
Abstract
Glucose activates the glucose-sensing receptor and induces rapid intracellular signals in pancreatic β-cells. When the glucose-sensing receptor is blocked by an inhibitor of T1R3 or deletion of the T1R3 gene, glucose-induced insulin secretion (GIIS) is significantly reduced. In perifusion system, both first and second phases of GIIS are attenuated by the inhibition of the glucose-sensing receptor. Collectively, the glucose-sensing receptor is involved in both rapid and sustained action of glucose. Indeed, activation of the receptor by either artificial sweeteners or nonmetabolizable glucose analog increases ATP levels in β-cells. Furthermore, inhibition of the glucose-sensing receptor attenuates glucose-induced increase in ATP. These results indicate that activation of the glucose-sensing receptor promotes glucose metabolism and thereby augments ATP production in β-cells. Thus, glucose first acts on the cell-surface glucose-sensing receptor and primes the metabolic pathway of glucose. Glucose then enters β-cells and is metabolized through already activated metabolic pathway. The receptor pathway and the metabolic pathway act coordinately to stimulate insulin secretion.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Introduction
Glucose is a principal fuel in the body that acts as a critical stimulator of insulin secretion from pancreatic β-cells. The mechanism by which glucose stimulates insulin secretion is an important issue from basic and clinical points of view, and it has been investigated for several decades by many researchers. It is now thought that glucose exerts its action though its metabolism [1]. Thus, glucose enters β-cells, is catalyzed by glucokinase and downstream glycolytic enzymes and in mitochondria, and the resultant increase in the ATP/ADP ratio causes closure of the ATP-sensitive potassium channel (KATP channel). Resultant depolarization leads to opening of the voltage-gated calcium channel, and Ca2+ enters the cell. These sequences of events are dependent on glucose metabolism, and when glucokinase is blocked by mannoheptulose, for example, both Ca2+ response and insulin secretion do not take place [1].
5.2 Sweet Taste Receptor in Pancreatic β-Cells
We found recently that glucose generates rapid intracellular signals in pancreatic β-cells, which are not dependent on glucose metabolism [2]. Thus, glucose evokes rapid Ca2+ signals that are not blocked by mannoheptulose, an inhibitor of glucokinase [3]. Also, these rapid Ca2+ signals are reproduced by adding 3-O-methylglucose (3OMG), a glucose analog not catalyzed in β-cells. Furthermore, these Ca2+ signals are attenuated by a Gq inhibitor and an inhibitor of phospholipase C (PLC) [3]. Likewise, glucose evokes a rapid elevation of cyclic AMP (cAMP), which is not affected by mannoheptulose but is reproduced by 3OMG. In addition, glucose-induced elevation of cAMP is attenuated by transduction of a gene encoding dominant-negative mutant of Gs [3]. All these results suggest that glucose activates a cell-surface receptor and produces rapid intracellular signals by activating G proteins. We named this receptor glucose-sensing receptor (GSR) [4].
What is the molecular nature of the GSR? In this regard, Niki and colleagues showed three decades ago that an inhibitor of sweet taste sensation, p-nitrophenyl-D-glucopyranoside (PNP-Glu), attenuated glucose-induced insulin secretion [5]. In contrast, PNP-Glu did not affect insulin secretion induced by a high concentration of potassium. Their results raised a possibility that the sweet taste receptor or related molecules may be expressed in pancreatic β-cells and mediates the action of glucose.
The sweet taste receptor is expressed in taste cells of the taste bud in the tongue. In 2001, the molecular nature of the sweet taste receptor was revealed by molecular cloning and functional analyses [6, 7]. It is now generally thought that the sweet taste receptor is a heterodimer comprised of the two members of the taste receptor-1 (T1R) family, type 2 and type 3 T1R (T1R2 and T1R3, respectively). Both T1R2 and T1R3 are members of the class C G protein-coupled receptor (GPCR) superfamily. We speculated that the sweet taste receptor subunits are expressed in pancreatic β-cells and mediate the action of glucose [2]. Indeed, reverse-transcription PCR (RT-PCR) revealed that mRNA for T1R2 and T1R3 is expressed in mouse pancreatic islets [8]. Also, in MIN6 cells, a glucose-responsive mouse β-cell line [9], mRNA for T1R2 and T1R3 has been detected. Furthermore, immunoreactive T1R3 has been detected in the core of mouse islets and in MIN6 cells [8]. Based on these data, we initially thought that the canonical sweet taste receptor, i.e., a heterodimer of T1R2 and T1R3 (T1R2/T1R3), is expressed in pancreatic β-cells [8]. The function of the sweet taste receptor can be assessed by activating it using artificial sweeteners. Indeed, when sucralose, a potent artificial sweetener, is added, insulin secretion is augmented [8]. Likewise, other sweeteners, for example, acesulfame-potassium, saccharin, and glycyrrhizin all increase insulin secretion [8]. Therefore, the “sweet taste receptor” in β-cells seems to be functional. It should be mentioned that relatively high concentrations of artificial sweeteners, for example, 10–20 mM, are needed to stimulate insulin secretion. We assessed the signal transduction pathways activated by the “sweet taste receptor” using MIN6 cells [8]. Because of the complex structure of the taste buds, the signal transduction pathway activated by the sweet taste receptor has not been investigated thoroughly in taste cells of the tongue. β-cells may provide a good system to study the signal transduction of the sweet taste receptor. In fact, when MIN6 cells are stimulated by sucralose, both cytoplasmic Ca2+ concentration ([Ca2+]c) and cytoplasmic cAMP concentration ([cAMP]c) are elevated [8]. Sucralose also activates protein kinase C (PKC) [8]. Consequently, both Ca2+ and cAMP messenger systems are activated by an artificial sweetener sucralose. This is an interesting feature of the “sweet taste receptor” in β-cells. It should be noted that the effect of sucralose on [Ca2+]c is attenuated by gurmarin [8], an inhibitor of the sweet taste receptor, and lactisole [10], an inhibitor of T1R3. Knockdown of T1R3 also attenuates the effect of sucralose [11]. Hence, sucralose exerts its effect by acting on the T1R3-containing receptor, presumably the “sweet taste receptor.” An intriguing aspect of the “sweet taste receptor” in β-cells is that it produces a variety of patterns of signals. When MIN6 cells are stimulated by other sweeteners, considerably different types of signals are produced compared to those evoked by sucralose [12]. For example, when cells are stimulated by saccharin, a classical artificial sweetener, [cAMP]c, is increased, whereas [Ca2+]c is not affected. In contrast, glycyrrhizin, a natural sweetener derived from licorice, increases [Ca2+]c but not [cAMP]c [12]. Five or more sweeteners examined so far induce different patterns of intracellular signals presumably by activating different transducers and/or effectors [12]. The signal transduction mechanism activated by the “sweet taste receptor” is quite unique: different types of agonists generate distinct patters of signals. In this regard, these ligands may act as biased agonists. In fact, the sweet taste receptor in the tongue is activated by numerous natural or artificial sweet substances with a variety of chemical structures (7). Those ligands have multiple binding sites in the receptor molecule [7]. Ligands binding to distinct binding sites may activate different sets of responses. It is also possible that the “sweet taste receptor” in β-cells is not a single receptor molecule but is comprised of multiple types of receptor molecules. Then, it is possible that multiple ligands produce multiple types of signals by binding to different subtypes of the receptor. In this situation, these receptor molecules should contain T1R3 since inhibition or deletion of T1R3 attenuates the receptor functions. If T1R3 is a component of the receptor, it is unlikely that five or more types of receptors exist. Collectively, at least some of the ligands act as biased agonists.
As mentioned above, GSR responds to physiological concentrations of glucose and produces immediate signals in β-cells [3]. A critical question is whether or not GSR is identical to the canonical sweet taste receptor expressed in the tongue, i.e., T1R2/T1R3 heterodimer. In other words, whether or not the canonical sweet taste receptor is activated by physiological concentrations of glucose in the plasma is a critical question. The answer may be No. In the taste buds, it is known that glucose is much less potent than sucrose in activating the sweet taste receptor [7]. It is therefore unlikely that the canonical sweet taste receptor, i.e., T1R2/T1R3, functions as GSR. An important point is that T1R3 is required for the function of GSR [3]. Consequently, it is reasonable to speculate that GSR may be a dimer containing T1R3, which is distinct from T1R2/T1R3. We therefore re-examined the expression of T1Rs in pancreatic β-cells in more detail. When we measured the expression of T1R2 and T1R3 by quantitative RT-PCR, the expression of T1R3 was abundant whereas the expression of T1R2 was far less [13]. In fact, when we measured the protein expression by either immunohistochemistry or immunoblotting, T1R3 was detected abundantly in β-cells while T1R2 was undetectable [13]. These results indicate that in pancreatic β-cells, the expression of T1R2 is negligible and the canonical sweet taste receptor, T1R2/T1R3, is therefore a very minor component, if any.
5.3 Glucose-Sensing Receptor in Pancreatic β-Cells
The above results indicate that the receptor activated by glucose and sweeteners in β-cells may be slightly different from the canonical sweet taste receptor, T1R2/T1R3. Then what is it? Since class C GPCR functions as a dimer [15], and T1R3 is a major T1R in β-cells, it is reasonable to speculate that the putative receptor may be T1R3-containing dimer other than T1R2/T1R3. Theoretically, candidate receptors are a homodimer of T1R3 (T1R3/T1R3) and a heterodimer of T1R3 and X, where X is a class C GPCR expressed in pancreatic β-cells. To this end, we first examined whether or not T1R3/T1R3 functions as GSR. As we showed previously, T1R3/T1R3 is able to function as a signaling receptor [14]. Using HEK cells stably expressing T1R3 (HEK-T1R3 cells), we examined whether or not glucose increases [Ca2+]c. As depicted in Fig. 5.1, basal [Ca2+]c is stable and a high concentration of glucose induces an immediate elevation of [Ca2+]c. This elevation is only transient and is followed by a small but sustained reduction of [Ca2+]c. Noteworthy is the fact that this pattern of [Ca2+]c response is quite similar to that observed in β-cells stimulated by nonmetabolizable glucose analog 3OMG [3]. T1R3/T1R3 is therefore capable of producing this pattern of [Ca2+]c response. This is an intriguing feature of T1R3/T1R3, and such responses are not observed in cells expressing ordinary class A GPCR. Interestingly, glucose-induced changes in [Ca2+]c are attenuated by a Gq inhibitor and an inhibitor of PLC, suggesting that both elevation and reduction of [Ca2+]c are coupled and are mediated by Gq and subsequent PLC. This is the reason reduction of [Ca2+]c is observed after the transient elevation of [Ca2+]c. A possible interpretation is that Ca2+ is taken up to ER, and simultaneously, Ca2+ entry is inhibited by unknown reasons. When various concentrations of glucose are tested, however, a relatively high concentration of glucose, for example 25 mM, is required to elicit [Ca2+]c responses in HEK-T1R3 cells. This means that although T1R3/T1R3 is an interesting candidate for GSR, glucose sensitivity is not high enough. Another candidate receptor needs to be considered.

Effect of glucose on [Ca2+]c in HEK-T1R3 cells. Fluo-8-loaded HEK-T1R3 cells were stimulated by 25 mM glucose and changes in [Ca2+]c were measured
As mentioned above, GSR could be a heterodimer of T1R3 and X, where X is a class C GPCR expressed in pancreatic β-cells. The most interesting candidate for X is the calcium-sensing receptor (CaSR), which is expressed abundantly in pancreatic β-cells [15]. CaSR is identified as a cell-surface receptor detecting the changes in extracellular Ca2+ concentrations. It is expressed in parathyroid cells, renal tubular cells, and bone cells, three major organs regulating calcium metabolism in the body. Subsequently, the molecular structure of CaSR has been identified by molecular cloning, and the results show that CaSR belongs to the class C GPCR family acting as a homodimer. It is a multifunctional receptor and is in fact activated not only by Ca2+ but also by many other compounds including various amino acids and cationic compounds [16]. We established a stable HEK cell line expressing CaSR [17] and examined whether or not glucose is able to activate the receptor. We found that many sweet substances including sugars and artificial sweeteners activated CaSR [17]. Indeed, an addition of glucose induced a rapid increase in [Ca2+]c. The rapid Ca2+ transient was followed by sustained reduction of [Ca2+]c [17]. This pattern of [Ca2+]c response is rather unique but resembles that induced by 3OMG in β-cells. Glucose-induced biphasic changes in [Ca2+]c were inhibited by inhibitors of Gq and PLC. Hence, the unique pattern of [Ca2+]c response is dependent on Gq-mediated activation of PLC. To our surprise, CaSR is very sensitive to glucose and as low as 5 mM of glucose-induced the maximal response. Thus, when ambient glucose concentration was raised from 3 to 5 mM, a marked transient of [Ca2+]c was observed, which was followed by sustained reduction of [Ca2+]c. CaSR homodimer is too sensitive to function as a physiological receptor for glucose. However, CaSR may be a good candidate receptor that dimerizes with T1R3.
We then examined the effect of glucose in stable HEK cell line expressing both T1R3 and CaSR (HEK-T1R3/CaSR cells). In these cells, formation of a heterodimer of T1R3 and CaSR was detected. As shown in Fig. 5.2, oscillation of [Ca2+]c was observed in an unstimulated condition. Addition of a high concentration of glucose induces biphasic response of [Ca2+]c in HEK-T1R3/CaSR cells: a rapid transient peak followed by a small but sustained decrease in [Ca2+]c. Again, this pattern of changes in [Ca2+]c is similar to that observed in β-cells stimulated by 3OMG [3]. In these cells, 7 to 8 mM of glucose is able to induce [Ca2+]c response. A heterodimer of T1R3/CaSR is therefore an interesting candidate for GSR. In MIN6 cells, NPS-2143, an inhibitor of CaSR, blocks the [Ca2+]c response induced by 3OMG. Similarly, lactisole, an inhibitor of T1R3 blocks [Ca2+]c, responses induced by 3OMG. Consequently, both T1R3 and CaSR are required for the action of 3OMG on [Ca2+]c. A heterodimer of T1R3 and CaSR is the best candidate for the GSR.

Effect of glucose on [Ca2+]c in HEK-T1R3/CaSR cells. Fluo-8-loaded HEK-T1R3/CaSR cells were stimulated by 25 mM glucose and changes in [Ca2+]c were measured
5.4 Role of GSR in Glucose-Induced Insulin Secretion
Glucose activates cell-surface GSR and evokes rapid signals including elevation of [Ca2+]c, activation of protein kinase C (PKC), and elevation of [cAMP]c. Elevation of Ca2+ concentration, especially in the subplasma membrane area, is a trigger for exocytosis of insulin granules, and both cAMP and diacylglycerol act as allosteric modulators of exocytosis and increase the Ca2+-sensitivity of exocytosis. It is thus quite likely that intracellular signals evoked by activation of GSR facilitate insulin secretion. In fact, inhibition of T1R3 by adding lactisole significantly reduces glucose-induced insulin secretion [10]. Since GSR signals are rapid, we initially speculated that the GSR signals are important for the first phase of glucose-induced insulin secretion. We tested this idea by perifusion experiments. When we compared glucose-induced insulin secretion in a perifusion system using islets obtained from normal and T1R3-knockout mice, the results were slightly different from what we expected. As shown in Fig. 5.3, both first and second phases of glucose-induced insulin secretion were significantly reduced in islets obtained from T1R3-knockout mice. These results are rather unexpected but coincide with the results obtained by measuring [Ca2+]c response [3]. Inhibition of T1R3 reduces not only the rapid response but also subsequent oscillatory [Ca2+]c responses observed several minutes after the stimulation by a high concentration of glucose in β-cells [3]. The oscillatory elevation of [Ca2+]c has been thought to be due to Ca2+ entry caused by the inhibition of KATP channel. These results indicate that GSR not only induces rapid signals, but it also modulates sustained action of glucose in β-cells. How does GSR modulate the long-term action of glucose? Since glucose metabolism is important for sustained action of glucose, we speculated that the GSR signals modulate glucose metabolism in β-cells. To test this idea, we monitored changes in intracellular ATP ([ATP]c) using luciferase-expressing MIN6 cells [11]. We first examined changes in [ATP]c induced by various concentration of glucose. As depicted in Fig. 5.4a, glucose induced biphasic elevation of [ATP]c in MIN6 cells, and the effect of glucose was dose-dependent. Conversely, [ATP]c was rapidly reduced by inhibition of mitochondrial function by either dinitrophenol or 2-cycrohexen-1-one, indicating that changes in [ATP]c in MIN6 cells could be monitored. Using this system, we first tested whether or not activation of GSR by sucralose affected [ATP]c. To our surprise, addition of sucralose markedly increased [ATP]c in MIN6 cells (Fig. 5.4b). This experiment was performed in the presence of 5.5 mM of glucose in the incubation medium. It should be noted that sucralose is an artificial sweetener and therefore does not enter β-cells nor serve as a fuel. Nevertheless, sucralose markedly elevated [ATP]c in MIN6 cells. The effect of sucralose was also confirmed by monitoring the ATP/ADP ratio by using Percival [11]. This effect of sucralose is not simply due to the elevation of [Ca2+]c or [cAMP]c induced by sucralose since addition of muscarinic agonist carbachol or glucagon-like peptide-1, which increases [Ca2+]c or [cAMP]c, respectively, did not affect [ATP]c [11]. Furthermore, the effect of sucralose on [ATP]c was observed even in the absence of ambient glucose. When MIN6 cells were incubated for 60 min in medium containing no glucose, the basal level of [ATP]c was reduced. However, sucralose was still able to increase [ATP]c in this condition. Since glycogen content, if any, should be severely reduced in this condition, the result suggests that sucralose is able to increase ATP by not utilizing glucose-6-phosphate as a substrate. This effect of sucralose is due to the activation of GSR since nonmetabolizable analog of glucose 3OMG reproduced the sucralose effect. Also, lactisole, an inhibitor of T1R3, attenuated the effect of sucralose [10]. GSR signals thus activate the metabolic pathway and increase [ATP]c. One of the sites of action of the GSR signals is promotion of the metabolism in mitochondria since sucralose and mitochondrial fuel methylsuccinate act synergistically to increase [ATP]c. As mentioned above, 3OMG increases [ATP]c. This means that glucose not only serves as a substrate but it also activates GSR as a ligand and promotes its own metabolism. In fact, the effects of glucose on [ATP]c and insulin secretion were attenuated by knocking down T1R3 (Fig 5.4c, d) [11]. Collectively, glucose exerts its action by two different mechanisms. Glucose first acts on the cell-surface GSR and activates the receptor. Activation of this receptor causes priming of the metabolic pathway and thereby facilitates glucose metabolism. Then glucose enters β-cells and is catalyzed through the already activated metabolic pathway (Fig. 5.5). Thus, glucose acts as a ligand and also a substrate in β-cells, and it activates two distinct pathways: the GSR pathway and the metabolic pathway. These two pathways merge inside the β-cells, leading to production of more ATP and subsequent inhibition of KATP channel. These two pathways act coordinately to stimulate insulin secretion [4]. This is a new model showing the mechanism of action of glucose in β-cells. This model is an extension of the “glucoreceptor hypothesis” [2, 4], but it is also includes the “metabolic hypothesis.” In fact, in this model, there exists an interaction of the “receptor hypothesis” and the “metabolic hypothesis,” and the receptor pathway positively regulates the metabolic pathway. These two pathways act coordinately to exert the action of glucose.

Effect of glucose on insulin secretion in islets from normal and T1R3 knockout mice. Islets obtained from normal (○) and T1R3-knockout (●) mice were stimulated by 16.7 mM glucose, and insulin secretion was measured in a perifusion system

Effect of glucose and sucralose on [ATP]c in luciferase- expressing MIN6 cells [11]. (a) Effects of various concentration of glucose on [ATP]c. Luciferase- expressing MIN6 cells were incubated in Krebs-Ringer bicarbonate buffer containing 5.5 mM glucose. Then glucose concentration was raised to 8.3, 16.7, and 25 mM glucose, and changes in [ATP]c were monitored. (b) Effect of sucralose on [ATP]c. Luciferase-expressing MIN6 cells were stimulated by 10 mM sucralose and changes in [ATP]c were monitored. (c) Effect of knock-down of T1R3 on glucose-induced increase in [ATP]c. Luciferase-expressing MIN6 cells and T1R3-knocked down MIN6 cells were stimulated by 25 mM glucose and changes in [ATP]c were monitored (d) Effect of knockdown of T1R3 on glucose-induced in [ATP]c. Experiments were done as in c and area under the curve from 0 to 5 and 5.5 to 30 min was calculated

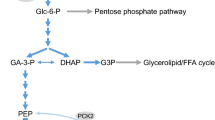
Action of glucose in pancreatic β-cells [11]. Glucose first activates the cell-surface glucose-sensing receptor (GSR) and primes the metabolic pathway. Glucose then enters β-cell and is metabolized through already activated metabolic pathway. The receptor pathway and the metabolic pathway act coordinately to stimulate insulin secretion
5.5 Future Direction of the GRS Research
Many questions still remain unanswered. First, which step(s) of the glucose metabolism is modulated by the GSR signals? In this regard, glucokinase is a rate-limiting enzyme in the glycolytic pathway, and it has been thought to act as a glucose sensor [18]. Whether or not glucokinase activity is modulated by GSR is an interesting question. There are many steps in the downstream of glucokinase, which could be a target(s) of the GSR. It is necessary to identify the step(s) modulated by the receptor signal. Second, how do the GSR signals stimulate the glucose metabolism? In other words, by what mechanisms does GSR modulate the glucose metabolism? Third, what is the physiological and pathophysiological significance of the GSR pathway?
With regard to the first question, GSR appears to modulate multiple steps in the metabolic pathway of glucose. Since activation of the GSR increases [ATP]c even in the absence of ambient glucose, GSR modulates the step(s) downstream of glucose-6-phosphate. Indeed, GSR acts synergistically with methylsuccinate to increase [ATP]c [11], indicating that GSR facilitates metabolism in mitochondria. In fact, the effect of sucralose on [ATP]c is inhibited by inhibition or deletion of the enzyme involved in the malate-aspartate shuttle. Also, metabolome analyses show that activation of the GSR increases the delivery of substrates to the glycolytic pathway from alanine and glycerol (unpublished observation). Collectively, GSR augments glucose metabolism by acting on the multiple steps in the metabolic pathways. Further studies are clearly needed to determine the precise steps and the regulatory mechanism responsible for promotion of the metabolism. Regarding the second question, we addressed the signal transduction pathways responsible for the elevation of [ATP]c [19]. Among the signaling pathways activated by the GSR, depolarization of the plasma membrane and resultant entry of Ca2+ are important for promotion of glucose metabolism [19]. This is not surprising because there are three dehydrogenases in the mitochondria regulating the TCA cycle. Also, the malate-aspartate shuttle is modulated by Ca2+. In addition, depolarization of the plasma membrane per se may facilitate the metabolism by a yet unknown mechanism [19]. It should be mentioned that the role of the GSR-mediated rapid signals is still unclear. Again, further studies are necessary to identify these mechanisms. Regarding the third question, the GSR signals are necessary for the full action of glucose since inhibition of the GSR function and deletion of the receptor subunit significantly reduce the insulin secretory response to glucose [2]. As mentioned above, both first and second phases of glucose-induced insulin secretion are inhibited without the GSR signal. Since GSR is required for the full action of glucose, it is possible that derangement of the GSR pathway is involved in the pathophysiology of type 2 diabetes. In fact, our results obtained in animal models of diabetes show that the expression of T1R3 is markedly reduced in diabetic animals [13]. This reduction of the expression is due at least partly to prolonged hyperglycemia. When hyperglycemia is corrected by treatment with insulin, the expression of T1R3 is recovered [13]. It is reasonable to speculate that impairment of insulin secretion observed in diabetes is partly due to down-regulation of the GSR by prolonged hyperglycemia. Reduction of the expression of GSR is one of the features of glucotoxicity in diabetes. Elucidation of the regulation of the expression level of the GSR would help with better understanding of the pathophysiology of type 2 diabetes.
References
Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:738–51.
Kojima I, Medina J, Nakagawa Y. Role of the glucose-sensing receptor in insulin secretion. Diabetes Obes Metab. 2018;19(Suppl 1):1954–62.
Nakagawa Y, Nagasawa M, Medina J, Kojima I. Glucose evokes rapid Ca2+ and cyclic AMP signals by activating the cell-surface glucose sensing receptor in pancreatic β-cells. PLoS One. 2015;10:e0144053.
Kojima I, Nakagawa Y, Ohtsu Y, Hamano K, Medina J, Nagasawa M. Return of the glucoreceptor: glucose activates the glucose-sensing receptor T1R3 and facifitates metabolism in pancreatic β-cells. J Diabetes Investig. 2015;6:256–63.
Niki A, Niki H, Hashioka T. Receptors of paraneurons, with special reference to glucoreceptors. Arch Histol Cytol. 1989;52:33–8.
Nelson G, Hoon MA, Chandrasekar J, Zhang Y, Ryba NJP, Zuker C. Mammalian sweet taste receptors. Cell. 2001;106:381–90.
Temussi P. The sweet taste receptor: a single receptor with multiple sites and modes of interaction. Adv Food Nutr Res. 2007;53:199–239.
Nakagawa Y, Nagasawa M, Yamada S, et al. Sweet taste receptor expressed in pancreatic β-cells activates the calcium and cyclic AMP signaling systems and stimulates insulin secretion. PLoS One. 2009;4:e5106.
Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K. Establishment of a pancreatic β cell line that retains glucose-inducible insulin secretion. Endocrinology. 1990;127:126–32.
Hamano K, Nakagawa Y, Ohtsu Y, Li L, Medina J, Tanaka Y, Masuda K, Komatsu M, Kojima I. Lactisole inhibits the glucosesensingreceptor in mouse pancreatic β-cells. J Endocrinol. 2015;226:57–66.
Nakagawa Y, Ohtsu Y, Nagasawa M, Shibata H, Kojima I. Glucose promotes its own metabolism by acting on the cell-surface glucosesensing receptor T1R3. Endocr J. 2014;61:119–31.
Nakagawa Y, Nagasawa M, Mogami H, Lohse M, Ninomiya Y, Kojima I. Multimodal function of the sweet taste receptor expressed in pancreatic β-cells: generation of diverse patterns of intracellular signals by sweet agonists. Endocr J. 2013;60:1191–206.
Medina A, Nakagawa Y, Ma J, et al. Expression of the glucose-sensing receptor T1R3 in pancreatic islet: changes in the expression levels in various nutritional and metabolic states. Endocr J. 2014;61:797–895.
Masubuchi Y, Nakagawa Y, Ma JH, Kojima I, Shibata H. Novel regulatory function of sweet taste-sensing receptor in adipogenic differentiation of 3T3-L1 cells. PLoS One. 2013;8:e54500.
Rasschaert J, Malaisse WJ. Expression of the calcium-sensing receptor in pancreatic islet B-cells. Biochem Biophys Res Commum. 1999;264:615–8.
Jensen AA, Bräuner-Osborne H. Allosteric modulation of the calcium-sensing receptor. Curr Neuropharmacol. 2007;5:180–6.
Medina J, Nakagawa Y, Nagasawa M, et al. Positive allosteric modulation of the calcium-sensing receptor by physiological concentrations of glucose. J Biol Chem. 2016;291(44):23126–35.
Matschinski FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic b-cells and hepatocytes. Diabetes. 1990;39:647–52.
Li LF, Ohtsu Y, Nakagawa Y, Masuda K, Kojima I. Sucralose, an activator of the glucose-sensing receptor, increases ATP by calcium-dependent and -independent mechanisms. Endocr J. 2016;63:715–25.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Nakagawa, Y., Medina, J. (2018). The Role of the Glucose-Sensing Receptor in Glucose-Induced Insulin Secretion in Pancreatic β-Cells. In: Kojima, I. (eds) Glucose-sensing Receptor in Pancreatic Beta-cells. Springer, Singapore. https://doi.org/10.1007/978-981-13-0002-8_5
Download citation
DOI: https://doi.org/10.1007/978-981-13-0002-8_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-0001-1
Online ISBN: 978-981-13-0002-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)