
Abstract
Notch signaling is a simple pathway in its mechanism and complex because it activates many genes at transcriptional level. Notch plays an imperative role in embryogenesis and progenitor/stem cell maintenance and critically preserves the balance between a cell proliferation, differentiation, and apoptosis. Mutation and deregulation of Notch will be a reason for many diseases including cancer. Aberrantly expressed Notch involves in the carcinogenesis of many cancers such as colorectal cancer (CRC), breast cancer, pancreatic cancer, prostate cancer, liver cancer, cervical cancer, Ewing sarcoma, Kaposi’s sarcoma, lung cancer, ovarian cancer, and lymphoma. Among these, Notch plays a major role in CRC from development to metastasis. CRC occurs when the lining of colon epithelial cells becomes neoplasm, and this happens when the poise is missing between the normal and cancerous condition due to the overexpression of Notch. In CRC Notch promotes the stemness and epithelial to mesenchymal transition (EMT) which are requiring in aberrant crypt formation, invasion, and metastasis, respectively. Moreover, overexpression of Notch in CRC is connected with poor prognosis and chemoresistance. Notch counteracts with Hippo/YAP, WNT, NFkB, PI3K/Akt, and EGFR pathways for tumor initiation and development. Tumors are dependent on angiogenic switch for development and invasion which is activated by Notch ligand Dll-4 by overexpressing vascular endothelial growth factor (VEGF). This review will be focusing on the uniqueness of Notch in CRC.
The original version of this chapter was revised. The book was inadvertently published without Abstracts and Keywords, which are now included in all the chapters. An erratum to this chapter can be found at https://doi.org/10.1007/978-981-10-6728-0_39
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
21.1 Introduction
Colorectal cancer (CRC) is the third most common cancer and reason for 610,000 deaths per year worldwide. The death in CRC is mostly due to liver metastasis, tumor recurrence, chemoresistance, and cancer stem cells (CSCs). Besides, the deregulated signaling pathways include WNT, Notch, EGFR, and TGF-β acting a key role in CRC development [26]. Among these, Notch pathways poise a pivotal role in CRC tumor initiation, tumor recurrence, metastasis, poor prognosis, chemoresistance, and maintains of CSCs [8]. Notch is a single transmembrane receptors composed of four receptors Notch 1–4, along with five canonical ligands, Jagged (Jag)1, Jag2, Delta like (DII) 1, Dll3, and Dll4. Notch plays an imperative role in embryogenesis and progenitor/stem cell maintenance and critically preserves the balance between a cell proliferation, differentiation, and apoptosis [8]. Mutation or deregulation of Notch signaling pathway leads to many diseases including cancer. Aberrantly expressed Notch involves in the carcinogenesis of many cancers such as CRC, breast cancer, pancreatic cancer, prostate cancer, liver cancer, cervical cancer, Ewing sarcoma, Kaposi’s sarcoma, lung cancer, ovarian cancer, and lymphoma [23]. Among these, Notch role in CRC is crucial from its initiation to metastasis. So understanding the role of Notch in CRC will pave a way to find new therapeutic targets for the treatment of CRC.
21.2 Notch Structure
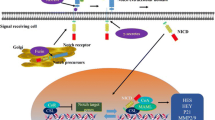
Notch is a single transmembrane juxtacrine-mediated signaling pathway. Notch family consists of four type-1 transmembrane receptors, Notch 1–4. These are synthesized in precursor form and undergo subsequent cleavages to form mature active Notch receptor. The first cleavage is by furin-like convertase at trans-Golgi apparatus and forms extracellular domain, intracellular domain, and transmembrane domain. The extracellular domain is made up of 36 EGF-like repeats. The C-terminal of the EGF repeats consists of three cysteine-rich lineage defective-12 (LIN-12) and Notch repeats (LNR) which prevents receptor activation in the absence of ligand. Following this, it consists of heterodimerization domain and transactivation domain. Intracellular domain consists of RBPJ-associated molecule (RAM) domain, two nuclear localization signals, 6 ankyrin (ANK) repeats, and a PEST (proline- glutamine-serine-threonine) sequences which are collectively called as Notch intracellular domain (NICD) (Fig. 21.1) [3, 11, 15]. The sequence of proteolytic cleavage, cleavage site, and enzymes involved in cleavage are given in Table 21.1.

Notch structure: EGF, epidermal growth factor: LNR, Lin12 Notch repeats; HD, heterodimerization domain: TMD, transmembrane domain:, ANK, ankyrin repeats: TAD, transactivation domain: and PEST, proline-glutamic acid-serine-threonine
21.3 Notch Mechanism
Notch is a single transmembrane receptor with extracellular, transmembrane, and intracellular regions, and it is activated by sequence of proteolytic cleavages. The pathway activation is initiated by binding of one of the five (Jag1 and Jag2, DLL-1, DLL-3, and DLL-4) ligands which leads to the conformational changes in Notch receptors. Subsequently, S2 cleavage site is exposed for ADAM to remove the extracellular region. Following this, S3 cleavage occurs by γ-secretase (complex of nicastrin and presenilin-1) which removes the transmembrane region and releases Notch intracellular domain (NICD). The NICD is accumulated in cytoplasm and then translocated to the nucleus by concentration gradient where it binds with transcriptional regulator termed as C-protein binding factor 1 [(CBF1) also known as RBP-Jk or CSL for CBF1/Su(H)/Lag1)] leading to the displacement of corepressor and brings coactivators such as MAML-1 to activate the expression of hairy and enhance of split (Hes) and Hes-related repressor protein (Hey) families [1, 6, 7] (Fig. 21.2).

Notch signaling pathway. 1. Notch receptor is cleaved by furin-like convertase in trans-Golgi to produce mature Notch receptor. 2. One of the five (Jag1, Jag2, Dll-1, Dll-3, and Dll-4) binds from instructing cell to activate signaling pathway. 3. Exposure of S2 cleavage site and cleaved by ADAM enzyme to extracellular region. 4. S3 cleavage carried out by γ-secretase to release Notch intracellular domain (NICD). 5. NICD is then translocated to nucleus and activates target gene expression
21.4 Notch and Transcription Factors
Once ligand binds to the Notch receptor, it can activate proteolytic cleavage mediated by gamma-secretase lead to release NICD. NICD is acting as a transcription coactivator, but it can’t bind to DNA, because it doesn’t have a zinc finger domain. Consequently, they can recruit RBP-J and MAML1–3 to activate transcription genes through binding with DNA. Once they are formed, it can recruit chromatin remodeling complex, which consists of histone deacetylase or histone acetylase proteins. In the absence of ligand or transcription coactivator of NICD, the RBP-J acts as a repressor. Notch activates many transcription factors directly and indirectly such as HES1, HEY1, TGFβ, NF-κB, snail, slug, PTEN, and cyclin D3 [14].
21.5 Notch in Colorectal Cancer
Colon is the part of digestive system is lining with absorptive and secretory epithelial cells interspersed with deep crypts which consists of 2000 cells with 5–10 stem cells [19]. CRC is identified with aberrant crypt formation intervened by Notch pathway. Notch signaling regulates the cell proliferation and differentiation of epithelial cells to absorptive or secretory cells, maintaining apoptosis in the colon. The deregulated Notch will fail to spot the poise and leads to the formation of cancer [26]. Notch-mediated tumor initiation mechanism in CRC is not well understood, but the possible reason will be overexpression of mutated Notch receptors, ligands, and Notch signaling interplay between other signaling pathways. Notch is highly expressed in the primary stage of CRC relatively than the later stage. Notch1 and Hes1 are highly expressed in primary and metastatic CRC than colonic mucosa [19]. The other reasons could be (i) mutation of E3 ubiquitin ligase which is responsible for NICD degradation; (ii) mutation in APC leads to the overexpression of NICD [28]; (iii) the decreased expression of atonal homolog1 (ATOH1) which is essential for intestinal secretory cell commitment; and (iv) Hes1, the direct target of Notch, is repressor for KLF4 which inhibits CRC cell proliferation when overexpressed [2]. Other than this Notch interplays with many signaling pathways in CRC such as WNT, NF-κB, EGFR, and TGFβ. Notch signaling can be an oncogenic or tumor suppressor; it depends on the context of cells in where cancer occurs [24].
21.5.1 Notch Interplay with Signaling Pathways in CRC
Wnt/β-catenin signaling is the essential pathway which is highly articulated in 80% of sporadic CRC, and it is the main reason for inherited familial adenomatous polyposis (FAP)-related CRC [23]. Wnt/β-catenin signaling is the key pathway involved in the transformation of epithelial cells into self-renewing CSCs for CRC development. Wnt/β-catenin signaling activates its target Jag1 that trigger the Notch and increase its expression in CRC. Most of the CRC carries the mutation of APC which is the component of Wnt signaling pathway. In Apc-mutated condition, Notch signaling is requisite for consequent development of CRC [4, 26, 27]. PI3/AKT pathway is upregulated in 40% of human CRC, and it is important for cell survival and growth. Notch activates its target HES1 which inhibits PTEN leading to the activation and overexpression of PI3/AKT pathway in CRC. Notch involves in colonic carcinogenesis by stimulating EGFR pathway. Notch signaling arbitrated activation of transcription factor NF-κB in CRC results in chemoresistance of CRC cells [10]. Notch also inhibits the activity of transcription factor TGF-β pathway which is important for tumor suppressor and cell growth inhibition. In CRC, TGF-β-mediated induction of Jag1 results in the overexpression of Notch [30]. Angiogenesis is the formation of new blood vessels which is an important characteristic of growing tumor. Inhibiting or disrupting angiogenesis will be the great therapeutic way to inhibit or treat cancer [13]. Jag1 Notch ligand activates the Notch-dependent angiogenesis by activating VEGF in CRC [21]. Notch signaling upregulates the expression of anti-apoptotic genes including Bcl2 and BclXL. With these Notch also upregulates the expression of cyclin D1 and p21/WAF1 which are involved in cell cycle regulation and cell proliferation [23].
21.5.2 Notch Interaction with EMT Pathway
An epithelial mesenchymal transition (EMT) pathway plays an important role in key biological process of embryogenesis and metastatic cancer development [31]. The basement-degraded epithelial cells migrate to form new types of mesenchymal cells in order to increase the motility and invasion [16]. During this EMT process, the epithelial cell loses the cell junction followed by the reorganization of the actin cytoskeleton to initiate the transition by the relocation of epithelial markers such as E-cadherins and integrins from cell membrane to the nucleus. These could gain the expression mesenchymal markers such as N-cadherins, vimentin, and fibronectin [30]. Moreover, the growing report indicates that cancer stemness and chemoresistant mechanism are achieved by high rates of EMT signaling pathway [8], especially its interaction with oncogenic signaling pathways like MMP-3, BCL9-2, EGFR, met, goosecoid, kaiso, TGF-β, FOXC2, GSK-3β, Smad-3, Pez, Snail1, Snail2, ILK, and Notch [30]. Interestingly, the recent report revealed that the growing incidence of colon cancer is due to overexpression of Notch and its regulation of EMT pathway [9]. Notch signaling can regulate Snail1 expression through the induction of hypoxia inducible factor 1α (HIF-1α). Subsequently the HIF-1α binds the promoter region of LOX (lysyl oxidase) for stabilization of Snail1 and its transcription. Likewise Notch interact Snail2 leads to repression of E-cadherin and β-catenin activation [25]. Moreover, targeting of Notch signal leads to downregulation of EMT and induction of p21 in human colon cancer cells [5].
21.5.3 Targeting Notch Signal for CRC Treatment
The gamma-secretase inhibitors (GSI) compounds treated CRC showed inhibition of the γ-secretase, which is a mandatory enzyme to activate Notch internal signaling, but it induces extracellular signal-regulated kinases (Erk) for continued cell proliferation. However, the GSI combined with cisplatin drug-treated cells exhibited the downregulation of Erk and activation of cell death [2]. The combined treatment of biphenolic compound and ionizing radiation (IR) treated in CRC results revealed the downregulation of the Notch, CRC stem cell protein marker DCLK1, and downstream target gene of Hes1. These downregulation genes can activate the apoptosis pathway. A similar result has observed in tumor xenograft animal model [22]. Interestingly the flavonoid compound epigallocatechin-3-gallate-treated cells has showed promising activity toward the inhibition of β-catenin, but quercetin showed weak inducer for downstream target genes reported in human colon carcinoma cell line HT29 [20]. The report of [12] indicates that the bioactive compound of Withaferin-A isolated from Withania somnifera-treated colon cancer cells exhibited downregulation of Notch signal by induction of c-Jun-NH2-kinase-dependent apoptosis. These mechanisms can reduce the expression of Akt/NF-κB/Bcl-2 and mammalian target of rapamycin signaling components (pS6K and p4E-BP1). Likewise, a Ƴ-secretase inhibitor can induce the differentiation of goblet cell from the proliferative crypt in mice carrying mutated gene for an Apc tumor suppressor. Similar result has seen in blocking of Notch signal with Ƴ-secretase inhibitor [29]. These reports further support the interaction of Wnt and Notch signaling pathways. Moreover, the Jagged1 was silenced by lentiviral Jagged1-shRNA resulted in decrease of cell viability. Besides, the knockdown studies showed the induction induced cell cycle arrest at G0/G1 phase with reduced expression of cyclin D1, cyclin E, and c-Myc in vitro colon cancer cell. However, the knockdown studies of xenograft mouse model have reflected the same results of in vitro [6]. The small molecular inhibitor FLLL32-treated colon cancer stem cell has seen the inhibition of STAT3 through the downregulation of survivin, Bcl-XL, and Notch signal. Moreover, the significant inhibition of tumorsphere formation and induction of cleaved caspase-3-mediated apoptosis observed in small molecule-treated cells are compared to curcumin-treated cells [17].
Curcumin analogue GO-Y030 treatment has shown the inhibition of activated STAT3 followed by reduction of cyclin D1, survivin, Bcl2, Notch, and p-RP in colon cancer cells. These reductions can facilitate the cleaved caspase-3-mediated apoptosis. Moreover, GO-Y030 treatment reduces the tumor growth in mouse xenografts with SW480 and HCT-116 colon cancer stem cells in dose-dependent studies [17]. However, Meng et al. [18] reported that the oxaliplatin, 5-fluorouracil (5-FU), and SN-38 compound-treated cell revealed the induction of the Notch-1 intracellular domain (NICD)-mediated chemoresistance toward cancer cell progression through the activation of Ƴ-secretase. Subsequently, silencing of Ƴ-secretase subunit nicastrin by small interfering RNA (siRNA) prevented activation of NICD after the treatment of oxaliplatin. Concurrently, the silencing or blocking of NICD by sulfonamide GSI (GSI34)-treated colon cancer cells sensitizes to chemotherapy compounds through downregulation of phosphoinositide kinase-3/Akt. The inhibition of Notch signaling leads to suppression of tumor invasion and intravasation in genetic depletion Aes in ApcD716 mice. These reports further confirm the inhibition of Notch potential target for control and treatment of metastasis colon cancer [28].
21.6 Conclusion
Notch plays a major role in normal development and cancer condition. Its role in CRC cancer is very important from cancer progression to metastasis. This review explained about Notch and its transcription factor role in CRC mainly in metastasis and targeting Notch pathway in CRC. Targeting Notch will be a great therapeutic source to treat the CRC.
References
Ables JL, Breunig JJ, Eisch AJ, Rakic P (2011) Not(ch) just development: notch signalling in the adult brain. Nat Rev Neurosci 12(5):269–283
Aleksic T, Feller SM (2008) Gamma-secretase inhibition combined with platinum compounds enhances cell death in a large subset of colorectal cancer cells. Cell Commun Signal 6:8
Andersson ER, Lendahl U (2014) Therapeutic modulation of notch signalling – are we there yet? Nat Rev Drug Discov 13(5):357–378
Bertrand FE, Angus CW, Partis WJ, Sigounas G (2012) Developmental pathways in colon cancer. Crosstalk between WNT, BMP, hedgehog and notch. Cell Cycle 11(23):4344–4351
Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I (2015) Targeting the translation machinery in cancer. Nat Rev Drug Discov 14(4):261–278
Dai Y, Wilson G, Huang B, Peng M, Teng G, Zhang D, Zhang R, Ebert MP, Chen J, Wong BC, Chan KW, George J, Qiao L (2014) Silencing of Jagged1 inhibits cell growth and invasion in colorectal cancer. Cell Death Dis 5:e1170
D’Souza B, Miyamoto A, Weinmaster G (2008) The many facets of notch ligands. Oncogene 27(38):5148–5167
Espinoza I, Miele L (2013) Deadly crosstalk: notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett 341(1):41–45
Fender AW, Nutter JM, Fitzgerald TL, Bertrand FE, Sigounas G (2015) Notch-1 promotes stemness and epithelial to mesenchymal transition in colorectal cancer. J Cell Biochem 116(11):2517–2527
Fernández-Majada V, Aguilera C, Villanueva A, Vilardell F, Robert-Moreno A, Aytés A, Real FX, Capella G, Mayo MW, Espinosa L, Bigas A (2007) Nuclear IKK activity leads to dysregulated notch-dependent gene expression in colorectal cancer. Proc Natl Acad Sci U S A 104(1):276–281
Haines N, Irvine KD (2003) Glycosylation regulates notch signalling. Nat Rev Mol Cell Biol 4(10):786–797
Koduru S, Kumar R, Srinivasan S, Evers MB, Damodaran C (2010) Notch-1 inhibition by Withaferin-A: a therapeutic target against colon carcinogenesis. Mol Cancer Ther 9(1):202–210
Kofler NM, Shawber CJ, Kangsamaksin T, Reed HO, Galatioto J, Kitajewski J (2011) Notch signaling in developmental and tumor angiogenesis. Genes Cancer 2(12):1106–1116
Kopan R (2002) Notch: a membrane-bound transcription factor. J Cell Sci 115(Pt 6):1095–1097
Leong KG, Karsan A (2006) Recent insights into the role of notch signaling in tumorigenesis. Blood 107(6):2223–2233
Lee JM, Dedhar S, Kalluri R, Thompson EW (2006) The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 172(7):973–981
Lin JA, Chen JH, Lee YW, Lin CS, Hsieh MH, Chang CC, Wong CS, Chen JJ, Yeh GC, Lin FY, Chen TL (2011) Biphasic effect of curcumin on morphine tolerance: a preliminary evidence from cytokine/chemokine protein array analysis. Evid Based Complement Alternat Med 452153:1–11
Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB, Schwartz GK (2009) Gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res 69(2):573–582
Miyamoto S, Rosenberg DW (2011) Role of notch signaling in colon homeostasis and carcinogenesis. Cancer Sci 102(11):1938–1942
Pahlke G, Ngiewih Y, Kern M, Jakobs S, Marko D, Eisenbrand G (2006) Impact of quercetin and EGCG on key elements of the Wnt pathway in human colon carcinoma cells. J Agric Food Chem 54(19):7075–7082
Peignon G, Durand A, Cacheux W, Ayrault O, Terris B, Laurent-Puig P, Shroyer NF, Van Seuningen I, Honjo T, Perret C, Romagnolo B (2011) Complex interplay between β-catenin signalling and notch effectors in intestinal tumorigenesis. Gut 60(2):166–176
Ponnurangam S, Mammen JM, Ramalingam S, He Z, Zhang Y, Umar S, Subramaniam D, Anant S (2012) Honokiol in combination with radiation targets notch signaling to inhibit colon cancer stem cells. Mol Cancer Ther 11(4):963–972
Qiao L, Wong BC (2009) Role of notch signaling in colorectal cancer. Carcinogenesis 30(12):1979–1986
Radtke F, Raj K (2003) The role of notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer 3(10):756–767
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U (2008) Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A 105(17):6392–6397
Sanchita R, Majumdar PNA (2012) Signaling in colon cancer stem cells. J Mol Signal 7:11
Sikandar SS, Pate KT, Anderson S, Dizon D, Edwards RA, Waterman ML, Lipkin SM (2010) NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res 70(4):1469–1478
Sonoshita M, Aoki M, Fuwa H, Aoki K, Hosogi H, Sakai Y, Hashida H, Takabayashi A, Sasaki M, Robine S, Itoh K, Yoshioka K, Kakizaki F, Kitamura T, Oshima M, Taketo MM (2011) Suppression of colon cancer metastasis by Aes through inhibition of notch signaling. Cancer Cell 19(1):125–137
van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, Clevers H (2005) Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435(7044):959–963
Wang Z, Li Y, Kong D, Sarkar FH (2010) The role of notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets 11(6):745–751
Yuan J, Liu M, Yang L, Tu G, Zhu Q, Chen M, Cheng H, Luo H, Fu W, Li Z, Yang G (2015) Acquisition of epithelialmesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: a new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and β1-integrin signaling pathway in tumor cells. Breast Cancer Res 17(1):69
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd
About this chapter

Cite this chapter
Rajendran, D.T., Subramaniyan, B., Ganeshan, M. (2017). Role of Notch Signaling in Colorectal Cancer. In: Nagaraju, G., Bramhachari, P. (eds) Role of Transcription Factors in Gastrointestinal Malignancies. Springer, Singapore. https://doi.org/10.1007/978-981-10-6728-0_21
Download citation
DOI: https://doi.org/10.1007/978-981-10-6728-0_21
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-6727-3
Online ISBN: 978-981-10-6728-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)