
Abstract
Horizontal gene transfer (HGT) has been proven to have a much stronger impact than mutation in the microbial world. Evidence of HGT is currently expressed as comparative sequence homology, whereas a substance with residing transfer initiative remains ambiguous. The simultaneous transfer of multiple genes could have more profound impacts than single-gene exchange on the evolutionary processes, but such data are still insufficient. Although three novel modes of HGT mechanisms have come to light—gene transfer agents (GTAs), membrane vesicles (MVs), and intercellular nanotubes (Ins)—the classic concepts of transformation, transduction, and conjugation must not be ignored.
In addition to the previously mentioned HGT phenomena, the author proposes another brand-new concept: broad-host-range vector particles (VPs) in the natural virus-like particle (VLP) assemblage. VPs are capable of transferring chromosomal genes, plasmids, and cytoplasmic material toward recipients with broad phylogenetic ranges, although the VP-production responsible gene(s) are unidentified to date. VPs exhibit similar morphological characteristics to double bilayered MVs and tailless virions. Accordingly, confidently discriminating between VPs, bona fide tailless virions, and MVs is impossible, even with electron microscopy; hence, the environmental VLP fraction should be inevitably reconsidered as a “continuous vector variety” composed of virus–VP–MV.
VPs can be characterized explicitly by the following: (1) low recipient lethality of ~10% irrespective of particle ultraviolet treatment; (2) transfer of the host chromosomal fragments (dsDNA, ~400 kb) toward broad phylogenic recipient range (Archaea-Bacteria-Eukarya), accompanied by a high generalized transduction frequency up to 1.16 × 10–2 CFU/VP; (3) progeny VP production from the VP-mediated transductant, in a phenomenon referred to as “serial transduction”; (4) spontaneous budding production that starts when the host attains the stationary phase without host cell lysis; (5) various discrete size distribution of progeny particles; (6) VPs that can only be revealed by electron microscopy by its budding from the regenerated VP-mediated transductant. The yield of a VP is strictly regulated by 3 ± 2 particles per cell, whereas introduction of the rel mutation would induce overproduction to bring forward a period of production to the host log-phase, which implies that a VP is an attribute of the host cell. VPs can transfer cytosolic substances, such as plasmid and protein, along with host chromosomal fragments; furthermore, both single genes and multiple genes for the host’s thermo- and psychro-tolerance enhance the host’s environmental adaptation.
Expertise: Molecular microbial genetics and microbial ecology of HGT media.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Broad-host range transfer
- Vector particle (VP)
- Serial transduction
- Virus-like particle or vesicle (VLP or VLV)
- Outer membrane vesicle (OMV)
- Membrane vesicle (MV)
7.1 Why Do We Need to Propose New Concepts of HGT Mediators?
The currently recognized horizontal gene transfer (HGT) vectors are summarized in Table 7.1. Gene transfer agents (GTAs) [17], DNA transfer by membrane vesicles (MVs) [30, 34], and intercellular nanotubes [19] have come to light as novel modes of HGT in bacteria (http://biobabel.wordpress.com/2012/01/24/novel-modes-of-lateral-gene-transfer-in-bacteria/) in addition to the three classic mechanisms: (1) transformation (in which naked DNA is taken up from the environment), (2) transduction (by which bacteriophages facilitate gene transfer by packaging host DNA as well as their own), and (3) conjugation (when plasmids encode a pilus by which they can be transferred from cell to cell) [49].
Some transduction-like unique mechanisms to mediate HGT between individual cells of the same bacterial population, termed GTAs (the specific names for individual GTAs are given by the abbreviation of genera and species of the original host as the prefix in principle), that are accomplished by a phage-like particle have already been reported for the α-proteobacteria Bartonella bacilliformis, Bartonella grahamii, Bartonella henselae, Bartonella vinsonii, Rhodobacter capsulatus (RcGTA), the δ-proteobacterium Desulfovibrio desulfuricans (Dd1), the spirochete Brachyspira hyodysenteriae (VSH-1), and the archaeon Methanococcus voltae (VTA) [50,51,52,53,54]. None of the packages have more than 14 kb of DNA, ranging in size from 4.4 to 13.6 kb; all of them take the form of small bacteriophages, although no commitment of a virus-like gene to the transportation of host DNA has been reported [50, 55]. Four known GTAs resemble “constitutive” generalized transducing phages [43]. All four GTA package double-stranded DNA (dsDNA) resemble “constitutive” generalized transducing phages, with a tailed phage structure and package of less DNA than would be expected to represent a complete tailed phage genome (except for Dd1). None of these agents appear to be particles with lytic activity. GTAs seem most likely to have been derived from bacteriophages that lost their ability to self-propagate [55]. Unfortunately, genetic information about the production of these particles is currently available only for RcGTA [55]. For a release of the RcGTA, cell lysis requires the genes rcc00555 and rcc00556, an endolysin and holin, respectively [56]. The production of RcGTA is regulated by host systems, including a putative histidine kinase (CckA) that is required for maximal expression of the holin gene (rcc00555) and for maturation of RcGTA to yield gene transduction-functional particles. VPs were observed to be distinct from GTAs and related entities from their susceptible recipient phylogenic range, cargo size, and budding production.
Although it has been recognized that MV production is a ubiquitous phenomenon found in many bacteria, the existence of a universal MV formation mechanism to explain it has not been clarified. The MV should not be collectively dealt with as one of the HGT mediators because it has several different functional aspects irrelevant to gene transfer. The MV is a universal cellular feature, common to the three domains of life [57]; however, the dimensions, morphology, and molecular composition of MVs in the environment are very comparable to those of some virions [58]. Despite this, molecular ecologists have not paid much attention to environmental MVs because their origin is ambiguous. Hence, the presence of MVs in natural environments has been mostly ignored; however, recent findings [59] demonstrating the abundance of bacterial MVs, comparable to that of VLPs, in marine ecosystems could change the situation. Biller et al. [59] observed an only negligible number of apparent tailed phages (or GTAs) in vesicle-rich ocean samples ranging from ~105 to ~106 MVs/mL, for which the MV isolating method was the same as that employed traditionally for the isolation of viruses. Hence, the isolation methods for viruses also result in the enrichment of MVs, so estimates of viral abundance might have been merged with MV abundance [60]. Therefore, complexity could be added to estimations of the environmental impact of either group of DNA contained in derived vesicles. Consequently, it is necessary to elucidate the releasing mechanisms of molecular contents to the environment for an understanding of the ecological impacts of MVs [61].
MV research has been performed mainly on functions and formation mechanisms. A cell under stress accumulates material, which is worsened upon damage of the regular housekeeping and stress-responsive mechanisms. The production of bacterial outer membrane vesicles (OMVs) that occurs under stress is an entirely independent, general envelope stress response that allows selective elimination of unwanted material cell by the preferential packaging of a misfolded protein. It has unknown how the released MV adheres to the cell and further fuses and transfers its contents [25]. Regarding the function of MVs, their involvement in pathogenicity has been studied; however, considering the presence of endogenous plant bacteria and intestinal bacteria, among others, the coordinated action of MVs on the host should also be considered sufficiently. Furthermore, little is known about the role of MVs in the environment. Considering that many MVs have been identified from biofilms, it is predicted that MVs are produced throughout the environment.
The MV induction mechanism differs depending on the environment; hence, MVs have different properties and functions depending on the environment being produced. Comprehensively identifying proteins contained in the MVs of P. aeruginosa revealed that MVs from free-living organisms contained toxins, whereas ones originating from biofilm contained a large number of siderophore proteins instead of toxins, distinct from free-living MVs. Although the role of MVs against infected hosts has been drawing attention because MVs produced by P. aeruginosa contain toxins such as protease, newly elucidated characteristics from the biofilm-originating MVs indicate an expanded role for environmental MVs [62].
DNA carrying MVs [38] represents only a small proportion (0.01–1%) of vesicles [22, 63], although some vesicles contain sufficient DNA to be visible after staining with SYBR fluorescent DNA dyes that are typically used to enumerate viruses. Thus, DNA is packaged heterogeneously within vesicle populations, and it appears that vesicles are likely to be a minor component of SYBR-visible particles in natural seawater compared with viruses [61]. Considering the content of the marine virus fraction that is mainly composed of bona fide viruses would be dependable, whereas employing an appropriate method to classify viruses from MVs, such as buoyant density, would be indispensable. Therefore, an argument concerning the environmental gene transfer frequency would be plausible to construct on reported VLP assemblages in the environment because the study of MV-mediated HGT is currently underway. Whether the adhesion of MVs to cells is selective or equally attached is of great interest to researchers. By elucidating this, the whole picture of MV formation, diffusion, and adhesion/fusion—that is, the whole bacterial membrane traffic—can be clarified and further application developments would be expected [37, 64].
Dubey and Ben-Yehuda [19] demonstrated the existence of tubular conduits forming between Bacillus subtilis cells. These nanotubes were shown to be able to mediate the exchange of proteins and non-conjugative plasmids. Nanotubes were also formed between both gram-positive B. subtilis and Staphylococcus aureus, and a thinner variety was formed between either of the gram-positive species and gram-negative E. coli. The plasmid transfer does not require any intrinsic plasmid elements, and a given cell can be either donor or recipient. Dubey and Ben-Yehuda suggested that the formation of “syncytium-like synergistic consortia” mediated by nanotube connections underlies many of the traits displayed by biofilms. How cargo is transported through the nanotubes remains unanswered. Whether the transport is active and requires energy or is passive and prompted by diffusion is also unknown. Both mechanisms possibly coexist, and utilization would depend on the delivered cargo. In eukaryotic cells, nanotubes are frequently associated with cytoskeletal and motor proteins, implying a role for active transport [65].
“Trans-kingdom gene transfer” has been reported [66,67,68,69], although the Ti plasmid is the sole molecular device found to be practically acting in situ to date [70, 71]. Bacterial cells generally restrict their acceptance of foreign genetic materials [72] by the restriction-modification systems, which interfere with flexible gene transfer between species of different genera [72,73,74,75,76,77]. Gene transfer is costly to the bacterium; the benefits provided by the acquisition of advantageous genes also act as a repair mechanism [78]. Studies of the genetic structure of bacterial populations have clearly revealed that accessory element (phages, plasmids, transposons, and insertion sequences) transfer has occurred during their recent evolutionary history [75, 79,80,81] and further conferred that some accessory elements have been transferred across much greater phylogenic differences [82].
Recently discovered Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) found in 40% of eubacterial and 90% of archaeal sequences prokaryotes [83] act as a kind of acquired immune system for plasmids and phages [84, 85]. A CRISPR-Cas system being considered to limit HGT [48]. Bacteria can acquire an entire chromosomal CRISPR-Cas system through transduction [48, 86, 87], whereas the protective effect from phage infection increases the HGT of phage-sensitive members of mixed populations [88]. Consequently, the overall ability of CRISPR-Cas promotes HGT through transduction with broader implications for microbial evolution [48, 88]; it is also involved in virus budding [89] in eukaryotes and archaea.
The host range of an HGT mediator is defined as the breadth of organisms capable of infecting and expressing its function, with limits on the host cell ascribed to the mediator, host, or environmental characteristics. Reflective of the mediator’s discipline, there is no unified definition for a “broad-host-range” mediator because the host of the mediator regulates the physicochemical interaction with the mediator to infect, replicate, and maintain [90]. Therefore, the author empirically defines the a broad-host-range VP as one capable of infecting, transferring, and budding reproduction in the recipient cell in excess of the phylogenetic family level from the original host.
In addition to the HGT phenomena, the author has proposed another brand-new concept of broad-host-range VPs, which are composed in part of natural virus assemblages. Viruses have been considered as the most abundant biological entities in the living world and the reservoir of most genetic diversity in the sea, which amounts to ~105 to ~109 virus-like particles/mL [91]. The barrier hindering the acceptance of virus-mediated gene transfer resides in the concept that most viruses have a narrow host-range [92,93,94], which reduces the likelihood of extensive gene transfer within a mixed bacterial population. It is also believed that the host range of bacterial viruses (bacteriophages) is restricted to specific bacterial strains or closely related species [95, 96]. A given type of virus usually has a restricted range of hosts—often a single species; however, some viruses infect only certain subspecies, whereas others may infect more than one related species or even genus [92]. Such viruses with extended host range exceeding one genus are called “broad-host-range viruses”, which likely comprise <0.5% of the total virus population [92].
Furthermore, viruses may exhibit a broader host range in deep waters than in surface waters [97]. The supposed host specificity of viruses, the mostly unknown species diversity and composition of marine microbial assemblages [98], and the quantitative evaluation of contact between marine viruses and hosts still involve much guesswork [99]. Hence, the contribution of virus-mediated gene transfer (transduction) has been considered a factor of minor importance for genetic diversity and evolution in the natural microbial community. Furthermore, transduction is a reductive process, in that the genetic donor is killed (lysis) in the process of producing a transducing phage particle [100]. However, because viruses are released in a free form and do not necessarily require cell contact to survive, transduction may represent an ideal method for dispersing genes in the environment [101].
HGT among organisms in natural environments is already a fact, not a “theory” as Koonin expressed [102]. Much like evolution itself, extensive HGT in the microbial world is not doubted anymore, as HGT is a dominant process in microbial evolution that generally occurs at a high rate. Rapid advancements in genome sequencing technology have provided evidence of HGT. The determination and analysis of complete genome sequences have led to the suggestion that HGT dominates microbial evolution, with the rate of gene gain and loss being comparable to the rate of spontaneous point mutations and much greater than the duplication rate [103]. Moreover, the relevance of “horizontal” as applied to gene flow is validated by strong evidence of the existence of a central vertical, tree-like trend in genome evolution. Thus, the focus of research has shifted toward the how and why of HGT, although much more remains to be done than has been accomplished already in these directions. The tree of life may be unresolvable because of the extent of HGT [104, 105], and that which we consider phylogeny may be defined in large part by HGT [106]. Many of these studies, however, rely on evidence that could be generated by forces other than gene transfer, including selection, variable evolutionary rates, and biased sampling [107].
Additionally, little information on the probable donor lineage and the likely time of evolution has been provided in most proposed cases. Even when donors and recipients have been proposed, there is rarely supporting evidence regarding the absence of the genes from relatives of the recipient lineage that diverged before the transfer. For a complete picture of any proposed case of HGT, it will be essential to have information regarding the vectors, what (if any) selective forces were involved in the transfer, and the extent of amelioration [107]. It is necessary to integrally consider these HGT phenomena as one of the elements constituting the mobilome, although there are only a few examples of the findings on the HGT for these elements. However, other than VP, the evidence for HGT has been studied primarily by using amplicon sequencings of plasmid, which would be transferred. To the author’s knowledge, no other system intensively accumulates findings by examining HGT in cooperation with the culture system other than VP. First, the author recognized the VP as a part of the lysogenic virus and studied it as a kind of transducing particle; hence, the following argument is mainly based upon a comparison with the virus-mediated transduction.
Biological features of the entity the author incidentally discovered are undoubtedly involved with such VLP-MV conflated assemblages with morphological similarity and residing DNA; however, they are entirely distinct from what has been reported. Hence, this novel concept is proposed for consideration as an overlooked biological information-transfer medium.
7.2 Curiosity as an Incentive to Thinking Led to Novel Findings
An attempt to screen restriction enzymes from marine isolates was offered by Prof U. Simidu, Ocean Research Institute, the University of Tokyo. Three type 2 restriction enzymes—FspI1604, FspI1611, and AspMD1—were consequently obtained from Flavobacterium sp. I 1604, F. sp. I 1611, and Alcaligenes sp. MD1, amongst approximately 150 species of marine bacteria [108, 109]. These Gram-negative strains were collected at approximately 2000 m deep in the central part of the Indian Ocean. They showed stiff resistance to streptomycin, harboring plasmid-like DNA, ultraviolet light, and/or mitomycin-inducible phages. The spontaneous release of phage-like particles (PLPs) into the culture medium without artificial induction attracted curiosity because no host-vector system had been developed for marine lysogenic phages. For microbial genetics to establish a host-vector system to elucidate somatic gene transfer mediated by transduction, the development of auxotrophic mutants is inevitable [110].
Unfortunately, the minimal medium essential for the marine bacterial genetics study has not been available since 1991 [111]. Therefore, an ability of dichlorophenoxyacetic acid (2,4-D) utilization was used as a marker for the gene transfer of the marine bacteria instead of the somatic gene mutant. 2,4-D is an anthropogenic persistent organic compound that did not originally have a natural origin; therefore, to be useful for the gene transfer index, previous studies used a decomposition of 2,4-D [112,113,114]. The bacterial 2,4-D biodegradation is plasmid-dependent, with properties that have been studied as a model of gene transfer in nature [115,116,117,118,119,120,121,122].
Contrary to prediction, 18 strains out of 25 ubiquinone-10 possessing marine isolates belonging to α-proteobacteria exhibited strong 2,4-D degradation that was superior to Escherichia coli JMP397. E. coli JMP397 is a transformant [123] harboring 2,4-D degradation pJP4 (80 kb); it exhibited higher 2,4-D degradation ability than the plasmid originating from Alcaligenes eutrophus JMP134 [118]. Currently, A. eutrophus has been reclassified as Ralstonia eutropha JMP 134 [124].
Extraction of plasmid-like elements (PLEs) from those high 2,4-D utilization marine strains (Alc 096, 233, 252, Agrobacterium kieliense IAM12618 [125]: basonym Ahrensia kielensis: [126]) and F. sp. I1604 was somewhat fastidious [127] by requiring repeated phenol treatment, with traits ascribed to tight coupling of DNA content with proteinaceous material to be encapsulated into the particle, as described in Fig. 7.2. The extracted PLEs were successfully generated transformants in the minimal medium supplemented with 2,4-D as a sole carbon and energy source from the recipient E. coli AB1157 (F−; thr-1 leuB6 thi-1 lacY1 galK2 ara-14 xyl-5 mtl-1 proA2 hisG4 argE3 rpsL31 tsx-33 supE44), with a higher frequency than authentic pJP4 (PLEs: 1.5~13.1 × 106 CFU/μg DNA; pJP4: 5.7 × 105 CFU/μg DNA); the amino acid requirements of prototrophic revertants were also generated [1]. Hence, PLE is considered to combine a 2,4-D degrading ability and somatic genes to complement the auxotroph. However, the conjugative transfer of 2,4-D assimilability from those marine strains to E. coli was not achieved (Chiura, Unpublished data). A report indicated the physiological situation of marine donor bacteria would become the essential key to the conjugative transfer to terrestrial bacteria because the accomplishment of the conjugation is severely affected by the ionic strength and type of ion required [140,141,142,143]. Conjugative transfer of pJP4 has reported toward strains belonging to a variety of genera of the α-, β-, γβ- and γ-classes of the Proteobacteria; however, only Pseudomonas putida and Delftia sp. strains were able to grow on 2,4-D as the sole carbon source [144]. The broad-host-range (BHR) plasmids were defined as those plasmids that can self-transfer themselves and can stably replicate and maintain in bacterial species from at least two subgroups within the Proteobacteria (e.g., between α- and β- Proteobacteria) [145, 146]. Accordingly, the definition of BHR is not uniform with the overall HGT mediators. The BHR plasmids typically have mosaic genomes that include two distinct regions [147]. The “plasmid backbone” genes encode proteins involved in the replication, maintenance, control, and conjugative transfer of the BHR plasmid. In the marine environment, the directly determined rates of HGT in the marine bacterial community were reported to be high, ranging from 2.3 × 10−6 to 2.2 × 10−4 transconjugants per recipient [148, 149]. The HGT performance of the above PLE is comparable.
Despite PLEs being susceptible to DNase I, attempts to draw restriction maps for those extracted PLEs was a wasted effort because restriction enzymes had been incapable of producing restriction fragments. As an avoidable attempt, the electron microscopic observation of the PLE samples showed an apparent existence of particles instead of free DNA strings. Those PLEs must have been intracellularly maturing virus particles extracted from the cell [1]. Accordingly, the PLE must have been an intracellular form of that spontaneously released VLP to the culture medium, which was capable of encapsulating a piece of a chromosomal gene to achieve prototrophic reversion of recipient auxotrophy as the plasmid responsible for 2,4-D degradation [150]. Hence, the object of attention changed to the spontaneously released VLPs to the culture medium.
The release of VLPs to the culture medium started when the host cells entered to the stationary phase, and after that without accompanying any reduction of the host cell abundance. Yields of the VLP at 100 h culture at 30 °C attained 6–70% of the host cell abundance, depending upon the strain [1]. An attempt to examine the capability of the broad-host-range HGT exceeding at least the family level using the above VLPs demonstrated successful gene transfer exceeding the difference of the family level [1].
Here, VLPs can potentially accomplish transduction-like gene transfer exceeding the family level (see Table 7.4), referred to as VP. A generated VP-mediated transductant is named by combining the VP donor initial, the recipient genus initial, and “trans.” Therefore, an A. kielensis originating VP is referred to as AkVP and a generated E. coli transductant is named AkEtrans. The transduction frequency is defined as the fraction of the generated colony number in each selected marker plate in the total applied particles. An overall average frequency of VLP-mediated gene transfer at a multiplicity of infection (MOI) of 0.1 was estimated to be between 2.62 × 10−3 and 3.58 × 10−5 per VP [2].
AkEtrans acquired VLP production also may transfer genes; namely, AKEVP was produced. 2,4-D transduction frequency/μg DNA was estimated as 2.1 × 106 CFU for AkVP and 2.1 × 106 CFU for AkEVP. At 6.4 × 106 CFU for FsI1604VP, it is four orders of magnitude more efficient than MVs derived from Acinetobacter baylyi demonstrated for drug resistance transfer [63].
The VPs of A. kielensis (AkVP: ρ25 = 1.4157 g/cm3; size ± SD, nm = 123 ± 2.6, n = 106) are especially characterized for their enveloped structure (see Fig. 7.2) with varying lengths (0.8–1.5 μm in diameter) being encapsulated one to several spherical particles (5.1 ± 2.8 particles/envelope; n = 60) [1]. This phenomenon is an uncommon feature for prokaryotic viruses, although some eukaryotic viruses, such as the influenza virus, have envelopes. RNA phage Ø6 of Pseudomonas is a particular example of an encapsulated prokaryotic virus whose particle is incorporated into the recipient cell during infection [151]. Remarkably, the generated transductants acquired VLP production other than prototrophic reversion of the amino acid requirement and 2,4-D utilization.
Transduction is a process of abortive infection. No progenies from infected viruses’ transducing particles are expected, as shown in the schematic diagram in Fig. 7.1, whereas the VLPs from the above marine bacteria were not the case. Therefore, new terms should be given to express this unusual phenomenon: the “serial transduction” and “serial transducing particle” as the broad-host-range VP.

A schematic representation of “ordinary” transduction and “serial transduction”. The left panel shows the life cycle of lysogens and the production of transducing particles. Note that transducing particles would not be capable of producing progeny from the transductant. The right panel shows the serial transduction with the aid of broad-host-range vector particle (VP) discussed in this article. Transductants generated with the VP acquire VP production without accompanying cell lysis, whose reproduced VP can carry out successive transduction as the serial transducing particle
E. coli transductant (AkEtrans) generated by AkVP acquired comparable VLP production (AkEtransVP: ρ25 = 1.4159 g/cm3; size ± SD, nm = 106 ± 4.3, n = 54). This again made it possible to transfer 2,4-D utilization and prototrophic reversion of amino acid auxotrophy to the recipient E. coli, which exhibited a comparable overall transduction frequency of 5.25 ± 2.85 × 10−4 CFU/particle, comparable to that of AkVP (2.31 ± 1.50 × 10−4 CFU/particle). The transducing frequencies of these VPs were found to be higher by 4–7 orders of magnitude than those reported for naturally isolated transducing phages [42, 45, 47].
The distribution of particle abundance in the sedimentation profile exhibited a similar continual increase with size—an apparent difference from findings concerning the size distribution of particles showing discrete bands, which were composed of several subpopulations. The trend curve showing the relationship between the distance of the ultracentrifuge tube from the bottom and buoyant density is given as:
Here, d is the distance from the bottom in cm. As an extension, particle diameter (in nm) and content as dsDNA (kbp) could be expressed as the following functions of buoyant density (ρ):
According to the empirical formula [6] above, AkVP could encapsulate 416 kb as the transfer cargo capacity, whereas the result of a nucleic acid type analysis for the encapsulated DNA species estimated by an in-situ lysis technique [152]and pulse field gel electrophoresis (PFGE) [153] was 198 kb. Hence, a single AkVP might have carried two segments of the 198 kb DNA cargo inside. AkEtrans produced particles without an envelope or vesicle structure (see Fig. 7.2), with a cargo capacity of 266 kb as dsDNA showed two types of DNA species estimated to be 174 and 195 kb [5]. It is unknown how and why the AKEVP was produced from E. coli transductant as a free single particle but not collectively packed in the envelope or vesicle. High coordinated marker transfer (i.e. linkage) was recorded to exceed 70% between every selected and unselected marker for both AKVP and AkEVP mediated transduction, even using low MOI.

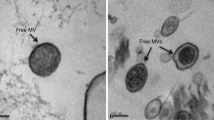
Electron micrography (EM) images of VPs. Kiel Bay originating Ahrensia kielensis produced VPs are characterized for their envelope structures, which range from single to several particles. A. kielensis VP-mediated Escherichia coli transductants acquire production of VPs without the envelope, whose size is slightly smaller than that of A. kielensis VP. FspI1604 VP from Flavobacterium sp. I1604 originating from the Indian Ocean. Size scale, 100 nm, is placed above. Upper right: A bacterium is bearing five electron-dense bodies (EDBs) in the cell corresponding to the yields of “virus” as five particles per cell. A proportion of such cells having an intracellular mature “virus” in the population stands for the frequency of visibly infected cells (FVIC [128]). In the case of the VP lysogens, FVIC should be read as the VP induction frequency because VP production takes budding from the host without accompanying lysis. Center: A lengthy sausage-shaped Aquifex sp. is under VP production; EDBs are seen in the left end of the cell. Superimposed panels indicate the process of the budding production of VP, referred to as STVP, released to the milieu. STVP exhibited trans-Domain HGT via the serial transduction of VP-mediated transductants towards Archaea and Eukarya. Another example of the trans-Domain HGT is given by the VP originating from a hyperthermophilic archaeon, Thermococcus kodakaraensis B41, to transfer thermo-resistance towards a mesophile enterobacterium, E. coli, to generate TkBEtrans. For details of VP budding transductants, see Tables 7.2 and 7.3
As the mode of gene transfer, the generalized transduction was carried out because preferential marker transfer was not observed. In the case of AkVP, multiple particle infections might have occurred for its architecture; however, AkEVP did not organize a multi-particle assembly in an envelope. The distance between markers deduced from the co-transfer frequency [154] was given as E. coli markers: his-arg, 203.2 kb and pro-arg, 232.2 kb; however, based upon the genome sequence data of E. coli AB1157 (Chiura, 2016, Unpublished data), it was given as his-arg, 139.1 kb and pro-arg, 322.2 kb. Although the results of coordinated marker transfer are not always represented as the distance of respective markers in the recipient and carried DNA species molecular mass, it is plausible that significant genetic traits are transferred to the recipient beyond generations in a phenomenon termed “serial transduction” [4, 7]. Concerning the cargo content of the vehicle, Hageman and colleagues [155] reported that MVs produced by A. kielensis DSM No. 5890 Strain B, whose outer membrane vesicle’s encapsulated genome size was 30.1 kb, encoded a prokaryotic sequence without a virus. Both strains used in these experiments are the same strain according to NCBI. Although a considerable discrepancy was found between the sizes carried in the AkVP and the MV derived from A. kielensis DSM, the contents of these vehicles’ cargo would consistently host chromosomal genes. Furthermore, A. kielensis encodes GTA in the genome (NCBI Reference Sequence: NZ_ARFW00000000.1 https://www.ncbi.nlm.nih.gov/genome/15060), which might have been contributing to MV biogenesis, although the MV genome would be exceeding the GTA capacity reported thus far [156].
7.2.1 Looking for the Environmental Broad-Host-Range Serial Transducing Particles: VPs
Some marine isolates may possibly produce VPs to carry out broad-host-range serial transduction [1, 2, 6, 7, 157, 158]. Hence, “virus” fractions collected from normal and thermal [8, 9, 130] environments were examined for the broad-host-range serial transducing particles: VPs. The phylogenic position of the recipient and the original VLP host bacteria are differently classified at the family level (see Table 7.4). These findings are not explained by the general concept of lytic or lysogenic cycles of viruses infectious to bacterial cells, suggesting that some VLPs do not fit the conventional molecular view of viral genetics.
Although it would be a herculean task to set up a protocol in the laboratory to reproduce transduction consistent with the environmental situation, the procedure was done according to the empirically studied method for phage-mediated transduction. Criteria for the VP judgement are achieved for a VLP or VLP producer to accomplish the serial transduction with the acquisition of budding particle production from the generated transductant belonging to at least the family level. The budding prokaryotic virus is quite scarce, with a strictly restricted host range. Budding is suggested as the mode of exit for pleomorphic archaeal and bacterial viruses [159]: pleolipovirus, plasmavirus, and the mycoplasma phage L2 and L172 [160,161,162,163]. Practically, prokaryotic virus budding has not been established because of the current lack of knowledge concerning the exit mechanism [159]; it relies on eukaryotic virus research, which is beyond the scope of this chapter.
To ensure their purity prior to running the transduction experiment, the recovered VLPs as a fraction of 0.2 μm > specimen > 30 kDa were treated with DNase I and RNase A to avoid the possibility of transformation. This was followed by equilibrium CsCl-density-gradient ultracentrifugation [164] at 175000 × g overnight at 15 °C. The bands formed were separately recovered and examined for the abundance, shape, and size of VLPs using electron microscopy (EM) [2], whereas the SYBR-epifluorescence microscopy method [165] was used on board the research cruise. The purified VLP was suspended in TBT (100 mM Tris-HCl, 100 mM NaCl, and 10 mM MgCl2, pH 7.4) buffer to make an appropriate dilution after CsCl removal.
For the recipient preparation, the physiological condition of the recipient should be reproducible to ensure the application of different VLP specimens from different origins. The recipient strains were grown to the mid-log phase. Then, the culture broth was substituted to 7% glycerine containing TBT buffer, dispensed to aliquots, frozen in liquid nitrogen, and kept in the deep freezer until use to endorse such requirement.
As the standardized protocol for transduction, 1 mL of thawed recipient suspension was mixed with the VLPs to obtain an appropriate MOI, and the mixture was left undisturbed at 30 °C for 15~30 min. Cells were washed with buffer and suspended in 1 mL of the same solution. Inactivation of the VLPs was attained by ultraviolet (UV) irradiation as previously described [2]. The four controls were as follows: (1) recipient cells tested with TBT buffer instead of the VLPs to determine the spontaneous revertant rate; (2) UV-inactivated VLPs and recipient cells; (3 and 4) VLPs with/without UV inactivation without the addition of recipient cells. For the transduction experiment, the recipients were incubated with or without VLPs, inoculated in LB medium, and kept at 30 °C for 2 days. In the transduction experiment, a portion of the sample was fixed with 2.5% glutaraldehyde; free particles and bacteria were enumerated by electron microscopy [166] to determine the practical MOI.
The growth profiles of generated transductants were examined, with specific reference to the particle budding production, the proportion of the particle producing population (VPIF; see below), the amount of the produced particle per cell, and the second VP-mediated transduction (see Fig. 7.1). Using this method, verification of the lethal effect on “VP infection,” the period of VP production, and the yields of VPs per cell can be obtained. The lethal effect caused by the virus is the mature virion release through the host cell rupture, with infection from cell death-inducing MVs as the microbial cellular response being equivocal. In addition, if the recipient fails to make an appropriate membrane fusion with VP at the VP-mediated transduction, plausible cytosol leakage of the recipient cell would occur and cause death (see Fig. 7.2).
The results obtained to date are summarized in Tables 7.2 and 7.3. The frequency of the marker transfer with the aid of VP-mediated generalized transduction depends upon the combination of donor and recipient, varying between 3.23 × 10−2 and 3.24 × 10−9, with a mean value of 1.14 × 10−3 and a median value of 9.22 × 10−6 CFU/VP. These values are considered to be high frequency among the values accounting for HGT [42, 44, 167]. A summary of the VP-mediated HGT occurrence between phylogenetically different organisms is given in Table 7.4, which shows that VPs are susceptible to a broad range of recipients. This fact implies that VPs should be quite prevalent on the planet (see Fig. 7.1 and 7.2).
7.2.2 VPs in the Environment
Cells undergoing viral induction can be recognized with transmission electron microscopy (TEM) [3, 168] in whole-cell TEM images of the induction; the proportion of the population with such intracellular particles is given as the frequency of visibly infected cells (FVIC) [128]. The method was first developed using thin-sectioning of picoplankton cells, which makes it possible to enumerate the fraction of cells infected within aquatic microbial consortia. This approach has demonstrated infection of several planktonic microorganisms in an environmental situation (in situ) and has been used to estimate the virioplankton production rate. Proctor and Fuhrman [169] demonstrated that intracellular viruses could be detected in cyanobacteria and heterotrophic bacteria collected from several marine environments, ranging from mesotrophic coastal sites to the oligotrophic ocean. The incidence of cells containing viruses [101] ranged from 0.9 to 4.3% of heterotrophic bacteria and 0.8 to 2.8% of cyanobacteria. In another study examining bacterial populations associated with sinking particles, a similar FVIC (0.7–3.7%) was recorded. Bacterial populations within this unique marine niche may also be controlled by viral lysis [170].
The FVIC within bacterioplankton was also determined by merely examining whole bacterial cells at a high accelerating voltage (80 kV) using TEM [171]. This method requires much less effort than the thin sectioning of bacterioplankton, but concern has been expressed over its accuracy [76]. Weinbauer [128] and colleagues found that whole-cell estimates of FVIC were on average 79% of the value of estimates based on thin sections [101]. Hence, whole-cell estimation consistently underestimates the level of virus-mediated mortality. FVIC values are inevitably converted to a proportion of the entire bacterioplankton community, estimating the overall level of virus-mediated lysis in situ; however, cells bearing mature viral particles would be ruptured according to the current concept. Proctor [172] and colleagues hypothesized that infected heterotrophic marine bacteria exhibit mature phages after 73% to 86% of the latent period has passed. Therefore, the total abundance of infected cells is expected to exceed the number of visibly infected cells by between 3.7- and 7.14-folds [101]. Based on FVIC data, a phages infecting average of 17% of the bacteria comprising bacterioplankton communities from a variety of aquatic environments [101].
Although VPs are distinct from common lytic viruses, this method can be applied for determining the extent of VP induction because the time of the particle production does not accompany the host cell disruption. Hence, the timing of the start of VP production can be judged from the time of the intracellular formation of electron-dense bodies (EDBs). Furthermore, GTA behaves the same as a virus, whereas the majority of MVs are considered to be “outer membrane vesicles,” with the relationship between EDBs and MVs still being uncertain. In the author’s experience, FVIC accounted for 6.02 ± 3.45% (n = 3591) in the marine environment (Pacific Ocean, Mediterranean Sea) and 6.92 ± 3.93% (n = 3925) in the thermal environment (geothermal vent, hydrothermal vent). Indeed, FVIC represents the total cell proportion engaging in virus and VP production. Estimates of VLP and cell abundance are 2.3 × 107 VLP/mL and 1.8 × 106 cells/mL in marine environments and 1.9 × 107 VLP/mL and 3.6 × 106 cells/mL in thermal environments, respectively. The somatic gene transfer frequency of VPs has been roughly estimated as ~4 × 10−4 CFU/VP for the marine environment and ~3.6 × 10−3 CFU/VP for the thermal environment (Chiura, Unpublished data). Based upon this frequency and VP yields per cell of 3, the number of cells being committed to VP production would be 3.0 × 103 cells/mL in the marine environment and 2.3 × 104 cells/mL in the thermal environment. Consequently, VP lysogens in the environmental microbial assemblage would be estimated as 0.2% and 0.6% of the population.
The term FVIC is not appropriate to express the extent of VP induction in VP lysogens, so the term “VP-induction frequency” (VPIF)” is used hereafter. Both natural and VP transductant strains generally start particle production by host cells becoming stationary. Furthermore, the yields of VPs per cell are in the range of three and five particles, whose period and productivity per cell appear to be strictly controlled to fit the cell phase. In most cases, the abundance of free (extracellular) VPs do not exceed the host cell abundance, although this was not the case for VP production from Vibrionaceae (Aliivibrio fischeri NCIMB1281T = ORI No.194: basonym, Vibrio fischeri, and Vibrio sp. FK01073). The number of free particles appeared to be associated with the number of VP-producing cells in the population—that is, the extent of the VPIF would determine the number of extracellular particles. Therefore, Vibrionaceae exhibited a higher frequency of VPIF. A. fischeri entered a logarithmic phase of growth for up to 12 h immediately after the initiation of culture, showing a generation time of approximately 1.84 h, and then entered a stationary phase to attain a stabilized cell abundance at approximately 1.2 × 109 cells/mL until the end of the culture. Initiation of the production of particles, referred to as AfVPs, by budding was determined from the particle per cell ratio and particle induction frequency to occur at approximately 15 h after initiation. The number of free particles continued to increase up to approximately 50 h after initiation, then stabilized at 1.20 × 1010 AfVP/mL. Three sharp peaks of induction (VPIF, period, %: 24 h = 15.8; 100 h = 19.1; 200 h = 23.8) were recorded, whereas no marked change in the cell population was observed to the end of culture. The average number of mature AfVPs per cell was approximately 3 (median ± SD, 3.0 ± 0.9, n = 475). Mucoidal substances heavily surrounded cells with budding particles and other particles. Budding particles from the cells showed a clear boundary of electron-dense bodies [6].
The oligotrophic West Mediterranean seawater (Calvi, Corsica, France; VFIC = 4.2 ± 2.5, n = 1800) originating VLP fraction exhibited ρ25 = 1.2684 − 1.4055 g/cm3, with a diameter ranging between 38 and 132 nm (diameter median ± SD = 67.6 ± 35.4 nm, n = 317). A portion (ρ25 = 1.3135 − 1.3304 g/cm3) of 118.3–126.7 nm VLPs were infected with E. coli AB1157 at MOI from 0.1 to 200 to examine the efficiency of plating (EOP) and the transduction frequency; this resulted in an EOP from 100 to 17.2% regardless of the VLP UV-irradiation. Transduction frequencies from 3.5 × 10−6 to 9.8 × 10−3 CFU/VP were observed. The generated transductants (CEtrans-F1) acquired budding particle production (produced particles per cell of approximately 3). A correlation between MOI and transduction frequency was observed, and the highest performance was recorded between MOI values of 5 and 20. Produced particles from CEtrans-F1 showed varied diameters ranging from 37.4 to 185.4 nm. Subsequently, the purified portion (ρ25=1.3285 − 1.3057 g/cm3) of 119.2–130.7 nm in diameter was infected at an MOI of 5.5 to E. coli AB1157. Consequently, a comparable EOP to the first experiment was observed, while the transduction frequency was reduced by approximately one order of magnitude from the first transduction. The generated second transductants (CEtrans-F2) acquired consistent particle production with CEtrans-F1. For the particle production from transductants, VPIF was between 15% and 20% during the stationary phase.
P. filamentus ATCC700397T, which was isolated from Arctic ice and cultured in 1/2 ZoBell at 10 °C, showed budding particle production. The produced particle size was distributed between 86 and 346 nm in diameter and contained approximately 150 kb as dsDNA (P. filamentus: 5 × 108 cells/mL, 2 × 109 VLP/mL, VP yields: 1.1 VP/cell). However, during the stationary phase, VPIF maintained an approximately 14% higher yield of free particles when approximately 120% of the cell population was given. The particles were purified in two bands by equilibrium CsCl density gradient ultracentrifugation to give 1.2893 ± 0.0102 g/cm3 as the buoyant density. The upper particles were infected with E. coli. Consequently, the recipient lethality was not observed. As for gene transfer, three transductants with complete marker reversion exhibiting a frequency at 5.0 ± 1.6 × 10−5 CFU/VP, referred to as PfEtrans, were obtained [138]. A generated E. coli transductant, PfEtrans, exhibited enhanced growth in 1/2 ZoBell at 10 °C, exceeding parental P. filamentus by attaining ~2 × 109 cells/mL. VP yields per cell were consistent with the parental strain at 1.1 and VPIF during the stationary phase kept recording as high as approximately 30%, while yields of free particles were suppressed to approximately 20% of the cell population. Therefore, the continual reincorporation of produced particles to the surrounding cells must have occurred.
Fluctuating particle abundance, like AfVP, was as well observed for a VP transductant, TYEtrans, which was generated with the aid of VPs collected at the Toyoha [173] mine (42°48’N, 141°2’E, Hokkaido, Japan). The parental recipient, E. coli AB1157, reached a stationary phase at approximately 9 h after initiation (1.7 × 109 cells/mL) at 30 °C, whereas TYEtrans entered the stationary phase at approximately 48 h (6.0 × 108 cells/mL, approximately 35% of the parental recipient population); the same level of population was observed until the end of culture (195 h). The minimum number of free particles (TYEVP) was recorded just after starting the culture (particle number: 1.13 × 104 particles/mL). The highest particle population was observed at approximately 48 h, amounting to 5.68 × 109 TYEVP/mL (VP/Cell = 9.50) when the transductant cells reached a stationary phase. Approaching 70 h, the free particle number decreased by three orders of magnitude and remained at the similar level, between 1 × 106 and 1 × 107 TYEVP/mL, up to 144 h. A second substantial increase of TYVP was observed at 170 h, which amounted to approximately 9.4 × 108 TYEVP/mL, followed by a decrease in the free particle number again to 1 × 106 TYEVP/mL at 195 h, exhibiting a similar trend as observed between 70 h and 144 h (VP/Cell = 0.01 and 0.05) [130].
A decrease of the free particle population without a change in cell numbers might have caused the reincorporation of free particles into the cells. The oscillating behavior of free particle abundance might reflect occasional repetition of the release from and reincorporation of VPs to the cell. Such reincorporated free particles must have been providing a template strand eligible for chromosomal mismatch repair [174], having the highest performance of the repair system.
An extraordinarily broad range of susceptible recipients was shown by a thermophile-originating particle collected from a hot spring called “Nakanoyu” (36°12’N, 137°36’E) in Nagano prefecture, Japan [8]. Sampling was occasionally done from 1993 to 1997, with 1997 having the highest water temperature during the period. The characteristic large sausage-shaped bacteria (see Fig. 7.2 [8]) of the sulfur-turf mats have been uncultivable to date. 16S rDNA cloning and sequencing studies have shown them to be members of the order Aquificales, which is the deepest-branching lineage of the domain Bacteria [175]. The cell population (2~6 × 105 cells/mL) was kept at the same order of magnitude, while VLP abundance was shown to be two orders of magnitude higher in the 1997 sampling (1.4 × 107 cells/mL, Aquificales cell No. ± SD, 95.7 ± 15.6%, n = 7) compared with other times (1~7 × 105 cells/mL, Aquificales ca. 6.4%). The thermophiles might have provided HGT with a relaxed broad-host-range. However, VLPs originating from the marine isolates of the most recent lineage of the domain Bacteria were incapable of transferring genes across the border of phyla [8], which could not have been accomplished by AKVP.
Both intra- and extra-cellular particles collected from the sulfur-turf cells exhibited comparable physicochemical parameters [8]. The second band of equilibrium CsCl density ultracentrifugation with the highest proportion (STVLP: ρ25, 1.2892 ± 0.0188; amount, 2.23 × 1011; particle size, 104.4 ± 9.2 nm in diameter; proportion, 40.1%; protein/nucleic acid ratio, 23.93) was applied to gene transfer experiments toward recipients of E. coli AB1157 and Bacillus subtilis PS9 (hisA metB5 thr-5 leuA8 trpCll lys21 purA non A-1/B-1 rfmR spβ−). Results indicated that STVLPs had a lethal effect on both recipients regardless of UV treatment, as well as a gene transfer capability [9] that resembled the case of those of marine origin [1, 2, 157, 158]. Consequently, VLPs originating from the “oldest” thermophile were capable of transferring genes across the borders of phyla. Hence, the existence of broad-host-range gene transporters among VLPs originating from thermophiles was confirmed.
It is notable that B. subtilis and E. coli transductants generated with the aid of STVLPs acquired the ability to produce particles [9]. The production of particles started from the host attaining the stationary phase, like that in the original host Aquifex strain, whose yield also was 3–5 particles per cell [8, 9]. Nucleic acid cargo in STVLP is susceptible to DNase, whose size as linear dsDNA was approximately 406.4 kb and RNA was not detected [8]. When the particles (STEVP) reproduced from E. coli transductants (STEtrans) were again infected with a B. subtilis auxotrophic mutant, they showed a lethal effect regardless of UV treatment of the particles, together with a gene transfer frequency between 2 × 10−8 and ×10−7 transductants/particle. The transduction frequency was slightly low and incomparable to that of the original particles from Aquificales used directly towards B. subtilis and E. coli.
Gene transfer from gram-positive to gram-negative bacteria does not take place because of the differences in applicable systems in the respective bacteria [94], with a few exceptions among artificial plasmid vectors with the natural transposon Tn916 [176]. Therefore, the finding is the first real evidence that VPs have the potential to enhance biodiversity among the bacterial community.
Furthermore, applications of STEVP to recipients of Archaeon—Sulfolobus acidocaldarius MR87 (his ura) [177, 178], Eukarya: Saccharomyces cerevisiae SEY499, SEY6120, YPH500, W303 (lys trp his leu) (provided by Takeuchi M, NAIST, 1999)—and the transductants were obtained with a frequency of 1 × 10−8~1 × 10−3 CFU/particle together with particle production from these transductants. S. acidocaldarius MR87 transductant produced particles ranging between 40 and 145 nm, whose productivity was reduced by approximately 22% of parental STEtrans. The S. cerevisiae SEY 6210 lys+ transductant, ScEtrans_Sc, produced two types of particles (ScEtrans VP, ρ25 = 1.2415 and 1.2968) that were again infected with E. coli AB1157 to produce transductants with particle production. The yield of particles per cell was estimated as 3 ± 2 (Fig. 7.2 [132]). The particles are likely to “infect” recipients belonging to different domains, which implies that at least some environmental VLPs share similar characteristics—a phenomenon that is not explained by the general features of viruses that infect microorganisms. A greater extent of recipient chromosomal marker transfer was recorded by several orders of magnitude compared with that reported for the marine environment [41, 73, 140].
Another example of trans-domain gene transfer was demonstrated by an archaeon-originating particle and the recipient E. coli. The survey area was extended to hydrothermal vents at Suiyo seamount located in Izu-Ogasawara arc. It showed the existence of a microbial population, even in the hyper-thermal fluid [3], with ~5 × 104 cells and ~3 × 104 VLPs/mL. A hyperthermophilic archaeon, Thermococcus kodakaraensis, was successfully obtained from an APSK06 (28°34.313’N, 140°38.617’E, 1386 m deep) boring core. The permissive growth temperature ranged between 70 °C and 90 °C and was recorded for an 80 °C culture with elemental sulfur, with 80% of particle abundance to the cell. A 70 °C culture with elemental sulfur gave the highest cell yields and 132% of particle abundance to the cell. Therefore, the strain was cultivated in the sulfur-supplemented medium at 70 °C. VLP production started when the stationary phase attained T. kodakaraensis, whose cell and particle abundances at 480 h of culture were 1.8 × 108 and 4.2 × 109, respectively [131, 179]. The median particle size was 125.19 ± 36.45 nm (n = 351) and yield per cell was approximately 1.5, with VPFI of approximately 19% during the stationary phase.
Gene transfer frequencies of particles from APSK07 fluid and T. kodakaraensis were between 2.6 × 10−4 CFU/particle and 3.0 × 10−6 CFU/particle. The production of particles from generated transductants was observed under TEM, with the particle yield per cell resembling that of the previously described marine strains and Aquifex sp. at 3–5 [131]. The size of the particles produced from the transductant, TkBEtrans, became larger (median ± SD = 132.55 ± 23.6, n = 272). Hence, a hyperthermophilic archaeon produced VP-transferred genetic traits towards mesophilic E. coli across the Domain border.
7.3 How Do VPs Produced in the Cell Release from the Host and Infect a New Host?
Electron microscopy observations suggest that membrane fusion occurs between the recipient cell and VPs (Fig. 7.3) at the initial stage of infection [6] because membrane fusion between the VP “capsid” and recipient cell was observed upon “infection.” Before membrane fusion, the VP pierces the recipient cell wall to create a place for membrane fusion [182]. Therefore, a membrane-fusion error upon the VP’s “infection” would cause cytoplasmic content efflux [183, 184]. VPs have shown an extraordinarily relaxed susceptible recipient range (Table 7.4). An interaction between the particle and recipient cell might determine the orientation for the recipient’s survival. As shown in Table 7.2, some VPs had a lethal effect on the recipients regardless of the strength of the UV treatment, reducing the EOP by up to one order of magnitude. VP infection of the recipient could be accomplished by the membrane fusion [6]. The lethal effect would be a consequence of a failure to form complete membrane fusion between the recipient cell and the particle, which must have lethally leaked the cytoplasmic content.

VP treatment of organic solvent, membrane vesicle (MV) production from DHlactrans, membrane fusion and translocation of DNA cargo to the recipient at VP infection, and pleomorphic size distribution of VPs after being experienced serial transduction. Electron micrography (EM) image of an organic solvent treated VP shows in (a). Purified VPs were dialysed against an up series of ethanol and chloroform and a descending series of ethanol and then treated VPs were placed on the EM grid [166] to observe. VP must have been contained in the lipid component for its coat because the content was extruded from the particle. In (b), extruding DNA is shown together with bead-like structure from a membrane vesicle from relA1 bearing transductant, DHlactrans, whose shape was not a string like Coliphage T4 but a tangled structure like a nucleosome observed for an archaeon [180]. (c, d) Membrane fusion and translocation of DNA cargo to the recipient at VP infection, DNA strand looks like an archaeon nucleosome structure [180, 181]. In (d), a schematic representation of (c) is shown. In (e), a relA1-bearing transductant produced MV together with VP, whose proportion was about 1/10 of the real VPs. In (f), the pleomorphic size distribution of VPs is shown after serial transduction. VPs showed production of several discretely different particle sizes, even a selected size VP employed for the transduction. Although the host strictly controls the VP yields per cell, an introduction of relA1 resulted in the turbulence of cellular control to produce many different sized empty MVs. Generally, the proportion of DNA bearing MVs is quite small; the DNA cargo size is also as small as 10 kb [22, 61, 63]. MVs produced by the relA1-bearing transductant must have been produced to encapsulate DNA cargo as intracellularly produced EDB
The contents of nucleic acid, carbohydrate, proteinase/peptidase, and glycanase activities were examined for particles and vesicles derived from STEtrans and DHlactrans (see Tables 7.2 and 7.3). The VP was predicted to have lipids as its surface component (Fig. 7.3), although an insufficient amount of samples prevented the lipid component from being analyzed. VPs are supposed to carry cellular cytoplasmic material as proteinaceous material and DNA; however, an RNA component has not been detected to date. A specific and strictly controlled VP cargo sorting and intracellular trafficking mechanism is expected to exist. However, it is currently difficult to infer an appropriate expression feasible for the strict control mechanism that produces 3 VPs/cell per generation. The results are given in Table 7.5. The surface structure of VPs must be composed of the multifunctional domain to accomplish infection of a variety of organisms.
The VP assemblage seems to carry all host genomic information almost exclusively, whereas the yields per cell are controlled strictly to be small unless a host control disorder is introduced, such as a rel mutation. Prior to the budding release of VPs, condensation of DNA cargo, which is likely a part of the host chromosome, makes EDS followed by intracellular trafficking in proximity of the cell membrane (Fig. 7.5). In general, transducing particles are formed when the phage packaging mechanism seizes upon host DNA, instead of concatemeric phage DNA, as a packaging substrate [191, 192]. The association of transducing host chromosomal DNA with a protease-sensitive component is estimated as 500 kDa of protein [46], with a substantial fraction (>75%) of the transduced DNA appearing to adopt a circular conformation maintained by an attached protein. Unfortunately, this protein has not been studied further, although the protein has been suggested to be a packaging enzyme that remains associated with the transducing DNA after completing cleavage [193]; this, however, remains to be established. There are two plausible hypotheses on how the DNA packaging mechanism makes transducing particles: one is a sequence-specific manner from DNA ends, whereas the other is a consequence of nonspecific cutting by packaging enzymes. The accumulated evidence clearly favors a pac site mechanism for P22 transducing particle formation, but a non-sequence-specific mechanism for the formation of P1 transducing phages [194, 195]. The findings would also apply to the case of VPs, as VP-encapsulated DNA seems to be heavily associated with a proteinaceous material (Figs. 7.3, 7.4, and 7.5).


VP cargo nucleic acid type, cargo production profile along with the cell growth, the difference between the recipient and transductants in the logarithmic phase, and transductant-specific membrane localized proteinaceous species. (a) CEtrans-F2 released from the first CEtrans-F1VP were again subjected to in situ lysis and obtained nucleic acids treated with DNase I and RNase A. DNase treatment showed no bands on the electrophoretogram; however, RNase treatment showed no change. STEtrans and STEVP were also treated in the same manner, of which VPs consisted of DNA [4]. (b) Intermittently withdrawn samples from the culture were subjected to in situ lysis (4 μg as nucleic acid per piece) and nucleic acids were examined. VP cargo DNA appeared in the cell after the stationary phase was attained. Although particle size showed pleomorphism, cargo DNA size was observed to be somewhat uniform throughout the producing period. Dalton markers: λ ladder and λ genome+λ/Hind III. VP encapsulated nucleic acid size for both CEtrans-F1 and CEtrans-F2 was estimated to be 368.5 ± 8.7 kbp (n = 49), which showed a resemblance to that of STEVP 372.09 ± 10.12 (n = 46). (c) The logarithmic phase cell provided a difference between transductants (CEtrans-F2, STEtrans) and parental strain AB1157. Broad-faint bands, whose size ± SD (n = 3) was 11.78 ± 7.87 kbp, were detected only for the transductants on the electrophoretogram. (d, e) VP production periods exhibited specific molecular species of membrane fraction only for the transductant, and relA1-bearing transductants showed different molecular species as above. Small DNA species found in the preceding period of VP production might have been related to proteinaceous species found explicitly for the transductants’ membrane fraction

Transductant cells in the process of budding. In (a) to (d), STE-trans was used as a reference; in (e) to (l), CEtrans-F1 has used a reference. (a) STEtrans under particle budding. (b) Before the budding, the electron-dense body (EDB) shifted to the vicinity of the cell wall, and then the EDB attached to the cell wall (c). Following this step, the phase change of membrane from bi- to monolayer was observed (d). CEtrans-F1 is shown around the budding particle (e). Another three EBDs can be seen in the cell. Thin sections of CEtrans-F1 demonstrated EDBs. (f) Different stages of EDB formation in the cells. (g) Particle under budding. A change of membrane phase is seen at the budding position. (h) The final stage of budding. Note “capsid” structure is different from the cell membrane. Epon-etched thin sections of the CEtrans-F1: (i–k) sections with central EDB and a dense cluster of gold-labelled DNA-antibodies. (l) Control: thin-section without EDB with sparse gold-labelled DNA antibodies. All sections belong to the same assay. Scale bars: a, d, 100 nm; b–d, f–l, 200 nm
Some MVs are known to inhabit a variety of enzymes [196]. The hydrolytic enzyme activities of VPs reside in an association of phage endolysins, which recalls the production process of some MVs [197]. Endolysin is a well-conserved enzyme that is typically involved in the release of dsDNA phages [198]. Endolysin opens holes in the cell wall, through which MVs are formed presumably due to turgor pressure and eventually released [28, 29]. Toyofuku et al. indicated that the simultaneous release of abundant MVs and the matrix in P. aeruginosa biofilm would occur by endolysin-induced explosive cell lysis in a subpopulation of the cells to supply public goods for the remaining cells [62]. Furthermore, endolysin was recently shown to induce MVs in Gram-positive bacteria through a distinct mechanism of explosive cell lysis [28, 29]. Hence, endolysin is a universal trigger for MV formation. Cell death is involved in MV formation, which renews the concept of cell death in microbial communities [199]; however, MVs were produced only from growing cells but not from lysing cells. From this standpoint, MV formation is considered to be a consequence of cell lysis that is distinct from the case of VPs.
MVs from a variety of organisms form the whole Domain of life and are called by several names, including membrane vesicles, outer membrane vesicles [OMVs], exosomes, and shedding microvesicles [57]. Bitto et al. demonstrated that chromosomal DNA is packaged into OMVs shed by bacteria during the exponential phase. Most of this DNA was present on the external surfaces of OMVs [35], with smaller amounts located internally. The DNA within the internal compartments of P. aeruginosa OMVs were consistently enriched in specific regions of the bacterial chromosome, encoding proteins involved in virulence, stress response, antibiotic resistance, and metabolism [200]. Roier et al. proposed a novel and highly conserved bacterial OMV biogenesis mechanism for Gram-negative bacteria based on phospholipid accumulation in the outer leaflet of the OM [201]. A proposed phospholipid transporter system of the VacJ/Yrb ABC (ATP-binding cassette) transport was shown to be involved in OMV formation [202]. However, the mechanism for intracellular trafficking of the cargo DNA in accounting for HGT is still insubstantial.
The conserved functions and mechanistic strategies of MV release are similar, including the use of eukaryotic endosomal sorting complexes required for transport (ESCRT) proteins and ESCRT protein homologues to facilitate these processes in archaea and eukaryotic microbes [57]. The ESCRT machinery is made up of cytosolic protein complexes: ESCRT-0, ESCRT-I, ESCRT-II, and ESCRT-III. Together with several accessory proteins, these ESCRT complexes enable a unique mode of membrane remodeling that results in membranes bending/budding away from the cytoplasm [203, 204]. These ESCRT components have been respectively studied in numerous eukaryotes, including yeast and humans [205].
The change in the intracellular structure before budding is illustrated in Figs. 7.4 and 7.5. DNA cargo must have been prepared around the center of the cell; however, how and what mechanism would proceed to select the DNA cargo content accompanied by budding production are unclear. OMV was reported to have continually budding production with a morphologically similar shape to VPs from γ-proteobacteria, Acinetobacter baylyi [63]. OMVs could be grouped into three size populations, with mean diameters ranging from 13 to 304 nm. OMVs were extracted from bacterial cultures and tested for their ability to vector gene transfer into populations of E. coli and A. baylyi, including naturally transformation-deficient mutants of A. baylyi, and determine the movement of DNA into OMVs using FITC-labelled and anti-dsDNA antibodies to track the movement of OMVs. Exposure to OMVs isolated from plasmid-containing donor cells resulted in HGT to A. baylyi and E. coli at transfer frequencies ranging from 1 × 10−6 to 1 × 10−8, with transfer efficiencies of approximately 1 × 103 and 1 × 102/μg of vesicular DNA, respectively. The intracellular production looked at several points in the cell, which was different from that observed for VP, as described below. It is uncertain whether this finding could be comparable to the content described previously because no DNA content per OMV was provided. Nonetheless, this report provides the only data on quantitatively estimated MV-mediated gene transfer. To the author’s knowledge, there have been no reports on MV-mediated “serial transduction” attempts to date.
DNA containing extracellular vesicles (see Table 7.1), referred to as ToEV, produced by budding was observed for T. onnurineus” NA1T. The DNA content in ToEV amounted 40 to 20 kb [22]. In comparing the encapsulated DNA in VP with such MVs, VP carried substantial DNA content, as shown in Table 7.2.
The VP production process was studied using ultra-thin sections to analyze features of the membrane fraction at the time of particle production. The materials CEtrans_F2 and STEVP (Table 7.1) were used because their morphological characteristics, production profiles of VP, and cargo size DNA (CEtrans_F2: 368.5 ± 8.7 kb, n = 49; STEVP: 372.09 ± 10.12 kb, n = 46) coincided with each other, even though the environmental situation of sampling sites differed [7, 8]. Consequently, the particle production initiated around the center of the cell by an adhering electron-dense material around the chromosome, following a hurricane-like structure formation. The shape was changed by condensation to create an EDB, which migrated to the vicinity of the cell membrane. Finally, budding took place. VP production in STEtrans was also consistently observed for intracellular trafficking of EDB from the cellular center towards the cell membrane for budding. The mode of VP production was found to be consistent between STEtrans and CEtrans. The acquisition of membrane vesicle production of VP-mediated transductant was observed when the rel-mutant was used for the recipient, whose population size was approximately 1/10 of that of “bona fide” VP particles.
To collect information on the cell membrane during VP production, 8 M urea dissolved the cell lysate membrane fraction of STEtrans, DHlactrans, and the parental E. coli and was applied to 5.2 M urea denatured-polyacrylamide gel electrophoresis (PAGE) (Fig. 7.4d). The denatured-PAGE showed that specific proteins in the membrane protein fraction corresponded to the period of particle production only for the transductant. The cytoplasmic extrachromosomal nucleic acid fraction before the VP budding release period was also examined for STEtrans, CEtrans_F2, and the recipient E. coli AB1157. Small DNA species specific for this period were observed, whereas the RNA fraction showed no difference (Fig. 7.4c). Such a VP transductant-specific molecular species must have been committed to intracellular EDB production. Successful intracellular trafficking from the central part towards budding proximity site might have been imitated in ESCRT of Archaea and Eukarya. Further biochemical studies are needed to elucidate the molecular mechanisms of VP production.
A standard feature of all VPs is that the particles reproduced from the transductant do not have a uniform size and are composed of varying types of particle species, even though particles of a specific size are selected for transduction. In the marine strain, approximately 20 species of VP were produced; a much smaller number of VP species was produced by the thermophile strain, whose trend for particle production of the transductant appeared to be inherited from the parental strain. Such pleomorphic particle production can be found in MV production; however, the molecular mechanism of MV depends upon the host cell belonging to a variety of phylogenic positions, and a commonly applicable pathway is still uncertain. Some similarity of pleomorphic particle production can be found among budding viruses. Usually, the capacity of a viral capsid is well regulated, and the packaging of a nucleic acid is performed by a head-full mechanism [94]. A eukaryotic paramyxovirus with an ssRNA genome (Myxoviridae) produces a varied particle size and comprises several pleomorphic viruses that may occur as irregular spheres of size, ranging from approximately 150 to nearly 300 nm in diameter [206, 207], or even as long filaments [207]. Although a prokaryotic budding virus is rare, the mode of exit is shared by pleomorphic archaeal and bacterial viruses (pleomorphiclipovirus, plasmavirus) [208] and mycoplasma phages [160] out of 70 lipid-containing viruses known to date (Archaea: 41; Bacteria, Gram-positive: 21, Gram-negative: 8; lipid enveloped: 9; Archaea 7; Gram-positive: 2) [159].
Genomic sequencing is currently in progress for recipient E. coli AB 1157, PfEtrans, VP source P. filamentus, STEtrans, and DHlactrans followed by comparative genomic analyses combining data with the generalized transducing phage Myoviridae P1, budding prokaryotic virus Plasmavirus Acholeplasma phage L2 [160], its host Acholeplasma laidlawii PG-8A, and the polymorphic virus Pleolipoviridae HGPV- 1, HHPV-1, HIS 2, HRPV-1, 2, 3, 6 [209]. Preliminary findings revealed that none of the complete gene set of viruses was discovered in the VP transductants’ genome; however, dnaB-like Phage P1 replicative helicase, Phage P1 DNA invertase, Phage P1 IS5 transposase family mobile element, Phage P1 ssDNA binding protein, and Phage P1 methyl-directed repair DNA adenine transferase were shared with all VP donor microorganisms and generated transductants.
7.4 Function-Related Gene Transfer
The VP-mediated HGT described previously is mainly for host somatic gene rescue; however, the simultaneous transfer of multiple genes might have impacts that are much more profound on the evolutionary processes. Some examples of VP-mediated function transfer are described in the following sections.
7.4.1 Nitrogen Fixation Acquisition of Escherichia coli by VPs Originating from Klebsiella pneumoniae subsp. rhinoscleromatis [139]
From the rhizosphere of the Sago palm tree, the Sago2 strain was isolated, which has a 16S DNA sequence with 99.93% similarity to Klebsiella pneumoniae subsp. rhinoscleromatis. Sago2 cultured in LB at 30 °C showed budding spherical particle (SGVP) production; the particles were purified by equilibrium CsCl density gradient ultracentrifugation. The particles had densities between 1.3183 and 1.2722 and diameters between 103.8 and 164.1 nm. They were used for transduction at a MOI of 2 by infecting the following recipients at 30 °C for 15 min: E. coli AB1157, DH5α [F−; ø80d, lacZ M15 endA1 recA1 hsdR17 (rk−, mk−) supE44 thi-1 λ− gyrA96 relA1 (lacZYA-argF)U169]; and JE6937 [F−; strR]. Irrespective of UV irradiation, SGVPs showed no lethal effect on the recipients.
The selection of transductants was made on N− agar plates supplemented with and without 20-μg/mL arginine and incubated anaerobically at 30 °C. The strains, which could form distinctive colonies within 5 days of incubation, were regarded as the transductants. SGVPs without UV inactivation were successfully transferred with N2 fixation to generate on N− agar plates. The results for DH5α were: four colonies (SGDHtrans), N2 fixation gene transfer frequency = 2.83 ± 3.24 × 10−7 CFU/SG-VP (n = 3). The results for the JE6937 recipient were: four colonies (SGEtrans), N2 fixation gene transfer frequency = 1.84 ± 0.80 × 10−8 CFU/SGVP (n = 3). To examine the N2 fixation ability of SGVP-mediated transductants, seven clones of SGDHtrans and four clones of SGEtrans were subjected to an acetylene reduction assay, with the parental recipients as the negative control and Sago2 as the positive control.
Consequently, anaerobically incubated Sago2 in LB and N− medium showed nitrogenase activity. All the SGDHtrans clones exhibited nitrogenase activity when anaerobically cultured in LB; however, two of them did not show nitrogenase activity in N− medium. As for SGEtrans, no clones exhibited nitrogenase activity in LB and N− medium. Hence, different genotypic variations of transductants could be generated by VPs; still, more acceptance of incorporating gene sets must have been affected by recipient genetic constitution.
7.4.2 Thermo- and Psychro-Tolerance Acquisition
P. filamentus ATCC700397T, which was isolated from Arctic ice, cultured in 1/2 ZoBell at 10˚C showed budding particle production. Produced particle size in diameter distributed between 86 and 346 nm that contained ca. 150 kb as dsDNA (P. filamentus: 5×108 cells/mL, 2×109 VLP/mL, VP yields: 1.1 VP/cell). Although VPIF during the stationary phase kept ca. 14% high yields of free particles by ca. 120% of the cell population was given. The particles were purified in two bands by equilibrium CsCl density gradient ultracentrifugation to give 1.2893 ± 0.0102 g/cm3 as the buoyant density. The upper particles were infected with E. coli AB1157. Consequently, the recipient lethality was not observed. As for gene transfer, three transductants with the complete marker reversion exhibiting the frequency at 5.0 ± 1.6 × 10-5 CFU/VP, referred to as PfEtrans, were obtained [138]. PfEtrans exhibited enhanced growth in 1/2 ZoBell at 10 °C, exceeding parental P. filamentus to attain ~2 × 109 cells/mL. A hypothesis was examined that the gene transfer due to VPs originating from P. filamentus lowers the range and optimum temperature of growth of the recipient E. coli. The optimum growth temperature of P. filamentus is 10 °C, and PfEtrans exhibited a cell population in the stationary phase that enhanced growth to the maximum of P. filamentus cell population at 10 °C. PfEtrans was examined in the still culture in 1/2 ZoBell in 3 days, consequently showing that the optimal temperature was reduced to 30 °C and it was possible to grow at 0 °C. Furthermore, culture in 1/2 ZoBell at 10 °C showed a maximum cell density of ~2 × 109 cells/mL with VP production, by which serial transduction was also demonstrated [138]. Genomic sequence analyses for P. filamentus with E. coli AB1157 clarified how PfEtrans acquire cold and high osmotic tolerance that resulted in the enhancement of genes relating to stress response and osmotic regulators (Chiura, Yoshizawa, Kogure, Unpublished). (Fig. 7.6).

Acquisition of heat resistance and change in the optimum and permissive growth range. E. coli AB1157 transductants generated by the VP originating from thermophiles provided thermo-resistance to the mesophilic enterobacterium E. coli recipient (a) [134]. VP originating from the psychrophile lowered the optimum and permissive growth range of E. coli (b) [138]
Numerous factors are assumed to control the temperature range and the optimum growth. Hence, the growth–temperature relationship has been examined. To determine whether thermo-resistance gene(s) could be transferred towards mesophilic recipients, Aquifex sp. and T. kodakaraensis B4 originating VP-mediated E. coli transductants, STEtrans and TkBEtrans, were grown in conditions exceeding the permissive temperature for E. coli (50, 56, and 70 °C) for 15 days. Consequently, the parental recipient, E. coli AB1157, decreased its population to less than a few percent of the inoculum by 48 h; then, the living cells became undetectable by LIVE/DEAD in all conditions [131, 134] (Fig. 7.6). It was known that heat-shock response would be activated when E. coli cells were transferred from a permissive state to a high temperature; however, the situation exceeding 50 °C could not be rescued even with such a response. Hence, prolonged treatment above 42 °C would cause the disorganization of cell structure, resulting in cell death. On the contrary, STEtrans and TkBEtrans treated under the same conditions exhibited different profiles. At the initiation of the treatments under the restricted condition for E. coli, the population decreased by 5–25% in 2–5 days because transductants were generated at 30 °C. After that, the population increased by 2- to 140-fold or at least maintained the initial inoculum size.
TkBEtrans was inoculated at an initial population size of 1.23 × 107 cells/mL. The cells decreased to 6.31 × 105 cells/mL (approximately 5% of the inoculant) at day 5, and then the cells grew to 7.48 × 107 cells/mL (approximately six-fold of the inoculant) at day 13 at 50 °C. The generation time of the transductant in the period of propagation was estimated to be approximately 28 h. At 56 °C, the cells decreased to approximately 3 × 106 cells/mL (~25%) at day 5, and then the cells grew to 1.95 × 107 cells/mL (~1.6-fold of the inoculant); the generation time in the growing period was estimated to be approximately 73 h. At 70 °C, the cells decreased to 1.53 × 106 cells/mL (~12% of the inoculant) at day 3, and then grew to 2.43 × 107 cells/mL (~2-fold of the inoculant) at day 13, of which generation time during the propagation was estimated to be approximately 266 h.
Change in the population under high-temperature incubation for STEtrans is also given in Fig. 7.6. For STEtrans (initial population size: 4.20 × 107 cells/mL), the population was observed to decrease to approximately 2 × 107 cells/mL (~50%) toward the second day. It maintained this level for another 4 days, after which the cells grew to approximately 6 × 109 cells/mL (~140-fold of the inoculant) in the following 2 days; finally, the population became stable. The generation time of the transductant during the growth was estimated to be approximately 6 h. At 56 °C, the cell number at day 2 was observed to be approximately 2 × 107 cells/mL (~50%), and its population became constant afterwards. At 70 °C, approximately 2 × 107 cells/mL (~50%) of cells were observed on day 2, and its population became constant afterwards.
This trans-phyla thermo-tolerance transfer is a consequence of broad-host-range transduction from a thermophile to a non-thermophilic enterobacterium. The transductants used above were first selected by the prototrophic reversion of the auxotrophic recipient at 30 °C and resided at the mesophilic temperature for several generations. Even after such a period when the transductants remained at a permissive temperature, the thermo-tolerance trait of the transductant suggesting VPs contributes to enhanced host adaptation.
Although it is often assumed that phages do not intrude on the genus barrier [92], this concept has been questioned. The host ranges of marine phages have been shown to vary by Wichels et al. [210]. Jensen et al. [211] disputed that narrow host-ranges are an isolation artefact. Substantial variability in host ranges was also reported by Suttle [212] for cyanophages. Viruses might exhibit a broader host range in deep waters than in surface waters [97]. However, viruses also can influence the stable stockpile of genes in prokaryotic species on ecological scales [213].
7.4.3 Relationship between Cell Population Density and Induction Frequency of VP Lysogen: A Marine-Originating VP-Mediated Transductant, CEtrans [214]
In a natural water column, FVIC was observed to be an equilibrium because of cell and virus interactions; however, there is no substantial evidence to interpret such a situation. VP lysogens started particle production when the host attained the stationary phase, whose VPIF is commonly quite low (the Pacific Ocean and Mediterranean Sea average, 6.92 ± 3.93%, n = 3925). Namely, less than 10% of the population would commit to particle production. Some VP lysogens, such as Vibrionaceae, produce VP exceeding the host population, which has been ascribed to their high VPIF [6]. One of the well-known characteristics of Vibrionaceae is its quorum-sensing (QS) behavior. However, the current understanding of QS is that the signal is not only solubilized in an aqueous environment but is also packaged in MVs and delivered in varying propensities to different bacteria [28, 29, 39, 40, 197]. As described in Sect. 7.6, VP transfers plasmid and cytoplasmic material; VP can transfer cytoplasmic material, so VP production must be intimately connected with the community situation. In this situation, a change in VPIF under different VP lysogen abundances around the host secreted diffusible substance. CEtrans_F1 was grown in minimal medium (MM) after Davis for approximately 2000 h and then the cells were removed to recover the supernatant, followed by a subsequent filter with 0.2 μm and 30 kDa cut-off membranes to collect the permeate. This permeate was used as the base material. The conditioned medium (CM) was prepared by adding the nutrient supplement as MM (Fig. 7.7).

Quorum sensing-like response of VP production with a specific reference to VPIF and VP yields per cell. The prolonged culture medium used as the material of the “conditioned”-medium and the low initiating population density induced growth suppression (a). The sever effect of the conditioned-medium to VPIF was observed for the low initiating population density (b). Furthermore, VP yields per cell were also enhanced by the low initiating population density (c). The findings suggest that the QS mechanism might have been involved in VP production
CEtrans_F1 was cultured as described above for 48 h, corresponding to the period to enter the stationary phase; then, centrifugally collected cells were washed with Davis salt. Two sets of 1 × 106 and 1 × 108 cells/mL as the initial cell abundance were prepared. For one set of 1 × 107 and 1 × 108 cells/mL, the specimen was cultured in the MM as the fresh medium (FM); another set of cells was cultured in the CM. The same conditions were used for both sets of specimens. Growth profiles were monitored. Subsamples were intermittently withdrawn to examine VPIF and the particle yield per cell by EM. As a result, VP abundance for the CM series was 10-fold higher than that of FM at the beginning; after 161 h of incubation, it increased to 100-fold higher than that of FM (Fig. 7.7a).
The initially high VPIF for every specimen was ascribed to the inoculant being started with VP production collected from the 48 h culture. The high initial cell abundance seemed to enhance VP induction; furthermore, the CM showed excessive VP induction. In the usual situation of small cell abundance, VPIF was usually suppressed less than 20%, while more than 80% of the population committed to VP production with a high cell abundance in the stationary phase (Fig. 7.7b). The particle yields per cell (2.7 ± 0.7. n = 96) were somewhat consistent with each other; however, the extended production period and higher VPIF resulted in high yields of produced VPs for the CM series (Fig. 7.7c). The result suggests that a diffusible substance was profoundly affected by the induction of VPs. A QS system is considered to be a bi-directional function towards lysogenized entities and their host. QS is a system for bacteria to communicate with each other with a diffusible substance [215].
7.5 Molecular Evidence for VP-Mediated Gene Transfer
Attempts have been made to perform polymerase chain reaction (PCR) for molecular evidence of VP-mediated gene transfer and construct a clone library from VP-derived DNA for gene determination [136]. STEVP was infected with E. coli DH5α (ΔlacZYA-argF relA1 recA) and selected by lactose utilization acquisition; arginine prototrophism resulted in the generation of E. coli DHlactrans. DHlactrans produced DHlacVP (size ± SD: 69.4 ± 2.1, 78.6 ± 1.1, and 93.4 ± 4.0 nm; n = 480) with smaller diameter and more compact encapsulated DNA (size ± SD: 74.9 ± 6.7, 108.5, ± 4.5, and 181.9 ± 23.4 kb; n = 4) than parental STEVP (size ± SD: 128.3 ± 2.3 nm, n = 1225; DNA size ± SD: 389.7 ± 6.5 kb, n = 49). Particle production commenced immediately after inoculation, accompanied by transparent membrane vesicles (DHlacMV), whose proportion to the VP with electron-dense content was approximately 10%; this can be ascribed to relA1 because a comparable phenomenon has been observed for STEVP-infected E. coli JM109 transductants (Chiura, Unpublished). Production of the membrane vesicle concomitantly changed with a change in particle production to maintain the MV/VP ratio (Fig. 7.4f).
The recA mutation is a point mutation caused by a substitution of glycine to aspartic acid (G160→D160), which prevents the homologous recombination of exogenous fragments from the host chromosome (“Strain-DH5a”; https://cogsc2.biology.yeal.edu/Strain.php?=43590). Hence, the cargo DNA strand stays in the extrachromosomal state after STEVP infection in a DH5α recipient. Lactose utilization made the selection of the transductant; the cargo content must have been only for the lactose metabolism with additional VP production-responsible genes. Generated DHlactrans reproduced DHlacVP to accomplish the serial transduction. Therefore, the cargo DNA size is postulated to decrease as a consequence of packaging the ΔlacZYA-argF frame of 78 kbp (Fig. 7.8b) to complement the host, with an additional 16 kbp of DNA encoding the VP production-responsible genes (PPRG) originating from STEVP. VP cargo size decreased, which supports the above hypothesis, as shown in Fig. 7.8a.

A change in VP cargo size depended on the transduction recipient, loci of target genes in E. coli chromosome, and PCR amplification of target genes in DH5α and DHlac trans. (a) After in situ lysis, the treated gel plugs were loaded on 1.0% agarose ME gel and run in a gel with 0.5× TBE buffer at 14 °C; switching time, 50–90 s; runtime, 22 h; angle, 120°, and voltage gradient, 6 V/cm. A higher yield of ~120 kb DNA fragments was gained in samples washed with salt. Lanes: λ ladder, size marker (48.5 kb – 1.2 Mb, FMC, USA); E. coli AB1157, the transduction recipient of STVP; STEVP, VP produced by STEtrans; STEtrans: a transductant of E. coli AB1157 generated with the aid of STVP-mediated transduction originated from Aquifex sp.; DHlacVP, VP produced by DHlactrans; DHlactrans, a transductant of E. coli DH5α generated with the aid of STEVP-mediated transduction originated from STEtrans. Note that the cargo size of DHlacVP decreased by 1/3 from STEVP. The recipient E. coli AB1157 is an auxotrophic mutant but has wild rel, whereas E. coli DH5α has a deletion of lacZ-argF as depicted above and mutations of relA1 and recA. The recipient with the relA mutation was found to decrease the cargo size of reproduced VPs from VP-mediated transduction. (b) PCR primers were designed to amplify genes shown as arrows. Sequence information was obtained from the database “Profiling of E. coli Chromosome” (PEC: http://www.shigen.nig.ac.jp/ecoli/pec/index.jsp). DH5α has a deletion frame over the lacZ-argF region (broken line). Genes yagX and yahF were located inside, whereas yagF and mhpB were located outside the deletion frame. The sharp end of the individual gene indicates the transcription direction. All genes described here are non-essential. (c) Amplification of total DNA prepared from the recipient E. coli DH5α (R) and the transductant DHlactrans (T) by using specific primers for marker genes, lacZ, argF, and mhpB. The gene mhpB locates ~7 kb downstream of lacZ in E. coli genome and therefore should be contained in both R and T DNA. The reaction mixture was directly loaded on 1.0% agarose gel. Template-primer pairs are shown as follows: Lane, T + lacZ template; R + lacZ template; −w/o addition of lacZ template; T + argF; R + argF; −w/o addition of argF template; T + mhpB template; R + mhpB template; −w/o addition of mhpB template. λ/Sty size marker: 19.33, 7.74, 6.22, 4.26, 3.47, 2.69, 1.88, 1.49, 0.93, 0.42, and 0.07 kbp. (d) DNA extracted from DHlactrans was amplified with primers (+) for six positional marker genes regarding non-template reactions (−). Lane, (+ and −) underneath respective maker gene stands for +, template added and −, without addition of the template. λ/Sty: size marker
Three-step PCR was used for verification of lacZ with lacZ-specific primers on VP-derived extrachromosomal DNA and total DNA extracted from DH5α and DHlactrans. Positive DNA amplification was observed in the VP transductant DHlactrans and VP-derived extrachromosomal DNA (Fig. 7.8c, d). Nonetheless, DHlacVP cargo content was found to be composed of the whole range of E. coli chromosomal genes (Kawarabayashi, personal communication). Hence, the above hypothesis is disproven. DHlactrans was subjected to a genome sequence together with STEtrans and E. coli AB1157. The sequence data of DH5α was obtained from the NCBI database (Escherichia coli strain DH5alpha chromosome, complete genome https://www.ncbi.nlm.nih.gov/nuccore/CP025520.1?report=genbank). The comparative genomic analysis is currently in progress (Table 7.6).
Preliminary findings revealed that the recA1 mutation in DH5α remains unchanged in DHlactrans and no introduction of wild recA was found from STEtrans. The total amino acid sequence identity between DH5α and DHlactrans is 99.39%. In all, 26 unique genes, of which 25 are hypothetical proteins, are exclusively found in DHlactrans, in addition to 79 non-synonymous substitutions. Genomic analysis revealed the genetic robustness of the recipient as a biological entity, together with the flexibility to adapt to environmental change. There must be an unknown scheme for genetic recombination upon intracellular VP production. Including the material from the experiment described in Sect. 7.4.2 on thermo- and psychro-tolerance acquisition, all host and recipient strains were found to share a generalized transducing phage P1 recombinase gene; therefore, such a recombinase might have been committed in recombination with host chromosomal fragment replication to produce the VP cargo. Thus, the gene set for the VP production apparatus may have been widely distributed among cellular organisms in advance throughout evolutionary history.
7.6 VPs Also Transfer Plasmid and Cytoplasmic Material
VPs from the marine strains exhibited 2,4D utilization and drug resistance transfer. Hence, cellular proteinaceous material and plasmids for drug resistance transfer to the recipient, mediated by VPs, in addition to the host chromosomal fragments examined. To survey the cytoplasmic substance transfer capability of VPs, conjugative plasmid pYFP-Bluescript (yfp::ampr) was constructed from BamHI-NotI double-digested fragments pEYFP-Mito (YFP) and Blue-script II kS (ampr) to generate fusion plasmid pYFP-Bluescript (yfp::ampr), applied to E. coli JM109 (recA1, endA1, gyrA96, thi, hsdR17 (rk−mk+), e14− (mcrA−), supE44, relA1, Δ(lac-proAB)/F’ [traD36, proAB+, lac lq, lacZΔM15]) and selected white colonies. The obtained transformants, E. coli JM109 (yfp::ampr), exhibited ampicillin resistance and yellow fluorescence under B excitation. Then, STEVP was infected with E. coli JM109 (yfp::ampr) at a MOI of 3.0 to generate the lactose utilization transductants (JMlactrans) with a transduction frequency of 1.0 × 10−5 CFU/particle [137].
JMlactrans acquired budding particle (JMlacVP) production together with empty vesicles starting immediately after the culture; like DHlactrans, this was ascribed to the relA1 mutation of the recipient. The abundance of of JMlacVPs in the 96-h culture was ~1 × 1011 particle/mL, with 1/10 abundance of the vesicles. Collected and purified JMlacVPs exhibited yellow fluorescence under B excitation. Consequently, VPs originating from the thermophile were demonstrated to encapsulate cellular proteinaceous material (YFP) in the particle. Applying JMlacVPs to an E. coli DH5α recipient at a MOI of 9.9 resulted in the generation of ampicillin-resistant E. coli DH5α with a transduction frequency of 1.9 × 10−6 CFU/particle.
Some novel properties of VP-mediated broad-host-range gene transfer originating from Aquifex sp. after several serial transductions showed transferability of cellular proteinaceous material and plasmids for drug resistance. Thus, the JMlacVP characterized above might be a novel pathway for gene and material flux in the environment, contributing to the dispersal of drug resistance among environmental microbes [4, 5, 9].
7.7 VP Production-Responsible Gene(s) Are Recombined with the Host Chromosome
The author defined VPs as being capable of infecting, transferring, and budding reproduction in a recipient cell and exceeding the phylogenetic family level from the original host, as previously described. Plentiful VLPs are found in the oceans that exceed the cell population by tenfold, but their original host is uncertain because a VLP is defined as a particle that is morphologically like a virus under electron microscopy. Other than the bona fide virus, VPs and MVs compose VLP assemblages, whose cargo is considered to be information substances. In other words, the VLP assemblage is an enormous gene pool composed of total organismal genes pursuing HGT in situ. The organismal range—of which individual VLPs can infect, propagate, and maintain—corresponds to the host range. The author has isolated VLPs from the natural environment originating from various hosts to verify as VPs by applying them to various recipients, whose hosts are phylogenetically distant from the family level. Although the first VP phenomenon was demonstrated by adopting VPs originating from a marine proteobacteria (AkVP), the VP is not capable of transferring genes toward exceeding phylum. An extreme example of a VP capable of transferring genes towards the entire Domain is produced by an Aquifex sp. (STVP). Thus, there must be a level of accessibility for the VP to the recipient cell that is still unknown. If VPs have a VP production gene (PPRG), then elucidation of the PPRG redefines the new HGT scheme establishment. Would PPRG be transferred exclusively by VP infection of the recipient? If PPRG is integrated with the chromosome, then conjugative transfer would be possible. If PPRG is like a plasmid, there must be an incompatibility group. If PPRG is like a bona fide virus, then there must be new category group. Thus, the possibility of the conjugative transfer of VP production must be considered.
A particular genetic subsystem for particle production must be inherited in the progeny transductant. Thus, the locus of the VP production-related gene, namely PPRG, was investigated by employing a classical microbial genetics method. First, STEVP was infected at a MOI of 0.29 in an E. coli W2252 (HfrC; metB λ− Sms) recipient and selected by met prototrophic reversion; met+ E. coli transductants were successfully obtained with a transduction frequency of 1.43 ± 0.82 × 10−7 CFU/particle (n = 3), referred to as Hfrmettrans. Then, Hfrmettrans (donor) and E. coli AB1157 (F−; leuB proA hisG argE Smr, recipient) were applied to conjugation at donor/recipient ratios of 1:30, 1:50, and 1:90. Combination HfrC without VP infection and the recipient F− was used as the control [135].
For arg and his markers of Hfrmettrans, conjugative transfer frequencies toward AB1157 were comparable to that of the E. coli W2252 control, whereas leu marker frequency decreased by four orders of magnitude; furthermore, pro marker frequency was undetectable. Moreover, about half the proportion of the generated Hfrmettrans-E. coli AB1157 transconjugants were incapable of particle production. Gene transfer of E. coli W2252 at conjugation begins at 13 min on the linkage map (thrA: 0 min) and proceeds in the counterclockwise direction [216]. Hence, pro and leu markers should be translocated to the recipient before arg and his markers from the donor. Namely, DNA originating from STEVP is likely to integrate near proA (5.6 min) and leuB (1.7 min) loci. Likewise, proA and leuB were recombined between hisG (45.0 min) and the terminal of conjugation (13 min) because the generated transductant showed prototrophic reversion for pro and leu markers. STEVP can transfer a dsDNA fragment of approximately 370 kb [130] and the distance between proA and leuB markers is 180.9 kb [217]. Therefore, STEVP would have the capacity for multiple recombinations at the transduction of E. coli W2252. PPRG would probably be transferred at the beginning of conjugation, being likely to locate near the conjugative terminal point because the coordinated marker transfer with leu, arg, and his markers was observed without the pro marker. VP-mediated generalized transduction has been observed [4]. Generalized recombination involves large regions of homologous DNA sequences; however, site-specific recombination involves considerably smaller segments of DNA, in which the recombination event occurs at a specific sequence—that is, the recognition sequence [218]. Based on this result, proA and leuB were integrated between 13 and 45 min on the linkage map of E. coli. This result indicates that the recombination in the VP-mediated transduction should not be ascribed to homologous recombination.
Furthermore, the existence of the recognition sequence or insertion sequence was postulated. From the above findings, the bacteriophage P22 (attP22: 5.6 min) attachment site is located within the region of 1.7–5.6 min on the E. coli linkage map [217, 219, 220], which would be associated with the PPRG integration region. VP is distinct from the P22 phage regarding shape, DNA molecular mass, host range, and so on. However, generalized transduction occurred by a wrapping choice mechanism [191, 192, 221], which might indicate that a comparable mechanism is involved in VP-mediated transduction. Indicated site-specific recombination may provide a reference to explain the genetic transfer processes in VP-mediated transductions.
Consequently, PPRG is likely to locate between 1.7 and 5.6 min on the transductant chromosome. A sulfur-reducing, hyperthermophilic archaeon, Thermococcus onnurineus NA1T, produces extracellular vesicles referred to as ToEV by budding [22], with a TEM image resembling that described for VP above. The nucleic acid retained in the ToEVs amounted 40 to 20 kb. Interestingly, the host T. onnurineus chromosomal region corresponding to 9.4 kb is not encapsulated in ToEV. The region encodes hydrogenase, formate- and alcohol-dehydrogenase, oxidoreductase, Fe and S binding protein; however, why these enzyme genes are not encapsulated in the particles is unknown. As described previously, the conjugation of E. coli AB1157 with HfrmetEtrans was not able to generate proAB transconjugant. The region of proAB encodes dehydrogenase, protein-kinase, and DNA binding protein. The lack of transconjugant generation for that region might imply that the sequence region in the chromosome somehow prevented transfer from the encapsulation to the particle. A comparative genome analysis for such a region might provide some insight.
7.8 Summary and Concluding Remarks
7.8.1 Discovery
Plasmid-like elements were extracted from marine isolates by 2,4 D utilization and prototrophic reversion of auxotrophic recipient by transformation. Electron microscopy observations of PLE revealed the existence of particles instead of DNA strings being postulated intracellularly as maturing phage-like particles. Thus, the term “plasmid-like elements” was changed to “phage-like particles.”
The spontaneous release of PLPs to the culture broth started when the host cell entered the stationary phase. Collected and purified PLPs accomplished transductant generation of the auxotrophic recipient beyond the family border. The spontaneous release without host cell disruption by PLPs from the transductant was observed, which was inconsistent with the concept of virus-mediated transduction. This curious and unusual manner of gene transfer was denoted as serial transduction, and the mediator for this gene transfer phenomenon is referred to as the broad-host-range VP.
7.8.2 Definition
VP-mediated transductants can produce new progeny particles by budding, which are again infectious to carry on the same transfer gene toward the next recipients. The resulting second-generation VP-mediated transductants again produce particles like the original donor, which again infect and induce successive transductants; this phenomenon is termed “serial transduction.” Under current technical restraints, confirmation of this phenomenon is exclusively provided by the budding particle production from the transductant.
7.8.3 Distribution
VPs and VP producers must be ubiquitous on this planet because they have been found in a variety of environments, including marine, thermal, and terrestrial.
7.8.4 Physico-Chemical Characteristics
Based on studies to date, the morphology of VPs takes a spherical shape with a diameter between 20 nm and 500 nm, resembling a spherical virus. The collection and purification of VPs may be achieved by a method used for environmental viruses because the VPs reside in a virus-like particle fraction (20 nm < VLPs <100 kDa) in the environment. The concentration of VPs in environmental samples and the culture supernatant of producers was found by the tangential flow technique followed by equilibrium CsCl density gradient ultracentrifugation. VPs are considered to have lipid components in their surface structure; the buoyant density of VPs is lighter than that of a bona fide virus.
VPs are composed of cargo DNA combined with proteinaceous material and cytoplasmic substances without RNA, whose surface must have embedded enzyme domains (proteinase/peptidase and glycanase) [222] to achieve membrane fusion the infection of the recipient. The production of a VP appears as an electron-dense body under electron microscopy, starting around the center of the producing cell by a spooling DNA strand covered with proteinaceous material to make a reel structure. Then, the reel structure is translocated to the vicinity of the cell membrane, from where the budding-like extrusion of the VP occurs. Finally, pinching releases the VP to the external environment. The yield of VPs per cell was found to be an average of three particles, so the host strictly controls the production of VPs. The produced VPs exhibit a pleomorphic discrete size distribution that is even used to select a distinct range of VP sizes. A VP’s cargo DNA could range from 5 kb to 2 Mb in size, whereas the volume of a VP does not always correspond to the molecular type of DNA estimated by PFGE. Large VPs may encapsulate multiple segments of somewhat similar sizes.
7.8.5 VPs as a Biological Function
No lethal effects (or a slight lethal effect) were observed for the VP on the recipient, whereas enhanced growth was found in some cases. A trend of lethal effects may be found in a smaller subset of close phylogenic positions between the producer and the recipient. Gene transfer was observed for a single gene locus as well as gene cluster, contributing to adaptations such as temperature and osmolality tolerance.
7.8.6 Resemblances and Differences of VPs with the Budding Virus and the MV
The host range of an HGT mediator is defined as the breadth of organisms that are capable of infecting and expressing their function, with limits on the host cell ascribed to the mediator, host, or environmental characteristics. However, no standard definition of a broad-host-range mediator currently exists because of the viewpoints of different disciplines.
The author empirically defines a broad-host-range vector particle as a VP that is capable of infecting, transferring, and budding reproduction in recipient cell and exceeds the phylogenetic family level from the original host.
VPs are characterized by a massive chromosomal gene transfer ability together with cytosolic substances, including plasmids and proteins. VPs are distinct from budding viruses and MVs. Although pleomorphic viruses have been recently conferred, pleomorphic viruses have a strict host range. None of the pleomorphic or budding virus genes are found in the VP-mediated transductants’ genomes (Chiura, 2018, Unpublished data). The production scheme of MVs is considered to be entirely different based on the DNA cargo size and yields per cell. The proportion of DNA containing MVs is reportedly quite small.
The molecular mechanism for VP production must be partly shared with budding viruses and MVs, whereas the scheme for VP production is considered to be unique. A VP production scheme would be an intermingled function taken from the virus and variety of the cells during the evolution of life to be maintained throughout the cell.
Considering that VP budding release is specific to the stationary phase, a particular purpose for the VP lysogenic host must have existed for the VP production. Focusing on Aquifex sp., the originating STVP encapsulated approximately 400 kb DNA species; the host strain’s genome size of 1.5 Mb corresponds to approximately 3.2 VPs yields per cell. STVP-mediated transduction toward E. coli generated VP lysogens of STEVP. Encapsulated DNA species amounted to the same as STVP, approximately 400 kb, whereas VP yields per cell were consistent with the original host. Three times more VP production would be necessary to cover the whole E. coli genome size of 4.6 Mb. To compensate for the shortage of the amount, STEVP should encapsulate three sets of 400-kb DNA cargo in one particle. As seen above, AkVP originating from A. kielensis is considered to carry plural segments of DNA cargo, which might have induced pleomorphism of the particle.
VP assemblage is considered to be the gene pool of VP lysogens because the DNA cargo in VPs consists of the host genome chromosome. Microorganisms in situ are continually suffering from environmental stresses, with the most severe impairment being chromosomal damage. Among the DNA repair systems, mismatch repair is the most effective system [174] to maintain the sequence fidelity; however, the system could not be achieved without the template strand. The most prominent role of VPs in the environment is likely to be the provider for the template strand to cure cells in need, in addition to carrying genetic information to be shared in the microbial assemblage to achieve environmental adaptation enhancements.
References
Chiura HX, Takagi J. Phage-like particles production and gene transfer by marine bacteria. Bull Jap Soc Microb Ecol. 1994;9:74–90.
Chiura HX. Generalized gene transfer by virus-like particles from marine bacteria. Aquat Microb Ecol. 1997;13:75–83.
Chiura HX. Marine lysogenic virus. Kaiyo. 2003;33:137–45.
Chiura HX. Novel broad-host range gene transfer particles in nature. Microbes Environ. 2004;19:249–64.
Chiura HX. Contribution of serial transduction for horizontal gene transfer in the natural environment. Actinomycetotologica. 2008;20:s13–4.
Chiura HX, Uchiyama N, Kogure K. Broad-host range gene transfer particle produced by Aliivibrio fischeri. Microbes Environ. 2009;24:322–9.
Chiura HX, Kogure K, Hagemann S, Ellinger A, Velimirov B. Evidence for particle-induced horizontal gene transfer and serial transduction between bacteria. FEMS Microbiol Ecol. 2011;76:576–91. https://doi.org/10.1111/j.1574-6941.2011.01077.x.
Chiura HX, Yamamoto H, Koketsu D, Naito H, Kato K. Virus-like particle derived from a bacterium belonging to the oldest lineage of the domain bacteria. Microbes Environ. 2002;17:48–52.
Chiura HX. Broad host range xenotrophic gene transfer by virus-like particles from a hot spring. Microbes Environ. 2002;17:53–8.
Cury J, Touchon M, Rocha EPC. Integrative and conjugative elements and their hosts: composition, distribution and organization. Nucleic Acids Res. 2017;45(15):8943–56. https://doi.org/10.1093/nar/gkx607.
Derbyshire KM, Gray TA. Distributive conjugal transfer: new insights into horizontal gene transfer and genetic exchange in Mycobacteria. Microbiol Spectr. 2014;2(1):61–79. https://doi.org/10.1128/microbiolspec.MGM2-0022-2013.
Waters VL. Conjugation between bacterial and mammalian cells. Nat Genet. 2001;29:375–6. https://doi.org/10.1038/ng779.
Nonaka L, Isshiki T, Suzuki S. Distribution of the oxytetracycline resistance determinant Tet 34 among bacterial isolated from diseased fish. Microbes Environ. 2002;17:26–31.
Nonaka L, Suzuki S. New Mg2+ dependent oxytetracycline resistance determinant Tet 34 in Vibrio isolates from marine fish intestinal contents. Antimicrob Agents Chemother. 2002;46:1550–2.
Nonaka L, Ikeno K, Suzuki S. Distribution of tetracycline resistance gene, tet(M), in gram-positive and gram-negative bacteria isolated from sediment and seawater at a coastal aquaculture site in Japan. Microbes Environ. 2007;22:355–64.
Lang AS, Beatty J. Importance of widespread gene transfer agent genes in α-proteobacteria. Trends Microbiol. 2007;15(2):54–62. https://doi.org/10.1016/j.tim.2006.12.001.
McDaniel L, Young E, Delaney J, Ruhnau F, Ritchie K, Paul J. High frequency of horizontal gene transfer in the oceans. Science. 2010;330(6000):50. https://doi.org/10.1126/science.1192243.
Stanton T. Prophage-like gene transfer agents—novel mechanisms of gene exchange for Methanococcus, Desulfovibrio, Brachyspira, and Rhodobacter species. Anaerobe. 2007;13(2):43–9. https://doi.org/10.1016/j.anaerobe.2007.03.004.
Dubey G, Ben-Yehuda S. Intercellular nanotubes mediate bacterial communication. Cell. 2011;144(4):590–600. https://doi.org/10.1016/j.cell.2011.01.015.
Yang F, Moss LG, Phillips GN Jr. The molecular structure of green fluorescent protein. Nat Biotechnol. 1996;14:1246–51.
Chatterjee SN, Chaudhuri K. Outer membrane vesicles of bacteria. Heidelberg: Springer; 2012.
Choi DH, Kwon YM, Chiura HX, Yang EC, Bae SS, Kang SG, Lee J-H, Yoon HS, Kim S-J. Extracellular vesicles of the hyperthermophilic archaeon Thermococcus onnurineus NA1T. Appl Environ Microbiol. 2015;81:4591–9. https://doi.org/10.1128/AEM.00428-15.
Kulp A, Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol. 2010;64:163–84. https://doi.org/10.1146/annurev.micro.091208.073413.
Lee EY, Choi DY, Kim DK, Kim JW, Park JO, Kim S, Kim SH, Desiderio DM, Kim YK, Kim KP, Gho YS. Gram-positive bacteria produce membrane vesicles: proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics. 2009;9:5425–36.
McBroom AJ, Kuehn MJ. Chapter 2.2.4, Outer membrane vesicles. In: Finlay BB, editor. EcoSal - Escherichia coli and Salmonella: cellular and molecular biology. Washington: ASM Press; 2005.
Rivera J, Cordero RJB, Nakouzi AS, Frases S, Nicola A, Casadevall A. Bacillus anthracis produces membrane-derived vesicles containing biologically active toxins. Proc Natl Acad Sci U S A. 2010;107:19002–7.
Soler N, Marguet E, Verbavatz J-M, Forterre P. Virus-like vesicles and extracellular DNA produced by hyperthermophilic archaea of the order Thermococcales. Res Microbiol. 2008;159:390–9. https://doi.org/10.1016/j.resmic.2008.04.015.
Toyofuku M, Cárcamo-Oyarce G, Yamamoto T, Eisenstein F, Hsiao M, Kurosawa C-C, Gademann K, Pilhofer M, Nomura N, Eberl L. Prophage-triggered membrane vesicle formation through peptidoglycan damage in Bacillus subtilis. Nat Commun. 2017;8:481. https://doi.org/10.1038/s41467-017-00492-w.
Toyofuku M, Morinaga K, Hashimoto Y, Uhl J, Shimamura H, Inaba H, Schmitt-Kopplin P, Eberl L, Nomura N. Membrane vesicle-mediated bacterial communication. ISME J. 2017;11:1504–9.
Yaron S, Kolling G, Simon L, Matthews K. Vesicle-mediated transfer of virulence genes from Escherichia coli O157:H7 to other enteric bacteria. Appl Environ Microbiol. 2000;66:4414–20. https://doi.org/10.1128/AEM.66.10.4414-4420.2000.
Beveridge T. Structures of gram-negative cell walls and their derived membrane vesicles. J Bacteriol. 1999;181:4725–33.
Brown L, Kessler A, Cabezas-Sanchez P, Luque- Garcia JL, Casadevall A. Extracellular vesicles produced by the gram-positive bacterium Bacillus subtilis are disrupted by the lipopeptide surfactin. Mol Microbiol. 2014;93:183–98.
Brown L, Wolf JM, Prados-Rosales R, Casadevall A. Through the wall: extracellular vesicles in gram-positive bacteria, mycobacteria and fungi. Nat Rev Microbiol. 2015;13(10):620–30. https://doi.org/10.1038/nrmicro3480.
Haurat MF, Elhenawy W, Feldman Mario F. Prokaryotic membrane vesicles: new insights on biogenesis and biological roles. Biol Chem. 2015;396:95. https://doi.org/10.1515/hsz-2014-0183.
Kadurugamuwa JL, Beveridge TJ. Membrane vesicles derived from Pseudomonas aeruginosa and Shigella flexneri can be integrated into the surfaces of other gram-negative bacteria. Microbiology. 1999;145:2051–60.
Kolling GL, Matthews KR. Export of virulence genes and Shiga toxin by membrane vesicles of Escherichia coli O157:H7. Appl Environ Microbiol. 1999;65:1843–8.
Li Z, Clarke AJ, Beveridge TJ. Gram-negative bacteria produce membrane vesicles which are capable of killing other bacteria. J Bacteriol. 1998;180:5478–83.
Renelli M, Matias V, Lo RY, Beveridge TJ. DNA-containing membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation potential. Microbiology. 2004;150:2161–9. https://doi.org/10.1099/mic.0.26841-0.
Tashiro Y, Ichikawa S, Nakajima-Kambe T, Uchiyama H, Nomura N. Pseudomonas quinolone signal affects membrane vesicle production in not only gram-negative but also gram-positive bacteria. Microbes Environ. 2010;25:120–5. https://doi.org/10.1264/jsme2.ME09182.
Tashiro Y, Uchiyama H, Nomura N. Multifunctional membrane vesicles in Pseudomonas aeruginosa. Environ Microbiol. 2012;14:1349–62. https://doi.org/10.1111/j.1462-2920.2011.02632.x.
Jiang SC, Kellogg CA, Paul JH. Characterization of marine temperate phage-host systems isolated from Mamala Bay, Oahu, Hawaii. Appl Environ Microbiol. 1998;64:535–42.
Jiang SC, Paul JH. Gene transfer by transduction in the marine environment. Appl Environ Microbiol. 1998;64:2780–7.
Lang AS, Beatty JT. The gene transfer agent of Rhodobacter capsulatus and “constitutive transduction” in prokaryotes. Arch Microbiol. 2001;175(4):241–9.
Morrison WD, Miller RV, Sayler GS. Frequency of F116-mediated transduction of Pseudomonas aeruginosa in a freshwater environment. Appl Environ Microbiol. 1978;36:724–30.
Ripp S, Ogunseitan OA, Miller RV. Transduction of a freshwater microbial community by a new Pseudomonas aeruginosa generalized transducing phage, UT1. Mol Ecol. 1994;3:121–6.
Sandri RM, Berger H. Bacteriophage P1-mediated generalized transduction in Escherichia coli: structure of abortively transduced DNA. Virology. 1980;106:30–40.
Saye DJ, Ogunseitan OA, Sayler GS, Miller RV. Transduction of linked chromosomal genes between Pseudomonas aeruginosa during incubation in situ in a freshwater habitat. Appl Environ Microbiol. 1990;56:140–5.
Watson BNJ, Staals RHJ, Fineran PC. CRISPR-Cas-mediated phage resistance enhances horizontal gene transfer by transduction. MBio. 2018;9:e02406-17. https://doi.org/10.1128/mBio.02406-17.
Arber W. Horizontal gene transfer among bacteria and its role in biological evolution. Life. 2014;4:217–24. https://doi.org/10.3390/life4020217.
Bertani G. Transduction-like gene transfer in the methanogen Methanococcus voltae. J Bacteriol. 1999;181:2992–3002.
Eiserling F, Pushkin A, Gingery M, Bertani G. Bacteriophage-like particle associated with the gene transfer agent of Methanococcus voltae PS. J Gen Virol. 1999;80:3305–8.
Humphrey SB, Stanton TB, Jensen NS, Zuerner RL. Purification and characterization of VSH-1, a generalized transducing bacteriophage of Serpulina hyodysenteriae. J Bacteriol. 1997;179:323.
Solioz M, Marrs B. The gene transfer agent of Rhodopseudomonas capsulata. Arch Biochem Biophys. 1977;181:300–7.
Wood AG, Whiteman WB, Konisky J. Isolation and characterization of an archaebacterial virus-like particle from Methanococcus voltae A3. J Bacteriol. 1989;171:93–8.
Lang AS, Beatty JT. Genetic analysis of a bacterial genetic exchange element: the gene transfer agent of Rhodobacter capsulatus. Proc Natl Acad Sci U S A. 2000;97:859–64.
Westbye AB, Leung MM, Florizone S, Taylor TA, Johnson JA, Fogg PC, Beatty JT. Phosphate concentration and the putative sensor-kinase protein CckA modulate cell lysis and release of the Rhodobacter capsulatus gene transfer agent (RcGTA). J Bacteriol. 2013;195(22):5025–40.
Deatherage BL, Cookson BT. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun. 2012;80:1948–57. https://doi.org/10.1128/IAI.06014-11.
Forterre P, Soler N, Krupovic M, Marguet E, Ackermann HW. Fake virus particles generated by fluorescence microscopy. Trends Microbiol. 2013;21:1–5. https://doi.org/10.1016/j.tim.2012.10.005.
Biller SJ, Schubotz F, Roggensack SE, Thompson AW, Summons RE, Chisholm SW. Bacterial vesicles in marine ecosystems. Science. 2014;343:183–6.
Soler N, Krupovic M, Marguet E, Forterre P. Membrane vesicles in natural environments: a major challenge in viral ecology. ISME J. 2015;9:793–6. https://doi.org/10.1038/ismej.2014.184.
Biller SJ, McDaniel LD, Breitbart M, Rogers E, Paul JH, Chisholm SW. Membrane vesicles in seawater: heterogeneous DNA content and implications for viral abundance estimates. ISME J. 2017;11:394–404. https://doi.org/10.1038/ismej.2016.134.
Toyofuku M, Roschitzki B, Riedel K, Eberl L. Identification of proteins associated with the Pseudomonas aeruginosa biofilm extracellular matrix. J Proteome Res. 2012;11:4906–15.
Fulsundar S, Harms K, Flaten GE, Johnsen PJ, Chopade BA, Nielsen KM. Gene transfer potential of outer membrane vesicles of Acinetobacter baylyi and effects of stress on vesiculation. Appl Environ Microbiol. 2014;80:3469–83. https://doi.org/10.1128/AEM.04248-13.
Mashburn LM, Whiteley M. Membrane vesicles traffic signals and facilitate group activities in a prokaryote. Nature. 2005;437:422–5. https://doi.org/10.1038/nature03925.
Davis DM, Sowinski S. Membrane nanotubes: dynamic long- distance connections between animal cells. Nat Rev Mol Cell Biol. 2008;9:431–43.
Hayashi T, Makino K, Ohnishi M, Kurokawa K, Ishii K, Yokoyama K, et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001;8:11–22.
Shaefer R, Hinnen R, Franklin RM. Bacteriophage of Halobacterium salinarium. Nature. 1974;243:681–3.
Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Appl Environ Microbiol. 1994;44:846–9.
Zambrynski P, Tempe J, Schell J. Transfer and function of T-DNA genes from Agrobacterium Ti and Ri plasmids in plants. Cell. 1989;56:193–201.
Schell J, Van Montagu M. The Ti-plasmid of Agrobacterium tumefaciens, a natural vector for the introduction of nif genes in plants? Basic Life Sci. 1977;9:159–79.
Stachel SE, Timmerman B, Zambryski P. Generation of single-stranded T-DNA molecules during the initial stages of T-DNA transfer from Agrobacterium tumefaciens to plant cells. Nature. 1986;322:706–12.
Straube WL, Deming WJ, Somerville CC, Colwell RR, Baross JA. Particulate DNA in smoker fluids: evidence for existence of microbial populations in hot hydrothermal system. Appl Environ Microbiol. 1990;56:1440–7.
Hennes KP, Simon M. Significance of bacteriophages for controlling bacterioplankton growth in a mesotrophic lake. Appl Environ Microbiol. 1995;61:333–40.
Hennes KP, Suttle CA. Direct counts of viruses in natural waters and laboratory cultures by epifluorescence microscopy. Limnol Oceanogr. 1995;40:1050–5.
Levy SB, Miller RV. Gene transfer in the environment. New York: McGraw-Hill; 1989.
Steward GF, Smith DC, Azam F. Abundance and production of bacteria and viruses in the Bering and Chukchi seas. Mar Ecol Prog Ser. 1996;131:287–300.
Mathias CB, Kirschner AKT, Velimirov B. Seasonal variations of virus abundance and viral control of the bacterial production in a backwater system of Danube river. Appl Environ Microbiol. 1995;61:3734–40.
Levin BR, Lenski RE. Coevolution in bacteria and their viruses and plasmids. In: Futuima DJ, Slatkin M, editors. Coevolution. Sunderland: Sinauer; 1983. p. 99–127.
Ohnishi M, Kurokawa K, Hayashi T. Diversification of Escherichia coli genomes: are bacteriophages the main contributors? Trends Microbiol. 2001;9:481–5.
Yeats S, McWilliam P, Zillig W. A plasmid in the archaebacterium Sulfolobus acidocaldarius. EMBO J. 1982;9:1035–8.
Young JPW, Wexler M. Sym plasmid and chromosomal genotypes are correlated in field population of Rhizobium leguminosarum. J Gen Microbiol. 1988;134:2731–9.
Paul JH. Microbial gene transfer. J Mol Microbiol Biotechnol. 1999;1:45–50.
Grissa I, Vergnaud G, Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. https://doi.org/10.1186/1471-2105-8-172.
Barrangou R. The roles of CRISPR-Cas systems in adaptive immunity and beyond. Curr Opin Immunol. 2015;32:36–41. https://doi.org/10.1016/j.coi.2014.12.008.
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–12. https://doi.org/10.1126/science.1138140.
Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–70. https://doi.org/10.1126/Science.1179555.
Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322(5909):1843–5. https://doi.org/10.1126/science.1165771.
Quemin ERJ, Chlanda P, Sachse M, Forterre P, Prangishvili D, Krupovic M. Eukaryotic-like virus budding in Archaea. mBio. 2016;7(5):e01439–16. https://doi.org/10.1128/mBio.01439-16.
Jouvenet N, Zhadina M, Bieniasz PD, Simon SM. Dynamics of ESCRT protein recruitment during retroviral assembly. Nat Cell Biol. 2011;13(4):394–401. https://doi.org/10.1038/ncb2207.
Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8:15–25. https://doi.org/10.1038/nrmicro2259.
Suttle CA. Marine viruses-major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–12. https://doi.org/10.1038/nrmicro1750.
Ackerman H-W, DuBow MS. Viruses of prokaryotes. In: General properties of bacteriophages, vol. 1. Boca Raton: CRC Press; 1987. p. 49–85.
Ambile-Cuevas CF, Chicurel ME. Bacterial plasmid and gene flux. Cell. 1992;70:189–99.
Birge EA. Bacterial and bacteriophage genetics. 3rd ed. New York: Springer; 1994.
Streips UN, Yasbin RE. Modern microbial genetics. New York: Wiley-Liss; 1991.
Fry JC, Day MJ. Overview of gene transfer in aquatic habitats. In: Guerrero R, Pedros-Alio C, editors. Trends in microbial ecology. Barcelona: Spanish Society for Microbiology; 1993. p. 315–8.
De Corte D, Sintes E, Winter C, Yokokawa T, Reinthaler T, Herndl GJ. Links between viral and prokaryotic communities throughout the water column in the (sub)tropical Atlantic Ocean. ISME J. 2010;4(11):1431–42.
Fuhrman JA, Ouverney CC. Marine microbial diversity studied via 16S rRNA sequences: cloning results from coastal waters and counting of native archaea with fluorescent single cell probes. Aquat Ecol. 1998;32:3–15.
Fuhrman JA. Marine viruses and their biogeochemical and ecological effects. Nature. 1999;399:541–8.
Miller RV, Ripp S, Relicon J, Ogunseitan OA, Kokjohn TA. Virus-mediated gene transfer in freshwater environment. In: Gauthier MJ, editor. Gene transfers and environment. Berlin: Springer; 1992. p. 51–62.
Wommack KE, Colwell RR. Virioplankton: viruses in aquatic ecosystems. Microbiol Mol Biol Rev. 2000;64:69–114.
Koonin EV. Horizontal gene transfer: essentiality and evolvability in prokaryotes, and roles in evolutionary transitions. F1000Res. 2016;5. https://doi.org/10.12688/f1000research.8737.1.
Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci U S A. 1998;95:9413–7.
Martin W. Mosaic bacterial chromosomes: a challenge en route to a tree of genomes. Bioassays. 1999;21:99–104.
McInerney J, Cummins C, Haggerty L. Goods-thinking vs. tree-thinking. Mob Genet Elem. 2011;1(4):304–43. https://doi.org/10.4161/mge.19153.
Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–9.
Eisen JA. Horizontal gene transfer among microbial genomes: new insights from complete genome analysis. Curr Opin Genet Dev. 2000;10(6):606–11.
Chiura H, Noro Y, Kanayama S, Ueda Y, Simidu U, Takagi J. Site-specific deoxyribonuclease produced by a marine bacterium, Flavobacterium I 16-04. Agric Biol Chem. 1988;52:2107–9.
Chiura HX, Kamiyama T, Hirano H, Futagami M, Watahiki M, Kobayashi K, Simidu U, Takagi J. Purification and characterization of AspMD1, an isoschizomer of Sau3AI, from a marine bacterium, Alcaligenes sp. MD1. Nucleic Acids Res. 1992;20:1996.
Chiura HX, Kato K, Takagi J. PLPs released by a marine bacterium. In: 4th international symposium on contamination of the environment by viruses and method of control, Die Universität für Bodenkultur Wien, Wien, Austria, August 1993.
Östling J, Goodman A, Kjelleberg S. Behaviour of IncP-1 plasmids and a miniMu transposon in a marine Vibrio sp.: isolation of starvation inducible lac operon fusions. FEMS Microbiol Ecol. 1991;86:83–94.
Chaudhry GR, Chapalamadugu S. Biodegradation of halogenated organic compounds. Microbiol Rev. 1991;55:59–79.
Reineke W, Knackmus H-J. Microbial degradation of haroaromatics. Annu Rev Microbiol. 1988;42:263–87.
van der Meer JR, de Vos WM, Harayama S, Zehnder AJB. Molecular mechanisms of genetic adaptation to xenobiotic compounds. Microbiol Rev. 1992;56:677–94.
Amy PS, Schulke W, Fraizer LM, Seidler RJ. Characterization of aquatic bacteria and cloning of genes specifying partial degradation of 2,4-dichlorophenoxyacetic acid. Appl Environ Microbiol. 1985;49:1237–45.
Chaudhry GR, Huang GH. Isolation and characterization of a new plasmid from a Flavobacterium sp. which carries the genes for degradation of 2,4-dichlorophenoxyacetic acid. J Bacteriol. 1988;170:3897–902.
Don RH, Pemberton JM. Properties of six pesticide degradation plasmids isolated from Alcaligenes paradoxus and Alcaligenes eutrophus. J Bacteriol. 1981;45:681–6.
Don RH, Weightman AJ, Timmis KN. Transposon mutagenesis and cloning analysis of the pathways for degradation of 2,4-dichlorophenoxyacetic acid and 3-chlorobenzonate in Alcaligenes eutrophus JMP134(pJP4). J Bacteriol. 1985;161:85–90.
Friello OA, Chakrabarty AM. Transposable mercury resistance. In: Stuttard C, Rozee KR, editors. Plasmids and transposons. New York: Academic Press; 1980. p. 249–60.
Mazodier P, Davies J. Gene transfer between distantly related bacteria. Annu Rev Genet. 1991;25:147–71.
Silver S, Misra TK. Plasmid-mediated heavy metal resistances. Annu Rev Microbiol. 1988;42:717–43.
Brown MJ, Lund PA, Nibhriain N. Mercury resistance in bacteria. In: Hapwwod DA, Chater KE, editors. Genetics of bacterial diversity. New York: Academic; 1998. p. 121–39.
DiGiovanni DG, Neilson JW, Pepper IL, Sinclair NA. Gene transfer of Alcaligenes eutrophus JMP134 plasmid pJP4 to indigenous soil recipients. Appl Environ Microbiol. 1996;62:2521–6.
Yabuuchi E, Kosako Y, Yano I, Hotta H, Nishiuchi Y. Transfer of two Burkholderia and an Alcaligenes species to Ralstonia gen. Nov.: proposal of Ralstonia pickettii (Ralston, Palleroni and Doudoroff 1973) comb. Nov., Ralstonia solanacearum (Smith 1896) comb. Nov. and Ralstonia eutropha (Davis 1969) comb. Nov. Microbiol Immunol. 1995;39(11):897–904. https://doi.org/10.1111/j.1348-0421.1995.tb03275.x.
Ahrens R. Taxonomische Untersuchungen an sternbildenden Agrobacterium - Arten aus der westlichen. Ostsee Kiel Meeresforsch. 1968;24:147–73.
Uchino Y, Hirata A, Yokota A, Sugiyama J. Reclassification of marine Agrobacterium species: proposals of Stappia stellulata gen. nov., comb. nov., Stappia aggregata sp. nov., nom. rev., Ruegeria atlantica gen. nov., comb. nov., Ruegeria gelatinovora comb. nov., Ruegeria algicola comb. nov., and Ahrensia kieliense gen. nov., sp. nov., nom. rev. J Gen Appl Microbiol. 1998;44(3):201–10.
Chiura HX, Rao Bhamidimarri SM, Yu PL. Plasmid for the degradation of 2,4-dichlorophenoxyacetic acid (2,4-D). In: Yu PL, editor. Fermentation technology: industrial application. London: Elsevier; 1990. p. 59–62.
Weinbauer MG, Höfle MG. Significance of viral lysis and flagellate grazing as factors controlling bacterioplankton production in a eutrophic lake. Appl Environ Microbiol. 1998;64:431–8.
Izumi A, et al. Broad-host-range vector particle-mediated horizontal drug resistance gene transfer. In: ISME 2006, Vienna, Austria, 20–25 August 2006.
Chiura HX, Umitsu M. Isolation and characterisation of broad-host range gene transporter particles from geo-thermal vent of the Toyoha mine. Microbes Environ. 2004;19:20–30.
Chiura HX, Umitsu M, Fukazawa Y, Nakata D, Nakamura K, Tomaru A, Okita N, Kawarabayashi Y, Hoaki T. Broad-host range vector-particle from thermal vents. In: The 6th international marine biotechnology conference (IMBC2003), Makuhari, Chiba, 21–27 September 2003.
Chiura HX, Imada M, Maruyama F, Yasuoka N. Transkingdom gene transfer using virus-like particles originating from thermophile. In: The 6th international marine biotechnology conference (IMBC2003), Makuhari, Chiba, 21–27 September 2003.
Chiura HX, Sakai A, Takasaki Y, Hoaki T. Are broad-host-range gene transfer particles ubiquitous existences in the environment? In: The annual meeting of the Japanese Society of Microbial Ecology, Tsu, Mie, 15–17 November 2002.
Sugitate T, Chiura HX. Functional gene transfer towards a broad range of recipients with the aid of vector particle originating from a thermophile. In: The proceedings of international symposium on extremophiles and their applications, vol. 2005. Yokosuka: Extremobiosphere Research Center, JAMSTEC; 2007. p. 141–7. http://www.jstage.jst.go.jp/article/isea/2005/0/141/.
Inoue K, Sugitate T, Chiura HX. Study of particle production responsible gene locus by bacterial conjugation. ISME 2006, Vienna, Austria, 20–25 August 2006.
Inokuchi Y, Chiura HX. Search for a broad-host-range gene transfer particle (VP) production responsible gene of VP-transductant: Escherichia coli DH-lac-trans. In: MBSJ 2006 Forum, Nagoya, Aichi, Nagoya Congress Centre 6–8 December 2006.
Sugitate T, Ito K, Chiura HX. Novel Characteristics of Broad-host range gene transfer particles from thermophiles. In: 8th international conference on thermophiles research, Gold Coast, Australia, 18–22 September 2005.
Chiura HX, Yoshizawa S, Kogure K. Vector particles originating from Polaribacter changed the minimum and optimum growth temperature of Escherichia coli. In: ISME 2012, Copenhagen, Denmark, 19–24 August 2012.
Chiura HX, Kogure K. Nitrogen fixation acquisition of Escherichia coli by vector particles originating from Klebsiella pneumonia subsp. rhinoscleromatisi. In: XV ISME, Seoul, Korea, 24–29 August 2014.
Gauthier MJ, Breittmayer VA. Gene transfer in marine environments. In: Fry JC, Day MJ, editors. Bacterial genetics in natural environments. London: Chapman and Hall; 1990. p. 100–10.
Stewart GJ, Carlson CA. The biology of natural transformation. Annu Rev Microbiol. 1986;40:211–35.
Stewart GJ, Carson CA, Ingraham JL. Evidence for an active role of donor cells in natural transformation of Pseudomonas stutzeri. J Bacteriol. 1983;156:30–5.
Hirsch PR. Factors limiting gene transfer in bacteria. In: Fry JC, Day MJ, editors. Bacterial genetics in natural environments. London: Chapman and Hall; 1990. p. 31–40.
Bathe S, Lebuhn M, Ellwart JW, Wuertz S, Hausner M. High phylogenetic diversity of transconjugants carrying plasmid pJP4 in an activated sludge-derived microbial community. FEMS Microbiol Lett. 2004;235:215–21. https://doi.org/10.1016/j.femsle.2004.04.038.
Sen D, Van der Auwera GA, Rogers LM, Thomas CM, Brown CJ, Top EM. Broad-host-range plasmids from agricultural soils have IncP-1 backbones with diverse accessory genes. Appl Environ Microbiol. 2011;77:7975–83. https://doi.org/10.1128/AEM.05439-11.
Szpirer C, Top EM, Couturier M, Mergeay M. Retrotransfer or gene capture: a feature of conjugative plasmids, with ecological and evolutionary significance. Microbiology. 1999;145:3321–9.
Thomas CM. Paradigms of plasmid organization. Mol Microbiol. 2000;37:485–91. https://doi.org/10.1046/j.1365-2958.2000.02006.x.
Dahlberg C, Bergström M, Andreasen M, Christensen BB, Molin S, Hermansson M. Interspecies bacterial conjugation by plasmids from marine environments visualized by gfp expression. Mol Biol Evol. 1998;15:385–90.
Dahlberg C, Bergström M, Hermansson M. In situ detection of high levels of horizontal plasmid transfer in marine bacterial communities. Appl Environ Microbiol. 1998;64:2670–5.
Lejeune P, Mergeay M, Van Gijsegem F, Faelen M, Gerits J, Toussaint A. Chromosome transfer and R-prime plasmid formation mediated by plasmid pULB113 (RP4::Mini-Mu) in Alcaligenes eutrophus CH34 and Pseudomonas fluorescens 6.2. J Bacteriol. 1983;155:1015–26.
Black LW. DNA packaging in dsDNA bacteriophages. In: Calendar R, editor. The bacteriophages, vol. 2. New York: Plenum Publishing Corp; 1988. p. 321–73.
Charvin M, Rastogi N, Lévy-Frébault VV. An easy and rapid method for isolation of entire mycobacterial genome for application in pulsed-field gel electrophoresis. Current Microbiol. 1991;22(5):327–31. https://doi.org/10.1007/BF02091963.
McEllistrem MC, Stout JE, Harrison LH. Simplified protocol for pulsed-field gel electrophoresis analysis of Streptococcus pneumoniae. J Clin Microbiol. 2000;38(1):351–3.
Yen H-C, Marrs B. Map of genes for carotenoid and bacteriochlorophyll biosynthesis in Rhodopseudomonas capsulata. J Bacteriol. 1976;126:619–29.
Hagemann S, Stoeger L, Kappelmann M, Hassl I, Ellinger A, Velimirov B. DNA-bearing membrane vesicles produced by Ahrensia kielensis and Pseudoalteromonas marina. J Basic Microbiol. 2013;54(10):1062–72.
Lang AS, Zhaxybayeva O, Beatty JT. Gene transfer agents: phage-like elements of genetic exchange. Nat Rev Microbiol. 2012;10(7):472–82.
Chiura HX, Kato K, Takagi J. Phage-like particles released by a marine bacterium. Wien Mitteil. 1995;128:149–57.
Chiura HX, Velimirov B, Kogure K. Virus-like particles in microbial population control and horizontal gene transfer in aquatic environments. In: Bell CR, Brylinsky M, Johnson-Green P, editors. Microbial biosystems: new frontiers. Halifax: Atlantic Canada Society for Microbial Ecology; 2000. p. 167–73.
Atanasova NS, Senčilo A, Pietilä MK, Roine E, Oksanen HM, Bamford DH. Chapter one. Comparison of lipid-containing bacterial and archaeal viruses. In: Maramorosch K, Mettenleiter T, editors. Advances in virus research, vol. 92. 1st ed. Amsterdam: Elsevier; 2015. p. 1–61. Table 1: 4–16. https://doi.org/10.1016/bs.aivir.2014.11.005.
Dybvig K, Nowak JA, Sladeck TL, Maniloff J. Identification of an enveloped phage, mycoplasma virus L172, that contains a 14-kilobase single-stranded DNA genome. J Virol. 1985;53:384–90.
Pietilä MK, Roine E, Paulin L, Kalkkinen N, Bamford DH. An ssDNA virus infecting archaea: a new lineage of viruses with a membrane envelope. Mol Microbiol. 2009;72:307–19. https://doi.org/10.1111/j.1365-2958.2009.06642.x.
Pietilä MK, Roine E, Sencilo A, Bamford DH, Oksanen HM. Pleolipoviridae, a newly proposed family comprising archaeal pleomorphic viruses with single-stranded or double-stranded DNA genomes. Arch Virol. 2016;161(1):249–56.
Putzrath RM, Maniloff J. Growth of an enveloped mycoplasma virus and establish-ment of a carrier state. J Virol. 1977;22:308–14.
Davis WR, Bostein D, Roth JR. A manual for advanced bacterial genetics. Cold Spring Harbor: Cold Spring Harbor Laboratory; 1980. p. 148–52.
Chen F, Lu JR, Binder B, Hodson RE. Enumeration of viruses in aquatic environments using SYBR gold stain: application of digital image analysis and flow cytometer. Appl Environ Microbiol. 2001;67:539–45.
Børsheim KY, Bratbak G, Heldal M. Enumeration and biomass estimation of planktonic bacteria and viruses by transmission electron microscopy. Appl Environ Microbiol. 1990;56:352–6.
Kenzaka T, Tani K, Nasu M. High-frequency phage-mediated gene transfer in freshwater environments determined at single-cell level. ISME J. 2010;4:648–59.
Weinbauer MG, Peduzzi P. Frequency, size and distribution of bacteriophages in different marine bacterial morphotypes. Mar Ecol Prog Ser. 1994;108:11–20.
Proctor LM, Fuhrman JA. Viral mortality of marine bacteria and cyanobacteria. Nature (London). 1990;343:60–2.
Proctor LM, Fuhrman JA. Roles of viral infection in organic particle flux. Mar Ecol Prog Ser. 1991;69:133–42.
Weinbauer MG, Fuks D, Peuzzi P. Distribution of viruses and dissolved DNA along a coastal trophic gradient in the northern Adriatic Sea. Appl Environ Microbiol. 1993;59:4074–82.
Proctor LM, Okubo A, Fuhrman JA. Calibrating estimates of phage-induced mortality in marine bacteria: ultrastructural studies of marine bacteriophage development from one-step growth experiments. Microb Ecol. 1993;25:161–82.
Nakagawa T, Hanada S, Maruyama A, Marumo K, Urabe T, Fukui M. Distribution and diversity of thermophilic sulfate-reducing bacteria within a Cu-Pb-Zn mine (Toyoha, Japan). FEMS Microbiol Ecol. 2002;41(3):199–209. https://doi.org/10.1111/j.1574-6941.2002.tb00981.x.
Iyer R, Pluciennik A, Burdett V, Modrich P. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106(2):302–23. https://doi.org/10.1021/cr0404794.
Yamamoto H, Hiraishi A, Kato K, Chiura HX, Maki Y, Shimizu A. Phylogenetic evidence for the existence of novel thermophilic bacteria in hot spring sulphur-turf microbial mats in Japan. Appl Environ Microbiol. 1998;64:1680–7.
Lorenz MG, Wackernagel W. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol Rev. 1994;58:293–316.
Grogan DW. Selectable mutant phenotypes of the extremely thermophilic archaebacterium Sulfolobus acidocaldarius. J Bacteriol. 1991;173:7725–7.
Grogan DW. Exchange of genetic markers at extremely high temperatures in the archaeon Sulfolobus acidocaldarius. J Bacteriol. 1996;178:3207–11.
Sugitate T, Inaba N, Kurusu Y, Hoaki T, Chiura HX. Thermo-resistance acquisition of a mesophilic bacterium with the aid of vector particles originating from thermophiles. In: American Geophysical Union Autumn Meeting, San Francisco, USA, 13–17 December 2004. https://adsubs.harvard.edu/?#abs/2004AGUFM.V41B1371S/abstract.
Shioda M, Sugimori K, Shiroya T, Takayanagi S. Nucleosomelike structures associated with chromosomes of the archaebacterium Halobacterium salinarium. J Bacteriol. 1989;171:4514–7.
Takayanagi S, Morimura S, Kusaoke H, Yokoyama Y, Kano K, Shioda M. Chromosomal structure of the halophilic archaebacterium Halobacterium salinarium. J Bacteriol. 1992;174:7207–16.
Moak M, Molineux IJ. Peptidoglycan hydrolytic activities associated with bacteriophage virions. Mol Microbiol. 2004;51:1169–83.
Dreiseikelmann B. Translocation of DNA across bacterial membranes. Microbiol Rev. 1994;58(3):293–316. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC372970/pdf/microrev00022-0011.pdf
Kivelä HM, Daugelavicius R, Hankkio RH, Bamford JK, Bamford DH. Penetration of membrane-containing double-stranded-DNA bacteriophage PM2 into Pseudoalteromonas hosts. J Bacteriol. 2004;186:5342–54.
Adams E. Biochem Z. 1942;310:384.
Kates M. Techniques of lipidology. Isolation, analysis and identification of lipids. Amsterdam: Elsevier; 1975. p. 83. Translated under the title Tekhnika lipidologii. Moscow: Mir; 1972.
Troll W, Cannan RK. A modified photometric ninhydrin method for the analysis of amino and imino acids. J Biol Chem. 1953;200:803–11.
Somogyi M. A new reagent for the determination of sugars. J Biol Chem. 1945;60:61–8.
Chiura HX, Kita-Tsukamoto K. Purification and characterisation of a novel agarase secreted by a marine bacterium, Pseudoalteromonas sp. strain CKT1. Microbes Environ. 2000;15:11–22.
Saito M, Sugitate T, Chiura HX. Study of broad-host range vector particle-host cell surface interaction upon initial stage of infection. In: The annual meeting of the Japanese Society of Microbial Ecology, Fukuoka, Fukuoka, 30 October–1 November 2005.
Ikeda H, Tomizawa J. Transducing fragments in generalized transduction by phage P1. I. Molecular origin of the fragments. J Mol Biol. 1965;14:85–109.
Ikeda H, Tomizawa J. Transducing fragments in generalized transduction by phage P1. II. Association of DNA and protein in the fragments. J Mol Biol. 1965;14:110–9.
Yarmolinsky MB, Sternberg N. Bacteriophage P1. In: Calendar R, editor. The bacteriophages, vol. 1. New York: Plenum Press; 1988. p. 291–438.
Schmieger H. The fate of the bacterial chromosome in P22-infected cells of Salmonella typhimurium. Mol Gen Genet. 1971;110:238–44.
Schmieger H, Buch U. Appearance of transducing particles and the fate of host DNA after infection of Salmonella typhimurium with P22-mutants with increased transducing ability (HT mutants). Mol Gen Genet. 1975;140:111–22.
Hasegawa Y, Futamata H, Tashiro Y. Complexities of cell-to-cell communication through membrane vesicles: implications for selective interaction of membrane vesicles with microbial cells. Front Microbiol. 2015;6:633. https://doi.org/10.3389/fmicb.2015.00633.
Toyofuku M, Nomura N. What will membrane vesicles (MVs) bring to bacterial communication? Microbes Environ. 2017;32(3):185–7. https://doi.org/10.1264/jsme2.ME3203rh.
Oliveira H, Melo DL, Santos SB, Nóbrega FL, Ferreira EC, Cerca N, Azeredo J, Kluskens LD. Molecular aspects and comparative genomics of bacteriophage endolysins. J Virol. 2013;87:4558–70.
Attar N. Biofilms: have exploding cells blown up MV dogma? Nat Rev Microbiol. 2016;14:334–5.
Bitto NJ, Chapman R, Pidot S, Costin A, Lo C, Choi J, D’Cruze T, Reynolds EC, Dashper SG, Turnbull L, Whitchurch CB, Stinear TP, Stacey KJ, Ferrero RL. Bacterial membrane vesicles transport their DNA cargo into host cells. Sci Rep. 2017;7(1):7072. https://doi.org/10.1038/s41598-017-07288-4.
Roier S, Zingl FG, Cakar F, Schild S. Bacterial outer membrane vesicle biogenesis- a new mechanism and its implications. Microb Cell. 2016;3(6):257–9. https://doi.org/10.15698/mic2016.06.508.
Roier S, Zingl FG, Cakar F, Durakovic S, Kohl P, Eichmann TO, Klug L, Gadermaier B, Weinzerl K, Prassl R, Lass A, Daum G, Reidl J, Feldman MF, Schild S. A novel mechanism for the biogenesis of outer membrane vesicles in gram-negative bacteria. Nat Commun. 2016;7:10515. https://doi.org/10.1038/ncomms10515.
Babst M. MVB vesicle formation: ESCRT-dependent, ESCRT-independent and everything in between. Curr Opin Cell Biol. 2011;23(4):452–7. https://doi.org/10.1016/j.ceb.2011.04.008.
Schmidt O, Teis D. The ESCRT machinery. Curr Biol. 2012;22(4):R116–20. https://doi.org/10.1016/j.cub.2012.01.028.
Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT machinery: it’s all in the neck. Nat Rev Mol Cell Biol. 2010;11(8):556–66. https://doi.org/10.1038/nrm2937.
Fenner F, McAuslan BR, Mims CA, Sambrook J, White DA. The biology of animal viruses. 2nd ed. New York: Academic; 1974.
Flint SJ, Enquist LW, Krug RM, Racaniello VR, Sakalka AM. Principle of virology. Washington: American Society Microbiology; 2000.
Senčilo A, Paulin L, Kellner S, Helm M, Roine E. Related haloarchaeal pleomorphic viruses contain different genome types. Nucleic Acids Res. 2012;40:5523–34. https://doi.org/10.1093/nar/gks215.
Luk AWS, Williams TJ, Erdmann S, Papke RT, Cavicchioli R. Viruses of Haloarchaea. Life. 2014;4:681–715.
Wichels A, Biel SS, Gelderblom HR, Brinkhoff T, Muyzer G, Schuett C. Bacteriophage diversity in the North Sea. Appl Environ Microbiol. 1998;64:4128–33.
Jensen E, Schrader H, Rieland B, Thompson T, Lee K, Nickerson K, Kokjohn T. Prevalence of broad-host-range lytic bacteriophages of Spherotilus natans, Escherichia coli, and Pseudomonas aeruginosa. Appl Environ Microbiol. 1998;64:575–80.
Suttle C. Cyanophages and their role in the ecology of cyanobacteria. In: Whitton B, Potts M, editors. The ecology of cyanobacteria. Dordrecht: Kluwer Academic Publishers; 2000. p. 563–89.
Weinbauer MG, Rassoulzadegan F. Are viruses driving microbial diversification and diversity? Environ Microbiol. 2003;6:1–11.
Chiura HX, Velimirov B, Kogure K. Relationship between cell population density and induction frequency of lysogen: studied for marine originating broad-host range gene transfer particle (VP)-mediated transductant. In: IUMS 2011 Sapporo. International Congress of Applied Microbiology, Sapporo, Hokkaido, October 2011.
Fuqua WC, Winans SC, Greenberg EP. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol. 1994;176:269–75.
Nishimura A, Iyama S. Catalogue of Escherichia coli genetic stocks in National Institute of Genetics. Shizuoka: National Institute of Genetics, Genetic Stocks Research Center; 1986.
Berlyn MB. Linkage map of Escherichia coli K-12, edition 10: the traditional map. Microbiol Mol Biol Rev. 1998;62:814–984.
Birge EA. Bacterial and bacteriophage genetics. 4th ed. New York: Springer; 2000.
Canchaya C, Proux C, Fournous G, Bruttin A, Brüssow H. Prophage genomics. Microbiol Mol Biol Rev. 2003;67:238–76.
Poteete RA. Bacteriophage P22. In: Calendar R, editor. The bacteriophages, vol. 2. New York: Plenum Press; 1988. p. 647–82.
Susskind MM, Botstein D. Molecular genetics of bacteriophage P22. Microbiol Rev. 1978;42:385–413.
Nishima W, Kanamaru S, Arisaka F, Kitao A. Screw motion regulates multiple functions of T4 phage protein gene product 5 during cell puncturing. J Am Chem Soc. 2011;133:13571–6. https://doi.org/10.1021/ja204451g.
Acknowledgments
The author is grateful to A Ellinger, M Fukui, A Hiraishi, H Hoaki, S Hagemann, J Ishibashi, K Kogure, S-J Kim, Y Kumagai, Y Kawarabayashi, A Maruyama, M Maruyama, T Nakagawa, RW Ridge, U Simidu, S Suzuki, T Urabe, B Velimirov, S Yoshizawa for their collaboration, discussion, and helpful comments. Sincere thanks to M Arata, K Hirota, E Ito, K Ito, K Inoue, M Imada, S Imazu, T Ichinose, Y Inokuchi, D Koketsu, D Kato, F Maruyama, S Morimoto, Y Morikawa, D Nakata, H Naito, K Nakamura, Late M Segawa, Y Suzuki, M Takeuchi, T Takemura, N Uchiyama-Arai and other Lab members at ICU and AORI, the University of Tokyo for important collaboration and assistance with the experiments, and R/V Tanseimaru, Shinseimaru, Shinryumaru, Toyoha Mines Co. Ltd. and Nikko Tankai Co. Ltd. for assistance with sampling. The research presented here was supported in part by Grant-in-Aid for Scientific Research Nos. 10490012, 12490009, 14208063, 14405016, 15310007, 16310031 Archaean Park Project, Int’l Study Programme 259 (1995-97), 15H0549 from the Japan Society for the Promotion of Science (JSPS), the Donations to Encourage Research by Kyowa Hakko Kogyo Ltd., Life Cycle Association Ltd., and Amano Enzyme Inc. Last and certainly not least, thanks are given to the author’s family.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Chiura, H.X. (2019). Overlooked Broad-Host-Range Vector Particles in the Environment. In: Nishida, H., Oshima, T. (eds) DNA Traffic in the Environment. Springer, Singapore. https://doi.org/10.1007/978-981-13-3411-5_7
Download citation
DOI: https://doi.org/10.1007/978-981-13-3411-5_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3410-8
Online ISBN: 978-981-13-3411-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)