resistance to oxidative damage were observed [216]. However, aging causes many oxidative changes in adrenocortical and testicular Leydig cells [217]. With age, the protective antioxidant system in cells weakens, causing excessive oxidative stress and decline of steroid production. It is not clear why the oxidative balance in steroidogenic cells cannot be maintained through expression of antioxidant enzymes. In many mammals, females live longer than males. For example, male Wistar rats have an average life span of 24 months, while females have average life span of 29 months; a similar gender difference exists in humans [218]. The difference seems to lie in the oxidative status of both species. Studies showed that levels of glutathione, a major cellular antioxidant, are approximately half in males compared to females. Levels of 8-oxo-deoxyguanosine (8-oxo-dG), an excellent indicator of oxidative damage, are fourfold higher in males than females [218].
Since the endocrine system is a major regulator of overall metabolism and growth, changes in steroid hormone levels are important contributors to overall aging. Little is known of the interrelations between oxidative stress, steroidogenesis, and aging at the molecular level. It is necessary to understand the molecular events that cause ROS generation and the mechanisms by which ROS reduce levels of proteins such as StAR and PBR/TSPO, which ultimately impact steroidogenesis. Precise roles of androgen and estrogen in aging, longevity, and quality of life need to be deciphered. Suitability of sex steroid therapy for reversing age-related accumulation of ROS needs to be explored. Therapy, however, should be directed specifically to target tissue(s) without interfering with vital physiological processes.
References
Zajacic-Rotkvic V, Kavur L, Maja C-B (2010) Hormones and aging. Acta Clin Croat 49:549–554
Horstman AM, Gerrits KH, Beltman MJ, Koppe PA, Janssen TW, de Haan A (2010) Intrinsic properties of the knee extensor muscles after subacute stroke. Arch Phys Med Rehabil 91:123–128
Zaidi KS, Shen W-Z, Azhar S (2012) Impact of aging on steroid hormone biosynthesis and Secretion. Open Longevity Sci 6:1–30
Eskin BA (1978) Sex hormones and aging. Adv Exp Med Biol 97:207–224
Miller WL, Auchus RJ (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 321:81–151
Kallen CB (2004) Steroid hormone synthesis in pregnancy. Obstet Gynecol Clin N Am 31:795–816
Arlt W, Stewart PM (2005) Adrenal corticosteroid biosynthesis, metabolism and action. Endocrinol Metab Clin N Am 34:293–313
Kendall B, Eston R (2002) Exercise-induced muscle damage and the potential protective role of estrogen. Sports Med 32:103–123
Goodman-Gruen D, Barrett-Connor E (2000) Sex differences in the association of endogenous sex hormone levels and glucose tolerance status in older men and women. Diabetes Care 23:912–918
Horstman AM, Dillon EL, Urban RJ, Sheffield-Moore M (2012) The Role of Androgens and Estrogens on Healthy Aging and Longevity. J Gerontol A Biol Sci Med Sci 67:1140–1152
Kumar S, Saradhi M, Chaturvedi NK, Tyagi RK (2006) Intracellular localization and nucleocytoplasmic trafficking of steroid receptors: an overview review. Mol Cell Endocrinol 246:147–156
Kumar S, Kumar S, Thakur K, Kumar S, Bagchi G, Tyagi RK (2016) Androgen receptor signaling by growth factors in androgen-independent prostate cancer: recent advances and emerging perspectives. In: Haldar C, Gupta S, Goswami S (eds) Updates on integrative physiology and comparative endocrinology. Press and Publication Division, Banaras Hindu University, Varanasi. ISBN: 81-85305-72-2
Tyagi RK, Lavrovsky Y, Ahn SC, Song CS, Chatterjee B, Roy AK (2000) Dynamics of intracellular movement and nucleocytoplasmic recycling of the ligand-activated androgen receptor in living cells. Mol Endocrinol 14:1162–1174
Htun H, Holth LT, Walker D, Davie JR, Hager GL (1999) Direct visualization of the human estrogen receptor alpha reveals a role for ligand in the nuclear distribution of the receptor. Mol Biol Cell 10:471–486
Roy AK, Tyagi RK, Song CS, Lavrovsky Y, Ahn SC, Oh TS, Chatterjee B (2001) Androgen receptor: structural domains and functional dynamics after ligand-receptor interaction. Ann NY Acad Sci 949:44–57
Dehm S, Tindall D (2011) Alternatively spliced androgen receptor variants. Endocrine Relat Cancer 18:R183–R196
Evans RM, Mangelsdorf DJ (2014) Nuclear receptors, RXR and the big bang. Cell 157:255–266
Ni L, Llewellyn R, Kesler CT, Kelley JB, Spencer A, Snow CL, Shank L, Paschal BM (2013) Androgen Induces a Switch from Cytoplasmic Retention to Nuclear Import of the Androgen Receptor. Mol Cell Biol 33:4766–4778
Cato L, Neeb A, Brown M, Cato ACB (2014) Control of steroid receptor dynamics and function by genomic actions of the cochaperones p23 and Bag-1L. Nucl Recept Signal 12:e005. https://doi.org/10.1621/nrs.12005
Rivera OJ, Song CS, Centonze VE, Lechleiter JD, Chatterjee B, Roy AK (2003) Role of the PML body in the dynamic interaction between the androgen receptor and steroid receptor coactivator-1 in living cells. Mol Endocrinol 17:128–140
Rosenfeld MG, Lunyak VV, Glass CK (2006) Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 20:1405–1418
Cai C, Yuan X, Balk SP (2013) Androgen receptor epigenetics. Transl Androl Urol 2:148–157
Zhang A, Zhao JC, Kim J, Chakravarti D, Mo YY, Yu J (2015) LncRNA HOTAIR enhances the androgen-receptor-mediated transcriptional program and drives castration-resistant prostate cancer. Cell Rep 13:209–221
Pascal LE, Wang Z (2014) Unzipping androgen action through ZIP9: a novel membrane androgen receptor. Endocrinol 155:4120–4123
Bagchi G, Wu J, French J, Kim J, Moniri NH, Daaka Y (2008) Androgens transduce the Gαs-mediated activation of protein kinase A in prostate cells. Cancer Res 68(9). https://doi.org/10.1158/0008-5472
Dagar M, Bagchi G (2015) Cross talk between protein kinase A and androgen signaling pathway. J Endocrinol Reprod 19:1–6
Zhou J, Richardson M, Reddy V, Menon M, Barrack ER, Reddy GP, Kim SH (2013) Structural and functional association of androgen receptor with telomeres in prostate cancer cells. Aging 5:3–17
Ma W-L, Lai H-C, Yeh S, Cai X, Chang C (2014) Androgen receptor roles in hepatocellular carcinoma, cirrhosis, and hepatitis. Endocr Relat Cancer 21:R165–R182
Lin H-Y, Yu I-C, Wang R-S, Chen Y-T, Liu N-C, Altuwaijri S, Hsu CL, Ma W-L, Jokinen J, Sparks JD, Yeh S, Chang C (2008) Increased hepatic steatosis and insulin resistance in mice lacking hepatic androgen receptor. Hepatology 47:1924–1935
Song CS, Jung MH, Kim SC, Hassan T, Roy AK, Chatterjee B (1998) Tissue-specific and androgen-repressible regulation of the rat dehydroepiandrosterone sulfotransferase gene promoter. J Biol Chem 273:21856–21866
Prakash CP, Zuniga B, Song CS, Jiang S, Cropper JD, Park S, Chatterjee B (2015) Nuclear receptors in drug metabolism, drug response and drug interactions. Nucl Recept Res 2:1–20
Kanda T, Yokosuka O (2015) The androgen receptor as an emerging target in hepatocellular carcinoma. J Hepatocell Carcinoma 2:91–99
Kemp CJ, Leary CN, Drinkwater NR (1989) Promotion of murine hepatocarcinogenesis by testosterone is androgen receptor-dependent but not cell autonomous. Proc Natl Acad Sci USA 86:7505–7509
Ma W-L, Hsu C-L, Yeh C-C, Wu M-H, Huang C-K, Jeng L-B, Hung Y-C, Lin T-Y, Yeh S, Chang C (2012) Hepatic androgen receptor suppresses hepatocellular carcinoma metastasis through modulation of cell migration and anoikis. Hepatology 56:176–185
Shi LH, Ko S, Kim S, Echchgadda I, Oh T, Song CS, Chatterjee B (2008) Reciprocal synamics of the tumor suppressor p53 and poly(ADP-ribose)polymerase PARP-1 regulates loss of androgen receptor in aging and oxidative stress. J Biol Chem 283:36474–36485
Song CS, Rao TR, Demyan WF, Mancini MA, Chatterjee B, Roy AK (1991) Androgen receptor messenger ribonucleic acid (mRNA) in the rat liver: changes in mRNA levels during maturation, aging, and calorie restriction. Endocrinol 128:349–356
Chatterjee B, Fernandes G, Yu BP, Song C, Kim JM, Demyan W, Roy AK (1989) Calorie restriction delays age-dependent loss in androgen responsiveness of the rat liver. FASEB J 3:169–173
Roy AK, Oh T, Rivera O, Mubiru J, Song CS, Chatterjee B (2002) Impacts of transcriptional regulation on aging and senescence. Ageing Res Rev 1:367–380
Supakar PC, Song CS, Jung MH, Slomczynska MA, Kim JM, Vellanoweth RL, Chatterjee B, Roy AK (1993) A novel regulatory element associated with age-dependent expression of the rat androgen receptor gene. J Biol Chem 268:26400–26408
Supakar PC, Jung MH, Song CS, Chatterjee B, Roy AK (1995) Nuclear factor кB functions as a negative regulator for the rat androgen receptor gene and NF-кB activity increases during the age-dependent desensitization of the liver. J Biol Chem 270:837–842
Ko SY, Shi LH, Song CS, Chatterjee B (2008) Interplay of NF-кB and B-myb in TNFα-controlled negative regulation of androgen receptor expression. Mol Endocrinol 22:273–286
Lavorvsky Y, Chatterjee B, Clark RA, Roy AK (2000) Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp Gerontol 35:521–532
Kraus WL, Lis JT (2003) PARP goes transcription. Cell 113:677–683
Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Guarente L, Auwerx J et al (2013) The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154:430–441
Gomes AP, Price NL, Ling AJY, Moslehi JJ, Montgomery MK, Rajman L, Bell EL, Sinclair DA et al (2013) Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155:1624–1638
Li J, Bonkowski MS, Moniot S, Zhang D, Hubbard BP, Ling AJY, Steegborn C, Sinclair DA et al (2017) A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science 355:1312–1317
Prins GS, Jung MH, Vellanoweth RL, Chatterjee B, Roy AK (1996) Age-dependent expression of the androgen receptor gene in the prostate and its implication in glandular differentiation and hyperplasia. Dev Genet 18:99–106
Cunha GR (1996) Growth factors as mediators of androgen action during male urogenital development. Prostate 6:22–25
Shi X, Gippi J, Dries M, Bushman W (2014) Prostate progenitor cells proliferate in response to castration. Stem Cell Res 13:154–163
Vignozzi L, Gacci M, Maggi M (2016) Lower urinary tract symptoms, benign prostatic hyperplasia and metabolic syndrome. Nat Rev Urol 13:108–119
Vital P, Castro P, Tsang S, Ittmann M (2014) The senescence-associated secretory phenotype promotes benign prostatic hyperplasia. Am J Pathol 184:721–733
Coppé J-P, Desprez P-Y, Krtolical A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Ann Rev Pathol 5:99–118
Begley L, Monteleon C, Shah RB, MacDonald JW, Macoska JA (2005) CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell:291–298
Harshman LC, Wang X, Nakabayashi M, Xie W, Valenca L, Werner L, Yu Y, Kantoff AM, Sweeney CJ, Mucci LA, Pomerantz M, Lee GM, Kantoff PW (2015) Statin use at the time of initiation of androgen deprivation therapy and time to progression in patients with hormone-sensitive prostate cancer. JAMA Oncol:0829
Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B et al (2011) Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res 71:6503–6513
de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr, Saad F, Staffurth JN, Mainwaring P, Harland S et al (2011) Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364:1995–2005
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Fléchon A et al (2012) Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 367:1187–1197
Nieschlag E, Behre HM, Nieschlag S. Andrology (2001) Male reproductive health and dysfunction, 3rd ed. Springer
Arianayagam R, Arianayagam M, McGrath S, Rashid P (2010) Androgen deficiency in the aging man. Aust Fam Physician 39:752–755
MacDonald PC. 1976. Origin of estrogen in men. In: Benign prostatic hyperplasia. DHEW Publication No. (NIH) 76–1113.
Harman SM, Metter EJ, Tobin JD, Pearson J, Blackman MR (2001) Longitudinal effects of aging on serum total and free testosterone levels in healthy men. Baltimore Longitudinal Study of Aging. J Clin Endocrin Metab 86:724–731
Chen W, Hunt DM, Lu H, Hunt RC (1999) Expression of antioxidant protective proteins in the rat retina during prenatal and postnatal development. Invest Ophthalmol Vis Sci 40:744–751
Zirkin BR, Chen H (2000) Regulation of Leydig cell steroidogenic function during aging. Biol Reprod 63:977–981
Stanworth RD, Hugh Jones T (2008) Testosterone for the aging male; current evidence and recommended practice. Clinical Interv Aging 3:25–44
Vermeulen A, Rubens R, Verdonck L (1972) Testosterone secretion and metabolism in male senescence. J Clin Endocr 34:730
Vermeulon A (1995) Declining androgens with age: an overview. In: Vermeulon A, Oddens BJ, International Health Foundation (eds) Androgens and the aging male. Parthenon Publishing, New York, pp 3–14
Simon D, Nahoul K, Charles MA. 1996. Sex hormones, aging, ethnicity and insulin sensitivity in men: an overview of the TELECOM study. In Vermeulon A, Oddens BJ, International Health Foundation (Eds), Androgens and the aging male (pp 85–102), Parthenon Publishing: New York.
Navarro D, Acosta A, Robles E, Díaz C (2012) Hormone profile of menopausal women in Havana. MEDICC Rev 14(2)
Liu PY, Iranmanesh A, Nehra AX, Keenan DM, Veldhuis JD (2005a) Mechanisms of hypoandrogenemia in healthy aging men. Endocrinol Metab Clin North Am 34:935–955
Liu PY, Swerdloff RS, Veldhuis JD (2004) The rationale, efficacy and safety of androgen therapy in older men: future research and current practice recommendations. J Clin Endocrinol Metab 89:4789–4796
Morley JE, Kaiser FE, Perry HM 3rd, Patrick P, Morley PM, Stauber PM, Vellas B, Baumgartner RN, Garry PJ. 1997. Longitudinal changes in testosterone, luteinizing hormone, and follicle-stimulating hormone in healthy older men. Metab Clin Exp 46: 410–413.
Gray A, Berlin JA, McKinlay JB, Longcope C (1991) An examination of research design effects on the association of testosterone and male aging: results of a meta-analysis. J Clin Epidemiol 44:671–684
Hamburger C, Halvorsen K, Pedersen J (1945) Assay of Androgenic substances in the urine of normal men and women. Acta Pharmacol Toxicol 1:129–140
Veldhuis JD (2008) Aging and hormones of the hypothalamo-pituitary axis: gonadotropic axis in men and somatotropic axes in men and women. Ageing Res Rev 7:189–208
Dahlman-Wright K, Cavailles V, Fuqua SA et al (2006) International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol Rev 58(4):773–781
Katzenellenbogen BS, Montano MM, Le Goff P et al (1995) Antiestrogens: mechanisms and actions in target cells. J Steroid Biochem Mol Biol 53:387–393
Heldring N, Pike A, Andersson S et al (2007) Estrogen receptors: how do they signal and what are their targets. Physiol Rev 87(3):905–931
Mendelsohn ME, Karas RH (2010) Rapid progress for non-nuclear estrogen receptor signaling. J Clin Investig 120:2277–2279
Thornton MJ (2013) Estrogens and aging skin. Dermato-endocrinology 5:264–270
Vina J, Sastre J, Pallardo FV, Gambini J, Borras C (2006) Role of mitochondrial oxidative stress to explain the different longevity between genders: protective effect of estrogens. Free Radic Res 40:1359–1365
Phillips SK, Rook KM, Siddle NC, Bruce SA, Woledge RC (1993) Muscle weakness in women occurs at an earlier age than in men, but strength is preserved by hormone replacement therapy. Clin Sci (Lond) 84:95–98
Greeves JP, Cable NT, Reilly T, Kingsland C (1999) Changes in muscle strength in women following the menopause: a longitudinal assessment of the efficacy of hormone replacement therapy. Clin Sci (Lond) 97(1):79–84
Naftolin F, Ryan KJ, Petro Z (1971) Aromatization of androstenedione by the diencephalon. J Clin Endocrinol Metab 33(2):368–370
Abney TO (1999) The potential roles of estrogens in regulating Leydig cell development and function: a review. Steroids 64(9):610–617
Campion JM, Maricic MJ (2003) Osteoporosis in men. Am Fam Physician 67:1521–1526
Riggs BL, Khosla S, Melton LJ 3rd. 1998. A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J Bone Miner Res 13(5):763-773.
Novotny R, Davis J (2015) Growth in bone and body size among Asian and white girls in the Female Adolescent Maturation (FAM) study. Arch Osteoporos 10:31
Albright F, Smith PH, Richardson AM (1941) postmenopausal osteoporosis. JAMA 116:2465–2474
Lindsay R, Aitkin JM, Anderson JB, Hart DM, MacDonald EB, Clarke AC (1976) Long-term prevention of postmenopausal osteoporosis by oestrogen. Lanceti:1038–1040
Genant HK, Cann CE, Ettinger B, Gordan GS (1982) Quantitative computed tomography of vertebral spongiosa: A sensitive method for detecting early bone loss after oophorectomy. Ann Intern Med 97:699–705
Riggs BL, Melton LJ (1986) Medical progress series: Involutional osteoporosis. N Engl J Med 314:1676–1686
Eriksen EF, Colvard DS, Berg NJ, Graham ML, Mann KG, Spelsberg TC, Riggs BL (1988) Evidence of estrogen receptors in normal human osteoblast-like cells. Science 241:84–86
Komm BS, Terpening CM, Benz DJ, Graeme KA, O’Malley BW, Haussler MR (1988) Estrogen binding receptor mRNA, and biologic response in osteoblast-like osteosarcoma cells. Science 241:81–84
Oursler MJ, Osdoby P, Pyfferoen J, Riggs BL, Spelsberg TC (1991) Avian osteoclasts as estrogen target cells. Proc Natl Acad Sci USA 88:6613–6617
Cosman F, Shen V, Xie F, Seibel M, Ratcliffe A, Lindsay R (1993) Estrogen protection against bone resorbing effects of parathyroid hormone infusion. Ann Intern Med 118:337–343
Heaney RP, Recker RR, Saville PD (1978) Menopausal changes in calcium balance performance. J Lab Clin Med 92:953–963
McKane WR, Khosla S, Burritt MF, Kao PC, Wilson DM, Ory SJ, Riggs BL (1995) Mechanism of renal calcium conservation with estrogen replacement therapy in women in early post- menopause-A clinical research center study. J Clin Endocrinol Metab 80:3458–3464
Gennari C, Agnusdei D, Nardi P, Civitelli R (1990) Estrogen preserves a normal intestinal responsiveness to 1,25-dihydroxyvitamin D3 in oophorectomized women. J Clin Endocrinol Metab 71:1288–1293
Heshmati HM, Khosla S, Robins SP, Geller N, McAlister CA, Riggs BL (2002) Endogenous residual estrogen levels determine bone resorption even in late post-menopausal women. J Bone Miner Res 17:172–178
Garnero P, Sornay-Rendu E, Chapuy M, Delmas PD (1996) Increased bone turnover in late postmenopausal women is a major determinant of osteoporosis. J Bone Miner Res 11:337–349
Lips P, Courpron P, Meunier PJ (1978) Mean wall thickness of trabecular bone packets in the human iliac crest: changes with age. Calcif Tissue Res 26:13–17
Marie PJ, de Vernejoul MC (1993) Proliferation of bone surface-derived osteoblastic cells and control of bone formation. Bone 14:463–468
Vermeulen A (1987) Nyctohemeral growth hormone profiles in young and aged men: correlation with somatomedin-C levels. J Clin Endocrinol Metab 64:884–888
Landin-Wilhelmsen K, Wilhelmsen L, Lappas G, Rosen T, Lindstedt G, Lundberg P, Bengtsson B (1994) Serum insulin-like growth factor I in a random population sample of men and women: relation to age, sex, smoking habits, coffee consumption and physical activity, blood pressure and concentrations of plasma lipids, fibrinogen, parathyroid hormone and osteocalcin. Clin Endocrinol 41:351–357
Ernst M, Heath JK, Rodan GA (1989) Estradiol effects on proliferation, messenger ribonucleic acid for collagen and insulin-like growth factor-I, and parathyroid hormone-stimulated adenylate cyclase activity in osteoblastic cells from calvariae and long bones. Endocrinology 125:825–833
Murphy S, Khaw KT, Cassidy A et al (1993) Sex hormones and bone mineral density in elderly men. Bone Miner 20:133–140
Rucker D, Ezzat S, Diamandi A et al (2004) IGF-I and testosterone levels as predictors of bone mineral density in healthy, community-dwelling men. Clin Endocrinol 60:491–499
Lorentzon M, Swanson C, Andersson N et al (2005) Free testosterone is a positive, whereas free estradiol is a negative, predictor of cortical bone size in young Swedish men: the GOOD study. J Bone Miner Res 20:1334–1341
Jackson JA, Riggs MW, Spiekerman AM (1992) Testosterone deficiency as a risk factor for hip fractures in men: a case-control study. Am J Med Sci 304:4–8
Khosla S, Melton LJ 3rd, Atkinson EJ, et al. 2001. Relationship of serum sex steroid levels to longitudinal changes in bone density in young versus elderly men. J Clin Endocrinol Metab 86: 3555–3561.
Smith EP, Boyd J, Frank GR et al (1994) Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 331:1056–1061
Carani C, Qin K, Simoni M et al (1997) Effect of testosterone and estradiol in a man with aromatase deficiency. N Engl J Med 337:91–95
Candow DG, Chilibeck PD (2005) Differences in size, strength, and power of upper and lower body muscle groups in young and older men. J Gerontol A Biol Sci Med Sci 60:148–156
Rosenberg IH (1997) Sarcopenia: origins and clinical relevance. J Nutr 127(suppl):990–991
Lexell J (1995) Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci 50:11–16
Goodpaster BH, Park SW, Harris TB et al (2006) The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci 61:1059–1064
Baum K, Hildebrandt U, Edel K et al (2009) Comparison of skeletal muscle strength between cardiac patients and age-matched healthy controls. Int J Med Sci 6:184–191
Kalyani RR, Corriere M, Ferrucci L (2014) Age-related and disease-related muscle loss: the effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol 2:819–829
Maggio M, Ceda GP, Lauretani F et al (2011) Gonadal status and physical performance in older men. Aging Male 14:42–47
Boxer RS, Kenny AM, Dowsett R, Taxel P (2005) The effect of 6 months of androgen deprivation therapy on muscle and fat mass in older men with localized prostate cancer. Aging Male 8:207–212
Bhasin S, Storer TW, Berman N et al (1996) The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med 335:1–7
Schroeder ET, He J, Yarasheski KE et al (2012) Value of measuring muscle performance to assess changes in lean mass with testosterone and growth hormone supplementation. Eur J Appl Physiol 112:1123–1131
Aloia JF, McGowan DM, Vaswani AN, Ross P, Cohn SH (1991) Relationship of menopause to skeletal and muscle mass. Am J Clin Nutr 53:1378–1383
Rolland YM, Perry HM III, Patrick P, Banks WA, Morley JE (2007) Loss of appendicular muscle mass and loss of muscle strength in young postmenopausal women. J Gerontol A Biol Sci Med Sci 62:330–335
Signorelli SS, Neri S, Sciacchitano S, Pino LD, Costa MP, Marchese G et al (2006) Behaviour of some indicators of oxidative stress in postmenopausal and fertile women. Maturitas 53:77–82
Baumgartner RN, Waters DL, Gallagher D, Morley JE, Garry PJ (1999) Predictors of skeletal muscle mass in elderly men and women. Mech Ageing Dev 107:123–136
Wolfe RR, Miller SL, Miller KB (2008) Optimal protein intake in the elderly. Clin Nutr 27:675–684
Lord C, Chaput JP, Aubertin-Leheudre M, Labonte M, Dionne IJ (2007) Dietary animal protein intake: association with muscle mass index in older women. J Nutr Health Aging 11:383–387
Hiona A, Leeuwenburgh C (2008) The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp Gerontol 43:24–33
DeLuca HF (2004) Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr 80(6 Suppl):1689S–1696S
Holick MF. 1999. Vitamin D: photobiology, metabolism, mechanism of action, and clinical applications. In: Favus MJ (ed) Primer on the metabolic bone diseases and disorders of mineral metabolism. Lippincott Williams & Wilkins, Philadelphia, pp 92–98
Zanello SB, Collins ED, Marinissen MJ, Norman AW, Boland RL (1997) Vitamin D receptor expression in chicken muscle tissue and cultured myoblasts. Horm Metab Res 29:231–236
Bischoff HA, Borchers M, Gudat F, Duermueller U, Theiler R, Stahelin HB et al (2001) In situ detection of 1,25-dihydroxyvitamin D3 receptor in human skeletal muscle tissue. Histochem J 33:19–24
Calmels P, Vico L, Alexandre C, Minaire P (1995) Cross-sectional study of muscle strength and bone mineral density in a population of 106 women between the ages of 44 and 87 years: relationship with age and menopause. Eur J Appl Physiol Occup Physiol 70:180–186
Sitnick M, Foley AM, Brown M, Spangenburg EE (2006) Ovariectomy prevents the recovery of atrophied gastrocnemius skeletal muscle mass. J Appl Physiol 100:286–293
Wiik A, Ekman M, Johansson O, Jansson E, Esbjornsson M (2009) Expression of both oestrogen receptor alpha and beta in human skeletal muscle tissue. Histochem Cell Biol 131:181–189
Ciana P, Raviscioni M, Mussi P, Vegeto E, Que I, Parker MG et al (2003) In vivo imaging of transcriptionally active estrogen receptors. Nat Med 9:82–86
Murphy LJ, Ghahary A (1990) Uterine insulin-like growth factor-1: regulation of expression and its role in estrogen-induced uterine proliferation. Endocr Rev 11:443–453
MacKay JD, Mensah GA (2004) The atlas of heart disease and stroke. WHO, Geneva
Jones RD, Pugh PJ, Hall J et al (2003) Altered circulating hormone levels, endothelial function and vascular reactivity in the testicular feminised mouse. Eur J Endocrinol 148:111–120
Jones RD, Nettleship JE, Kapoor D et al (2005) Testosterone and atherosclerosis in aging men: purported association and clinical implications. Am J Cardiovasc Drugs 5:141–154
Hak AE, Witteman JC, de Jong FH et al (2002) Low levels of endogenous androgens increase the risk of atherosclerosis in elderly men: the Rotterdam study. J Clin Endocrinol Metab 87:3632–3639
Haffner SM, Mykkanen L, Valdez RA et al (1993) Relationship of sex hormones to lipids and lipoproteins in nondiabetic men. J Clin Endocrinol Metab 77:1610–1615
Van Pottelbergh I, Braeckman L, De Bacquer D et al (2003) Differential contribution of testosterone and estradiol in the determination of cholesterol and lipoprotein profile in healthy middle-aged men. Atherosclerosis 166:95–102
Lichtenstein MJ, Yarnell JW, Elwood PC et al (1987) Sex hormones, insulin, lipids, and prevalent ischemic heart disease. Am J Epidemiol 126:647–657
Khaw KT, Barrett-Connor E (1988) Blood pressure and endogenous testosterone in men: an inverse relationship. J Hypertens 6:329–332
Schirmer SH, Buschmann IR, Jost MM, Hoefer IE, Grundmann S, Andert JP, Ulusans S, Bode C, Piek JJ, van Royen N (2004) Differential effects of MCP-1 and leptin on collateral flow and arteriogenesis. Cardiovasc Res 64:356–364
Kontoleon PE, Anastasiou-Nana MI, Papapetrou PD et al (2003) Hormonal profile in patients with congestive heart failure. Int J Cardiol 87:179–183
Tappler B, Katz M (1979) Pituitary-gonadal dysfunction in low-output cardiac failure. Clin Endocrinol 10:219–226
Noirhomme P, Jacquet L, Underwood M et al (1999) The effect of chronic mechanical circulatory support on neuroendocrine activation in patients with end-stage heart failure. Eur J Cardiothorac Surg 16:63–67
Ahmed SA, Penhale WJ, Talal N (1985) Sex hormones, immune responses, and autoimmune diseases. Mechanisms of sex hormone action. Am J Pathol 121:531–551
Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J (2002) Writing group for the women’s health initiative investigators. JAMA 288:321–333
Hodis HN, Mack WJ (2002) Atherosclerosis imaging methods: assessing cardiovascular disease and evaluating the role of estrogen in the prevention of atherosclerosis. Am J Cardiol 89:19E–27E. discussion 27E
Colditz GA, Willett WC, Stampfer MJ, Rosner B, Speizer FE, Hennekens CH (1987) A prospective study of age at menarche, parity, age at first birth, and coronary heart disease in women. Am J Epidemiol 126:861–870
Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, Vittinghoff E (1998) Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 280:605–613
Genazzani AR, Petraglia F, Purdy RH (1996) The brain: source and target for sex steroid hormones. The Parthenon Publishing Group, Italy
Hebert LE, Beckett LA, Scherr PA, Evans DA (2001) Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis Assoc Disord 15:169–173
Sloane PD, Zimmerman S, Suchindran C, Reed P, Wang L, Boustani M, Sudha S (2002) The public health impact of Alzheimer’s disease, 2000–2050: potential implication of treatment advances. Annu Rev Public Health 23:213–231
Schumacher M, Akwa Y, Guennoun R, Robert F, Labombarda F, Desarnaud F, Robel P, De N, Baulieu EE (2000) Steroid synthesis and metabolism in the nervous system: trophic and protective effects. J Neurocytol 29:307–326
Ott A, Breteler MM, van Harskamp F, Stijnen T, Hofman A (1998) Incidence and risk of dementia: the Rotterdam Study. Am J Epidemiol 147:574–580
Amouyel P (2002) Genetic susceptibility to aging-associated diseases. C R Biol 325:741–745
Schumacher M, Weill-Engerer S, Liere P, Robert F, Franklin RJM, GarciaSegura LM, Lambert JJ, Mayo W, Melcangi RC, Parducz A et al (2003) Steroid hormones and neurosteroids in normal and pathological aging of the nervous system. Prog Neurobiol 71:3–29
Baulieu EE (1997) Neurosteroids: of the nervous system, by the nervous system, for the nervous system. Recent Prog Horm Res 52:1–32
Kruijver FP, Fernandez-Guasti A, Fodor M, Kraan EM, Swaab DF (2001) Sex differences in androgen receptors of the human mammillary bodies are related to endocrine status rather than to sexual orientation or transsexuality. J Clin Endocrinol Metab 86:818–827
Beyer C (1999) Estrogen and the developing brain. Anat Embryol 199:379–390
Sherwin BB (1997) Estrogen effects on cognition in menopausal women. Neurology 48:S21–S26
Alonso-Soleis R, Abreu P, Leopez-Coviella I, Hernandez G, Fajardo N (1996) Gonadal steroid modulation of neuroendocrine transduction: a transynaptic view. Cell Mol Neurobiol (3):357–382
Keefe D, Garcia-Segura LM, Naftolin F (1994) New insights into estrogen action on the brain. Neurobiol Aging 15:495–497
Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS (1997) Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology 138:4613–4621
Gruber CJ, Tschugguel W, Schneeberger C, Huber JC (2002) Production and actions of estrogens. N Engl J Med 346:340–352
Falkenstein E, Wehling M (2000) Nongenomically initiated steroid actions. Eur J Clin Invest 30(Suppl 3):51–54
Kuppers E, Ivanova T, Karolczak M, Lazarov N, Fohr K, Beyer C (2001) Classical and nonclassical estrogen action in the developing midbrain. Horm Behav 40:196–202
Phillips SM, Sherwin BB (1992a) Effects of estrogen on memory function in surgically menopausal women. Psychoneuroendocrinology 17:485–495
Simerly RB, McCall LD, Watson SJ (1988) Distribution of opioid peptides in the preoptic region: immunohistochemical evidence for a steroidsensitive enkephalin sexual dimorphism. J Comp Neurol 276:442–459
Craft RM, Mogil JS, Aloisi AM (2004) Sex differences in pain and analgesia: the role of gonadal hormones. Eur J Pain 8:397–411
Genazzani AR, Stomati M, Bernardi F, Luisi S, Casarosa E, Puccetti S, Genazzani AD, Palumbo M, Luisi M (2004) Conjugated equine estrogens reverse the effects of aging on central and peripheral allopregnanolone and beta-endorphin levels in female rats. Fertil Steril 81:757–766
Genazzani AR, Pluchino N, Luisi S, Luisi M (2007) Estrogen, cognition and female ageing. Hum Reprod Update 13:175–187
Genazzani AR, Petraglia F, Facchinetti F et al (1984) Increase of proopiomelanocortin-related peptides during subjective menopausal flushes. Am J Obstet Gynecol 149:775–779
Linghtman SL, Jacobs HS, Maguire AK (1981) Climateric flushing: clinical and endocrine response to infusion of naloxone. Br J Obstet Gynecol 88:919–924
Adler MW (1980) Minireview: opioid peptides. Life Sci 26:496–510
Reiter RJ (1991) Pineal melatonin: cell biology of its synthesis and of its physiological interactions. Endocr Rev 12:151–180
Reiter RJ (1998) Oxidative damage in the central nervous system: protection by melatonin. Prog Neurobiol 56:359–384
Morioka N, Okatani Y, Wakatsuki A (1999) Melatonin protects against age-related DNA damage in the brains of female senescence-accelerated mice. J Pineal Res 27:202–209
Lewy AJ, Sack RL (1997) Exogenous melatonin’s phase-shifting effects on the endogenous melatonin profile in sighted humans: a brief review and critique of the literature. J Biol Rhythms 12:588–594
Zhdanova IV, Wurtman RJ (1997) Efficacy of melatonin as a sleep-promoting agent. J Biol Rhythms 12:644–650
Birau N, Peterssen U, Meyer C, Gottschalk J (1981) Hypotensive effect of melatonin in essential hypertension. IRSC Med Sci 9:906
Okatani Y, Morioka N, Wakatsuki A (2000) Changes in nocturnal melatonin secretion in peri-menopausal women. Correlation with endogenous estrogen concentrations. J Pineal Res 28:111–118
Alonso R, Abreu P, Fajardo N (1995) Ovarian hormones regulate α1- and β-adrenoceptor interactions in female rat pinealocytes. Neuroreport 6:345–348
Ponholzer A, Temml C, Mock K et al (2005) Prevalence and risk factors for erectile dysfunction in 2869 men using a validated questionnaire. Eur Urol 47:80–85; discussion 85–86.
Shabsigh R, Anastasiadis AG (2003) Erectile dysfunction. Annu Rev Med 54:153–168
Kratzik CW, Schatzl G, Lunglmayr G et al (2005) The impact of age, body mass index and testosterone on erectile dysfunction. J Urol 174:240–243
Gray PB, Singh AB, Woodhouse LJ et al (2005) Dose-dependent effects of testosterone on sexual function, mood, and visuospatial cognition in older men. J Clin Endocrinol Metab 90:3838–3846
Aversa A, Isidori AM, Spera G et al (2003) Androgens improve cavernous vasodilation and response to sildenafil in patients with erectile dysfunction. Clin Endocrinol (Oxf) 58:632–638
Shabsigh R, Kaufman JM, Steidle C et al (2004) Randomized study of testosterone gel as adjunctive therapy to sildenafil in hypogonadal men with erectile dysfunction who do not respond to sildenafil alone. J Urol 172:658–663
Inoue T, Miki Y, Abe K, Hatori M, Hosaka M, Kariya Y et al (2011) The role of estrogen-metabolizing enzymes and estrogen receptors in human epidermis. Mol Cell Endocrinol 344:35–40
Brincat M, Versi E, Moniz CF, Magos A, de Trafford J, Studd JW (1987) Skin collagen changes in postmenopausal women receiving different regimens of estrogen therapy. Obstet Gynecol 70:123–127
Brincat M, Moniz CJ, Studd JW, Darby A, Magos A, Emburey G et al (1985) Long-term effects of the menopause and sex hormones on skin thickness. Br J Obstet Gynaecol 92:256–259
Affinito P, Palomba S, Sorrentino C, Di Carlo C, Bifulco G, Arienzo MP et al (1999) Effects of postmenopausal hypoestrogenism on skin collagen. Maturitas 33:239–247
Shah MG, Maibach HI (2001) Estrogen and skin. An overview. Am J Clin Dermatol 2:143–150
Emmerson E, Hardman MJ (2012) The role of estrogen deficiency in skin ageing and wound healing. Biogerontology 13:3–20
Ashcroft GS, Dodsworth J, van Boxtel E, Tarnuzzer RW, Horan MA, Schultz GS et al (1997) Estrogen accelerates cutaneous wound healing associated with an increase in TGF-beta1 levels. Nat Med 3:1209–1215
Lobo RA (2017) Hormone-replacement therapy: current thinking. Nat Rev Endocrinol 13:220–231
Siregar MFG (2015) Hormonal therapy for aging process in women. Int J Adv Med 2:83–87
Schwartz E, Holtorf K (2011) Hormone replacement therapy in the geriatric patient: current state of the evidence and questions for the future. Estrogen, progesterone, testosterone, and thyroid hormone augmentation in geriatric clinical practice: part 1. Clin Geriatr Med (4):542–559
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–1217
Hulbert AJ, Pamplona R, Buffenstein R, Buttemer WA (2007) Life and death: metabolic rate, membrane composition, and life span of animals. Physiol Rev 87:1175–1213
Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39:359–407
Beckman KB, Ames BN (1998) Free radical theory of aging matures. Physiol Rev 78:547–581
Bokov A, Chaudhuri A, Richardson A (2004) The role of oxidative damage and stress in aging. Mech Ageing Devel 125:811–826
Hanukoglu I (2006) Antioxidant protective mechanisms against reactive oxygen species (ROS) generated by mitochondrial P450 systems in steroidogenic cells. Drug Metab Rev 38:171–196
Nebert D, Dalton TP (2006) The role of cytochrome P450 enzymes in endogenous signaling pathways and environmental carcinogenesis. Nat Rev Cancer 6:947–960
Jackson MJ, Papa S, Bolaños J et al (2002) Antioxidants, reactive oxygen and nitrogen species, gene induction and mitochondrial function. Mol Aspects Med 23:209–285
Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247
Stowe DF, Camara AKS (2008) Mitochondrial reactive oxygen species production in excitable cells: Modulators of mitochondrial and cell function. Antioxid Redox Signal 11:1373–1414
Hornsby PJ (1989) Steroid and xenobiotic effects on the adrenal cortex: mediation by oxidative and other mechanisms. Free Radic Biol Med 6:103–115
Azhar S, Cao L, Reaven E (1995) Alteration of the adrenal antioxidant defense system during aging in rats. J Clin Invest 96:141424
Cao L, Leers-Sucheta S, Azhar S (2004) Aging alters the functional expression of enzymatic and non-enzymatic anti-oxidant defense systems in testicular rat Leydig cells. J Steroid Biochem Mol Biol 88:61–67
Vina J, Borras C, Gambini J, Sastre J, Pallardo FV (2005) Why females live longer than males: control of longevity by sex hormones. Sci Aging Knowl Environ (23):pe17
Acknowledgments
The RKT laboratory has been supported by research grants from the Government of India agencies including UPE-II, UGC (major), CSIR, DST, and ICMR. Central grants from ICMR-CAR, UGC-SAP, and DST-PURSE are also acknowledged. GB was supported by BioCaRE grant from DBT, India. Research in the BC laboratory was supported by Federal grants in the USA (VA-1I01BX000280 and VA-RCS (IK6BX004207); NIH-R01AG-10486; DOD-W81XWH-14-1-0606) and a Foundation grant from Morrison Trust, San Antonio, Texas.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Bagchi, G. et al. (2019). Sex Steroids, Cognate Receptors, and Aging. In: Rath, P. (eds) Models, Molecules and Mechanisms in Biogerontology. Springer, Singapore. https://doi.org/10.1007/978-981-13-3585-3_13
Download citation
DOI: https://doi.org/10.1007/978-981-13-3585-3_13
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3584-6
Online ISBN: 978-981-13-3585-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)