
Abstract
The links between hormonal signalling and lifespan have been well documented in a range of model organisms. For example, in C. elegans or D. melanogaster, lifespan can be modulated by ablating germline cells, or manipulating reproductive history or pregnenolone signalling. In mammalian systems, however, hormonal contribution to longevity is less well understood. With increasing age human steroid hormone profiles change substantially, particularly following menopause in women. This article reviews recent links between steroid sex hormones and ageing, with special emphasis on the skin and wound repair. Estrogen, which substantially decreases with advancing age in both males and females, protects against multiple aspects of cellular ageing in rodent models, including oxidative damage, telomere shortening and cellular senescence. Estrogen’s effects are particularly pronounced in the skin where cutaneous changes post-menopause are well documented, and can be partially reversed by classical Hormone Replacement Therapy (HRT). Our research shows that while chronological ageing has clear effects on skin wound healing, falling estrogen levels are the principle mediator of these effects. Thus, both HRT and topical estrogen replacement substantially accelerate healing in elderly humans, but are associated with unwanted deleterious effects, particularly cancer promotion. In fact, much current research effort is being invested in exploring the therapeutic potential of estrogen signalling manipulation to reverse age-associated pathology in peripheral tissues. In the case of the skin the differential targeting of estrogen receptors to promote healing in aged subjects is a real therapeutic possibility.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Evolutionary conservation
The links between hormones and ageing, principally identified through the study of model organisms, are numerous. The most prominent example would be the correlation between lifespan and insulin signalling, which is conserved across numerous species. Indeed, the importance of endocrine signalling in lower animal species was initially demonstrated for nematode (Caenorhabditis elegans) DAF-2, a homologue of the mammalian insulin and insulin-like growth factor (IGF) receptors, mutations in which lengthened lifespan (Kimura et al. 1997; Kenyon et al. 1993; Larsen et al. 1995). Mutations in numerous genes in the insulin and IGF signalling pathway have also been shown to prolong the lifespan of Drosophila melanogaster (Tatar et al. 2001). Mice and higher vertebrates, unlike worms and flies, have separate insulin and IGF receptors. Interestingly, mice heterozygous for IGF-1 receptor live ~30% longer than wild-type mice (Holzenberger et al. 2003), while mice null for the downstream mediators, insulin receptor substrate (IRS)-1 and -2, also have extended lifespan (Selman et al. 2008; Taguchi et al. 2007). Such extensive cross-species conservation confirms the importance of this pathway in regulating lifespan (Liang et al. 2003). In fact, decreasing insulin/IGF signalling remains one of the few interventions that does robustly extend murine longevity.
Reproductive state and reproductive system signals can also influence lifespan. For example, germline precursor cell ablation in C. elegans and D. melanogaster extends lifespan by ~60% (Hsin and Kenyon 1999; Flatt et al. 2008; Arantes-Oliveira et al. 2002), a process thought to be regulated via DAF-2 (Hsin and Kenyon 1999), DAF-9 [a homologue of mammalian cytochrome P450] (Gerisch et al. 2001; Gerisch and Antebi 2004; Jia et al. 2002) and DAF-12 (Broue et al. 2007), amongst others (Fig. 1a). The hormone repertoire of model organisms most commonly used for ageing research, such as C. elegans and D. melanogaster, is limited, yet they do express hormone precursors present in higher animals, such as pregnenolone and essential enzymes necessary for conversion, such as cytochrome P450 (aromatase) (Broue et al. 2007; Motola et al. 2006). Signalling via such precursors extends lifespan in C. elegans (Yamawaki et al. 2010; Broue et al. 2007; Mak and Ruvkun 2004) and D. melanogaster (Yamawaki et al. 2010; Simon et al. 2003). For example, pregnenolone extends lifespan in nematodes, via DAF-9 (Broue et al. 2007) (Fig. 1b). Surprisingly, the relevance of steroid precursors, such as pregnenolone, in mammalian longevity is largely unknown. Interestingly, aged ovariectomised (Ovx) mice transplanted with the ovaries of young mice exhibit significantly increased life expectancy (Mason et al. 2009; Cargill et al. 2003), an effect potentially mediated through the estrogen receptors and/or the IGF-1R (Fig. 1c).

Association between steroid sex hormones and lifespan. a Germline cell ablation in C. elegans and D. melanogaster extends lifespan via DAF-2, -9 and -12. b Pregnenolone signals via DAF-9 and extends lifespan in C. elegans. c Hormone transfer by ovary transplant from young mice to aged mice extends lifespan, an effect presumably mediated by estrogen and the ERs or IGF-1R
Ageing in humans, the menopause and pathology
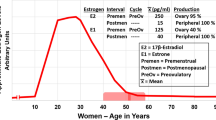
With increasing age the human endocrine system undergoes substantial change, particularly with respect to hormones of adrenal origin. The steroid precursor dehydroepiandrosterone (DHEA), its sulphate, DHEA-S, and the precursor androstendione decline substantially in both males and females from age 20 (Labrie et al. 1997). Interestingly, serum concentrations of pregnenolone (discussed above) also decrease with age in both males and females (Havlikova et al. 2002; Labrie et al. 1997). In women levels of sex steroid estrogens begin to fall from approximately 35 years of age and follicle stimulating hormone (FSH) production is increased in an effort to stimulate ovarian function (Chakravarti et al. 1976). While 17β-estradiol, the most potent form of estrogen, decreases with age serum estrone concentrations remain fairly constant (Labrie et al. 1997). The most pronounced changes occur when women enter the menopause, a permanent cessation of menstruation resulting from the loss of ovarian follicular activity, which occurs at an average age of 51 years in the developed world (Stanford et al. 1987). Post-menopause the majority of circulating sex steroids actually originate from circulating adrenally-derived DHEA (Labrie et al. 2011). In aged males estrogen levels also decrease, although not as rapidly as in females. By contrast, testosterone and dihydrotestosterone (DHT) fluctuate but remain fairly constant with age in both sexes (Labrie et al. 1997). Of note, the enzyme 11β-hydroxysteroid hydrogenase (HSD), which is responsible for the conversion of cortisone to cortisol, is markedly increased with age (Vukelic et al. 2011; Tiganescu et al. 2011).
Increasing life expectancy now means that the average woman in the developed world spends one-third of her life in the post-menopausal period (Kligman and Koblenzer 1997). It is widely accepted that decreased systemic hormones in the post-menopausal state is associated with increased risk for a range of age-associated pathologies (Table 1).
Estrogen deficiency as a general mechanism of ageing
Arguably the most profound hormonal change in ageing is the post-menopause reduction in 17β-estradiol. Moreover, a substantial body of literature links estrogen decline to the distinct cellular ageing mechanisms of oxidative damage and cellular senescence.
Oxidative damage
Free radicals are strongly implicated in the cellular damage that accompanies ageing and age-associated disease (Harman 1956; Beckman and Ames 1998), part of the ‘mitochondrial theory of ageing’ (Miquel et al. 1980). The link between mitochondria and longevity comes from the observation that by reducing mitochondrial peroxidase production lifespan can be extended (Lopez-Torres et al. 1993; Ku et al. 1993). Pertinent to this review is the observation that both peroxidase production and mitochondrial DNA damage are significantly (40–80%) higher in male rats than in age-matched females, and this is directly attributed to differences in estrogen levels (Borras et al. 2003; Pinto and Bartley 1968, 1969). Mitochondrial glutathione, a biological marker of ageing and age-associated damage (Hazelton and Lang 1984; Sastre et al. 2000) is significantly higher in male rats than females (Vina et al. 2005). Conversely, expression of 16S ribosomal RNA, a biological marker of youth (Calleja et al. 1993), is substantially higher in female rats than males (Borras et al. 2003; Vina et al. 2005). Although, Sanz et al. (2007) report no difference in oxidative stress, age-related damage or lifespan between male and female C57Bl6 mice, Ali et al. (2006) show that females, the reported shorter lived gender, exhibit enhanced reactive oxygen species (ROS) production with age.
Estrogen deficiency post-menopause is also strongly linked to altered oxidative state, with estrogen a potent direct antioxidant and indirect inducer of antioxidant enzymes. In ovariectomised (Ovx) female rats oxidised glutathione, lipid peroxidation and mitochondrial DNA damage are significantly increased, and can be reversed by estrogen replacement or phytoestrogen treatment (Baeza et al. 2010). In vitro both estradiol and the phytoestrogen genistein reduce the production of hydrogen peroxide in MCF-7 cells (Borras et al. 2005, 2006). Additionally, keratinocytes isolated from aged female rats exhibit increased oxidative stress (lipoperoxides) and apoptosis (caspases 3 and 8), which can be reversed by estrogen treatment (Tresguerres et al. 2008). Finally, it should be noted that while gender-specific effects are common across many species, including man (Gurwitz 2005), marsupials (Humphries and Stevens 2001) and non-human primates (Herndon et al. 1999) this is not the case in all animal species. Some species lack gender difference in lifespan (Sanz et al. 2007; Wich et al. 2004) while in a limited number of species males actually live longer (Asdell and Joshi 1976; McCulloch and Gems 2003).
Senescence
Significant telomere shortening is associated with replicative cell senescence and tissue deterioration (Allsopp et al. 1992; Harley 1991), and prevented by the enzyme telomerase (Morin 1989). The expression of the human telomerase reverse transcriptase (hTERT) gene and telomerase activity (TA) is upregulated in hepatocytes in vitro in response to estradiol, leading to maintained telomere length (Sato et al. 2004). Similarly, estrogen dose-dependently prevents cellular senescence and increases telomerase activity in endothelial progenitor cells (Imanishi et al. 2005b), vascular smooth muscle cells (Ling et al. 2006) and leukocytes (Aviv et al. 2006) in vitro. In endothelial progenitor cells estrogen prevents angiotensin-II-mediated oxidative stress and senescence (Imanishi et al. 2005a). Estrogen deficiency significantly inhibits TERT expression and telomerase activity in the mouse adrenal gland, an effect reversed by estrogen replacement (Bayne et al. 2008). It has thus been speculated that gender specific differences in longevity are, at least in part, due to hormonal regulation of telomere function (Aviv et al. 2005; Stindl 2004). One method of detecting senescent cells in vitro is a modified beta-galactosidase (β-gal) assay (Dimri et al. 1995). Interestingly, the soy-derived phytoestrogen genistein suppresses UVB-induced expression of β-gal in primary human dermal fibroblasts (Wang et al. 2010), while soybean extract protects against cellular senescence in HaCaT cells (Chiu et al. 2009).
Skin ageing and estrogens
With increasing age a combination of intrinsic and extrinsic factors (primarily UV exposure) lead to skin deterioration. Aged skin has altered structure and reduced function, particularly loss of elasticity, wrinkling, thinning and fragility. While there are differences in the etiology of intrinsic and extrinsic ageing (Tsoureli-Nikita et al. 2006) the majority of resultant changes are similar (Tsoureli-Nikita et al. 2006; Pillai et al. 2005; Ashcroft et al. 1997e; Varani et al. 2001; Lavker 1979). Specifically, intrinsic ageing involves thinning of the epidermis and dermis, reduced epidermal proliferation and turnover and reduced vascularity (Gilchrest et al. 1982b; Lavker 1979). Collagen production is reduced (Shuster et al. 1975) and distribution is altered (Richard et al. 1993), while the production of matrix degrading enzymes (MMPs) is increased with intrinsic ageing (Ashcroft et al. 1997e). Extrinsic ageing or photoageing involves dermal elastosis (Mitchell 1967; Braverman and Fonferko 1982), reduced Langerhans cell numbers, altered melanocyte distribution (Thiers et al. 1984; Gilchrest et al. 1982a) and increased MMP activity (Pillai et al. 2005). With advancing age reduced sebaceous gland secretion leads to skin xerosis, an event that involves corticotropin-releasing hormone (CRH) and coincides with the onset of menopause in women (Pochi et al. 1979; Zouboulis et al. 2002).
The effects of ageing can be readily observed in skin cells in vitro, where dermal fibroblasts and epidermal keratinocytes from aged donors have reduced proliferative capacity and display premature senescence (Stanulis-Praeger and Gilchrest 1989; Gilchrest 1983; Schneider and Mitsui 1976; Mets et al. 1983). In ageing human skin senescent cells accumulate through a combination of growth arrest and increased resistance to apoptosis (Dimri et al. 1994; Wang 1995). In the principal skin cell types (keratinocytes/fibroblasts) telomeres shorten an average of 9 and 11 base pairs per year, respectively, throughout life (Sugimoto et al. 2006; Krunic et al. 2009). Interestingly, telomere shortening is reportedly unchanged in sun-exposed (extrinsically aged) versus sun-protected (intrinsically aged) skin (Sugimoto et al. 2006). Instead, large deletions of the mitochondrial genome are believed to be involved in UV-induced skin photoageing (Schroeder et al. 2008). In vitro, repetitive exposure of human dermal fibroblasts to UVA has been shown to induce deletions of up to 5000 base pairs, causing a partial loss of the mitochondrial genome (Berneburg et al. 1999). Moreover, in a model where mtDNA deletions were induced without irradiation, gene expression mirrored that observed in photoaged skin (Schroeder et al. 2008).
While laboratory animals provide excellent tractable models, some aspects of skin ageing, e.g., changes in epidermal thickness, are contentious and appear species and strain dependent. The majority of mouse studies find that with age epidermal thickness is reduced (Iversen and Schjoelberg 1984; Haratake et al. 1997; Bhattacharyya and Thomas 2004; Argyris 1983) and epidermal keratinocyte proliferation is decreased (Cameron 1972). However, other studies have reported increased epidermal thickness with age (Hill 1988). Epidermal thickness in aged rats has been reported to increase (Bhattacharyya et al. 2005; Thomas 2005), decrease (Morris et al. 1990) or remain unchanged (Giangreco et al. 2008; Monteiro-Riviere et al. 1991). Interestingly, Ishibashi rats (progeny of Wistar and wild rats) reportedly undergo qualitatively similar skin changes to humans. From 12 weeks of age wrinkling of the skin occurs, consistent with a reduction in elastin (Sakuraoka et al. 1996). Calorie restriction (CR), which extends lifespan in numerous model organisms (S. cerevisiae, C. elegans, D. melanogaster) (Lin et al. 2002; Klass 1977; Hosono et al. 1989; Mair et al. 2003; Pletcher et al. 2002) and higher mammals, such as mice and non-human primates (Lane et al. 1995; McCay et al. 1935; Rezzi et al. 2009; Ramsey et al. 2000), prevents age-related skin changes when compared to age-matched ad libitum controls (Thomas 2005; Bhattacharyya et al. 2005). Unfortunately, neither of these studies specifies the sex of the experimental animals.
Historic studies provide the first evidence of estrogen’s cutaneous effects: topically applied follicular hormone, now known to be estrogen, was found to locally improve acne and eczema (Loeser 1937). It has also long been known that the symptoms of skin disorders such as psoriasis improve during pregnancy, an observation now directly attributed to increased circulating estrogen (Dunna and Finlay 1989; Boyd et al. 1996) Moreover, the oral contraceptive pill is often prescribed to treat severe acne (Arowojolu et al. 2009). During the menopause, skin undergoes major changes which include reduced epidermal and dermal thickness, a decrease in collagen content (Brincat et al. 1987), reduced elasticity (Sumino et al. 2004), dryness and fragility (Brincat et al. 1985). Crucially, the majority of these changes can be reversed by either topical or systemic hormone replacement therapy (HRT) (Table 2). Not surprisingly, topical estrogen treatment has little effect on young skin (Goldzieher 1949). The effects of estrogen are predominantly mediated through the estrogen receptors (see “Estrogen receptors and SERMs” section). Estrogen also protects against photoageing, with mortality rates from both melanoma (Miller and Mac Neil 1997) and non-melanoma (Weinstock 1994) skin cancer lower in women than men. In experimental animal studies estrogen deprivation enhances sensitivity to UV skin damage and accelerates photoageing, measured as wrinkling, a loss of elasticity and damage to elastic fibres (Tsukahara et al. 2001; Tsukahara et al. 2004), while estrogen treatment increases skin collagen content in rats and guinea pigs (Smith and Allison 1966; Henneman 1968), increases hyaluronic acid synthesis (Sobel et al. 1965) and promotes epidermal thickening in mice (Bullough 1947). A potential link between ageing and estrogen involves IGF-1. Estradiol is known to be able to signal through the IGF-1R in the skin, in a non-genomic manner, strongly implicating it’s involvement in the regulation of cutaneous ageing (Makrantonaki et al. 2008; Surazynski et al. 2003).
In reality, the skin is not only an estrogen target, but also a major synthetic organ with the capacity to release and produce a wide range of hormones, including estrogens. Skin cells are able to synthesise locally acting estrogens from the precursors cholesterol and DHEA (Fig. 2) (Simpson et al. 1997; Bulun et al. 1999; Chen et al. 2002; Chen et al. 1998; Thiboutot et al. 1998; Hughes et al. 1997; Sawaya and Penneys 1992; Dumont et al. 1992; Eicheler et al. 1995; Thiboutot et al. 2000). Additionally, local production of 11β-HSD by epidermal keratinocytes, dermal fibroblasts and hair follicles (Tiganescu et al. 2011) increases cortisol conversion and has been speculated to control the inflammatory response (Vukelic et al. 2011). In aged individuals peripheral hormone production and local hormone signalling are likely to be particularly important, in light of the considerable reduction in systemic hormone production (Longcope 1971). Surprisingly, the effects of ageing on peripheral hormone synthesis/conversion are virtually unknown.

Skin contains all the components for peripheral steroid hormone synthesis. The precursors cholesterol and dehydroepiandrosterone (DHEA) can be converted to steroid hormones by aromatase (the product of the CYP19 gene), 3β-hydroxysteroid dehydrogenase (3β-HSD), 17β-hydroxysteroid dehydrogenase (17β-HSD) and 5α-reductase. 17α-estradiol has been shown to influence the conversion of testosterone to 17β-estradiol and androstenedione. Double boxes represent functionally important non-steroid hormone synthesis
Wound repair with age
Cutaneous wound healing is a complex tightly orchestrated response to injury, carefully regulated at temporal and spatial levels (reviewed in Shaw and Martin 2009). In young individuals the innate inflammatory response is first initiated, followed by a widespread proliferative phase, involving fibroblasts, keratinocytes and endothelial cells. Keratinocyte migration restores the skin’s barrier, fibroblast-mediated contraction aids wound closure, while matrix remodelling leads to a mature scar. With the passage of time skin becomes fragile and prone to trauma, while cellular ageing leads to aberrant healing. Platelet function (Boldt et al. 2001; Fukaya et al. 2000) and platelet-derived growth factor (PDGF) expression (Ashcroft et al. 1997d) is altered with age. The inflammatory response becomes disrupted, with excessive neutrophil influx, altered endothelial cell adhesion, prolonged macrophage recruitment and delayed resolution (Ashcroft et al. 1998). This leads to over-production of matrix degrading enzymes, classically elastase and MMPs (Ashcroft et al. 1997e; Ashcroft et al. 1997f; Herrick et al. 1997), and under-expression of tissue inhibitors of metalloproteinases (TIMPs) (Ashcroft et al. 1997b). In vivo, re-epithelialisation is delayed in aged humans and murine models (Ashcroft et al. 1997a). Corroborative in vitro studies show that keratinocytes from young donors proliferate at a faster rate (Stanulis-Praeger and Gilchrest 1986) and exhibit less sensitivity to epidermal growth factor (EGF) and keratinocyte growth factor (KGF) (Gilchrest 1983) than those from old aged donors. Angiogenesis is delayed in aged humans and mice (Ashcroft et al. 1997c; Sadoun and Reed 2003) and collagen deposition is reduced (Lenhardt et al. 2000; Ashcroft et al. 1997c). Ultimately, in elderly subjects scar strength is reduced (Lindstedt and Sandblom 1975; Sandblom et al. 1953; Mendoza et al. 1970), however, scar quality is improved (Ashcroft et al. 1999).
Repair, ageing and hormones
While chronological age is a clear risk factor for poor healing our recent studies suggest that estrogen deprivation is the major factor controlling delayed healing in elderly humans. In our recent microarray study 78% of genes differentially expressed between wounds from young and elderly men were estrogen-regulated, while only 3% were age-associated, strongly implicating reduced estrogen, and not known gerontogenes, as the primary regulator of delayed healing in aged subjects (Hardman and Ashcroft 2008). In post-menopausal women HRT improves healing (Ashcroft et al. 1997a), while topical estrogen treatment improves healing in elderly subjects of either gender (Ashcroft et al. 1999). Estrogen’s cellular effects include dampening excessive neutrophil recruitment, preventing disproportionate elastase production, and increasing fibronectin and collagen deposition. Crucially, two independent studies reveal that HRT protects post-menopausal women from developing venous leg ulcers or pressure ulcers (Margolis et al. 2002; Berard et al. 2001).
Animal models have provided key insight into estrogen’s role in wound healing (summarised in Fig. 3). Estrogen replacement accelerates cutaneous healing in Ovx female mice and rats (Ashcroft et al. 1997a; Ashcroft et al. 2003; Emmerson et al. 2010; Hardman et al. 2008) and male rats (Rajabi and Rajabi 2007). Systemic treatment with the sex steroid precursor DHEA accelerates wound healing in young Ovx female mice and old male mice, a result attributed to the local conversion of DHEA to estrogen (Mills et al. 2005). Of interest, androgens are detrimental to healing. The testosterone metabolite DHT retards repair (Gilliver et al. 2009), while castration or androgen receptor blockade improves healing in rodents (Gilliver et al. 2003; Gilliver et al. 2006; Ashcroft and Mills 2002).

Mechanism of estrogen activity in older people. Estrogen aids healing through effects on fibroblasts, keratinocytes and inflammatory cells
At the cellular level estrogen down-regulates neutrophil-expressed L-selectin, preventing the excessive neutrophil accumulation and neutrophil-derived elastase production characteristic of aged healing (Ashcroft et al. 1999), and improves neutrophil phagocytic ability (Magnusson and Einarsson 1990). Estrogen dampens expression of numerous pro-inflammatory cytokines, including MIF, TNFα, MCP-1, IL-1β and IL-6 (Kovacs et al. 1996; Hu et al. 1988). The reduction in Macrophage Migration Inhibitory Factor (MIF), in particular, plays a major role in the beneficial effects of estrogen on wound repair (Ashcroft et al. 2003; Hardman et al. 2005; Emmerson et al. 2009). Of interest, plasma MIF levels, which inversely correlate with systemic estrogen (Aloisi et al. 2005), are increased post-menopause and fall following HRT (Hardman et al. 2005). Estrogen is also a keratinocyte mitogen (Verdier-Sevrain et al. 2004), promoting migration across an artificial scratch in vitro (Emmerson et al. 2009; Campbell et al. 2010) and wound re-epithelialisation in Ovx mice (Emmerson et al. 2010; Hardman et al. 2008). Moreover, impaired wound re-epithelialisation post-menopause can be reversed to pre-menopause levels by just 3 months of HRT (Ashcroft et al. 1997a). Estrogen directly stimulates dermal fibroblast migration in vitro (Emmerson et al. 2009; Campbell et al. 2010) and indirectly via stimulating macrophage platelet derived growth factor (PDGF), a key fibroblast mitogen and stimulator of wound contraction (Battegay et al. 1994). The role of estradiol in stimulating angiogenesis is somewhat contentious. PDGF directly stimulates angiogenesis, while estradiol increases endothelial cell capillary-like structure formation upon reconstituted basement membrane (Morales et al. 1995). However, conflicting in vitro studies report either no change or decreased angiogenesis following estrogen treatment (Nyman 1971; Lundgren 1973).
Estrogen receptors and SERMs
Estrogen signals via two nuclear hormone receptors, ERα and ERβ. ERα, the first receptor to be identified and cloned (Walter et al. 1985), predominates in reproductive tissues and is strongly associated with cancer (Zou and Ing 1998; Kuiper et al. 1997; Ali and Coombes 2000). The second receptor, ERβ, identified over a decade later (Kuiper et al. 1996; Ogawa et al. 1998), is more highly expressed in peripheral, non-reproductive tissues (Kuiper et al. 1997; Onoe et al. 1997; Brandenberger et al. 1997; Couse et al. 1997). Both receptors are reportedly expressed in human facial skin (Miller and Mac Neil 1997), scalp (Thornton et al. 2003a, b; Mosselman et al. 1996) and upper arm skin (Reed et al. 2005). Several studies suggest that ERβ predominates in keratinocytes of human scalp skin (Mosselman et al. 1996; Thornton et al. 2003a, b) while others identify both receptors in neonatal foreskin-derived keratinocytes (Verdier-Sevrain et al. 2004). In mouse skin both receptors are widely expressed (Campbell et al. 2010; Cho et al. 2008).
Our recent data indicate that delayed healing in Ovx female mice can be reversed by stimulating signalling through ERβ alone (using the ERβ-specific agonist DiarylPropioNitrile [DPN]) (Campbell et al. 2010). Conversely, signalling through ERα alone (using the ERα agonist Propyl Pyrazole Triol [PPT]) entirely fails to improve healing in Ovx mice. To support this finding, we find that estrogen replacement in Ovx mice lacking functional ERβ (ERβ−/−) actually further delays healing beyond the Ovx wild-type phenotype. Moreover, epidermal specific ERβ null mice (K14-cre/ERβL2/L2) phenocopy ERβ−/− mice, i.e., estrogen replacement again impairs healing. This would suggest that the beneficial effects of estrogen on cutaneous repair are predominantly mediated via epidermal ERβ (Campbell et al. 2010). Of note, the beneficial effects of 17β-estradiol on outcome in a skin flap necrosis model is reportedly mediated via ERα (Toutain et al. 2009) while Ovx rats treated with PPT exhibit reduced wound tensile strength (Gal et al. 2010). A crucial link between our mouse data and aged human healing is provided by the observation that polymorphisms in the human ERβ gene are significantly associated with venous ulceration in the Caucasian population (Ashworth et al. 2005, 2008).
Most pathological states in the elderly involve a single ER isoform, for example in cancers of the reproductive system ERα predominates (Herynk and Fuqua 2004; Yang et al. 2008; Cai et al. 2003), while in colon cancers ERβ predominates (Arai et al. 2000; Fiorelli et al. 1999; Foley et al. 2000; Qiu et al. 2002). The clear differential roles of the two estrogen receptors in cutaneous repair (Campbell et al. 2010; Gal et al. 2010; Toutain et al. 2009) suggest that pharmacological manipulation may be a viable therapeutic option. Compounds termed Selective Estrogen Receptor Modulator (SERMs) are being developed to treat pathologies such as osteoporosis and breast cancer by exploiting natural estrogen signalling to confer tissue specific estrogenic or anti-estrogenic effects.
SERMs have been developed to provide estrogen-like beneficial effects, to treat disorders such as osteoporosis, an approach that should bypass systemic estrogen risks, including breast cancer (Matsumoto 2006). Arguably, the best characterised and commonly used SERMs are tamoxifen and raloxifene. Both are considered ER antagonists in the breast, but have the potential to act as ER agonists in other tissues (Frasor et al. 2004). The dietary phytoestrogen genistein, is also becoming increasingly popular for the treatment of post-menopausal pathology (Atteritano et al. 2008; D’Anna et al. 2007). Despite use of SERMs in age- and menopause-associated pathologies (reviewed in Pickar et al. 2010) knowledge of SERM activity in the skin is severely lacking. In vitro both tamoxifen and raloxifene stimulate fibroblast proliferation, but fail to promote fibroblast migration (Stevenson et al. 2009). Preliminary studies in post-menopausal skin indicate increased elasticity and collagen content following raloxifene treatment (Sumino et al. 2009) and improved dermal vascularisation and increased epidermal thickness following genistein treatment (Moraes et al. 2009). Topical tamoxifen reportedly improves the appearance of keloid scars in acute burns patients, through dampening of fibroblast proliferation and collagen synthesis (Mousavi et al. 2010; Gragnani et al. 2009). Our recent studies indicate that the SERMs tamoxifen, raloxifene and genistein all substantially benefit healing in the estrogen-deprived Ovx mouse, promoting re-epithelialisation and contraction, and reducing inflammation, an effect that is most likely mediated via ERβ (Emmerson et al. 2010; Hardman et al. 2008), while a separate study provides evidence that genistein accelerates murine healing by modulating TGFβ1 (Marini et al. 2010). Tamoxifen, raloxifene and genistein are anti-inflammatory in other pathologies, including systemic lupus erythematosus (SLE) (Sthoeger et al. 2003), stroke (Tian et al. 2009), UV-associated cutaneous damage (Brand and Jendrzejewski 2008; Shyong et al. 2002; Widyarini et al. 2001), colitis (Seibel et al. 2009), ileitis (Sadowska-Krowicka et al. 1998) and multiple sclerosis (MS) (De Paula et al. 2008). An important and on-going goal of our research is to evaluate cutaneous healing in post-menopausal women prescribed topical SERM treatment.
Conclusions and further perspectives
Despite links between estrogen and ageing being suggested many years ago, only over the last decade has estrogen emerged as a key determinant of ageing in peripheral non-reproductive tissues, particularly bone, skin and brain. Indeed, the endocrine theory of ageing, underpinned by germline cell ablation experiments in model organisms, states that chronological changes in hormones levels accelerate the cellular effects of ageing. In skin particularly, it is only over the last few years that we have begun to understand the relative contributions of hormones and ageing to pathological healing. Indeed, although our knowledge of estrogen’s beneficial effects on wound repair has greatly expanded over recent years there is still much that is not understood. The prospect of manipulating estrogen signalling to alleviate poor healing in the elderly is an exciting one, but we have yet to fully understand how such ER-mediated signalling changes with normal ageing and how this impacts upon cutaneous pathology post-menopause. Importantly, how do the protective effects of estrogen on cellular ageing (i.e., prevention of telomere shortening and oxidative stress) directly contribute to improved healing? A clearer detailed mechanistic understanding will be of relevance to a range of peripheral estrogen-target tissues and aid the development of targeted hormone-based therapies, to promote cutaneous repair in the elderly.
References
Aertgeerts J (1974) Effect of oral estrogens on the skin of the woman in post-menopause or after castration. Ther Ggw 113(10):1739–1748
Ali S, Coombes RC (2000) Estrogen receptor alpha in human breast cancer: occurrence and significance. J Mammary Gland Biol Neoplasia 5(3):271–281
Ali SS, Xiong C, Lucero J, Behrens MM, Dugan LL, Quick KL (2006) Gender differences in free radical homeostasis during aging: shorter-lived female c57bl6 mice have increased oxidative stress. Aging Cell 5(6):565–574
Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, Greider CW, Harley CB (1992) Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA 89(21):10114–10118
Aloisi AM, Pari G, Ceccarelli I, Vecchi I, Ietta F, Lodi L, Paulesu L (2005) Gender-related effects of chronic non-malignant pain and opioid therapy on plasma levels of macrophage migration inhibitory factor (mif). Pain 115(1–2):142–151
Arai N, Strom A, Rafter JJ, Gustafsson JA (2000) Estrogen receptor beta mRNA in colon cancer cells: growth effects of estrogen and genistein. Biochem Biophys Res Commun 270(2):425–431
Arantes-Oliveira N, Apfeld J, Dillin A, Kenyon C (2002) Regulation of life-span by germ-line stem cells in caenorhabditis elegans. Science 295(5554):502–505
Argyris TS (1983) The effect of aging on epidermal mass in balb/c female mice. Mech Ageing Dev 22(3–4):347–354
Arowojolu AO, Gallo MF, Lopez LM, Grimes DA, Garner SE (2009) Combined oral contraceptive pills for treatment of acne. Cochrane Database Syst Rev (3):CD004425
Asdell SA, Joshi SR (1976) Reproduction and longevity in the hamster and rat. Biol Reprod 14(4):478–480
Ashcroft GS, Mills SJ (2002) Androgen receptor-mediated inhibition of cutaneous wound healing. J Clin Invest 110(5):615–624
Ashcroft GS, Dodsworth J, van Boxtel E, Tarnuzzer RW, Horan MA, Schultz GS, Ferguson MW (1997a) Estrogen accelerates cutaneous wound healing associated with an increase in tgf-beta1 levels. Nat Med 3(11):1209–1215
Ashcroft GS, Herrick SE, Tarnuzzer RW, Horan MA, Schultz GS, Ferguson MW (1997b) Human ageing impairs injury-induced in vivo expression of tissue inhibitor of matrix metalloproteinases (timp)-1 and -2 proteins and mrna. J Pathol 183(2):169–176
Ashcroft GS, Horan MA, Ferguson MW (1997c) Aging is associated with reduced deposition of specific extracellular matrix components, an upregulation of angiogenesis, and an altered inflammatory response in a murine incisional wound healing model. J Invest Dermatol 108(4):430–437
Ashcroft GS, Horan MA, Ferguson MW (1997d) The effects of ageing on wound healing: immunolocalisation of growth factors and their receptors in a murine incisional model. J Anat 190(Pt 3):351–365
Ashcroft GS, Horan MA, Herrick SE, Tarnuzzer RW, Schultz GS, Ferguson MW (1997e) Age-related differences in the temporal and spatial regulation of matrix metalloproteinases (mmps) in normal skin and acute cutaneous wounds of healthy humans. Cell Tissue Res 290(3):581–591
Ashcroft GS, Kielty CM, Horan MA, Ferguson MW (1997f) Age-related changes in the temporal and spatial distributions of fibrillin and elastin mrnas and proteins in acute cutaneous wounds of healthy humans. J Pathol 183(1):80–89
Ashcroft GS, Horan MA, Ferguson MW (1998) Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab Invest 78(1):47–58
Ashcroft GS, Greenwell-Wild T, Horan MA, Wahl SM, Ferguson MW (1999) Topical estrogen accelerates cutaneous wound healing in aged humans associated with an altered inflammatory response. Am J Pathol 155(4):1137–1146
Ashcroft GS, Mills SJ, Lei K, Gibbons L, Jeong MJ, Taniguchi M, Burow M, Horan MA, Wahl SM, Nakayama T (2003) Estrogen modulates cutaneous wound healing by downregulating macrophage migration inhibitory factor. J Clin Invest 111(9):1309–1318
Ashworth JJ, Smyth JV, Pendleton N, Horan M, Payton A, Worthington J, Ollier WE, Ashcroft GS (2005) The dinucleotide (ca) repeat polymorphism of estrogen receptor beta but not the dinucleotide (ta) repeat polymorphism of estrogen receptor alpha is associated with venous ulceration. J Steroid Biochem Mol Biol 97(3):266–270
Ashworth JJ, Smyth JV, Pendleton N, Horan M, Payton A, Worthington J, Ollier WE, Ashcroft GS (2008) Polymorphisms spanning the on exon and promoter of the estrogen receptor-beta (erbeta) gene esr2 are associated with venous ulceration. Clin Genet 73(1):55–61
Atteritano M, Pernice F, Mazzaferro S, Mantuano S, Frisina A, D’Anna R, Cannata ML, Bitto A, Squadrito F, Frisina N, Buemi M (2008) Effects of phytoestrogen genistein on cytogenetic biomarkers in postmenopausal women: 1 year randomized, placebo-controlled study. Eur J Pharmacol 589(1–3):22–26
Aviv A, Shay J, Christensen K, Wright W (2005) The longevity gender gap: are telomeres the explanation? Sci Aging Knowledge Environ 2005(23):pe16
Aviv A, Valdes A, Gardner JP, Swaminathan R, Kimura M, Spector TD (2006) Menopause modifies the association of leukocyte telomere length with insulin resistance and inflammation. J Clin Endocrinol Metab 91(2):635–640
Baeza I, Fdez-Tresguerres J, Ariznavarreta C, De la Fuente M (2010) Effects of growth hormone, melatonin, oestrogens and phytoestrogens on the oxidized glutathione (gssg)/reduced glutathione (gsh) ratio and lipid peroxidation in aged ovariectomized rats. Biogerontology 11(6):687–701
Barrett-Connor E (1995) Postmenopausal estrogen and heart disease. Atherosclerosis 118(Suppl):S7–S10
Battegay EJ, Rupp J, Iruela-Arispe L, Sage EH, Pech M (1994) Pdgf-bb modulates endothelial proliferation and angiogenesis in vitro via pdgf beta-receptors. J Cell Biol 125(4):917–928
Bayne S, Jones ME, Li H, Pinto AR, Simpson ER, Liu JP (2008) Estrogen deficiency leads to telomerase inhibition, telomere shortening and reduced cell proliferation in the adrenal gland of mice. Cell Res 18(11):1141–1150
Beckman KB, Ames BN (1998) The free radical theory of aging matures. Physiol Rev 78(2):547–581
Berard A, Kahn SR, Abenhaim L (2001) Is hormone replacement therapy protective for venous ulcer of the lower limbs? Pharmacoepidemiol Drug Saf 10(3):245–251
Berneburg M, Grether-Beck S, Kurten V, Ruzicka T, Briviba K, Sies H, Krutmann J (1999) Singlet oxygen mediates the uva-induced generation of the photoaging-associated mitochondrial common deletion. J Biol Chem 274(22):15345–15349
Bhattacharyya TK, Thomas JR (2004) Histomorphologic changes in aging skin: observations in the cba mouse model. Arch Facial Plast Surg 6(1):21–25
Bhattacharyya TK, Merz M, Thomas JR (2005) Modulation of cutaneous aging with calorie restriction in Fischer 344 rats: a histological study. Arch Facial Plast Surg 7(1):12–16
Boldt J, Huttner I, Suttner S, Kumle B, Piper SN, Berchthold G (2001) Changes of haemostasis in patients undergoing major abdominal surgery—is there a difference between elderly and younger patients? Br J Anaesth 87(3):435–440
Borras C, Sastre J, Garcia-Sala D, Lloret A, Pallardo FV, Vina J (2003) Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic Biol Med 34(5):546–552
Borras C, Gambini J, Gomez-Cabrera MC, Sastre J, Pallardo FV, Mann GE, Vina J (2005) 17beta-oestradiol up-regulates longevity-related, antioxidant enzyme expression via the erk1 and erk2[mapk]/nfkappab cascade. Aging Cell 4(3):113–118
Borras C, Gambini J, Gomez-Cabrera MC, Sastre J, Pallardo FV, Mann GE, Vina J (2006) Genistein, a soy isoflavone, up-regulates expression of antioxidant genes: involvement of estrogen receptors, erk1/2, and nfkappab. Faseb J 20(12):2136–2138
Boyd AS, Morris LF, Phillips CM, Menter MA (1996) Psoriasis and pregnancy: hormone and immune system interaction. Int J Dermatol 35(3):169–172
Brand RM, Jendrzejewski JL (2008) Topical treatment with (−)-epigallocatechin-3-gallate and genistein after a single uv exposure can reduce skin damage. J Dermatol Sci 50(1):69–72
Brandenberger AW, Tee MK, Lee JY, Chao V, Jaffe RB (1997) Tissue distribution of estrogen receptors alpha (er-alpha) and beta (er-beta) mrna in the midgestational human fetus. J Clin Endocrinol Metab 82(10):3509–3512
Braverman IM, Fonferko E (1982) Studies in cutaneous aging: I. The elastic fiber network. J Invest Dermatol 78(5):434–443
Brincat MP (2000) Hormone replacement therapy and the skin. Maturitas 35(2):107–117
Brincat M, Moniz CJ, Studd JW, Darby A, Magos A, Emburey G, Versi E (1985) Long-term effects of the menopause and sex hormones on skin thickness. Br J Obstet Gynaecol 92(3):256–259
Brincat M, Versi E, Moniz CF, Magos A, de Trafford J, Studd JW (1987) Skin collagen changes in postmenopausal women receiving different regimens of estrogen therapy. Obstet Gynecol 70(1):123–127
Broue F, Liere P, Kenyon C, Baulieu EE (2007) A steroid hormone that extends the lifespan of caenorhabditis elegans. Aging Cell 6(1):87–94
Bullough HF (1947) Epidermal thickness following oestrone injections in the mouse. Nature 159(4029):101
Bulun SE, Zeitoun K, Sasano H, Simpson ER (1999) Aromatase in aging women. Semin Reprod Endocrinol 17(4):349–358
Cai Q, Shu XO, Jin F, Dai Q, Wen W, Cheng JR, Gao YT, Zheng W (2003) Genetic polymorphisms in the estrogen receptor alpha gene and risk of breast cancer: results from the shanghai breast cancer study. Cancer Epidemiol Biomarkers Prev 12(9):853–859
Calleja M, Pena P, Ugalde C, Ferreiro C, Marco R, Garesse R (1993) Mitochondrial DNA remains intact during drosophila aging, but the levels of mitochondrial transcripts are significantly reduced. J Biol Chem 268(25):18891–18897
Calmels P, Vico L, Alexandre C, Minaire P (1995) Cross-sectional study of muscle strength and bone mineral density in a population of 106 women between the ages of 44 and 87 years: relationship with age and menopause. Eur J Appl Physiol Occup Physiol 70(2):180–186
Cameron IL (1972) Cell proliferation and renewal in aging mice. J Gerontol 27(2):162–172
Campbell L, Emmerson E, Davies F, Gilliver SC, Krust A, Chambon P, Ashcroft GS, Hardman MJ (2010) Estrogen promotes cutaneous wound healing via estrogen receptor beta independent of it’s anti-inflammatory activities. J Exp Med 207(9):1825–1833
Carcenac G, Herard ME, Kergoat MJ, Lajeunesse Y, Champoux N, Barsauskas A, Kergoat H (2009) Assessment of visual function in institutionalized elderly patients. J Am Med Dir Assoc 10(1):45–49
Cargill SL, Carey JR, Muller HG, Anderson G (2003) Age of ovary determines remaining life expectancy in old ovariectomized mice. Aging Cell 2(3):185–190
Castelo-Branco C, Duran M, Gonzalez-Merlo J (1992) Skin collagen changes related to age and hormone replacement therapy. Maturitas 15(2):113–119
Chakravarti S, Collins WP, Forecast JD, Newton JR, Oram DH, Studd JW (1976) Hormonal profiles after the menopause. Br Med J 2(6039):784–787
Chen W, Zouboulis CC, Fritsch M, Blume-Peytavi U, Kodelja V, Goerdt S, Luu-The V, Orfanos CE (1998) Evidence of heterogeneity and quantitative differences of the type 1 5alpha-reductase expression in cultured human skin cells—evidence of its presence in melanocytes. J Invest Dermatol 110(1):84–89
Chen W, Thiboutot D, Zouboulis CC (2002) Cutaneous androgen metabolism: basic research and clinical perspectives. J Invest Dermatol 119(5):992–1007
Chiu TM, Huang CC, Lin TJ, Fang JY, Wu NL, Hung CF (2009) In vitro and in vivo anti-photoaging effects of an isoflavone extract from soybean cake. J Ethnopharmacol 126(1):108–113
Cho JL, Allanson M, Domanski D, Arun SJ, Reeve VE (2008) Estrogen receptor-beta signaling protects epidermal cytokine expression and immune function from uvb-induced impairment in mice. Photochem Photobiol Sci 7(1):120–125
Colditz GA, Willett WC, Stampfer MJ, Rosner B, Speizer FE, Hennekens CH (1987) Menopause and the risk of coronary heart disease in women. N Engl J Med 316(18):1105–1110
Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS (1997) Tissue distribution and quantitative analysis of estrogen receptor-alpha (eralpha) and estrogen receptor-beta (erbeta) messenger ribonucleic acid in the wild-type and eralpha-knockout mouse. Endocrinology 138(11):4613–4621
Creidi P, Faivre B, Agache P, Richard E, Haudiquet V, Sauvanet JP (1994) Effect of a conjugated oestrogen (premarin) cream on ageing facial skin. A comparative study with a placebo cream. Maturitas 19(3):211–223
D’Anna R, Cannata ML, Atteritano M, Cancellieri F, Corrado F, Baviera G, Triolo O, Antico F, Gaudio A, Frisina N, Bitto A, Polito F, Minutoli L, Altavilla D, Marini H, Squadrito F (2007) Effects of the phytoestrogen genistein on hot flushes, endometrium, and vaginal epithelium in postmenopausal women: a 1-year randomized, double-blind, placebo-controlled study. Menopause 14(4):648–655
De Paula ML, Rodrigues DH, Teixeira HC, Barsante MM, Souza MA, Ferreira AP (2008) Genistein down-modulates pro-inflammatory cytokines and reverses clinical signs of experimental autoimmune encephalomyelitis. Int Immunopharmacol 8(9):1291–1297
Dimri GP, Hara E, Campisi J (1994) Regulation of two e2f-related genes in presenescent and senescent human fibroblasts. J Biol Chem 269(23):16180–16186
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O et al (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92(20):9363–9367
Dumont M, Luu-The V, Dupont E, Pelletier G, Labrie F (1992) Characterization, expression, and immunohistochemical localization of 3 beta-hydroxysteroid dehydrogenase/delta 5-delta 4 isomerase in human skin. J Invest Dermatol 99(4):415–421
Dunna SF, Finlay AY (1989) Psoriasis: improvement during and worsening after pregnancy. Br J Dermatol 120(4):584
Eicheler W, Dreher M, Hoffmann R, Happle R, Aumuller G (1995) Immunohistochemical evidence for differential distribution of 5 alpha-reductase isoenzymes in human skin. Br J Dermatol 133(3):371–376
Emmerson E, Campbell L, Ashcroft GS, Hardman MJ (2009) Unique and synergistic roles for 17beta-estradiol and macrophage migration inhibitory factor during cutaneous wound closure are cell type specific. Endocrinology 150(6):2749–2757
Emmerson E, Campbell L, Ashcroft GS, Hardman MJ (2010) The phytoestrogen genistein promotes wound healing by multiple independent mechanisms. Mol Cell Endocrinol 321(2):184–193
Evans WJ (2010) Skeletal muscle loss: cachexia, sarcopenia, and inactivity. Am J Clin Nutr 91(4):1123S–1127S
Evans JR, Schwartz SD, McHugh JD, Thamby-Rajah Y, Hodgson SA, Wormald RP, Gregor ZJ (1998) Systemic risk factors for idiopathic macular holes: a case-control study. Eye (Lond) 12(Pt 2):256–259
Fiorelli G, Picariello L, Martineti V, Tonelli F, Brandi ML (1999) Functional estrogen receptor beta in colon cancer cells. Biochem Biophys Res Commun 261(2):521–527
Flatt T, Min KJ, D’Alterio C, Villa-Cuesta E, Cumbers J, Lehmann R, Jones DL, Tatar M (2008) Drosophila germ-line modulation of insulin signaling and lifespan. Proc Natl Acad Sci USA 105(17):6368–6373
Foley EF, Jazaeri AA, Shupnik MA, Jazaeri O, Rice LW (2000) Selective loss of estrogen receptor beta in malignant human colon. Cancer Res 60(2):245–248
Frasor J, Stossi F, Danes JM, Komm B, Lyttle CR, Katzenellenbogen BS (2004) Selective estrogen receptor modulators: discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res 64(4):1522–1533
Fuchs KO, Solis O, Tapawan R, Paranjpe J (2003) The effects of an estrogen and glycolic acid cream on the facial skin of postmenopausal women: a randomized histologic study. Cutis 71(6):481–488
Fukaya S, Iwamoto T, Kin K, Takasaki M (2000) Factors influencing platelet function in elderly patients with chronic phase thrombotic diseases. Nippon Ronen Igakkai Zasshi 37(8):619–626
Gal P, Novotny M, Vasilenko T, Depta F, Sulla I, Tomori Z (2010) Decrease in wound tensile strength following post-surgical estrogen replacement therapy in ovariectomized rats during the early phase of healing is mediated via er-alpha rather than er-beta: a preliminary report. J Surg Res 159(1):e25–e28
Gates BJ, Sonnett TE, Duvall CA, Dobbins EK (2009) Review of osteoporosis pharmacotherapy for geriatric patients. Am J Geriatr Pharmacother 7(6):293–323
Gerisch B, Antebi A (2004) Hormonal signals produced by daf-9/cytochrome p450 regulate c. Elegans dauer diapause in response to environmental cues. Development 131(8):1765–1776
Gerisch B, Weitzel C, Kober-Eisermann C, Rottiers V, Antebi A (2001) A hormonal signaling pathway influencing c. Elegans metabolism, reproductive development, and life span. Dev Cell 1(6):841–851
Giangreco A, Qin M, Pintar JE, Watt FM (2008) Epidermal stem cells are retained in vivo throughout skin aging. Aging Cell 7(2):250–259
Gilchrest BA (1983) In vitro assessment of keratinocyte aging. J Invest Dermatol 81(1 Suppl):184s–189s
Gilchrest BA, Murphy GF, Soter NA (1982a) Effect of chronologic aging and ultraviolet irradiation on langerhans cells in human epidermis. J Invest Dermatol 79(2):85–88
Gilchrest BA, Stoff JS, Soter NA (1982b) Chronologic aging alters the response to ultraviolet-induced inflammation in human skin. J Invest Dermatol 79(1):11–15
Gilchrest BA, Szabo G, Flynn E, Goldwyn RM (1983) Chronologic and actinically induced aging in human facial skin. J Invest Dermatol 80 Suppl:81s–85s
Gilliver SC, Wu F, Ashcroft GS (2003) Regulatory roles of androgens in cutaneous wound healing. Thromb Haemost 90(6):978–985
Gilliver SC, Ashworth JJ, Mills SJ, Hardman MJ, Ashcroft GS (2006) Androgens modulate the inflammatory response during acute wound healing. J Cell Sci 119(Pt 4):722–732
Gilliver SC, Ruckshanthi JP, Hardman MJ, Zeef LA, Ashcroft GS (2009) 5alpha-dihydrotestosterone (dht) retards wound closure by inhibiting re-epithelialization. J Pathol 217(1):73–82
Goldzieher JW (1949) The direct effect of steroids on the senile human skin. J Gerontol 4(2):104–112
Gragnani A, Warde M, Furtado F, Ferreira LM (2009) Topical tamoxifen therapy in hypertrophic scars or keloids in burns. Arch Dermatol Res 302(1):1–4
Gurwitz JH (2005) The age/gender interface in geriatric pharmacotherapy. J Womens Health (Larchmt) 14(1):68–72
Haratake A, Uchida Y, Mimura K, Elias PM, Holleran WM (1997) Intrinsically aged epidermis displays diminished uvb-induced alterations in barrier function associated with decreased proliferation. J Invest Dermatol 108(3):319–323
Hardman MJ, Ashcroft GS (2008) Estrogen, not intrinsic aging, is the major regulator of delayed human wound healing in the elderly. Genome Biol 9(5):R80
Hardman MJ, Waite A, Zeef L, Burow M, Nakayama T, Ashcroft GS (2005) Macrophage migration inhibitory factor: a central regulator of wound healing. Am J Pathol 167(6):1561–1574
Hardman MJ, Emmerson E, Campbell L, Ashcroft GS (2008) Selective estrogen receptor modulators accelerate cutaneous wound healing in ovariectomized female mice. Endocrinology 149(2):551–557
Harley CB (1991) Telomere loss: mitotic clock or genetic time bomb? Mutat Res 256(2–6):271–282
Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11(3):298–300
Havlikova H, Hill M, Hampl R, Starka L (2002) Sex- and age-related changes in epitestosterone in relation to pregnenolone sulfate and testosterone in normal subjects. J Clin Endocrinol Metab 87(5):2225–2231
Hazelton GA, Lang CA (1984) Glutathione levels during the mosquito life span with emphasis on senescence. Proc Soc Exp Biol Med 176(3):249–256
Hederstierna C, Hultcrantz M, Collins A, Rosenhall U (2007) Hearing in women at menopause. Prevalence of hearing loss, audiometric configuration and relation to hormone replacement therapy. Acta Otolaryngol 127(2):149–155
Hederstierna C, Hultcrantz M, Collins A, Rosenhall U (2010) The menopause triggers hearing decline in healthy women. Hear Res 259(1–2):31–35
Henneman DH (1968) Effect of estrogen on in vivo and in vitro collagen biosynthesis and maturation in old and young female guinea pigs. Endocrinology 83(4):678–690
Herndon JG, Tigges J, Anderson DC, Klumpp SA, McClure HM (1999) Brain weight throughout the life span of the chimpanzee. J Comp Neurol 409(4):567–572
Herrick S, Ashcroft G, Ireland G, Horan M, McCollum C, Ferguson M (1997) Up-regulation of elastase in acute wounds of healthy aged humans and chronic venous leg ulcers are associated with matrix degradation. Lab Invest 77(3):281–288
Herynk MH, Fuqua SA (2004) Estrogen receptor mutations in human disease. Endocr Rev 25(6):869–898
Hill MW (1988) Influence of age on the morphology and transit time of murine stratified squamous epithelia. Arch Oral Biol 33(4):221–229
Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y (2003) Igf-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421(6919):182–187
Hosono R, Nishimoto S, Kuno S (1989) Alterations of life span in the nematode caenorhabditis elegans under monoxenic culture conditions. Exp Gerontol 24(3):251–264
Hsin H, Kenyon C (1999) Signals from the reproductive system regulate the lifespan of c. Elegans. Nature 399(6734):362–366
Hu SK, Mitcho YL, Rath NC (1988) Effect of estradiol on interleukin 1 synthesis by macrophages. Int J Immunopharmacol 10(3):247–252
Hughes SV, Robinson E, Bland R, Lewis HM, Stewart PM, Hewison M (1997) 1,25-dihydroxyvitamin d3 regulates estrogen metabolism in cultured keratinocytes. Endocrinology 138(9):3711–3718
Humphries S, Stevens DJ (2001) Reproductive biology. Out with a bang. Nature 410(6830):758–759
Imanishi T, Hano T, Nishio I (2005a) Estrogen reduces angiotensin ii-induced acceleration of senescence in endothelial progenitor cells. Hypertens Res 28(3):263–271
Imanishi T, Hano T, Nishio I (2005b) Estrogen reduces endothelial progenitor cell senescence through augmentation of telomerase activity. J Hypertens 23(9):1699–1706
Iversen OH, Schjoelberg AR (1984) Age-related changes of epidermal cell kinetics in the hairless mouse. Virchows Arch B Cell Pathol Incl Mol Pathol 46(1–2):135–143
Jacobsen BK, Nilssen S, Heuch I, Kvale G (1997) Does age at natural menopause affect mortality from ischemic heart disease? J Clin Epidemiol 50(4):475–479
Jia K, Albert PS, Riddle DL (2002) Daf-9, a cytochrome p450 regulating c. Elegans larval development and adult longevity. Development 129(1):221–231
Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R (1993) A c. Elegans mutant that lives twice as long as wild type. Nature 366(6454):461–464
Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G (1997) Daf-2, an insulin receptor-like gene that regulates longevity and diapause in caenorhabditis elegans. Science 277(5328):942–946
Klass MR (1977) Aging in the nematode caenorhabditis elegans: major biological and environmental factors influencing life span. Mech Ageing Dev 6(6):413–429
Kligman AM, Koblenzer C (1997) Demographics and psychological implications for the aging population. Dermatol Clin 15(4):549–553
Kovacs EJ, Faunce DE, Ramer-Quinn DS, Mott FJ, Dy PW, Frazier-Jessen MR (1996) Estrogen regulation of je/mcp-1 mrna expression in fibroblasts. J Leukoc Biol 59(4):562–568
Krunic D, Moshir S, Greulich-Bode KM, Figueroa R, Cerezo A, Stammer H, Stark HJ, Gray SG, Nielsen KV, Hartschuh W, Boukamp P (2009) Tissue context-activated telomerase in human epidermis correlates with little age-dependent telomere loss. Biochim Biophys Acta 1792(4):297–308
Ku HH, Brunk UT, Sohal RS (1993) Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species. Free Radic Biol Med 15(6):621–627
Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA (1996) Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA 93(12):5925–5930
Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138(3):863–870
Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, van Belle G, Jolley L, Larson EB (2002) Dementia and alzheimer disease incidence: a prospective cohort study. Arch Neurol 59(11):1737–1746
Labrie F, Belanger A, Cusan L, Gomez JL, Candas B (1997) Marked decline in serum concentrations of adrenal c19 sex steroid precursors and conjugated androgen metabolites during aging. J Clin Endocrinol Metab 82(8):2396–2402
Labrie F, Martel C, Balser J (2011) Wide distribution of the serum dehydroepiandrosterone and sex steroid levels in postmenopausal women: role of the ovary? Menopause 18(1):30–43
Lane MA, Ball SS, Ingram DK, Cutler RG, Engel J, Read V, Roth GS (1995) Diet restriction in rhesus monkeys lowers fasting and glucose-stimulated glucoregulatory end points. Am J Physiol 268(5 Pt 1):E941–E948
Larsen PL, Albert PS, Riddle DL (1995) Genes that regulate both development and longevity in caenorhabditis elegans. Genetics 139(4):1567–1583
Lavker RM (1979) Structural alterations in exposed and unexposed aged skin. J Invest Dermatol 73(1):59–66
Lenhardt R, Hopf HW, Marker E, Akca O, Kurz A, Scheuenstuhl H, Sessler DI (2000) Perioperative collagen deposition in elderly and young men and women. Arch Surg 135(1):71–74
Liang H, Masoro EJ, Nelson JF, Strong R, McMahan CA, Richardson A (2003) Genetic mouse models of extended lifespan. Exp Gerontol 38(11–12):1353–1364
Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L (2002) Calorie restriction extends saccharomyces cerevisiae lifespan by increasing respiration. Nature 418(6895):344–348
Lindblad U, Langer RD, Wingard DL, Thomas RG, Barrett-Connor EL (2001) Metabolic syndrome and ischemic heart disease in elderly men and women. Am J Epidemiol 153(5):481–489
Lindstedt E, Sandblom P (1975) Wound healing in man: tensile strength of healing wounds in some patient groups. Ann Surg 181(6):842–846
Ling S, Zhou L, Li H, Dai A, Liu JP, Komesaroff PA, Sudhir K (2006) Effects of 17beta-estradiol on growth and apoptosis in human vascular endothelial cells: influence of mechanical strain and tumor necrosis factor-alpha. Steroids 71(9):799–808
Loeser AA (1937) The resorption and action of follicular hormone rubbed into the skin. J Obstet Gynaecol Br Emp 44:710
Lokkegaard E, Jovanovic Z, Heitmann BL, Keiding N, Ottesen B, Pedersen AT (2006) The association between early menopause and risk of ischaemic heart disease: influence of hormone therapy. Maturitas 53(2):226–233
Longcope C (1971) Metabolic clearance and blood production rates of estrogens in postmenopausal women. Am J Obstet Gynecol 111(6):778–781
Lopez-Torres M, Perez-Campo R, Rojas C, Cadenas S, Barja G (1993) Maximum life span in vertebrates: relationship with liver antioxidant enzymes, glutathione system, ascorbate, urate, sensitivity to peroxidation, true malondialdehyde, in vivo H2O2, and basal and maximum aerobic capacity. Mech Ageing Dev 70(3):177–199
Lundgren D (1973) Influence of estrogen and progesterone on exudation, inflammatory cell migration and granulation tissue formation in preformed cavities. Scand J Plast Reconstr Surg 7(1):10–14
Lye M, Donnellan C (2000) Heart disease in the elderly. Heart 84(5):560–566
Magnusson U, Einarsson S (1990) Effects of exogenous oestradiol on the number and functional capacity of circulating mononuclear and polymorphonuclear leukocytes in the sow. Vet Immunol Immunopathol 25(3):235–247
Mair W, Goymer P, Pletcher SD, Partridge L (2003) Demography of dietary restriction and death in drosophila. Science 301(5640):1731–1733
Mak HY, Ruvkun G (2004) Intercellular signaling of reproductive development by the c. Elegans daf-9 cytochrome p450. Development 131(8):1777–1786
Makrantonaki E, Vogel K, Fimmel S, Oeff M, Seltmann H, Zouboulis CC (2008) Interplay of igf-i and 17beta-estradiol at age-specific levels in human sebocytes and fibroblasts in vitro. Exp Gerontol 43(10):939–946
Margolis DJ, Knauss J, Bilker W (2002) Hormone replacement therapy and prevention of pressure ulcers and venous leg ulcers. Lancet 359(9307):675–677
Marini H, Polito F, Altavilla D, Irrera N, Minutoli L, Calo M, Adamo EB, Vaccaro M, Squadrito F, Bitto A (2010) Genistein aglycone improves skin repair in an incisional model of wound healing: a comparison with raloxifene and oestradiol in ovariectomized rats. Br J Pharmacol 160(5):1185–1194
Mason JB, Cargill SL, Anderson GB, Carey JR (2009) Transplantation of young ovaries to old mice increased life span in transplant recipients. J Gerontol A Biol Sci Med Sci 64(12):1207–1211
Matsumoto T (2006) Selective estrogen receptor modulators (serms). Clin Calcium 16(9):100–105
McCay CM, Crowell MF, Maynard LA (1935) The effect of retarded growth upon the length of the lifespan and upon the ultimate body size. J Nutr 10:63–79
McCulloch D, Gems D (2003) Evolution of male longevity bias in nematodes. Aging Cell 2(3):165–173
Melton LJ III, Chrischilles EA, Cooper C, Lane AW, Riggs BL (1992) Perspective. How many women have osteoporosis? J Bone Miner Res 7(9):1005–1010
Mendoza CB Jr, Postlethwait RW, Johnson WD (1970) Veterans administration cooperative study of surgery for duodenal ulcer. Ii. Incidence of wound disruption following operation. Arch Surg 101(3):396–398
Mets T, Bekaert E, Verdonk G (1983) Similarity between in vitro and in vivo cellular aging. Mech Ageing Dev 22(1):71–78
Miller JG, Mac Neil S (1997) Gender and cutaneous melanoma. Br J Dermatol 136(5):657–665
Mills SJ, Ashworth JJ, Gilliver SC, Hardman MJ, Ashcroft GS (2005) The sex steroid precursor dhea accelerates cutaneous wound healing via the estrogen receptors. J Invest Dermatol 125(5):1053–1062
Miquel J, Economos AC, Fleming J, Johnson JE Jr (1980) Mitochondrial role in cell aging. Exp Gerontol 15(6):575–591
Mitchell RE (1967) Chronic solar dermatosis: a light and electron microscopic study of the dermis. J Invest Dermatol 48(3):203–220
Monteiro-Riviere NA, Banks YB, Birnbaum LS (1991) Laser Doppler measurements of cutaneous blood flow in ageing mice and rats. Toxicol Lett 57(3):329–338
Moraes AB, Haidar MA, Soares JMJ, Simoes MJ, Baracat EC, Patriarca MT (2009) The effects of topical isoflavones on postmenopausal skin: double-blind and randomized clinical trial of efficacy. Eur J Obstet Gynecol Reprod Biol 146:188–192
Morales DE, McGowan KA, Grant DS, Maheshwari S, Bhartiya D, Cid MC, Kleinman HK, Schnaper HW (1995) Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation 91(3):755–763
Morin GB (1989) The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes ttaggg repeats. Cell 59(3):521–529
Morris GM, van den Aardweg GJ, Hamlet R, Whitehouse E, Hopewell JW, Franke H, Loeffler M (1990) Age-related changes in the cell kinetics of rat foot epidermis. Cell Tissue Kinet 23(2):113–123
Morrison JH, Brinton RD, Schmidt PJ, Gore AC (2006) Estrogen, menopause, and the aging brain: how basic neuroscience can inform hormone therapy in women. J Neurosci 26(41):10332–10348
Mosselman S, Polman J, Dijkema R (1996) Er beta: identification and characterization of a novel human estrogen receptor. FEBS Lett 392(1):49–53
Motola DL, Cummins CL, Rottiers V, Sharma KK, Li T, Li Y, Suino-Powell K, Xu HE, Auchus RJ, Antebi A, Mangelsdorf DJ (2006) Identification of ligands for daf-12 that govern dauer formation and reproduction in c. Elegans. Cell 124(6):1209–1223
Mousavi SR, Raaiszadeh M, Aminseresht M, Behjoo S (2010) Evaluating tamoxifen effect in the prevention of hypertrophic scars following surgical incisions. Dermatol Surg 36(5):665–669
Nyman S (1971) Studies on the influence of estradiol and progesterone on granulation tissue. J Periodontal Res Suppl 7:1–24
Ogawa S, Inoue S, Watanabe T, Hiroi H, Orimo A, Hosoi T, Ouchi Y, Muramatsu M (1998) The complete primary structure of human estrogen receptor beta (her beta) and its heterodimerization with er alpha in vivo and in vitro. Biochem Biophys Res Commun 243(1):122–126
Onoe Y, Miyaura C, Ohta H, Nozawa S, Suda T (1997) Expression of estrogen receptor beta in rat bone. Endocrinology 138(10):4509–4512
Owsley C, McGwin G, Scilley K, Meek GC, Dyer A, Seker D (2007) The visual status of older persons residing in nursing homes. Arch Ophthalmol 125(7):925–930
Pickar JH, Macneil T, Ohleth K (2010) Serms: progress and future perspectives. Maturitas 67(2):129–138
Pierard-Franchimont C, Letawe C, Goffin V, Pierard GE (1995) Skin water-holding capacity and transdermal estrogen therapy for menopause: a pilot study. Maturitas 22(2):151–154
Pillai S, Oresajo C, Hayward J (2005) Ultraviolet radiation and skin aging: roles of reactive oxygen species, inflammation and protease activation, and strategies for prevention of inflammation-induced matrix degradation—a review. Int J Cosmet Sci 27(1):17–34
Pinto RE, Bartley W (1968) Changes in glutathione reductase and glutathione peroxidase activities in rat liver related to age and sex. Biochem J 109(3):34P
Pinto RE, Bartley W (1969) The effect of age and sex on glutathione reductase and glutathione peroxidase activities and on aerobic glutathione oxidation in rat liver homogenates. Biochem J 112(1):109–115
Pletcher SD, Macdonald SJ, Marguerie R, Certa U, Stearns SC, Goldstein DB, Partridge L (2002) Genome-wide transcript profiles in aging and calorically restricted drosophila melanogaster. Curr Biol 12(9):712–723
Pochi PE, Strauss JS, Downing DT (1979) Age-related changes in sebaceous gland activity. J Invest Dermatol 73(1):108–111
Punnonen R (1971) On the effect of castration and peroral estrogen therapy on the skin. Acta Obstet Gynecol Scand 9(Suppl 9):32
Punnonen R, Vaajalahti P, Teisala K (1987) Local oestriol treatment improves the structure of elastic fibers in the skin of postmenopausal women. Ann Chir Gynaecol Suppl 202:39–41
Qiu Y, Waters CE, Lewis AE, Langman MJ, Eggo MC (2002) Oestrogen-induced apoptosis in colonocytes expressing oestrogen receptor beta. J Endocrinol 174(3):369–377
Rajabi MA, Rajabi F (2007) The effect of estrogen on wound healing in rats. Pak J Med Sci 23(3):349–352
Ramsey JJ, Colman RJ, Binkley NC, Christensen JD, Gresl TA, Kemnitz JW, Weindruch R (2000) Dietary restriction and aging in rhesus monkeys: the University of Wisconsin study. Exp Gerontol 35(9–10):1131–1149
Reed GA, Tawfik O, Casparian JM (2005) Immunohistochemical analysis of steroid hormone receptors and structural proteins in skin: effects of aging and hormone status. J Appl Res 5(2):371–377
Rezzi S, Martin FP, Shanmuganayagam D, Colman RJ, Nicholson JK, Weindruch R (2009) Metabolic shifts due to long-term caloric restriction revealed in nonhuman primates. Exp Gerontol 44(5):356–362
Richard S, Querleux B, Bittoun J, Jolivet O, Idy-Peretti I, de Lacharriere O, Leveque JL (1993) Characterization of the skin in vivo by high resolution magnetic resonance imaging: water behavior and age-related effects. J Invest Dermatol 100(5):705–709
Sadoun E, Reed MJ (2003) Impaired angiogenesis in aging is associated with alterations in vessel density, matrix composition, inflammatory response, and growth factor expression. J Histochem Cytochem 51(9):1119–1130
Sadowska-Krowicka H, Mannick EE, Oliver PD, Sandoval M, Zhang XJ, Eloby-Childess S, Clark DA, Miller MJ (1998) Genistein and gut inflammation: role of nitric oxide. Proc Soc Exp Biol Med 217(3):351–357
Sakuraoka K, Tajima S, Seyama Y, Teramoto K, Ishibashi M (1996) Analysis of connective tissue macromolecular components in ishibashi rat skin: role of collagen and elastin in cutaneous aging. J Dermatol Sci 12(3):232–237
Sandblom P, Petersen P, Muren A (1953) Determination of the tensile strength of the healing wound as a clinical test. Acta Chir Scand 105(1–4):252–257
Sanz A, Hiona A, Kujoth GC, Seo AY, Hofer T, Kouwenhoven E, Kalani R, Prolla TA, Barja G, Leeuwenburgh C (2007) Evaluation of sex differences on mitochondrial bioenergetics and apoptosis in mice. Exp Gerontol 42(3):173–182
Sastre J, Pallardo FV, Vina J (2000) Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB Life 49(5):427–435
Sato R, Maesawa C, Fujisawa K, Wada K, Oikawa K, Takikawa Y, Suzuki K, Oikawa H, Ishikawa K, Masuda T (2004) Prevention of critical telomere shortening by oestradiol in human normal hepatic cultured cells and carbon tetrachloride induced rat liver fibrosis. Gut 53(7):1001–1009
Savvas M, Bishop J, Laurent G, Watson N, Studd J (1993) Type iii collagen content in the skin of postmenopausal women receiving oestradiol and testosterone implants. Br J Obstet Gynaecol 100(2):154–156
Sawaya ME, Penneys NS (1992) Immunohistochemical distribution of aromatase and 3b-hydroxysteroid dehydrogenase in human hair follicle and sebaceous gland. J Cutan Pathol 19(4):309–314
Schmidt JB, Binder M, Macheiner W, Kainz C, Gitsch G, Bieglmayer C (1994) Treatment of skin ageing symptoms in perimenopausal females with estrogen compounds. A pilot study. Maturitas 20(1):25–30
Schmidt JB, Binder M, Demschik G, Bieglmayer C, Reiner A (1996) Treatment of skin aging with topical estrogens. Int J Dermatol 35(9):669–674
Schneider EL, Mitsui Y (1976) The relationship between in vitro cellular aging and in vivo human age. Proc Natl Acad Sci USA 73(10):3584–3588
Schroeder P, Gremmel T, Berneburg M, Krutmann J (2008) Partial depletion of mitochondrial DNA from human skin fibroblasts induces a gene expression profile reminiscent of photoaged skin. J Invest Dermatol 128(9):2297–2303
Seibel J, Molzberger AF, Hertrampf T, Laudenbach-Leschowski U, Diel P (2009) Oral treatment with genistein reduces the expression of molecular and biochemical markers of inflammation in a rat model of chronic tnbs-induced colitis. Eur J Nutr 48(4):213–220
Selman C, Lingard S, Choudhury AI, Batterham RL, Claret M, Clements M, Ramadani F, Okkenhaug K, Schuster E, Blanc E, Piper MD, Al-Qassab H, Speakman JR, Carmignac D, Robinson IC, Thornton JM, Gems D, Partridge L, Withers DJ (2008) Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J 22(3):807–818
Shaw TJ, Martin P (2009) Wound repair at a glance. J Cell Sci 122(Pt 18):3209–3213
Shuster S, Black MM, McVitie E (1975) The influence of age and sex on skin thickness, skin collagen and density. Br J Dermatol 93(6):639–643
Shyong EQ, Lu Y, Lazinsky A, Saladi RN, Phelps RG, Austin LM, Lebwohl M, Wei H (2002) Effects of the isoflavone 4′,5,7-trihydroxyisoflavone (genistein) on psoralen plus ultraviolet a radiation (puva)-induced photodamage. Carcinogenesis 23(2):317–321
Siesky BA, Harris A, Patel C, Klaas CL, Harris M, McCranor LJ, Lauer J, Kaplan B (2008) Comparison of visual function and ocular hemodynamics between pre- and post-menopausal women. Eur J Ophthalmol 18(2):320–323
Simon AF, Shih C, Mack A, Benzer S (2003) Steroid control of longevity in drosophila melanogaster. Science 299(5611):1407–1410
Simonen O, Mikkola T (1991) Senile osteoporosis and femoral neck fractures in long-stay institutions. Calcif Tissue Int 49 Suppl:S78–S79
Simpkins JW, Yang SH, Wen Y, Singh M (2005) Estrogens, progestins, menopause and neurodegeneration: basic and clinical studies. Cell Mol Life Sci 62(3):271–280
Simpson ER, Zhao Y, Agarwal VR, Michael MD, Bulun SE, Hinshelwood MM, Graham-Lorence S, Sun T, Fisher CR, Qin K, Mendelson CR (1997) Aromatase expression in health and disease. Recent Prog Horm Res 52:185–213 (discussion 213–184)
Smith QT, Allison DJ (1966) Changes of collagen content in skin, femur and uterus of 17-beta-estradiol benzoate-treated rats. Endocrinology 79(3):486–492
Sobel H, Lee KD, Hewlett MJ (1965) Effect of estrogen on acid glycosaminoglycans in skin of mice. Biochim Biophys Acta 101(2):225–229
Son ED, Lee JY, Lee S, Kim MS, Lee BG, Chang IS, Chung JH (2005) Topical application of 17beta-estradiol increases extracellular matrix protein synthesis by stimulating tgf-beta signaling in aged human skin in vivo. J Invest Dermatol 124(6):1149–1161
Stanford JL, Hartge P, Brinton LA, Hoover RN, Brookmeyer R (1987) Factors influencing the age at natural menopause. J Chronic Dis 40(11):995–1002
Stanulis-Praeger BM, Gilchrest BA (1986) Growth factor responsiveness declines during adulthood for human skin-derived cells. Mech Ageing Dev 35(2):185–198
Stanulis-Praeger BM, Gilchrest BA (1989) Effect of donor age and prior sun exposure on growth inhibition of cultured human dermal fibroblasts by all trans-retinoic acid. J Cell Physiol 139(1):116–124
Stevenson S, Sharpe DT, Thornton MJ (2009) Effects of oestrogen agonists on human dermal fibroblasts in an in vitro wounding assay. Exp Dermatol 18(11):988–990
Sthoeger ZM, Zinger H, Mozes E (2003) Beneficial effects of the anti-oestrogen tamoxifen on systemic lupus erythematosus of (nzbxnzw)f1 female mice are associated with specific reduction of igg3 autoantibodies. Ann Rheum Dis 62(4):341–346
Stindl R (2004) Tying it all together: telomeres, sexual size dimorphism and the gender gap in life expectancy. Med Hypotheses 62(1):151–154
Sugimoto M, Yamashita R, Ueda M (2006) Telomere length of the skin in association with chronological aging and photoaging. J Dermatol Sci 43(1):43–47
Sumino H, Ichikawa S, Abe M, Endo Y, Ishikawa O, Kurabayashi M (2004) Effects of aging, menopause, and hormone replacement therapy on forearm skin elasticity in women. J Am Geriatr Soc 52(6):945–949
Sumino H, Ichikawa S, Kasama S, Takahashi T, Kumakura H, Takayama Y, Kanda T, Murakami M, Kurabayashi M (2009) Effects of raloxifene and hormone replacement therapy on forearm skin elasticity in postmenopausal women. Maturitas 62(1):53–57
Surazynski A, Jarzabek K, Haczynski J, Laudanski P, Palka J, Wolczynski S (2003) Differential effects of estradiol and raloxifene on collagen biosynthesis in cultured human skin fibroblasts. Int J Mol Med 12(5):803–809
Suthers K, Kim JK, Crimmins E (2003) Life expectancy with cognitive impairment in the older population of the United States. J Gerontol B Psychol Sci Soc Sci 58(3):S179–S186
Taguchi A, Wartschow LM, White MF (2007) Brain irs2 signaling coordinates life span and nutrient homeostasis. Science 317(5836):369–372
Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS (2001) A mutant drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 292(5514):107–110
Thiboutot D, Martin P, Volikos L, Gilliland K (1998) Oxidative activity of the type 2 isozyme of 17beta-hydroxysteroid dehydrogenase (17beta-hsd) predominates in human sebaceous glands. J Invest Dermatol 111(3):390–395
Thiboutot D, Bayne E, Thorne J, Gilliland K, Flanagan J, Shao Q, Light J, Helm K (2000) Immunolocalization of 5alpha-reductase isozymes in acne lesions and normal skin. Arch Dermatol 136(9):1125–1129
Thiers BH, Maize JC, Spicer SS, Cantor AB (1984) The effect of aging and chronic sun exposure on human langerhans cell populations. J Invest Dermatol 82(3):223–226
Thomas JR (2005) Effects of age and diet on rat skin histology. Laryngoscope 115(3):405–411
Thornton MJ, Taylor AH, Mulligan K, Al-Azzawi F, Lyon CC, O’Driscoll J, Messenger AG (2003a) The distribution of estrogen receptor beta is distinct to that of estrogen receptor alpha and the androgen receptor in human skin and the pilosebaceous unit. J Investig Dermatol Symp Proc 8(1):100–103
Thornton MJ, Taylor AH, Mulligan K, Al-Azzawi F, Lyon CC, O’Driscoll J, Messenger AG (2003b) Oestrogen receptor beta is the predominant oestrogen receptor in human scalp skin. Exp Dermatol 12(2):181–190
Tian DS, Liu JL, Xie MJ, Zhan Y, Qu WS, Yu ZY, Tang ZP, Pan DJ, Wang W (2009) Tamoxifen attenuates inflammatory-mediated damage and improves functional outcome after spinal cord injury in rats. J Neurochem 109(6):1658–1667
Tiganescu A, Walker EA, Hardy RS, Mayes AE, Stewart PM (2011) Localization, age- and site-dependent expression, and regulation of 11beta-hydroxysteroid dehydrogenase type 1 in skin. J Invest Dermatol 131(1):30–36
Toutain CE, Brouchet L, Raymond-Letron I, Vicendo P, Berges H, Favre J, Fouque MJ, Krust A, Schmitt AM, Chambon P, Gourdy P, Arnal JF, Lenfant F (2009) Prevention of skin flap necrosis by estradiol involves reperfusion of a protected vascular network. Circ Res 104(2):245–254 (212p following 254)
Tresguerres JA, Kireev R, Tresguerres AF, Borras C, Vara E, Ariznavarreta C (2008) Molecular mechanisms involved in the hormonal prevention of aging in the rat. J Steroid Biochem Mol Biol 108(3–5):318–326
Trichopoulos D, MacMahon B, Cole P (1972) Menopause and breast cancer risk. J Natl Cancer Inst 48(3):605–613
Tsoureli-Nikita E, Watson RE, Griffiths CE (2006) Photoageing: the darker side of the sun. Photochem Photobiol Sci 5(2):160–164
Tsukahara K, Moriwaki S, Ohuchi A, Fujimura T, Takema Y (2001) Ovariectomy accelerates photoaging of rat skin. Photochem Photobiol 73(5):525–531
Tsukahara K, Nakagawa H, Moriwaki S, Kakuo S, Ohuchi A, Takema Y, Imokawa G (2004) Ovariectomy is sufficient to accelerate spontaneous skin ageing and to stimulate ultraviolet irradiation-induced photoageing of murine skin. Br J Dermatol 151(5):984–994
Varani J, Spearman D, Perone P, Fligiel SE, Datta SC, Wang ZQ, Shao Y, Kang S, Fisher GJ, Voorhees JJ (2001) Inhibition of type i procollagen synthesis by damaged collagen in photoaged skin and by collagenase-degraded collagen in vitro. Am J Pathol 158(3):931–942
Varila E, Rantala I, Oikarinen A, Risteli J, Reunala T, Oksanen H, Punnonen R (1995) The effect of topical oestradiol on skin collagen of postmenopausal women. Br J Obstet Gynaecol 102(12):985–989
Verdier-Sevrain S, Yaar M, Cantatore J, Traish A, Gilchrest BA (2004) Estradiol induces proliferation of keratinocytes via a receptor mediated mechanism. FASEB J 18(11):1252–1254
Vina J, Borras C, Gambini J, Sastre J, Pallardo FV (2005) Why females live longer than males? Importance of the upregulation of longevity-associated genes by oestrogenic compounds. FEBS Lett 579(12):2541–2545
Vukelic S, Stojadinovic O, Pastar I, Rabach M, Krzyzanowska A, Lebrun E, Davis SC, Resnik S, Brem H, Tomic-Canic M (2011) Cortisol synthesis in epidermis is induced by IL-1 and tissue injury. J Biol Chem. doi:10.1074/jbc.M110.188268
Waetjen LE, Ye J, Feng WY, Johnson WO, Greendale GA, Sampselle CM, Sternfield B, Harlow SD, Gold EB (2009) Association between menopausal transition stages and developing urinary incontinence. Obstet Gynecol 114(5):989–998
Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, Staub A, Jensen E, Scrace G, Waterfield M et al (1985) Cloning of the human estrogen receptor cdna. Proc Natl Acad Sci USA 82(23):7889–7893
Wang E (1995) Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 55(11):2284–2292
Wang YN, Wu W, Chen HC, Fang H (2010) Genistein protects against uvb-induced senescence-like characteristics in human dermal fibroblast by p66shc down-regulation. J Dermatol Sci 58(1):19–27
Weinstein BE, Ventry IM (1982) Hearing impairment and social isolation in the elderly. J Speech Hear Res 25(4):593–599
Weinstock MA (1994) Epidemiologic investigation of nonmelanoma skin cancer mortality: the Rhode Island follow-back study. J Invest Dermatol 102(6):6S–9S
Wich SA, Utami-Atmoko SS, Setia TM, Rijksen HD, Schurmann C, van Hooff JA, van Schaik CP (2004) Life history of wild sumatran orangutans (pongo abelii). J Hum Evol 47(6):385–398
Widyarini S, Spinks N, Husband AJ, Reeve VE (2001) Isoflavonoid compounds from red clover (trifolium pratense) protect from inflammation and immune suppression induced by UV radiation. Photochem Photobiol 74(3):465–470
Williams ME, Pannill FC III (1982) Urinary incontinence in the elderly: physiology, pathophysiology, diagnosis, and treatment. Ann Intern Med 97(6):895–907
Yamawaki TM, Berman JR, Suchanek-Kavipurapu M, McCormick M, Maria Gaglia M, Lee SJ, Kenyon C (2010) The somatic reproductive tissues of c. Elegans promote longevity through steroid hormone signaling. PLoS Biol 8(8):e1000468
Yancik R, Ganz PA, Varricchio CG, Conley B (2001) Perspectives on comorbidity and cancer in older patients: approaches to expand the knowledge base. J Clin Oncol 19(4):1147–1151
Yang JZ, O’Flatharta C, Harvey BJ, Thomas W (2008) Membrane eralpha-dependent activation of pkcalpha in endometrial cancer cells by estradiol. Steroids 73(11):1110–1122
Zou K, Ing NH (1998) Oestradiol up-regulates oestrogen receptor, cyclophilin, and glyceraldehyde phosphate dehydrogenase mRNA concentrations in endometrium, but down-regulates them in liver. J Steroid Biochem Mol Biol 64(5–6):231–237
Zouboulis CC, Seltmann H, Hiroi N, Chen W, Young M, Oeff M, Scherbaum WA, Orfanos CE, McCann SM, Bornstein SR (2002) Corticotropin-releasing hormone: an autocrine hormone that promotes lipogenesis in human sebocytes. Proc Natl Acad Sci USA 99(10):7148–7153
Acknowledgments
Dr Emmerson is the David Hammond Charitable Trust Research Associate (The Healing Foundation) and recipient of the BSRA 2010 Korenchevsky award. Dr Hardman is the Edmund de Rothschild Senior Fellow in Ageing Research (Research Into Ageing; Age UK).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Emmerson, E., Hardman, M.J. The role of estrogen deficiency in skin ageing and wound healing. Biogerontology 13, 3–20 (2012). https://doi.org/10.1007/s10522-011-9322-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-011-9322-y