
Abstract
Polyphosphazenes are inorganic-organic high polymers with a backbone of alternating phosphorus and nitrogen atoms and two organic or organometallic side groups attached to each phosphorus. Most of these polymers are synthesized by macromolecular substitution reactions carried out on poly(dichlorophosphazene), (NPCl2)n. The chlorine substitution reactions involve alkoxides, aryloxides, primary or secondary amines, or a range of organometallic reagents. Structural variations are accomplished via the use of one, two, or more different nucleophiles and substituents along the polymer chain and by the employment of reagent size and reactivity to control polymer properties and emphasize specific uses. Applications have been developed for these polymers as elastomers, thermoplastics, biostable or bioerodible medical materials, fire-resistant lithium battery electrolytes, films, or foams, and gas and liquid separation membranes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
1 Introduction
Numerous advantages exist for the incorporation of inorganic elements into polymers. The inorganic elements broaden the range of structures and properties that are accessible. They also allow the use of nonclassical synthesis reactions and utilize starting materials other than oil or natural products. Moreover, the presence of inorganic elements in polymers provides access to materials that are resistant to extreme conditions. Examples of these advantages are provided by the polyphosphazene platform – a group of several hundred different polymers with a wide range of properties that in many cases cannot be generated from classic hydrocarbon-based macromolecules. These advantages have led to advances in elastomer technology, biomedical materials, non-flammable lithium battery electrolytes, gas transmission membranes, coatings, and textiles [1].
Polyphosphazenes are high molecular weight polymers with a backbone of alternating phosphorus and nitrogen atoms with two organic inorganic or organometallic side groups attached to every phosphorus atom (Fig. 10.1). The synthesis of most of these polymers is almost unique since it involves the use of macromolecular substitution, a process in which a broad range of different side groups and combinations of different groups are linked to the polyphosphazene chain after the polymerization process has been completed. Thus, one of the main features of this system is the use of the polyphosphazene backbone as a platform for the introduction of a wide variety of useful side group structures into a polymer system.

The basic polyphosphazene structure
2 History
The origins of this field can be traced to the early 1800s when Liebig and Rose [2, 3] reported that phosphorus pentachloride and ammonia or ammonium chloride react to yield crystalline solids with the composition NPCl2. Sporadic studies in Europe were followed by the work of H. N. Stokes in the USA in the 1890s [4], who proposed that these compounds are ring systems of formula (NPCl2)3, 4, 5 … with alternating single and double phosphorus-nitrogen bonds. Stokes also found that if any of these ring systems was heated to ~250 °C, the molten material gelled to a rubbery elastomer. However, the elastomer was insoluble in all solvents and slowly decomposed on exposure to atmospheric moisture – a characteristic of phosphorus-chlorine bonds. Moreover, with modern insight, it is clear that its insolubility was a strong indication of a cross-linked structure.
We began our studies by showing that the chlorine atoms in the cyclic trimer, (NPCl2)3, could be replaced by organic groups to produce water-stable derivatives, and this raised the question of whether Stokes’ high polymeric elastomer would react in a similar manner. Unfortunately, although some chlorine replacement occurred, the substitution was incomplete, and the insoluble cross-linked structure remained. As a result, it became clear that the preparation of stable, high polymeric polyphosphazenes from poly(dichlorophosphazene) depends on avoiding the cross-linking process. Thus, the breakthrough experiment was the demonstration that careful purification of the cyclic trimer and protection of it from the atmosphere, followed by heating at 200–250 °C, resulted in the formation of a polymer that was soluble in benzene, toluene, tetrahydrofuran (THF), and other solvents. Once this had been accomplished, the path was open to replacement of all the approximately 2000–30,000 chlorine atoms in each high polymer molecule by organic nucleophiles. This remains today the main method for the synthesis of several hundred different poly(organophosphazenes) [5,6,7].
3 Polyphosphazene Synthesis
The conventional synthesis of classical organic polymers is accomplished by addition, condensation, or ring-opening polymerization processes [8, 9] in which the side groups destined for the final polymer are already covalently linked to the monomer. A number of poly(organophosphazenes) have been synthesized by similar methods, especially by condensation reactions [10,11,12,13,14,15] or by the ring-opening polymerization of partially organic-substituted small-molecule phosphazene rings. The ring-opening method is effective only if three or four of the six chlorine atoms remain linked to the cyclic trimer. However, the molecular weights tend to be much lower than those obtained via the polymerization of (NPCl2)3 followed by halogen replacement reactions carried out on poly(dichlorophosphazene). Thus, this last method has evolved as the preferred route to exploit the broad scope of this system. Condensation routes to poly(dichlorophosphazene) are an alternative to the (NPCl2)3 ring-opening polymerization process but have a more specialized utility [10,11,12,13,14,15,16,17]. For example, the living cationic polymerization route to (NPCl2)n is a vehicle for the synthesis of block copolymers [16, 18].
The macromolecular substitution route (Fig. 10.2) [5,6,7] places few limits on the types of side groups or the combinations of different side groups that can be incorporated into the polymers, and this allows a broader range of properties and potential uses than in almost any other existing synthetic macromolecular system. Moreover, the main chain lengths are very high – typically from 1000 to 15,000 repeating units depending on polymerization conditions. Each different side group arrangement gives a polymer with different properties and potential uses.

The polyphosphazene macromolecular substitution approach
Thus, to summarize, this synthesis protocol is a two-step process in which a small-molecule cyclic phosphazene with chlorine side groups is first thermally melt-polymerized to a reactive polymer, and the chlorine atoms in this high molecular weight macromolecule are then replaced by a wide variety of different nucleophiles such as alkoxides, amines, or organometallic reagents either to give single-substituent or mixed-substituent polymers. Many of these side groups would not survive the polymerization process if they were present initially on the cyclic trimer, or they would inhibit the polymerization process. For some phosphazene polymers, a third step is also possible in which deprotection chemistry is subsequently carried out on the organic side groups to introduce specific functional units such as hydroxyl, carboxylic acid, amino, or organometallic units. An idea of the scope of this approach can be gauged by the abbreviated list of side groups that have been linked to a polyphosphazene chain shown in Table 10.1.
4 Challenges in the Macromolecular Substitution Process
The macromolecular substitution route to poly(organophosphazenes) is feasible mainly because of the high reactivity of the phosphorus-chlorine bonds in poly(dichlorophosphazene). However, this advantage must be balanced by several restrictions. First, by the end of the macromolecular substitution process, all the chlorine atoms must have been replaced by organic groups. Failure to accomplish this would leave the polymer liable to at least partial hydrolytic decomposition as residual P-Cl groups were hydrolyzed to P-OH units, which could then react to form cross-links or trigger hydrolytic cleavage at various sites along the chain. Perhaps the most remarkable thing about this synthesis is the fact that thousands of substitution reactions can be carried out efficiently on each macromolecule. Full substitution usually requires that a small excess of reagent is needed to ensure complete chlorine replacement.
Second, di- or multifunctional reagents cannot be employed directly for the macromolecular substitution reactions. The presence of more than one functional group in each nucleophile would allow it to react with two or more polyphosphazene chains to cross-link the system before halogen replacement is complete. This limitation can be circumvented by using protection techniques for some of the reactive units on a multifunctional reagent, followed by deprotection once the organic group has been linked to the main chain [19]. For example, the alkyl ester of p-hydroxy sodium benzoate reacts cleanly with poly(dichlorophosphazene) to yield a polymer with aryloxy ester side groups. Subsequent hydrolysis of the ester groups to carboxylic acid units generates the functional polymer [NP(OC6H4COOH)2]n. This could not have been accomplished by the direct reaction of p-hydroxybenzoic acid with poly(dichlorophosphazene). Protection-deprotection techniques have been used extensively in our laboratory for the linkage of biologically useful organic groups to the polymer chain [19].
Third, macromolecular SN2 substitution reactions are sensitive to steric inhibition if bulky nucleophiles such as steroids [20], iron porphyrins [21], or carboranes [22] are employed. However, in such systems typically ~10 to 50% chlorine substitution by bulky nucleophiles can be achieved, and this can then be followed by replacement of all the remaining chlorine atoms by less-hindered reagents. Some of the most interesting polymer produced in our laboratory in recent years have utilized this technique to yield mixed-substituent polymers with unexpected and useful properties [23,24,25,26,27,28]. Inherent in this technique is the question of the pattern of chlorine replacement (Fig. 10.3) and the effect of the substitution pattern on properties.

Alternative substitution sites along the polyphosphazene chain following the first chlorine replacement reaction
For example, following the replacement of the first chlorine atom somewhere along the polyphosphazene chain, the possibility exists that the second substitution will occur either at that same phosphorus atom or at a distant location along the chain. Steric hindrance will play a role in this process, which should (and does) become evident with the bulkiest nucleophiles. Alternatively, strong electron withdrawal by that first side group could favor the second substitution at the same phosphorus atom (geminal) or at a nearby phosphorus atom. Identifying these substitution patterns with a high polymer is a major challenge that can sometimes be approached through the study of 31P NMR spectra as the reaction proceeds.
In spite of these limitations, more than 250 different organic side groups have been linked to the polyphosphazene chain, to give one of the largest known synthetic polymer systems.
5 Broader Perspective
The differences between this polymer chemistry and traditional macromolecular science will be clear from the preceding discussion. However, it is necessary to view the polyphosphazene field further from two related but different points of view. First, there are inherent properties that arise because of the presence of inorganic elements in the backbone. Second, there are properties and design opportunities that result from the backbone being used as a carrier for specific side groups. The properties uniquely depend on both the backbone and the side groups, and only in a few cases, where the backbone is buried beneath massive side group structures, is the influence of the backbone relatively unimportant. The following discussion is organized into these two aspects.
6 Properties Generated by the Inorganic Backbone
The following special properties are a consequence of the phosphorus-nitrogen backbone.
-
1.
High skeletal flexibility. Unlike organic polymers such as polyacetylene that have conjugated electronic unsaturation along the main chain, the polyphosphazene skeleton has a high flexibility based on an unusually low barrier to torsion of the P-N bonds. This becomes manifest in low glass transition temperatures in the range of −100 to −60 °C, provided the side groups are themselves flexible or have small dimensions. For example, with flexible side groups such as ethoxy, propoxy, butoxy, etc., Tg’s in the −90 to −100 °C are accessible [29]. Even with fluoroalkoxy side groups, the Tg’s are generally below −40 °C [30]. Only when rigid, bulky side groups are linked to the chain do the Tg values start to climb. Poly(diphenoxyphosphazene) has a Tg near 0 °C [31], and the related polymer with p-phenylphenoxy side groups has a Tg near 90 °C [32]. 2-Naphthoxy side groups raise the Tg to ~40 °C. In the simple valence bond model, the skeletal flexibility is attributed to the presence of P-N-P three-center “islands” of dπ-pπ character (Fig. 10.4) rather than the stiff, broadly delocalized pπ-pπ chains as found in organic semiconductors. In the polyphosphazene chain, there is a mismatch of orbital signs at every phosphorus atom, which restricts electron delocalization to just three skeletal atoms.
Fig. 10.4 
Highly simplified bonding scheme for polyphosphazenes in which a mismatch of orbital signs at the 3d orbitals of phosphorus prevents long-range electron delocalization. (a) Electrons beyond those required for the sigma-bonded skeleton, which may interact in highly unusual ways to stabilize the P-N bonds. (b) The pi-bond “island” model proposed to explain why long-range delocalization of pi-electrons is apparently absent. (c) Illustration of why the barrier to torsion of the backbone bonds is so low, because the nitrogen lone pair orbitals can overlap different d-orbitals as the bond undergoes torsion
-
2.
Radiation stability. The polyphosphazene backbone is transparent throughout the visible region of the spectrum and into the 220 nm region of the ultraviolet, again a consequence of limited electron delocalization and a large band gap. This transparency provides resistance to photolytic decomposition in sunlight. Moreover, the free radical skeletal decomposition pathways that are common in classical polymers appear to be absent in this system.
-
3.
Nonmetallic appearance. Polymers with broadly delocalized electrons in the main chain, such as polyacetylene or polythiazyl, have a metallic appearance. Polyacetylene resembles either silver or gold in appearance and polythiazyl is gold. No polyphosphazenes synthesized to date have this characteristic, which is a strong indicator of a high band gap and poor electron delocalization along the backbone.
-
4.
Fire resistance. One of the most serious defects associated with many classical organic polymers is their combustibility, due to the thermo-oxidative free radical sensitivity of C-C and C-H bonds. Attempts to confer fire resistance on classical polymers are often made by the addition of organophosphorus small molecules. These small molecules can migrate in the solid or volatilize from the material over time. However, many phosphorus-nitrogen-based polymers have been shown to be inherently fire resistant throughout their lifetime [33,34,35].

7 Properties Generated by the Organic Side Groups
The sensitivity of P-Cl bonds to nucleophilic substitution is well known for small-molecule phosphorus compounds such as PCl3 and PCl5, and this is also the key to the macromolecular substitution pathways on which most of the chemistry of polyphosphazenes is based. Thus, this inorganic backbone is an excellent platform for the linkage of a broad range of different organic substituents that confer special properties on the polymers. In this way the characteristics of the final polymers can be controlled by the broad range of available organic nucleophiles, including properties that result from the permutations of two or more different side groups along the same chain. The following observations summarize some of the main trends. Specific examples are shown in Table 10.1.
-
1.
Alkoxy side groups. Short, non-fluorinated alkoxy side groups have a high torsional mobility, and this, combined with the flexibility of the backbone, leads to the formation of polymers with low glass transition temperatures (Tg). For example, the following Tg values are associated with polyphosphazenes that bear side chains such as ethoxy (−84 °C), propoxy (−99 °C), n-pentoxy (−102 °C), n-hexoxy (−104 °C), and n-octoxy (−104 °C) [36]. Ethoxy side groups impart hydrolytic sensitivity [37], a property that suggests biomedical uses since the hydrolysis products are an innocuous mixture of phosphate, ammonia, and ethanol.
-
2.
Cycloalkanoxy side groups. This series of polymers [38] (Fig. 10.5) provides an excellent example of how an increase in the dimensions of side groups linked to a polyphosphazene chain affects the ease of chlorine replacement and the glass transition temperatures. The glass transition temperatures rise steadily from −60 °C when all the side groups are cyclo-butanoxy to +40 °C for cyclo-octanoxy groups. Cyclobutanoxide through cyclohexanoxide anions react with poly(dichlorophosphazene) to give transparent, hydrophobic, film-forming polymers. However, the increased steric hindrance along this series requires increasingly more forceful conditions to replace all the chlorine atoms. The reaction of poly(dichlorophosphazene) with the cyclohexanoxy anion is one of the slowest reactions encountered in all the macromolecular substitution reactions studied, and forcing conditions (high temperatures, a large excess of nucleophile, and prolonged reaction times) are needed to ensure complete chlorine replacement. However, only the cyclo-octanoxy anion failed to replace all the chlorine atoms along the chain even under the most forcing reaction conditions. Molecular modeling of this system [38] illustrates clearly how the remaining P-Cl bonds are effectively buried within the electron density of the organic side groups already present. Nevertheless, separation of these cyclic structures from the reactive site by the introduction of methylene spacer groups allows even the most hindered of these rings to be linked to all of the reactive sites along the polymer chain. Partial substitution with cycloalkanoxy groups leaves P-Cl units that can be replaced by other less-hindered groups, such as trifluoroethoxy groups.
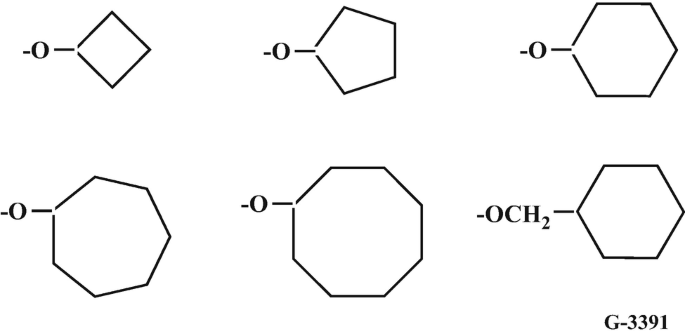
-
3.
Fluoroalkoxy side groups. Fluoroalkoxy side groups such as trifluoroethoxy, CF3CH2O-, or octafluoropentoxy (HCF2(CF2)3CH2O- have proved to be some of the most useful side units in the polyphosphazene series [1, 5, 6, 39, 40]. The polymer with trifluoroethoxy side groups is a hydrophobic, opalescent, microcrystalline, and film and fiber former (Tg = −66 °C, Tm = 242 °C) with numerous intermediate phase transitions, whereas the counterpart with octafluoropentoxy side groups is a soft, transparent film former (Tg = −64 °C). Combined as a 50-50 ratio in one polymer, these side groups yield amorphous, gum-like materials, but when lightly cross-linked, they are converted to elastomers [40]. These mixed-substituent, cross-linked polymers have been commercialized under the trade names such as PN-F® or Eypel-F® and have been utilized both for severe environment aerospace applications and as dental materials. Research is currently ongoing to optimize them as cardiovascular elastomers.
-
4.
Aryloxy groups. Numerous poly(organophosphazenes) with aryloxy side groups have been reported. As mentioned above, non-fluorinated phenoxy side groups, with their moderately bulky profile, have the effect of raising the glass transition temperatures to −8 °C [15, 41, 42]. They yield flexible fiber- and film-forming materials rather than soft elastomers. Polyphosphazenes with substituted phenoxy side groups have been studied in detail [41]. Larger aryloxy units such as biphenyloxy (Tg = 93 °C), triphenyleneoxy, tetra-phenyleneoxy, etc. progressively reduce the chain mobility and raise the glass transition temperatures. With linear tri- and tetra-phenyloxy groups, 100% replacement of the chlorine atoms in (NPCl2)n is not possible due to steric hindrance, although partial substitution below 50% is feasible [24,25,26]. The remaining chlorine atoms can be replaced by smaller side groups such as trifluoroethoxy. Noncrystalline elastomers are formed from normally microcrystalline polymers when ~5 to 8% of the side groups are bulky co-substituents.

Fluoroaryloxy groups have two influences on polymer properties. First, they withdraw electrons from the skeleton, which is a stabilizing effect. 4-Fluorophenoxy side groups increase hydrophobicity but have little effect on the Tg (−6.6 °C). However, as the level of side group fluorination increases beyond this point, the Tg values rise and the solubility in organic solvents falls. Pentafluorophenoxy side groups can replace all the chlorine atoms in (NPCl2)n but only under forcing reaction condition. Moreover, the molecular rigidity of this system is such that the detection of Tg and Tm transitions becomes difficult. These side groups confer special properties on the polymers including resistance to nonpolar solvents. Trifluoromethyl side groups linked to the phenoxy units have only a marginal effect on the chlorine replacement reactions but have a marked effect through an increase in hydrophobicity and general chemical stability.
-
5.
Alkyl ether side groups. Oligo-ethyleneoxy-type side groups can be readily linked to a polyphosphazene chain via the sodium salts of the appropriate alcohols [43,44,45]. For example, complete chlorine replacement occurs with nucleophiles such as Na0CH2CH2OCH2CH2OCH3 and its branched isomers. The resultant polymers are water-soluble and water-stable and form hydrogels when lightly cross-linked by exposure to gamma irradiation. In the anhydrous state, these polymers are excellent solid ionic conductors for lithium ions, with conductivities of ~10−5 S/cm in the absence of etheric solvents, but have values higher than 10−3 S/cm when small amounts (~10%) of etheric solvents are present. Experimental non-flammable lithium ion batteries with either graphite or metallic lithium electrodes and these electrolytes have been fabricated and tested in our program.
-
6.
Alkylamino and arylamino groups. As shown in Fig. 10.2, chlorine replacement in (NPCl2)n is also possible using primary or secondary amines as nucleophiles [7]. In most reactions a tertiary amine such as triethylamine is present to capture and insolubilize the hydrogen chloride formed in these processes. These reactions are particularly important for the linkage of biologically interesting side groups to the polyphosphazene skeleton. Typical amines that have been used in these reactions include ethylamine, propylamine, etc.; amino acid esters such as the ethyl esters of glycine, alanine, and phenylalanine [19]; and a range of bioactive molecules with NH functional units. Hydrogen bonding between NH bridging units within the polymer matrix is believed to be responsible for higher Tg values than in the case of similar side groups linked to the skeleton through alkoxy or aryloxy bonds.
-
7.
Organometallic side groups. A variety of different organometallic side groups such as metal carbonyls or metallocenes have been linked to the phosphazene skeleton. These substitutions are somewhat more challenging than the reactions of oxo- or nitrogen nucleophiles with poly(dichlorophosphazene) because some organometallic nucleophiles can coordinate to the polymer backbone nitrogen atoms via their lone pair orbitals. Thus, the following special techniques were developed to circumvent this problem.
First, small-molecule model reactions were explored to optimize reaction conditions [46,47,48].
Second, an avoidance of organometallic side reactions was accomplished by the use of an alternative strategy, using the following logic. Because coordination of metallo species to the backbone nitrogen lone pair orbitals leads to side reactions, withdrawal of those electrons from nitrogen by the use of fluorophosphazenes should lead to halogen replacement rather than nitrogen coordination. The linkage of ferrocenyl side groups to a polyphosphazene chain was accomplished by an unusual variation of the polyphosphazene substitution route. Thus ferrocene was first linked to a fluorophosphazene cyclic trimer, either through a single linkage or through a transannular structure, and that trimer was thermally polymerized [48]. The remaining fluorine atoms in the resultant polymer were then replaced by trifluoroethoxy groups. Ring strain in the transannular trimer provides an extra driving force for the ring-opening polymerization.
Polymerization of (NPF2)3 to (NPF2)n, followed by replacement of fluorine, in the manner discussed for chlorophosphazenes, is not possible because the high polymer is insoluble in all except a few rare fluorocarbon solvents. Thus, this pathway has not been developed. However, the cyclic trimer N3P3F5C6H5 does polymerize to an organic soluble polymer, and this then allows direct replacement of P-F bonds by organic or organometallic anions.
-
8.
Metal coordination side groups. Coordination of metals to a polyphosphazene can be accomplished either through the skeletal nitrogen atoms or through nitrogen, sulfur, or phosphine ligands in the side group structure. Thus, pendent macrocyclic rings, thioethers, phosphine, or amino ligands provide a facile means for the immobilization of metal atoms or clusters [49]. Some of these are of interest as immobilized catalysts.
-
9.
Substituent exchange. Replacement of all the chlorine atoms along a polyphosphazene chain by organic groups is not necessarily the final process since some organic side groups already present may be displaced by other alkoxides [50, 51]. For example, the aryloxyphosphazene polymer [NP(OC6H5)2]n in solution can be modified by exposure to nucleophiles such as sodium trifluoroethoxide. Trifluoroethoxy groups have been displaced in solution-state reactions by longer-chain fluoroalkoxy groups to give mixed-substituent species. Of additional interest is the possibility that such side group exchange reactions can take place selectively at the surface of a solid polymer fiber or film leaving the interior polymer intact. Thus, a hydrophobic polymer may be modified by surface exchange to have a hydrophilic interface. Such reactions become important for the modification of bio-implantable polymers in the body, where the deposition of biomolecules on the surface is required for maximum biocompatibility.
An alternative approach for surface modification is to use an environmental oxygen or nitrogen plasma [52] to implant hydrophilic amino, oxo, or hydroxy groups into the surface of a hydrophobic material. The relative stability of the inorganic polymer backbone to plasmas reduces the possibility of general degradation.
8 Design Opportunities Connected with the Introduction of Bulky Side Groups
As mentioned earlier, the dimensions of the reactants influence the progress of nucleophilic substitutions, even when two small molecules are involved in the process. Steric influences become even more important when a small-molecule nucleophile reacts with a macromolecule. The twisting and coiling of a polymer molecule in solution present a hindered target for the nucleophile especially when many of the reactive sites on the polymer are shielded by substituents already present.
This is certainly the case for the reactions of poly(dichlorophosphazene) with bulky aryloxides or amines. In many cases the steric hindrance will be inconsequential at the start of the chlorine replacement process, but it will become increasingly restrictive as fewer and fewer P-Cl units remain along the chain. Thus, studies with bulky nucleophiles, such as those shown in Fig. 10.5, assume a special significance if these groups are designed to confer special properties on the polymers. Such groups include multi-ring aromatic chromophores, transition metal catalysts, or photonic species. Large amino acid esters or steroids and other biologically interesting units are bulky structures that are attractive side groups for linkage to a polyphosphazene chain.

Examples of bulky side groups that have been linked to a polyphosphazene chain. The largest nucleophiles typically replace fewer than 50% of the available reaction sites, but the remaining chlorine atoms can be replaced by less-hindered side groups such as trifluoroethoxy groups
Recent work in our program has revealed another aspect of this principle – the design and synthesis of new polyphosphazene elastomers based on what appear to be the intermolecular interactions between bulky side groups. This becomes manifest in polyphosphazenes that contain minor percentages of bulky side groups interspersed among a majority of smaller side groups. Some of the bulky side groups shown in the polymers in Fig. 10.5 illustrate this principle.
A striking example of the influence of bulky side groups is where the polyphosphazene with all trifluoroethoxy side groups is a microcrystalline, film-, or fiber-forming polymer. It can be oriented by stretching, but it is not elastomeric. However, the presence of roughly 5–8% of bulkier side groups (such as the two shown at the top of Fig. 10.5) eliminates the crystallinity and converts the material to a rubbery elastomer, without chemical cross-linking [23,24,25,26].
Polyphosphazenes that bear end groups or side groups that associate with other molecules via a “lock and key” arrangement have also been studied [53]. Thus, polyphosphazenes that bear hydrophobic adamantane units linked to the ends or middle units of a chain form host-guest complexes with beta-cyclodextrin units arrayed along an organic polymer. These complexes form a variety of structures such as “palm tree” or reversible cross-linked arrangements.
9 Practical Applications of Polyphosphazenes
The ability to use the polyphosphazene chain as a carrier for different side groups opens numerous avenues for addressing practical problems, and many of these possibilities remain to be exploited. Two summaries that address this topic were published in 2003 [1, 54]. The following are a few examples that illustrate both the earlier and more recent initiatives.
-
1.
Biomedical Applications. First, the coupling of biologically useful side groups to the phosphazene polymer chain can be utilized for the controlled delivery of drugs either for subcutaneous implantation or as soluble molecules or nanospheres or microspheres that circulate in the blood. Polymers have been also designed to self-destruct by hydrolysis to release the drug following changes in pH or other stimuli [55,56,57,58].
Second, pH- or ion strength-responsive alkyl ether-substituted polyphosphazene membranes have been fabricated for the controlled release of drug molecules, or for the incorporation of enzymes, the permeability of which can be controlled by pH variations in the body [59].
Third, an appropriate choice of amino acid ester side groups can yield a polymer that serves a structural role in the body before bioeroding by hydrolysis to harmless products in what is called a tissue engineering procedure. Such polymers can in principle be used as resorbable surgical sutures, woven surgical mesh, or bone regeneration matrices. Bone regeneration has been a particular target in our collaborative program with the group of C. Laurencin at the University of Connecticut [60,61,62,63,64].
In another medical application, gold nanospheres coated with a polyphosphazene with ionically cross-linkable functional groups (PCPP) have been utilized by our collaborators at the University of Pennsylvania both for the imaging and identification of diseased tissue or for targeted drug delivery [55].
Dentistry is an area of biomedical materials development that has long utilized polyphosphazene elastomers [65]. The elastomers in question are mixed-substituent fluoroalkoxy polymers of the “PN-F” type (see earlier) which are incorporated into an interpenetrating network with a second polymer such as ethylene glycol dimethacrylate or trimethylolpropane trimethacrylate. The composite material has advantages over other materials because of its antifungal properties and its ability to absorb energy without instant rebound. It is also considered to be a favorable material for maxillofacial prosthetics.
Finally, cardiovascular devices and microspheres have been coated with the hydrophobic [NP(OCH2CF3)2]n or similar fluorophosphazenes to retard detrimental blood/materials interactions. This application is in semi-commercial use for drug delivery applications and for coating implanted anti-thrombogenic treated devices [66].
-
2.
Battery and Solar Cell Electrolytes. As mentioned earlier, the MEEP family of polyphosphazene (with alkyl ether side groups) has been investigated in some detail for rechargeable lithium battery electrolyte applications. These polymers are good solid or gel solvents for lithium salts, and the gel forms provide conductivities above 10−3 C/cm. The fire-retardant characteristics of these polymers are particularly interesting in view of the known flammability of most organic liquid electrolytes [67, 69].
-
3.
Hydrophobic and Super-hydrophobic Films, Fibers, and Foams. The combination of water repellency and limited organic solvent solubility means that fluoroalkoxy and fluoroaryloxy polyphosphazenes have some advantages over classical fluorocarbon polymers. In particular, nanofiber mats made by electrospinning or surface coatings or wire coatings applied from solution have potential uses in aircraft anti-icing surfaces and (as mentioned above) in biomedical devices. The solubility of [NP(OCH2CF3)2]n in supercritical carbon dioxide has facilitated the fabrication of expanded hydrophobic foams for possible use as fire-resistant flotation constructs [70].
-
4.
Traditional Photographic Applications. Patents have been issued by Fujii Photo Film, Eastman Kodak, and Konica for the use of alkyl ether- or phenyl alkyl ether-substituted polyphosphazenes as antistatic agents in silver halide films and as components in processing solutions [71,72,73]. It appears that these applications are based on the cation coordination properties of the polymers and the amphilicity.
-
5.
Fire-Resistant Applications. Small-molecule cyclic phosphazenes with aryloxy side groups have been offered commercially for several years as fire retardants additives for conventional organic polymers. An application for the polymers is as fire retardants for polyurethanes in which the polyphosphazene becomes chemically bonded into the polyurethane network [74].
-
6.
Optical and Photonic Applications. The control of refractive index by side group variations is being considered for a number of optical applications [75, 76]. Moreover the linkage of nonlinear optical side groups suggests applications in photonic switches. Dye molecules linked to the polyphosphazene chain have been proposed as non-light-scattering primary color filters for imaging sensors [28]. In earlier work polyphosphazenes with liquid crystalline and photochromic side groups have been studied in some detail [76]. Light-emitting polymers with phosphazene components have also been described [77].
-
7.
High-Performance Elastomers. One of the earliest uses for fluoroalkoxy or aryloxy polyphosphazenes was as energy-absorbing, solvent- and fire-resistant elastomers for uses in aircraft, satellites, or fuel lines in automobiles. This interest continues as one of the major applications of polyphosphazenes [1].
-
8.
Membranes. Numerous technological membrane applications for both gas and liquid separations have been studied [78, 79]. The most recent interest is in polyphosphazenes for carbon dioxide separations as part of the CO2 sequestration program for counteracting global warming [79]. As with many other phosphazene applications, the ease of manipulating properties by side group variations is considered to be a major advantage compared to other polymers.
10 The Future
The broad span of side groups and skeletal architectures currently known for the polyphosphazene platform already exceeds the scope of most other synthesized macromolecular systems. However, this is just a fraction of the number of different polymers and architectures that will probably become accessible in the future based on an expansion of current knowledge. Moreover, the practical applications of these polymers are only just beginning to be investigated, and this aspect also foretells a promising prospect in the coming years. The diversity of the polyphosphazene platform is also an indicator of what can be expected for other, still undeveloped polymers and materials that combine the attributes of the inorganic and organic elements.
References
Allcock HR (2003) Chemistry and applications of polyphosphazenes. John Wiley & Sons, Hoboken
Liebig J (1834) A Compound of Phosphorus with Nitrogen (Supplement). Ann Chem Pharm 11:139
Rose H (1834) A Compound of Phosphorus with Nitrogen. Ann Chem Pharm 11:129
Stokes HN (1897) On the Chloronitrides of Phosphorus (II). Amer Chem J 19:782
Allcock HR, Kugel RL (1965) Synthesis of High Polymeric Alkoxy- and Aryloxyphosphonitriles. J Am Chem Soc 87:4216–4217
Allcock HR, Kugel RL, Valan KJ (1966) High Molecular Weight Poly[alkoxy- and aryloxyphosphazenes]. Inorg Chem 5:1709–1715
Allcock HR, Kugel RL (1966) High Molecular Weight Poly(diamino-phosphazenes). Inorg Chem 5:1716–1718
Allcock HR, Lampe FW, Mark JE (2003) Contemporary polymer chemistry, 3rd edn. Prentice-Hall, Englewood Cliffs
Odian G (2004) Principles of polymerization, 4th edn. John Wiley & Sons, Hoboken
Flindt EP, Rose H (1977) Trivalent-pentavalente Phosphosverbindi-dungen/Phosphazene. IV. Z Anorg Allg Chem 428:204
Wisian-Neilson P, Neilson RH (1980) Poly(dimethylphosphazene), (Me2)PN)n. J Am Chem Soc 102:2848
Neilson RH, Wisian-Neilson P (1988) Poly(alkyl/aryl phosphazenes). Chem Rev 88:54
Wisian-Neilson P, Jung J-H, Potluri SK (2006) Cyclic and Polymeric Alky/Aryl Phosphazenes in Modern Aspects of Main Group Chemistry. ACS symp. series 917, Washington, DC, pp 335–346
Montague RA, Matyjaszewski K (1990) Synthesis of Poly[bis(trifluoroethoxy)-phosphazene] Under Mild Conditions Using a Fluoride Initiator. J Am Chem Soc 112:6721
Matyjaszewski K, Moore MM, White ML (1993) Synthesis of Polyphosphazene Block Copolymers Bearing Alkoxyethoxy and Trifluoroethoxy Groups 26:6741–6748
(a) Honeyman CH, Manners I, Morrissey CT, Allcock HR (1995) Ambient Temperature Synthesis of Poly(dichlorophosphazene) with Molecular Weight Control. J Am Chem Soc 117:7035–7036. (b) Allcock HR, Reeves SD, Nelson JM, Crane CA, Manners I (1997) Polyphosphazene Block Copolymers via the Living Cationic, Ambient Temperature Polymerization of Phosphoranimines. Macromolecules 30:2213–2215
De Jaeger R, Potin P (2004) Ch.2. In: Gleria M, De Jaeger R (eds) Synthesis and characterization of poly(organophosphazenes). Nova Publishers, New York
Allcock HR (2004) Ch. 3. In Gleria M, De Jaeger R (eds) Synthesis and characterization of poly(organophosphazenes). Nova Publishers, New York
Allcock HR, Morozowich NL (2012) Bioerodible Polyphosphazenes and their Medical Potential. RSC Polym Chem 3:578–590
Allcock HR, Fuller TJ (1980) Phosphazene High Polymers with Steroidal Side Groups. Macromolecules 13:1338–1345
Allcock HR, Greigger PP, Gardner JE, Schmutz JL (1979) Water Soluble Polyphosphazenes as Carrier Molecules for Iron(III) and Iron(II) Porphyrins. J Am Chem Soc 101:606–611
Allcock HR, Scopelianos AG, O’Brien JP, Bernheim MY (1981) Synthesis and Structure of Carborane-Substituted Cyclic and Polymeric Phosphazenes. J Am Chem Soc 103:350–357
Modzelewski T, Allcock HR (2014) An Unusual Polymer Architecture for the Generation of Elastomeric Properties in Fluorinated Polyphosphazenes. Macromolecules 47:6776–6782
Modzelewski T, Wilts E, Allcock HR (2015) Elastomeric Polyphosphazenes with Phenoxy-cyclotriphosphazene Side Groups. Macromolecules 48:7543–7549
Li Z, Chen C, Tian Z, Modzelewski T, Allcock HR (2016) Polyphosphazenes with Cyclotetraphosphazene Side Groups: Synthesis and Elastomeric Properties. J Inorg Organomet Mater Polym 26:667–674
Modzelewski T, Wonderling NM, Allcock HR (2015) Polyphosphazene Elastomers Containing Interdigitated Oligo-p-phenyleneoxy Side Groups: Synthesis, Mechanical Properties and X-ray Scattering Studies. Macromolecules 48:4882–4890
Tong C, Tian Z, Chen C, Li Z, Modzelewski T, Allcock HR (2016) Synthesis and Characterization of Trifluoroethoxy Polyphosphazenes Containing POSS Side Groups. Macromolecules 49:1313–1320
Li Z, Allcock HR (2015) Polyphosphazenes with Immobilized Dyes as Potential Color Filter Materials. Appl Mater Interface 1:13518–13523
Weikel AL, Lee D, Krogman NR, Allcock HR (2010) Phase Changes of Poly(alkoxyphosphazene) and their Behavior in the Presence of Oligoisobutylene. J Polym Eng Sci 92A:114–125
Tian Z, Liu X, Manseri A, Ameduri B, Allcock HR (2013) Limits to Expanding the PN-F Series of Polyphosphazene Elastomers. Polym Eng Sci 54:1827–1832
Singler RE, Schneider NS, Hagnauer GL (1975) Polyphosphazenes: Synthesis-Properties-Applications. Polym Eng Sci 51:321–338
Allcock HR, Mang MN, Dembek AA, Wynne KJ (1989) Poly(aryloxyphosphazenes) with Phenylphenoxy and Related Bulky Side Groups. Synthesis, Thermal Transition Behavior and Optical Properties. Macromolecules 22:4179–4190
Reed CA, Taylor GP, Guigley KS, Kully KS, Bernheim KA, Coleman MM, Allcock HR (2000) Polyurethane/Poly[bis(carboxylato-phenoxy)phosphazene] Blends and their Potential as Flame Retardant Materials. J Polym Sci Eng 40:465–472
Chen C, Liu X, Tian Z, Allcock HR (2012) 2,2,2-Trichloroethoxy-Substituted Polyphosphazenes: Synthesis, Characterization, and Properties. Macromolecules 45:9085–9091
Weikel AL, Owens SG, Fushimi T, Allcock HR (2012) Synthesis and Characterization of Methionine- and Cysteine-Substituted Phosphazenes. Macromolecules 4:5205–5210
Weikel AL, Lee D, Krogman NR, Allcock HR (2010) Phase Changes of Poly(alkoxyphosphazenes) and Their Behavior in the Presence of Oligoisobutylene. J Polym Sci Eng 92A:114–125
Nichol JL, Hotham IT, Allcock HR (2014) Ethoxyphosphazene Polymers and their Hydrolytic Behavior. Polym Degrad Stab 109:92–96
Tian Z, Hess A, Fellin CR, Nulwala H, Allcock HR (2015) Phosphazene High Polymers and Models with Cyclic Aliphatic Side Structure-Property Relationships. Macromolecules 48:4301–4311
Kojima M, Magill J (1985) Phase Transitions in Polyphosphazene Films: Poly[bis(trifluoroethoxy)phosphazene]. Macromol Chem Phys 186:649–663
Rose SH, Cable J (1969) U.S. Govt Research Report, AD 693,28
Schneider NS, Desper CR, Singler RE (1976) The Thermal Transition Behavior of Polyorganophosphazenes. J Appl Polym Sci 20:3087–3103
Allcock HR, Kim C (1990) Liquid Crystalline Phosphazenes Bearing Biphenyl Mesogenic Groups. Macromolecules 23:3881–3887
Blonsky PM, Shriver DF, Austin PE, Allcock HR (1984) Polyphosphazene Solid Electrolytes. J Am Chem Soc 106:6854–6855
Allcock HR, Austin PE, Neenan TX, Sisko JT, Blonsky PM, Shriver DF (1986) Polyphosphazenes with Etheric Side Groups: Prospective Biomedical and Solid Electrolyte Polymers. Macromolecules 19:1508–1512
Allcock HR, Kwon S, Riding GH, Fitzpatrick RJ, Bennett JL (1988) Hydrophilic Polyphosphazenes as Hydrogels: Radiation Crosslinking and Hydrogel Characteristics of Poly[bis(methoxyethoxyethoxy)phosphazene]. Biomaterials 19:509–513
Greigger PP, Allcock HR (1979) A Spirocyclophosphazene with Iron-Phosphorus Bonds and a P-Fe-Fe Three-Membered Ring. J Am Chem Soc 101:2492
Allcock HR, Manners I, Mang MN, Parvez M (1990) Transition Metal Derivatives of Phosphinophosphazenes: X-Ray Crystal Structures of N3P3Cl4PhPh2, N3P3Cl4PhPPh4,Cr(Co)5 and N3P3Cl4PhPPh2.Ru3(CO)11. Inorg Chem 29:522–529
Manners I, Riding GH, Dodge JA, Allcock HR (1989) Role of Ring Strain and Steric Hindrance in a New Method for the Synthesis of Macrocyclic and High Polymeric Phosphazenes. J Am Chem Soc 111:3067–3069
Diefenbach U, Cannon AM, Stromberg BE, Olmeijer DL, Allcock HR (2000) Synthesis and Metal Coordination of Thioether-Containing Cyclo- and Poly(organophosphazenes). J Appl Polym Sci 78:650–661
Liu X, Breon J, Chen C, Allcock HR (2012) Substituent Exchange Reactions of Linear Oligomeric Aryloxy Phosphazenes with 2,2,2-Trifluoroethoxide. Inorg Chem 51(21):11910–11916
Liu X, Breon JP, Chen C, Allcock HR (2012) Substituent Exchange Reactions of Trimeric and Tetrameric Arylocyclophosphazenes with Sodium 2,2,2-Trifluoroethoxide. Roy Soc Chem Dalton Trans 41:2100–2109
Allcock HR, Steely L, Kim S, Kang B (2007) Plasma Surface Functionalization of Poly[bis(2,2,2-trifluoroethoxy)phosphazene] Films and Nanofibers. Langmuir 23:8103–8107
Tian Z, Chen C, Allcock HR (2014) Ethoxyphosphazene Polymers and their Hydrolytic Behavior. Macromolecules 47:1065–1072
Gleria M, De Jaeger R (eds) (2003) Applicative aspects of poly(organophosphazenes). Nova Publishers, New York
Chhour P, Gallo N, Cheheltani R, Williams D, Al-Zaki A, Paik T, Nichol JL, Tian Z, Naha PC, Allcock HR, Murray CB, Sourkas TA, Cormode DP (2014) Nano-Disco Balls: Control over Surface versus Core Loading of Active Nanocrystals into Polymer Nanoparticles. ACS Nano 8(9):9143–9153
Liu X, Tian Z, Chen C, Allcock HR (2013) UV-Cleavable Unimolecular Micelles: Synthesis and Characterization Toward Photocontrolled Drug Release Carriers. Polym Chem 4:1114–1125
Liu X, Zhang H, Tian Z, Sen A, Allcock HR (2012) Preparation of Quaternized Organic-Inorganic Hybrid Brush Polyphosphazene-co-poly-[2-(dimethylamino)ethylmethacrylate] Electrospun Fibers and their Antibacterial Properties. Polym Chem 3:2082–2091
Tian Z, Zhang Y, Chen C, Guiltinan MJ, Allcock HR (2013) Biodegradable Polyphosphazenes Containing Antibiotics: Synthesis, Characterization, and Hydrolytic Release Behavior. Polymer 4:1826–1835
Allcock HR, Kwon S (1989) An Ionically-Crosslinkable Polyphosphazene: Poly[di(carboxylatophenoxy)phosphazene] and its Hydrogels and Membranes. Macromolecules 22:75–79
Peach MS, James R, Toti US, Deng M, Morozowich NL, Allcock HR, Laurencin CT, Kumbar SG (2012) Polyphosphazene Functionalized Polyester Fiber Matrices for Tendon Tissue Engineering: In Vitro Evaluations of Human Mesenchymal Stem Cells. Biomed Mater 7:1–13
Nichol JL, Morozowich NL, Allcock HR (2013) Biodegradable Alanine and Phenylalanine Alkyl Ester Polyphosphazenes as Potential Ligament and Tendon Tissue Scaffolds. RSC Polym Chem 4:600–606
Peach MS, Kumbar SG, James R, Toti US, Balasubramaniam D, Deng M, Ulery B, Maxxocca AD, McCarthy MB, Morozowich NL, Allcock HR, Laurencin CT (2012) Design and Optimization of Polyphosphazenes Functionalized Fiber Matrices for Soft Tissue Regeneration. J Biomed Nanotechnol 8:107–124
Deng M, Kumbar SG, Nair LS, Weikel AL, Allcock HR, Laurencin CT (2011) Biomimetic Structures: Biological Implications of Dipeptide-substituted Polyphosphazene-Polyester Blend Nanofiber Matrices for Load-Bearing Bone Regeneration. Adv Funct Mater 21:2641–2651
Deng M, Kumbar SG, Wan Y, Toti US, Allcock HR, Laurencin CT (2010) Polyphosphazene Polymers for Tissue Engineering: An Analysis of Materials Synthesis, Characterization, and Applications. Soft Matter 6:3119–3132
Gettleman L (2003) Ch. 2. In: Gleria M, De Jaeger R (eds) Applicative aspects of poly(organophosphazenes). Nova Publishers, New York
Celonova Bio-Sciences (2010) Peachtree, Georgia, promotional material (2017) U.S. Patent US7922764
Fei S-T, Lee S-HA, Pursel SM, Bashem J, Hess A, Grimes CA, Horn MW, Mallouk TE, Allcock HR (2011) Electrolyte Infiltration in Phosphazene-Based Dye-Sensitized Solar Cells. J Power Sources 196:5223–5230
Fei S-T, Allcock HR (2010) Methoxyethoxyethoxyphosphazenes as Ionic Fire Retardant Additives for Lithium Battery Systems. Power Sources 195(7):2082–2088
Fei S-T, Allcock HR (2009) Recent Progress with Ethyleneoxy Phosphazenes as Lithium Battery Electrolytes. Mater Res Soc Symp. 1127-T01-05
Steely LB, Li Q, Badding JV, Allcock HR (2008) Foam Formation with Fluorinated Polyphosphazenes by Liquid CO2 Processing. Polym Sci Eng 48:683–686
Mukunoki Y, Kubota T (1992) Fuji Photo Film, Japan, U.S. Patent 5,135,846
Fukuwatari N, Ueda E, Kurachi Y (1998) Konica Corp., Japan, U.S. Patent 5,840,471
Ishikawa W, Fukuwatari N. Konica Corp., Japan, European Patent Application
Reed CA, Taylor JP, Guigley KS, Kully KS, Bernheim KA, Coleman MM, Allcock HR (2000) Polyurethane/Poly[bis(carboxylato-phenoxy)phosphazene] Blends and their Potential as Flame Retardant Materials. Report to U.S. Federal Aviation Administration
Olshavsky M, Allcock HR (1997) Polyphosphazenes with High Refractive Indices: Optical Dispersion and Molar Reactivity. Macromolecules 30:4179–4183
Allcock HR, Bender JD, Chang Y, McKenzie M, Fone MM (2003) Controlled Refractive Index Polymers; Polyphosphazenes with Chlorinated- and Fluoroinated-Aryloxy and Alkoxy Side Groups. Chem Mater 15:473–477
Allcock HR, Chang Y, Stone DA (2006) Control of the Conjugation Length and Solubility in Electroluminescent Polymers. J Polym Sci 44:69–76
Stewart FF, Luther TA, Harrup MK, Orme CJ (2003) In: Gleria M, De Jaegar R (eds) Applicative aspects of poly(organophosphazenes). Nova Publishers, New York. Chapter 10
Venna SR, Spore A, Tian Z, Marti AM, Albenze EJ, Nulwala HB, Rosi NL, Luebke DR, Hopkinson DP, Allcock HR (2017) Polyphosphazene Polymer Development for Mixed Matrix Membranes Using SIFSIX-Cu-2i as Performance Enhancement Filler Particles. J Membr Sci 535:103–112
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Allcock, H.R. (2019). Polyphosphazenes as an Example of the Element-Blocks Approach to New Materials. In: Chujo, Y. (eds) New Polymeric Materials Based on Element-Blocks. Springer, Singapore. https://doi.org/10.1007/978-981-13-2889-3_10
Download citation
DOI: https://doi.org/10.1007/978-981-13-2889-3_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-2888-6
Online ISBN: 978-981-13-2889-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)