
Abstract
Pancreatic β-cells in the islets of Langerhans secrete insulin in response to blood glucose levels. Precise control of the amount of insulin secreted is of critical importance for maintaining systemic carbohydrate homeostasis. It is now well established that glucose induced production of ATP from ADP and the KATP channel closure elevate cytosolic Ca2+, triggering insulin exocytosis in β-cells. However, for full activation of insulin secretion by glucose, other mechanisms besides Ca2+ elevation are needed. These mechanisms are the targets of current research and include intracellular metabolic pathways branching from glycolysis. They are metabolic pathways originating from the TCA cycle intermediates, the glycerolipid/free fatty acid cycle and the pentose phosphate pathway. Signaling effects of these pathways including degradation (removal) of protein SUMOylation, modulation of insulin vesicular energetics, and lipid modulation of exocytotic machinery may converge to fulfill insulin secretion, though the precise mechanisms have yet to be elucidated. This mini-review summarize recent advances in research on metabolic coupling mechanisms functioning in insulin secretion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The International Diabetes Federation reported that approximately 537 million adults were suffering from diabetes, while more than 1.2 million children and adolescents were living with type 1 diabetes, in 2021 [1]. Now increasing numbers of young people are being diagnosed with type 2 diabetes, especially in developing countries such as those in the Middle East [2]. Etiologically, diabetes is very complex disease, but recent studies on the pathophysiology of all types of diabetes have focused on pancreatic β-cells [3,4,5]. Therefore, a precise and comprehensive understanding of β-cell biology would provide the basis for diabetes prevention, treatment and management, including diet therapy and the development of new drugs.
This issue of Diabetology International presents two mini-reviews aimed at providing readers with the most recent core information on the mechanisms of insulin secretion from pancreatic β-cells. In this first article, how elevation of blood glucose is transduced to signals for exocytosis will be summarized. The second article, by Nagao and colleagues, will discuss how exocytosis is regulated in β-cells.
MA Lane described being struck by his finding of two distinct cell types in the islets of Langerhans in the autumn of 1905 [6]. This is the first recognition of different cell types in pancreatic islets of Langerhans, 36 years after the discovery of this mini-organ in 1869. Since then, the mechanisms of regulating insulin secretion have attracted the attention of researchers. The goals are to determine how the β-cell recognizes blood glucose concentrations and how it then secrets an appropriate amount of insulin back into the bloodstream. The existence of a cell membrane-associated glucoreceptor was suggested decades ago [7]. Although glucose may play a role via interactions with plasma membrane molecules [8], glucose metabolism is a prerequisite for glucose-stimulated insulin secretion (GSIS).
Medical students learn about the “excitation–contraction coupling” in skeletal muscles in their early physiology courses [9, 10]. They also study “stimulus-secretion coupling” in nerve endings and neuroendocrine cells [11]. During these physiological phenomena, cells transduce external information into transmittable forms and convey it to other cells. In pancreatic β-cells, blood glucose concentrations are information transduced into the amounts of insulin secreted into the blood stream. The central mechanism is a special form of “stimulus-secretion coupling”, namely “metabolism-secretion coupling” as introduced by Wollheim and colleagues [12]. As to the mechanisms of “excitation–contraction coupling” and “stimulus-secretion coupling”, reception of the external information at the cell surface membrane immediately induces ionic events governed mainly by Ca2+. In the β-cell, transduction of the blood glucose concentrations into ionic events is much more complex. In the past quarter century, owing to the development of novel molecular cell biology techniques, including small interfering RNA and CRISPR/Cas9, major progresses have been achieved. This mini-review presents a summary of the generation of intracellular signals for insulin exocytosis evoked by glucose.
Overview of glucose-stimulated insulin secretion (GSIS)
As in other excitable cells, elevation of cytosolic Ca2+ is the initial trigger for insulin exocytosis from β-cells. Thus, glucose metabolism raises ATP and decreases ADP levels, thereby increasing the ATP/ADP ratio which in turn results in closure of the ATP-dependent K+ (KATP) channel at the plasma membrane. Major ions distributed across the plasma membrane are K+, Na+, and Cl− and intracellular and extracellular concentrations of these ions determine membrane potential, according to the Goldman–Hodgkin–Katz equation, with different relative contributions defined by channel status (open or closed). Thus, closure of KATP channels evoke membrane depolarization and opening of voltage-dependent Ca2+ channels, which then induce cytosolic Ca2+ elevation. Since pharmacological and molecular inhibition of store-operated Ca2+ entry suppresses GSIS [13], Ca2+ mobilization from the intracellular store also contributes to β-cell response to elevated glucose concentrations.
Although glucose induced intracellular elevation of Ca2+ triggers insulin secretion, in the early 1990s, two studies [14, 15] demonstrated that glucose can augment insulin secretion on top of the effects of elevated Ca2+. This means that glucose not only raises intracellular Ca2+ via membrane depolarization, through KATP channel closure, but also generates signals for insulin secretion independently of this KATP channel closure. The former mechanism plays an important role in the initiation of GSIS, and is thus referred to as the “triggering pathway”. The latter mechanism is called the “amplifying pathway” and two-thirds of the insulin secretion evoked by glucose is due to the latter pathway. Therefore, in the past quarter century, considerable research attention has been given to this mechanism.
Special feature of glycolysis in β-cells
Glucose is the fundamental fuel for living organisms and every cell is equipped with the glycolysis pathway. For β-cells, glucose is not only a fuel molecule but also is a molecule conveying information of systemic carbohydrate metabolism. Thus, glucose sensing β-cells have the special feature of glycolysis (Fig. 1). The glucose transport rate far exceeds the rate of glucose utilization. Therefore, modulations of glucose transport capacities do not affect insulin secretion, while the glucose phosphorylation step governed by glucokinase determines the overall rate of glucose utilization [16]. The MODY2 study provides clinical evidence for the importance of glucokinase [17]. Glucokinase is a distinct isoform of the hexokinase family. It has a high km value suitable for responding to physiological glucose changes and is not inhibited by the product, glucose-6-phosphate. Another glycolysis-related distinct characteristic of the β-cell is low expression of lactate dehydrogenase (LDH) and monocarboxylate transporters (MCTs) [18]. Thus, only a small fraction of the pyruvate generated through glycolysis is metabolized to lactate while a major fraction is efficiently transported to the mitochondria. In addition, low expressions of LDH and MCTs render β-cells selectively responsive to glucose but not lactate [19, 20]. Lactate serves as an energy source in other cells, such as neurons.

Pancreatic β-cell glycolysis and metabolic pathways stemming from glycolysis. Arrow width represents the relative amount of metabolic flux mediated by each enzyme. The TCA cycle initiated by pyruvate entry, the glycerolipid/free fatty acid cycle boosted by glycerol-3-phosphate and the pentose phosphate pathway originating from glucose-6-phoshtae, mediate generation of metabolism-secretion coupling factors
Recently, further modifications of glycolysis at a distal step have been discovered. Phosphoenolpyruvate (PEP) is provided not only from the upper half of glycolytic system but also from mitochondria via mitochondrial PEP carboxykinase/PCK2 at high glucose concentrations (Fig. 1) [21]. PCK2 knockout (KO) mice reportedly show reduced insulin secretion [22]. In their β-cells, some fraction of pyruvate kinase (PK) is localized beneath the plasma membrane and localized ADP consumption and ATP generation by PK is sufficient to raise the ATP/ADP ratio for closure of KATP channels [21]. These features of glycolysis make glucose the strongest known secretagogue.
In addition to these special features of glycolysis in the β-cell, there is the additional interesting feature regarding the connection between glycolysis and mitochondrial metabolism. In β-cells, pyruvate carboxylase (PC) expression is higher than other tissues [23]. A high level PC and the resulting increase in anaplerosis has been implicated in GSIS. However, the high level of PC expression in β-cells was reported to be restricted to rodents, i.e. it was not observed in human β-cells [24].
On the basis of β-cell glycolysis with these special features, downstream branches of metabolic pathways generate signals for insulin secretion: they are (1) pathways originating from metabolites of the mitochondrial tricarboxylic acid (TCA) cycle, (2) the glycerolipid/free fatty acid cycle, and (3) the pentose phosphate pathway.
Mitochondria as an organelle generating coupling factors for insulin exocytosis
Experimental data and clinical observations have established the importance of mitochondria in GSIS. One line of evidence comes from studies of mitochondrial DNA- depleted insulin secreting cells [25, 26]. Discovery of mitochondrial diabetes also highlight the importance of mitochondria for β-cell function [27]. Glucose activates the TCA cycle, several intermediates of which are points of origin for generation of coupling factors/second messengers, as discussed in detail below.
Citrate and isocitrate as sources of cytosolic NADPH
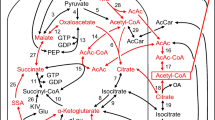
One of the most intensively studied candidate second messengers is NADPH, since it is a high energy compound produced upon cellular exposure to glucose [28]. NADPH is formed through metabolic pathways originating from citrate, isocitrate and malate. Malate exported from the mitochondria was suggested to be a source of cytosolic NADPH which is produced by Malic enzyme 1 (ME1). However, the naturally occurring ME1-KO mouse [29] is not glucose intolerant. Thus, the role of ME1 in the generation of NADPH is apparently not critical. As illustrated in Fig. 2, citrate exits to the cytosol through the citrate isocitrate carrier (CIC) and is metabolized by Aconitase 1 (ACO1) to isocitrate. TCA cycle isocitrate is also exported through CIC to the cytosol and, together with isocitrate produced from citrate by ACO1, is converted to α-ketoglutarate with reduction of NADP+ to NADPH by Isocitrate dehydrogenase 1 (IDH1). Important roles of IDH1 has been demonstrated by siRNA- or shRNA expression-mediated IDH1 knockdown experiments. IDH1 suppression resulted in reduced NADPH content and GSIS in INS-1 832/13 cells and rat islets [30]. Furthermore, CIC knockdown in INS-1 832/13 cells and rat islets resulted in reduced insulin secretion, supporting the possible role of citrate and isocitrate exiting the mitochondria in GSIS [31].

Coupling pathways originating from TCA cycle intermediates (citrate, isocitrate, α-ketoglutarate, oxoaloacetate). Citrate and isocitrate are directly exported to the cytosol. α-ketoglutarate is metabolized to glutamate in the mitochondria or in the cytosol. Mitochondrial PCK2 produces PEP from oxaloacetate. PEP is then exported back to the glycolysis. Subsequent metabolism of these molecules amplify insulin secretion. SCSG, GTP-specific succinate synthetase, GSSG, glutathione disulfide
Furthermore, a novel mechanism for boosting cytosolic NADPH generation was recently postulated [32]. By using isotomer analysis, α-ketoglutarate in the mitochondria was demonstrated to be metabolized, in the direction opposite that of the normal TCA flux, generating isocitrate via the mitochondrial isoform of NADP+-dependent IDH (IDH2). This is thought to be the pathway by which citrate and isocitrate are regenerated from α-ketoglutarate produced in the cytosol. GSIS from INS-1 832/13 cells treated with siRNA targeting IDH2 was reportedly reduced [32]. However, a different research group found that chronic inhibition of IDH2 by shRNA stable expression in INS-1 832/13 cells did not alter GSIS [33].
How is NADPH production linked to exocytosis of insulin granules? NADPH produced by IDH1 was thought to reduce glutathione (GSH) by glutathione disulfide reductase (GSR), and the resulting reduction in GSH in turn would activates glutaredoxin (GRX) [28]. Further studies revealed that GRX reduces cysteine disulfides of sentrin/SUMO-specific protease 1 (SENP1), thereby enhancing its deSUMOylation activity [34] (Fig. 2). SUMOylation is a post-translational modification of cellular proteins and serves to regulate protein stability, localization and function. [35]. Stimulatory glucose increases SENP1 activity and triggers the deSUMOylation of several proteins implicated in granule trafficking, including the Ca2+-sensing protein synaptotagmin VII [36].
Despite these elegant studies, however, CIC-KO mice have, very interestingly, been reported to show normal ability to secret insulin in response to glucose [37]. In the CIC- KO mice, NADPH increases are completely blocked in the islets. Furthermore, IDH1 knockdown suppresses GSIS in the wild type mice but not CIC-KO mice, suggesting accumulation of citrate due to IDH1 knockdown in the cytosol to be responsible [38]. Citrate accumulation may suppress glycolysis.
α-Ketoglutarate as a precursor of glutamate
Glutamate was first postulated to be a metabolic coupling factor based on experiments using permeabilized INS-1 cells [38]. In this cell preparation, INS-1 cells were permeabilized with α-toxin, and intracellular ATP and Ca2+ was clamped at sufficiently high levels for insulin secretion but otherwise remains normal [12]. Addition of glutamate, but not other mitochondrial intermediates augmented insulin secretion [12, 38]. In β-cells, α-ketoglutarate is metabolized to glutamate via glutamate dehydrogenase (GDH) in the mitochondria. Glutamate can also be generated from cytosolic α-ketoglutarate by aspartate aminotransferase (AST1) (Fig. 2). β-cell from mice with specific KO of GDH were reported to show reduced cellular glutamate content and insulin secretion in response to glucose [39]. In addition, mitochondrial glutamate transporter GC1 knockdown in INS-1E cells and rat islets reduced GSIS [40]. It was reported that GLP-1 potentiates GSIS through cAMP-activated glutamate uptake into insulin containing granules [41]. A similar mechanism might operate in glucose-activated β-cells, since glucose itself generates cAMP [42], although glucose-induced cAMP production is modest compared to that induced by incretins. How is glutamate production linked to exocytosis of insulin granules? It has been suggested that glutamate uptakes into insulin granules may modulate their pH and membrane potential, regulating insulin granule fusion with the plasma membrane [38, 41]. However, there is still much debate [43, 44] and the precise mechanisms have yet to be clarified.
Oxaloacetate as a precursor of phosphoenolpyruvate (PEP)
As mentioned earlier, it was recently discovered that mitochondrial PCK2 plays an important role of strengthening glycolysis in GSIS [21]. Pyruvate-derived oxaloacetate is metabolized via PCK2 to PEP, which is exported back to the cytosol, constituting a circular pathway, referred to the PEP cycle (Fig. 2). GTP produced in the mitochondria contributes to activation of this cycle. There are 2 isoforms of succinyl-CoA synthetase (SCS). SCS-ATP produces succinyl-CoA and ATP and another SCS-GTP produces succinyl-CoA and GTP. In INS-1 832/13 cells and rat islets, knockdown of SCS-ATP was observed to increase GSIS, while suppression of SCS-GTP reduced insulin secretion [45], suggesting that the mitochondrial GTP generated by SCS-GTP serves as a co-factor for mitochondrial PCK2 to enhance signal generation associated with insulin secretion.
Glycerolipid/free fatty acid cycle
Lipid signaling originating from glycolysis-derived glycerol-3-phosphate is also regarded as contributing to glucose-stimulated amplification of insulin secretion (Fig. 3). Malonyl-CoA generated from mitochondria-derived citrate plays an important role as a switch produced through glucose metabolism. Citrate exported to the cytosol is metabolized to acetyl CoA by ATP citrate lyase (ACLY). Acetyl-CoA in the cytosol is also produced from acetoacetate generated in the mitochondria of human islets [24]. This acetyl-CoA is then metabolized to malonyl-CoA by acyl-CoA carboxylase 1 (ACC1). Malonyl-CoA is an inhibitor of carnitine palmitoyltransferase 1 (CPT1) which catalyzes entry of fatty acyl-CoA (FA-CoA) into the mitochondria. Thus, inhibition of CPT1 suppresses β-oxidation of FA-CoA in the mitochondria under conditions of glucose abundance and facilitates FA-CoA proceeding through the glycerolipid/free fatty acid cycle, which is boosted by glycerol-3-phophate generated in the glycolysis (Fig. 3). An important role of malonyl-CoA production in this mechanism is supported by evidence that islets from β-cells specific ACC1-KO mice exhibit reduced GSIS [46]. In addition, mammalian glycerol-phosphate phosphatase was recently discovered, and demonstrated to degrade glycerol-3-phosphate to glycerol [47]. Overexpression of this enzyme reduced GSIS and shRNA-mediated knockdown augmented GSIS, indicating an importance of glycerol-3-phosphate provided by glycolysis in GSIS [47].

Glycerolipid/free fatty acid cycle, and pentose phosphate pathway. Glycerolipid metabolism initiated by glycerol-3-phosphate and pentose phosphate pathway originating from glucose-6-phosphate generate signals for activation of insulin exocytosis. ADSS, adenylosuccinate synthase, LPA, lysophosphatidic acid, DAG, diacylglycerol, TG, triglyceride
Conjugation of free fatty acid to glycerol-3-phosphate initiates glycerolipid/free fatty acid cycle. Triglycerides are formed and their lipolysis then generates monoacylglycerol (MAG), which activates Munc13-1. Munc13-1 is shown to be important for GSIS [48, 49]. In β-cells, the predominant MAG lipase, which hydrolyses MAG to glycerol and free fatty acid, is α/β-Hydrolase domain containing 6 (ABHD6). It has been reported that ABHD6-deficient and ABHD6-overexpressing β-cells showed enhanced and reduced GSIS, respectively, clearly indicating that ABHD6 plays an important role in augmentation of GSIS by the Glycerolipid/free fatty acid cycle [50].
Pentose-phosphate pathway
In addition to mitochondria-related pathways, recent studies revealed that pentose phosphate pathway could participate in GSIS (Fig. 3). This discovery arose from comprehensive metabolomics analysis of INS-1 832/13 cells [51]. These study uncovered significant increases in metabolites of the pentose phosphate pathway. This pathway generate NADPH, thus can be participate in the NADPH-deSUMOylation pathway as described earlier. Furthermore, cellular nucleotide profiling in INS-1 832/13 cells identified an increase in adenylosuccinate (S-AMP). Since S-AMP is generated from inosine monophosphate (IMP) by adenylosuccinate synthase (ADSS), this enzyme is postulated to play a regulatory role in S-AMP synthesis and β-cell glucose sensing (Fig. 3). It was reported that siRNA targeting to ADSS isoforms suppressed GSIS [52]. Interestingly, a candidate mechanism by which S-AMP increase insulin secretion involves the deSUMOylation protein SENP1. It is noteworthy that S-AMP-mediated insulin exocytosis is inhibited by siRNA targeting SENP1. Further analyses are anticipated to clarify how S-AMP activates insulin exocytosis via SENP1 activation.
Concluding remarks
Molecular cell biology experiments and metabolome analysis in engineered insulin secreting cell lines and rodent models as well as human islets in the past quarter century have obtained a large volume of information regarding how β-cells regulate the amounts of insulin secreted in response to demand for optimal glucose homeostasis. However, contradictory data have been reported. For example, whether or not PC is important remains uncertain [23, 24]. Several issues still under debate have been discussed in recent review articles [53, 54]. While such data are initially seemed challenging and even frustrating, they were soon proven to be useful for refining models or theories as well as for shaping our understanding of β-cell signal transduction. It is hoped that continued research efforts will fully clarify the interconnected molecular mechanisms underlying GSIS, and thereby ultimately lead to identification of target molecules for treatment, as well as novel strategies for managing, and perhaps even curing, diabetes.
References
IDF Diabetes Atlas. Diabetes around the world in 2021. https://diabetesatlas.org.
Ahmed SM, Haris B, Saraswathi S, Elawwa A, Khalifa A, et al. The epidemiology, clinical, biochemical, immunological and radiological features of youth onset type 2 diabetes mellitus in the state of Qatar. Diabet Int. 2021. https://doi.org/10.1007/s13340-021-00548-9.
Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese type II diabetic patients. Diabetologia. 2002;45:85–96.
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–10.
Spracklen CN, Horikoshi M, Kim YJ, Lin K, Bragg F, et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature. 2020;582:240–5.
Lane MA. The cytological characters of the areas of Langerhans. Am J Anatomy. 1907;7:409–22.
Hedeskov CJ. Mechanism of glucose-induced insulin secretion. Physiolo Rev. 1980;60:442–509.
Kojima I, Medina J, Nakagawa Y. Role of the glucose-sensing receptor in insulin secretion. Diabetes Obes Metab. 2017;19(Suppl 1):54–62.
Endo M. Reiji Natori, Setsuro Ebashi, and excitation-contraction coupling. Prog Biophys Mol Biol. 2011;105:129–33.
Calderón JC, Bolaños P, Caputo C. The excitation-contraction coupling mechanism in skeletal muscle. Biophys Rev. 2014;6:133–60.
Penner R, Neher E. The role of calcium in stimulus-secretion coupling in excitable and non-excitable cells. J Exp Biol. 1988;139:329–45.
Maechler P, Kennedy ED, Pozzan T, Wollheim CB. Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic beta-cells. EMBO J. 1997;16:3833–41.
Kono T, Tong X, Taleb S, Bone RN, Iida H, et al. Impaired store-operated calcium entry and STIM1 loss lead to reduced insulin secretion and increased endoplasmic reticulum stress in the diabetic β-cell. Diabetes. 2018;67:2293–304.
Sato Y, Aizawa T, Komatsu M, Okada N, Yamada T. Dual functional role of membrane depolarization/Ca2+ influx in rat pancreatic B-cell. Diabetes. 1992;41:438–43.
Gembal M, Gilon P, Henquin JC. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J Clin Invest. 1992;89:1288–95.
Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, et al. Overexpression of hexokinase I but not GLUT1 glucose transporter alters concentration dependence of glucose-stimulated insulin secretion in pancreatic beta-cell line MIN6. J Biol Chem. 1994;269:3081–7.
Vionnet N, Stoffel M, Takeda J, Yasuda K, Bell GI, et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356:721–2.
Sekine N, Cirulli V, Regazzi R, Brown LJ, Gine E, et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells potential low in nutrient sensing. J Biol Chem. 1994;269:4895–902.
Ishihara H, Wang H, Drewes LR, Wollheim CB. Overexpression of monocarboxylate transporter and lactate dehydrogenase alters insulin secretory responses to pyruvate and lactate in beta cells. J Clin Invest. 1999;104:1621–9.
Marqard J, Welters A, Bushmann T, Barthlen W, Vogelgesang S, et al. Association of exercise-induced hyperinsulinemic hypoglycemia with MCT1-expressing insulinoma. Diabetologia. 2013;56:31–5.
Zhao X, Capozzi ME, de Souza AH, Jahan I, Thomas CJ, et al. Pyruvate kinase controls signal strength in the insulin secretory pathway. Cell Metab. 2020;32:736–50.
Abulizi A, Cardone RL, Stark R, Lewandowski SL, Zhao X, et al. Multi-tissue acceleration of the mitochondrial phosphoenolpyruvate cycle improves whole-body metabolic health. Cell Metab. 2020;32:751–66.
MacDonald MJ. Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets further implication of cytosolic NADPH in insulin secretion. J Biol Chem. 1995;270:20051–8.
MacDonald MJ, Longacre MJ, Stoker SW, Kendrick M, Thonpho A, et al. Differences between human and rodent pancreatic islets. Low pyruvate carboxylase and pyruvate carboxylation and high glucose-stimulated acetoacetate in human pancreatic islets. J Biol Chem. 2011;286:18383–96.
Soejima A, Inoue K, Takai D, Kaneko M, Ishihara H, et al. Mitochondrial DNA is required for regulation of glucose-stimulated insulin secretion in a mouse pancreatic beta cell line, MIN6. J Biol Chem. 1996;271:26194–9.
Silva JP, Köhler M, Graff C, Oldfors A, Magnuson MA, et al. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet. 2000;26:336–40.
van den Ouweland JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, de Vijlder MF, et al. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet. 1992;1:368–71.
Ivarsson R, Quintens R, Dejonghe S, Tsukamoto K, Veld P, et al. Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes. 2005;54:2132–42.
Ronnebaum SM, Jensen MV, Hohmeier HE, Burgess SC, Zhou YP, et al. Silencing of cytosolic or mitochondrial isoforms of malic enzyme has no effect on glucose-stimulated insulin secretion from rodent islets. J Biol Chem. 2008;283:28909–17.
Ronnebaum SM, Ilkayeva O, Burgess SC, Joseph JW, Lu D, et al. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose- stimulated insulin secretion. J Biol Chem. 2006;281:30593–602.
Joseph JW, Jensen MV, Ilkayeva O, Palmieri F, Alárcon C, et al. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J Biol Chem. 2006;281:35624–32.
Zhang GF, Jensen MV, Gray SM, El K, Wang Y, et al. Reductive TCA cycle metabolism fuels glutamine- and glucose- stimulated insulin secretion. Cell Metab. 2021;33:804–17.
MacDonald MJ, Brown LJ, Longacre MJ, Stoker SW, Kendrick MA, et al. Knockdown of both mitochondrial isocitrate dehydrogenase enzymes in pancreatic beta cells inhibits insulin secretion. Biochem Biophys Acta. 2013;1830:5104–11.
Ferdaoussi M, Dai X, Jensen MV, Wang R, Peterson BS, et al. Isocitrate- to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional beta cells. J Clin Invest. 2015;125:3847–60.
Li N, Zhang S, Xiong F, Eizrik DL, Wang CY. SUMOylation, a multifaceted regulatory mechanism in the pancreatic beta cells. Semin Cell Dev Biol. 2020;103:51–8.
Dai XQ, Plummer G, Casimir M, Kang Y, Hajkrle C, et al. SUMOylation regulates insulin exocytosis downstream of secretory granule docking in rodents and humans. Diabetes. 2011;60:838–47.
Bauchle CJ, Rohli KE, Boyer CK, Pal V, Rocheleau JV, et al. Mitochondrial efflux of citrate and isocitrate is fully dispensable for glucose-stimulated insulin secretion and pancreatic islet β-cell function. Diabetes. 2021;70:1717–28.
Maechler P, Wollheim CB. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature. 1999;402:685–9.
Carobbio S, Ishihara H, Fernandez-Pascual S, Bartley C, Martin-Del-Rio R, Maechler P. Insulin secretion profiles are modified by overexpression of glutamate dehydrogenase in pancreatic islets. Diabetologia. 2004;47:266–76.
Casimir M, Lasorsa FM, Rubi B, Caille D, Palmieri F, et al. Mitochondrial glutamate carrier GC1 as a newly identified player in the control of glucose-stimulated insulin secretion. J Biol Chem. 2009;284:25004–14.
Gheni G, Ogura M, Iwasaki M, Yokoi N, Minami K, et al. Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Rep. 2014;23:661–73.
Dyachok O, Idevall-Hagren O, Sagetorp J, Tian G, Wuttke A, et al. Glucose-induced cAMP oscillations regulate pulsatile insulin secretion. Cell Metab. 2008;8:26–37.
Gammelsaeter R, Coppola T, Marcaggi P, Storm-Mathisen J, Chaudhry FA, et al. A role for glutamate transporters in the regulation of insulin secretion. PLoS ONE. 2011;6:e22960.
Zhou Y, Waanders LF, Holmseth S, Guo C, Berger UV, et al. Proteome analysis and conditional deletion of the EAAT2 glutamate transporter provide evidence against a role of EAAT2 in pancreatic insulin secretion in mice. J Biol Chem. 2014;2(89):1329–44.
Jesinkey SR, Madiraju AK, Alves TC, Yarborough OH, Cardone RL, et al. Mitochondrial GTP links nutrient sensing to β cell health, mitochondrial morphology, and insulin secretion independent of OxPhos. Cell Rep. 2019;28:759–72.
Cantley J, Davenport A, Vetterli L, Nemes NJ, Whitworth PT, et al. Disruption of beta cell acetyl-CoA carboxylase-1 in mice impairs insulin secretion and beta cell mass. Diabetologia. 2019;62:99–111.
Mugabo Y, Zhao S, Seifried A, Gezzar S, Al-Mass A, et al. Identification of a mammalian glycerol-3-phosphate phosphatase: role in metabolism and signaling in pancreatic beta-cells and hepatocytes. Proc Natl Acad Sci U S A. 2016;113:E430–9.
Kwan EP, Xie L, Sheu L, Nolan CJ, Prentki M, et al. Munc13-1 deficiency reduces insulin secretion and causes abnormal glucose tolerance. Diabetes. 2006;55:1421–9.
Kang L, He Z, Xu P, Fan J, Betz A, et al. Munc13-1 is required for the sustained release of insulin from pancreatic beta cells. Cell Metab. 2006;3:463–8.
Zhao S, Mugabo Y, Iglesias J, Xie L, Delghingaro-Augusto V, et al. α/β-Hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell Metab. 2014;19:993–1007.
Spegel P, et al. Time-resolved metabolomics analysis of beta-cells implicates the pentose phosphate pathway in the control of insulin release. Biochem J. 2013;450:595–605.
Gooding JR, et al. Adenylosuccinate is an insulin secretagogue derived from glucose- induced purine metabolism. Cell Rep. 2015;13:157–67.
Prentki M, Corkey BE, Madiraju SRM. Lipid-associated metabolic signaling networks in pancreatic beta cell function. Diabetologia. 2020;63:10–20.
Campbell JE, Newgard CB. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat Rev Mol Cell Biol. 2021;22:142–58.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
HI received a research grant from Astellas and donations from Boehringer Ingelheim, Daiichi Sankyo, Mitsubishi Tanabe, Eli-Lilly, and lecture fees from Novo Nordisk and Merck Sharp and Dohme.
Ethics statement
This article does not contain any studies with human or animal subjects performed by the author.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Ishihara, H. Metabolism-secretion coupling in glucose-stimulated insulin secretion. Diabetol Int 13, 463–470 (2022). https://doi.org/10.1007/s13340-022-00576-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13340-022-00576-z