
Abstract
Notch signaling pathway is evolutionarily conserved in mammals, which plays an important role in cell development and differentiation. In recent years, increasing evidence has shown that aberrant activation of Notch is associated with tumor process. Aberrant activation of Notch signaling pathway has been found in many different solid tumors can induce cell proliferation, metastasis and epithelial-mesenchymal transition. Notch receptor and its ligand are both single transmembrane protein, and Notch is activated when it binds to the Notch ligand of neighbor cells. The signal transduction of Notch signaling pathway is only between cells that are in contact with each other, which is independent of second messengers. Thus, Notch needs to cross talk with other signaling pathways, including PI3K/AKT, NF-κB, integrin and miRNAs, to precisely regulate cell fate. In this review, we summarize the roles of Notch signaling pathway in tumor metastasis and its regulatory mechanisms and discuss the current treatment strategies targeting Notch signal pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is a major public health problem in the word. The latest statistics report in 2017 indicates that lung cancer, colon cancer, breast cancer, and pancreas cancer are still the most common causes of cancer death according to the American Cancer Society, and these four cancers account for 46% of all cancer deaths [1]. The main reason for the high mortality of cancer is due to the highly invasive behavior of cancer cells, which usually results in cancer progression and metastasis. Metastasis is responsible for the vast majority of cancer-related death, so it is clear that the mechanism of cancer metastasis deserves urgent attention. The process of tumor metastasis is extremely complicated, which must (1) detach from the primary tumor, (2) adhere to extracellular matrix (ECM) and degrade it, (3) invade into bloodstream or lymphatic system, (4) tumor angiogenesis, (5) proliferate at the secondary site to form metastases [2]. Metastasis is a characteristic of malignant behavior of the tumor and is responsible for the failure of clinical treatment. Therefore, finding targets to treat or prevent tumor metastasis is an important therapeutic priority [3]. Progression toward a metastatic phenotype requires a concerted effort between different molecules that have been implicated in advancing of metastasis. It is a complex process controlled by multi-genes. Accumulated evidences show that Notch gene is aberrantly activated in many human malignancies [4,5,6,7]. Overexpression of Notch and its ligands has been shown to be associated with poor prognosis in breast cancer [4, 8, 9].
Notch signaling pathway is evolutionarily conserved which was first identified to play important role in diverse development progress. In recent decades, there are considerable studies suggesting that Notch signaling pathway is implicated in the development progress of cancer. In this review, we summarize the Notch signaling and its cross talk networks in tumor metastasis.
Notch structure and function
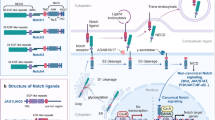
Notch was discovered by appearance of a notch in the wing of fruit fly [10], which was subsequently found to have an important role in embryonic development. Notch receptor is type I single-pass transmembrane protein, which is encoded by 2370 amino acids [11]. Notch signaling pathway consists of three parts: Notch receptor, Notch ligand and DNA binding sequence CSL (CBF1/Su(H)/Lag-1). In mammals, there are four isoforms of Notch receptors (Notch-1, Notch-2, Notch-3 and Notch-4 [12]) and five Notch ligands (Dll-1, Dll-3, Dll-4, Jagged-1 and Jagged-2 [13]). All the Notch receptors exhibit the same overall structures: 36 homologous epidermal growth factor (EGF)-like tandem repeats and three Lin-12/Notch repeats (LNR) in the extracellular domain; a CSL-binding domain RAM, a PEST sequence, and seven ankyrin-like repeat (ANK) in the intracellular domain [14]. Notch ligands are also type I transmembrane proteins, containing extracellular EGF-like repeats, a DSL (Delta/Serrate/Lag-2) motif accounting for Notch interaction and short and quite divergent intracellular domains [15]. Notch signaling pathway will be activated, when ligand binds to Notch receptor between two adjacent cells, to determine cell fate and regulate organ formation and morphogenesis. Classic Notch activation requires three steps of proteolytic cleavage (Fig. 1): First step, Notch single-stranded precursors are cleaved by Furin proteases in Golgi complex to form a large fragment containing extracellular domain and a small fragment containing transmembrane regions and intracellular regions, which are combined by Ca2+-dependent non-covalent bond to form a mature heterodimer receptor and transferred to cell membrane. When the mature receptor binds to the ligand, it undergoes a second cleavage by TACE or Kuz, which belongs to ADAM (A Disintegrin and Metalloprotease) metalloproteinase family, to release the extracellular fragment. The remainder fragment, containing transmembrane domain and intracellular domain, is cleaved for the third time by γ-secretase to release soluble Notch intracellular domain (NICD) and transferred to the nucleus. The NICD binds to transcription factor CSL in nucleus, leading to downstream gene transcription [16]. Besides CSL-dependent activation, Notch signaling pathway also has CSL-independent activation [17], but most Notch target genes have CSL binding site [18]. The Notch signal transduction is second messenger independent, which directly accepts the adjacent cell signaling and transfers to the nucleus, initiating the downstream transcription factor expression. Notch signaling conduction cannot amplify the signal, but it is necessary for precise control of cell differentiation in the initiation process. Notch target genes vary in different tissue and different cells, such as HES and HEY families, MMP-2, MMP-9, cyclin D1, Her2, c-Myc, p21 and cell apoptosis-associated genes. Notch precisely regulates cell differentiation, proliferation and tumorigenesis might by modulating these genes expression.

Schematic diagram of the Notch signaling pathway. Notch single precursors are cleaved by furin in Golgi apparatus to form mature Notch receptors and translocate to plasmic membrane. Notch is activated when Notch ligands on adjacent cells bind to it, resulting in the second and the third cleavage by ADAM and γ-secretes, releasing Notch intracellular domain NICD, which translocates into the nucleus and binds to CSL to initiate the expression of downstream genes
Notch signaling pathway in tumorigenesis
Notch signaling pathway was found to maintain the undifferentiated state of cells through paracrine inhibition and induce cell differentiate under appropriate stimulation at early stage. Subsequent studies had found that Notch not only regulates cell differentiation, but also participates in the development of various tissues. Accumulated evidence showed that dysregulated Notch signaling is involved in tumorigenesis and cancer development. Notch signaling directly leads to not only tumorigenesis, but also cross talks with other signaling pathways to induce carcinoma [19].
The carcinogenicity of Notch signaling pathway was initially identified in human T cell acute lymphoblastic leukemia (T-ALL), where Notch-1 was found to be activated, which was caused by the release of Notch-1 intracellular domain (N1ICD) [20]. Notch-1 signaling pathway was found to be aberrantly activated in more than 50% T-ALL patients [21], to activate NF-κB by inhibiting ubiquitin carboxyl-terminal hydrolase (CYLD) [22]. Subsequent studies indicated that Notch family were aberrantly activated in the tumorigenesis of many cancers. e.g., breast cancer [23], pancreatic cancer [24] and cervical cancer [25].
Notch is known to function as both oncogene and tumor suppressive gene, which depends on the tumor cell type and tissue type. As an oncogene, Notch is overexpressed in breast cancer [23], gastric cancer [26], pancreatic cancer [24] and colon cancer [27]. However, Notch expression is down-regulated in skin cancer [28], liver cancer [29], prostate cancer [30], non-small cell lung cancer [31] and some breast cancers [32], where Notch acts as a tumor suppressor gene. Moreover, Notch also plays a different role in cancer cells at different times. Notch-1 promotes tumor growth at early stage of cervical cancer, while it inhibits tumor growth at the late stage [33]. Whether Notch acts as a tumor suppressor gene or an oncogene is determined by the microenvironment. The microenvironment factors includes the type of Notch receptors, cell type, Notch activation state, and other signaling pathways which Notch is cross talking with.
Notch signaling in tumor metastasis
Tumor metastasis is the leading reason to the poor prognosis of various cancers. There are several theoretical models trying to decipher the mechanism of tumor metastasis, among which the commonly accepted mechanism of metastasis is the epithelial-mesenchymal transition (EMT) theory. During the process of tumor metastasis, tumor cells in primary tumor lose cell–cell adhesion and invade into the blood through intravasation to form circulating tumor cells (CTCs). These CTCs re-adherent and exit the bloodstream to form micrometastases at the other places suitable for the growth. In these processes, these cells loss the mesenchymal characteristics and transited into epithelial cells (MET). Thus, EMT and MET form the initiation and completion of the invasion-metastasis cascade [34]. EMT process is regulated by a variety of signaling pathways. Growth factors, such as HGF, FGF, TGF-β, initiate downstream signaling transduction through RTK, cMET and ERK/MAPK cascade signaling pathways. The PI3K/AKT and MAPK signaling pathways can directly induce the transcription of EMT-related genes (snail, slug and twist, etc.), thereby inducing EMT [35]. Accumulated evidence has confirmed that Notch is a key regulator for EMT. NICD can directly bind to the promoter of snail-1, a critical regulator of EMT, to upregulate snail-1 expression. Additionally, overexpression of NICD also inhibits the expression of E-cadherin to induce EMT [36].
Hypoxia/HIF-inducing EMT is a well-known phenomenon which has been implicated in numerous cancers [37]. Tumor cell migration and invasion in hypoxia are significantly accelerated. Notch-1 can replace hypoxia to induce cell migration and invasion in ovarian cancer [36]. What is more, overexpression of Notch-1 enhances cell invasion in breast cancer [38] and lung cancer [39] by promoting EMT. Notch signaling pathway has been found to upregulate the expression of Lysyl oxidase (LOX), which was implicated in stabilizing Snail protein and hypoxia-induced tumor invasion [40, 41], leading to the increase in snail-1 protein in hypoxia [36].
During EMT, tumor cell skeleton undergoes rearrangement, cell stiffness, and cell–cell connectivity decreases, facilitating tumor cell migration and invasion [42]. Cell mechanical properties depend on cytoskeletal fibrous structures, including actin filaments, microtubules and intermediate filaments [43]. Thus, down-regulating Vimentin, an intermediate filamentous protein, leads to cell motility inhibition. It has been found that Notch receptors are linked to myosin and affect cell motility [44].
The re-adherent cells need to extravasate vascular endothelial cells into tissue, which involves the degradation of ECM and the formation of pseudopods. Pseudopods are specific regions of the cell membrane, containing adhesion and proteolytic enzymes, to help cell adhesion, migration and extracellular proteolysis. Therefore, inhibiting the ability of cancer cells to form pseudopodia can limit tumor metastasis and invasion. Hypoxia can induce the pseudopodia formation of cancer cells in vitro, accompanied with F-actin aggregation at the leading edge of invasive cells. Diaz, B., et al. had found that blocking Notch-1 signaling pathway abrogated hypoxia-induced pseudopodia formation and F-actin aggregation [45]. Heparin-binding EGF-like growth factor (HB-EGF) [46] is synthesized as a membrane-anchored growth factor (pro-HB-EGF) [47]. The extracellular domain of pro-HB-EGF cleaved by metalloproteases releases a soluble form of HB-EGF, which potentiates tumor growth and angiogenesis [48]. ADAM12, a hypoxia effector, is regulated by Notch-1 and also a shedding enzyme of HB-EGF. Hypoxia-induced invasion pseudopodia formation through HB-EGF-activated EGFR signaling pathway is Notch-1 dependent. Studies have shown that hypoxia increases ADAM12 expression through the activation of Notch-1, thereby promoting the invasion pseudopodia formation induced by HB-EGF [45].
Matrix metalloproteinase (MMP) is an essential regulator of ECM degradation and is important for cancer cell metastasis. Among the MMP family, MMP-2 and MMP-9 could specifically degrade collagen type IV, which are abundant in ECM [49]. It has been reported that Notch-1 activation induces the MMP-2 and MMP-9 expression and activation, while down-regulation of Notch-1 decreases the MMP-2 and MMP-9 activation to inhibit cell metastasis in breast cancer cells and pancreatic cancer cells [50, 51].
These studies demonstrated that Notch signaling pathway plays an important role in every step of tumor metastasis process. Although Notch signaling pathway does not have second messenger to amplify signals, it precisely regulates the cell fate through cross talking with other signaling pathways (Fig. 2).

The Notch signaling pathway cross talking network in regulating cell metastasis. Notch signaling pathway is aberrant activated in tumor cells, which is associated with NF-κB and AKT hyperactivation. Notch signaling pathway is regulated by many other pathways, including CCN1, IL-6/IL-6R, Cav-1 and integrins
Notch signaling and NF-κB
Nuclear factor Kappa-light-chain-enhancer of activated B cells (NF-κB) is a transcriptional complex, being involved in physiological processes by regulating cell immune, differentiation, apoptosis, migration and invasion. A considerable number of studies have shown that aberrantly activated NF-κB promotes tumorigenesis and leads to tumor malignancy by inducing the expression of genes associated with survival, metastasis and carcinogenesis [52, 53].
Activated NF-κB upregulates the expression of Jagged-1, a ligand for Notch signaling pathway, in breast cancer stem cells, suggesting that NF-κB activation promotes the production of breast cancer stem cells by activating Notch signaling pathway [54]. More evidence has shown that Notch signaling pathway leads to NF-κB activation through IKK phosphorylation [19]. Notch-1 activation can induce AKT phosphorylation, resulting in Wnt/β-catenin and NF-κB activation to facilitate the migration and invasion of glioblastoma [55]. In breast cancer cells, suppressed Notch-1 activity decreases cell proliferation, migration and invasion by down-regulating AKT and NF-κB activity [56]. Notch also inhibits PI3K/AKT dephosphorylation by inhibiting PP2A and PTEN activation and consequently promotes cancer malignant process [26, 56, 57].
These findings suggest that there is an interaction between Notch and NF-κB pathways, but the mechanisms of action vary in different cells. The complicated interaction between Notch and NF-κB might depend on the exact pathophysiological context [58].
Notch signaling pathway and integrin
Integrins are transmembrane receptors that mediate cell–cell interaction and cell–ECM connection [59]. Integrin receptors are heterodimers constituted with α subunits and β subunits. There are 18 known α-subunits and 8 known β-subunits that can combine in various ways to form 24 functional integrins [60]. Integrin ligand has a special binding site containing an Arg-Gly-Asp (RGD) sequence [61]. MAGP2, an extracellular matrix protein, contains an RGD domain, which was found to suppress Notch activation by binding to integrin β1 and integrin β3 [62]. Moreover, Notch signaling can enhance cell adhesion by activating integrin β1 through small GTP-binding protein R-Ras when cancer cells enter the blood circulation system [63, 64].
It was revealed that CCN1 knockdown resulted in significant down-regulation of Notch ligand Jagged-1 [65]. Additionally, knockdown of integrin αv and β5 also inhibited Jagged-1 expression [65]. Activated αvβ5, which is downstream of CCN1, can activate NF-κB. CCN1 induces Jagged-1 expression through αvβ5/NF-κB pathway, which is essential for cholangiocyte proliferation [65]. CCN1 enhances the stability of Notch-1 intercellular domain (N1ICD) by preventing the proteasomal degradation events in pancreatic cancer cells. Moreover, the functional role of CCN1 can be mediated by the interaction with αvβ3 integrin receptor [66].
Integrin-linked kinase (ILK), an integrin-interacting protein with serine/threonine kinase, phosphorylates AKT, GSK3β and integrin β1/β3 [67]. It was found that overexpression of ILK facilitated the phosphorylation of AKT and also induced Notch-1 activation, associating with the increased mammosphere formation of tumor cells in 3D culture [68]. It has been reported that interleukin-6 (IL-6) can cross talk with Notch signaling pathway [69]. IL-6-inducing Notch activation is mediated by ILK, for that ILK regulates the assembly of γ-secretase complex [70]. IL-6 can promote breast cancer bone metastasis through Notch-1 [69] and induces mammosphere formation through Notch-3 in breast cancer cells [71]. Recent studies showed that IL-6 induced Notch-1 activation, which is dependent on the caveolin-1 [70]. Caveolin-1, a major component of caveolae, participates in various cellular processes, including lipid transport, membrane trafficking and tumorigenesis [72]. These findings suggested that Notch signaling pathway can be cross talking with caveolin-1.
The αvβ6 integrin, a receptor for fibronectin, tenascin-C and TGF-β1, is upregulated in several epithelial cancers and associated with poor prognosis of these cancers. Further research indicated that the cytoplasmic domain of β6 modulates the composition of intermediate filaments, thus regulating EMT. Truncation of the β6 cytoplasmic tail decreased RBP-Jκ expression by 75%, thereby also regulating the Notch pathway, indicating that Notch pathway is under the control of signals provided by the β6 cytoplasmic domain [73].
Notch activation can induce the expression of integrin αvβ5 in myeloma cells companied with enhanced cells adhesion on vitronectin. However, only integrins αv and β5, but not β1 and β3, were found increased upon Notch activation [74]. Notch downstream effector Hes-1 can directly bind to the promoter regions of N-cadherin and α9-integrin genes [75]. In rhabdomyosarcoma (RMS), Notch signaling inhibition significantly reduced N-cadherin and integrin α9 expression, but no changes were observed in the amount of integrin β1, the partner of α9 [75]. The evidence suggests that the cross talking between Notch and integrin depends on the subunit of integrin and the microenvironment in which they are located.
Notch signaling pathway and miRNAs
MicroRNAs (miRNAs) are short, conserved, noncoding RNA molecules that modulate mRNA translation and degradation, which were implicated in various cellular processes including metabolism and immunoregulation [76, 77]. In the recent years, increasing miRNAs have implicated human diseases including cancers. Many Notch signaling pathway-related molecules are identified to be the posttranscriptional targets of miRNAs. Accumulating evidence shows that the interaction between Notch signaling pathway and miRNAs contributes to tumor development [78]. MiR-223 plays an important role in regulating cell survival and is implicated in chronic inflammatory diseases [79, 80]. Notch and NF-κB are able to increase miR-223 expression. A conservative RBP-Jκ and NF-κB overlapping binding site has been found in the promoter of miR-223 [81]. Notch-1, Notch-3 and NF-κB are directly recruited to the promoter region of miR-233 to promote miR-233 expression [82]. miR-233 silencing resulted in cell cycle arrest and apoptosis [79], which is in accordance with the result that Notch and NF-kB signaling pathway inhibition led to cell cycle arrest at G1 phase and increased apoptosis [50].
miR-200 has been implicated in EMT and tumor motility. The EMT activator ZEB1 controls central cellular processes and states by inhibiting the expression of miR-200 family [83]. Furthermore, ZEB1 also promotes Notch expression. Jagged-1 is a predicted target of miR-200 family members. Overexpression of miR-200 leads to the reduced expression of Jagged-1. It has been reported that EMT inducer ZEB1 can trigger Notch signaling in cancer cells by stabilizing the expression of Notch pathway components, such as Jagged-1, Maml-2 and Maml-3, through inhibiting miR-200 expression [84]. Studies also found that tumor cell EMT and metastasis are dependent upon Jagged-2 and Notch-3, which promote EMT by decreasing miR-200 expression. [85, 86]. In addition, miR-200 interacting with Notch signaling pathway is also found in cancer stem cells (CSCs), such as breast CSC [87] and pancreatic CSC [88].
miR-598, which is down-regulated in colorectal cancer tissues, is implicated in colorectal cancer metastasis. It regulates EMT by directly suppressing its downstream target gene Jagged-1 to inactivate Notch signaling pathway [89]. Additionally, tumor suppressor miR-206 inhibiting cell proliferation and migration is reported by down-regulating Notch-3 expression [90]. These researches demonstrated that Notch signaling pathway is regulated by microRNAs, suggesting that targeting these microRNAs might be an effective strategy of therapeutics for cancers.
Notch signaling as a therapeutic target
Notch signaling pathway plays a critical role in development and tissue homeostasis, which is recapitulated in different forms of cancer. Dysregulated Notch signaling due to mutation or amplification or overexpression of ligands and/or receptors is implicated in a number of malignancies. Due to the important role of Notch signaling in tumorigenesis and metastasis, blocking Notch signaling pathway could be a potential therapy for cancer treatment.
High level of Notch signaling pathway molecules has been found in many types of cancer, resulting in Notch signaling enhancement, and promoting tumor cell survival. High expression of Notch-1 and Jagged-1 is associated with poor prognosis in breast cancer, promoting breast epithelial cell transformation. Aberrant expression of Notch-1, Notch-4 or Jagged-1 is usually present in breast ductal carcinoma and lobular carcinoma. In estrogen receptor-positive (ERα+) cells, estradiol suppresses Notch-1 activity by inhibiting the nuclear translocation of N1ICD. This effect can be reversed by tamoxifen and raloxifene to reactivate Notch-1. It was revealed that inhibition of Notch-1 signaling pathway decreased the expression of cyclin A and cyclin B1, causing G2 arrest in human breast carcinoma MDA-MB-231 cells (ERα-) and T47D:A18 cells (ERα+) [91]. Furthermore, Notch-1 inhibition enhanced the anti-tumor effects of tamoxifen. In addition, tamoxifen in combination with γ-secretase inhibitor (GSI) significantly inhibited the growth of MDA-MB-231 cells in vitro [91].
The N1ICD level is associated with the xenogeneic transplantation ability of triple-negative breast cancer, and the expression of Hes-4, a direct target gene of Notch, is related to the prognosis of triple-negative breast cancer patients. Notch signaling inhibitors MRK-003 markedly inhibited the proliferation of triple-negative breast cancer cell lines [92]. Hyperactivation of Notch-1 can be found in a variety of other solid tumors, including adenoid cystic carcinoma, pancreatic cancer and lung cancer. However, adenoid cystic carcinomas with activated Notch-1 mutations or overexpressed N1ICD are sensitive to GSI, whereas adenoid cystic carcinomas with low N1ICD expression or without Notch-1 mutations are insensitive to GSI [93]. In addition, although GSI can effectively inhibit the growth of pancreatic cancer, breast cancer and lung cancer xenograft model in preclinical research, it has various side effects in vivo. The chronic administration of GSI LY-411575 significantly changes the structure of intestinal tract and increases the number of goblet cells and mucin secretion, causing inflammatory cells to invade the lamina propria, all of which is due to that Notch-1 and Notch-2 are essential in these organs, but GSI blocks the signal transduction of all Notch family members, suggesting that more drugs specifically targeting Notch are needed. Recent studies have confirmed that the combination of GSI and glucocorticoid can reduce their side effects. Combination treatment of GSI with glucocorticoid induced a synergistic anti-leukemic effect in human T-ALL. Treatment of glucocorticoid dexamethasone could reverse GSI-induced gastrointestinal toxicity by inhibiting the transformation of goblet cells in vivo [94]. This enhances the capability that GSI can be successfully used in combination with glucocorticoid in adjuvant chemotherapy.
Notch-1 and its ligands Jagged-1 and Jagged-2, Dll-4 are overexpressed in clear cell renal cell carcinoma (CCRCC). Treatment with LY3039478, a small molecular GSI, inhibited the growth of CCRCC cells [95]. The data from a phase I study (NCT01695005) in breast cancer, adenoid cystic carcinoma and leiomyosarcoma suggest that LY3039478 is modestly effective against a range of advanced or metastatic cancers [96]. However, LY3039478 showed high efficacy (73% disease control rate) in adenoid cystic carcinoma with a manageable safety profile in a phase II study (NCT02518113).
Bristol-Myers Squibb (BMS) has developed a Notch receptor inhibitor, a small molecular compound named BMS-906024, which inhibits both leukemia (T-ALL-1) and triple-negative breast cancer cell proliferation [97]. In a clinical trial of BMS-906024 (NCT01363817), relapsed/refractory early T cell progenitor acute lymphoblastic leukemia patients received a treatment with BMS-906024 combined with dexamethasone. All the patients had complete hematologic response without complication. Next-generation sequencing and RNA sequence showed that high levels of activated Notch-1 and multiple Notch target genes were repressed by GSI [98]. Up to date, the BMS-906024 is in phase I clinical trial in advanced or metastatic solid tumors and acute lymphocytic leukemia.
Although there are more than ten GSIs under clinical trials, many of them failed due to modest efficacy and adverse events, except LY3039478 and BMS-906024. GSIs inhibit the activation of all Notch receptors and lead to gastrointestinal toxicity, limiting their clinical efficacy in patients. Combination with other drugs might be an effective strategy to limit the toxicity of GSI.
Future considerations
Accumulating studies have elucidated the role of the Notch signaling pathway in development and tissue homeostasis. Increasing experimental evidence suggests that Notch signaling pathway contributes to the proliferation of tumor cells and promotes EMT, drug resistance and tumor metastasis. Notch signaling pathway cross talks with other signaling pathways to form a complicated net in regulation of cancer development. It is important to clarify the role of Notch target genes in the process of cancer occurrence and metastasis. Further development in specific anti-tumor drug that acts on Notch signaling pathway will benefit for cancer patients, and the combination of Notch targeted drugs with other drugs is also an important direction for future research.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi:10.3322/caac.21387.
Charpentier M, Martin S. Interplay of stem cell characteristics, EMT, and microtentacles in circulating breast tumor cells. Cancers. 2013;5(4):1545–65. doi:10.3390/cancers5041545.
Andersson M, Olsen JH. Malignant mesotheliomas in Denmark 1943–1980. Cancer statistics 9. Ugeskr Laeger. 1984;146(14):1085–7.
Zardawi SJ, Zardawi I, McNeil CM, Millar EK, McLeod D, Morey AL, et al. High Notch1 protein expression is an early event in breast cancer development and is associated with the HER-2 molecular subtype. Histopathology. 2010;56(3):286–96. doi:10.1111/j.1365-2559.2009.03475.x.
Danza G, Di Serio C, Ambrosio MR, Sturli N, Lonetto G, Rosati F, et al. Notch3 is activated by chronic hypoxia and contributes to the progression of human prostate cancer. Int J Cancer. 2013;133(11):2577–86. doi:10.1002/ijc.28293.
Ai Q, Ma X, Huang Q, Liu S, Shi T, Zhang C, et al. High-level expression of Notch1 increased the risk of metastasis in T1 stage clear cell renal cell carcinoma. PLoS ONE. 2012;7(4):e35022. doi:10.1371/journal.pone.0035022.
Yang Y, Ahn Y-H, Gibbons DL, Zang Y, Lin W, Thilaganathan N, et al. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200—dependent pathway in mice. J Clin Investig. 2011;121(4):1373–85. doi:10.1172/jci42579.
Liu H, Wang J, Liu Z, Wang L, Liu S, Zhang Q. Jagged1 modulated tumor-associated macrophage differentiation predicts poor prognosis in patients with invasive micropapillary carcinoma of the breast. Medicine. 2017;96(16):e6663. doi:10.1097/MD.0000000000006663.
Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65(18):8530–7. doi:10.1158/0008-5472.CAN-05-1069.
Chance O. Value of statistics in the study of cancer of the uterine cervix. Comptes rendus de la Societe francaise de gynecologie. 1951;21(7):305–11.
Dotta JS, Delporte TV. Statistics on the treatment of prostatic cancer. Revista argentina de urologia. 1951;20(9–11):255–7.
Mumm J. Notch signaling: from the outside in. Dev Biol. 2000;228(2):151–65. doi:10.1006/dbio.2000.9960.
Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131(5):965–73. doi:10.1242/dev.01074.
Cesarani F, Garbagnoli E. Local recurrence and lymphatic and osseous metastases following surgery of breast cancer; radiotherapy department statistics for 1944-50. Athena; rassegna mensile di biologia, clinica e terapia. 1951;17(7–8):189–92.
Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–33. doi:10.1016/j.cell.2009.03.045.
Weinmaster G. Notch signal transduction a real rip and more. Curr Opin Genet Dev. 2000;10:363–9.
Nahum AM. Biting the bullet: minimum standards for reporting cancer treatment statistics. Head Neck Surg. 1979;1(3):201.
Upton AC. Survey: reporting practices for cancer treatment statistics. Head Neck Surg. 1979;1(6):500.
Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11(5):338–51. doi:10.1038/nrc3035.
Ellisen LW, Bird J, West PC, et al. TAN+1, the human hormolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastie neoplasms. Cell. 1991;66(4):649–61.
Weng AP, Ferrando AA, Lee W, Morris JP IV, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Thomas Look A, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–71. doi:10.1126/science.1102160.
Espinosa L, Cathelin S, D’Altri T, Trimarchi T, Statnikov A, Guiu J, et al. The Notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell. 2010;18(3):268–81. doi:10.1016/j.ccr.2010.08.006.
Wu F, Stutzman A, Mo YY. Notch signaling and its role in breast cancer. Front Biosci. 2007;12:4370–83.
Ma YC, Shi C, Zhang YN, Wang LG, Liu H, Jia HT, et al. The tyrosine kinase c-Src directly mediates growth factor-induced Notch-1 and Furin interaction and Notch-1 activation in pancreatic cancer cells. PLoS ONE. 2012;7(3):e33414. doi:10.1371/journal.pone.0033414.
Song LL, Peng Y, Yun J, Rizzo P, Chaturvedi V, Weijzen S, et al. Notch-1 associates with IKKalpha and regulates IKK activity in cervical cancer cells. Oncogene. 2008;27(44):5833–44. doi:10.1038/onc.2008.190.
Zhou W, Fu XQ, Zhang LL, Zhang J, Huang X, Lu XH, et al. The AKT1/NF-kappaB/Notch1/PTEN axis has an important role in chemoresistance of gastric cancer cells. Cell Death Dis. 2013;4:e847. doi:10.1038/cddis.2013.375.
Zhang Y, Li B, Ji ZZ, Zheng PS. Notch1 regulates the growth of human colon cancers. Cancer. 2010;116(22):5207–18. doi:10.1002/cncr.25449.
Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007;21(5):562–77. doi:10.1101/gad.1484707.
Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C, et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med. 2011;208(10):1963–76. doi:10.1084/jem.20110198.
Gupta A, Wang Y, Browne C, Kim S, Case T, Paul M, et al. Neuroendocrine differentiation in the 12T-10 transgenic prostate mouse model mimics endocrine differentiation of pancreatic beta cells. Prostate. 2008;68(1):50–60. doi:10.1002/pros.20650.
Konishi J, Yi F, Chen X, Vo H, Carbone DP, Dang TP. Notch3 cooperates with the EGFR pathway to modulate apoptosis through the induction of bim. Oncogene. 2010;29(4):589–96. doi:10.1038/onc.2009.366.
Parr C, Watkins G, Jiang WG. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int J Mol Med. 2004;14(5):779–86.
PRESENTATION of results in the treatment of cancer. V. World Health Organization Expert Committee on Health Statistics. Br J Radiol. 1951;24(282):311–314. doi:10.1259/0007-1285-24-282-311.
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–64. doi:10.1126/science.1203543.
Li S, Zhang J, Yang H, Wu C, Dang X, Liu Y. Copper depletion inhibits CoCl2-induced aggressive phenotype of MCF-7 cells via downregulation of HIF-1 and inhibition of Snail/Twist-mediated epithelial-mesenchymal transition. Sci Rep. 2015;5:12410. doi:10.1038/srep12410.
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci. 2008;105(17):6392–7. doi:10.1073/pnas.0802047105.
Kao SH, Wu KJ, Lee WH. Hypoxia, epithelial-mesenchymal transition, and TET-mediated epigenetic changes. J Clin Med. 2016;. doi:10.3390/jcm5020024.
Ahmad A, Wang Z, Kong D, Ali R, Ali S, Banerjee S, et al. Platelet-derived growth factor-D contributes to aggressiveness of breast cancer cells by up-regulating Notch and NF-kappaB signaling pathways. Breast Cancer Res Treat. 2011;126(1):15–25. doi:10.1007/s10549-010-0883-2.
Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH, et al. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem. 2012;113(5):1501–13. doi:10.1002/jcb.24019.
Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440(7088):1222–6. doi:10.1038/nature04695.
Peinado H, Portillo F, Cano A. Switching on-off Snail: LOXL2 versus GSK3beta. Cell Cycle. 2005;4(12):1749–52. doi:10.4161/cc.4.12.2224.
Yuan XW, Wang DM, Hu Y, Tang YN, Shi WW, Guo XJ, et al. Hepatocyte nuclear factor 6 suppresses the migration and invasive growth of lung cancer cells through p53 and the inhibition of epithelial-mesenchymal transition. J Biol Chem. 2013;288(43):31206–16. doi:10.1074/jbc.M113.480285.
Ramms L, Fabris G, Windoffer R, Schwarz N, Springer R, Zhou C, et al. Keratins as the main component for the mechanical integrity of keratinocytes. Proc Natl Acad Sci USA. 2013;110(46):18513–8. doi:10.1073/pnas.1313491110.
Mierke CT. The fundamental role of mechanical properties in the progression of cancer disease and inflammation. Rep Prog Phys. 2014;77(7):076602. doi:10.1088/0034-4885/77/7/076602.
Diaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201(2):279–92. doi:10.1083/jcb.201209151.
Higashiyama S, Abraham JA, Miller J, Fiddes JC, Klagsbrun M. A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science. 1991;251(4996):936–9.
Higashiyama S, Iwamoto R, Goishi K, Raab G, Taniguchi N, Klagsbrun M, et al. The membrane protein CD9/DRAP 27 potentiates the juxtacrine growth factor activity of the membrane-anchored heparin-binding EGF-like growth factor. J Cell Biol. 1995;128(5):929–38.
Miyamoto S, Hirata M, Yamazaki A, Kageyama T, Hasuwa H, Mizushima H, et al. Heparin-binding EGF-like growth factor is a promising target for ovarian cancer therapy. Cancer Res. 2004;64(16):5720–7. doi:10.1158/0008-5472.CAN-04-0811.
Jezierska A, Motyl T. Matrix metalloproteinase-2 involvement in breast cancer progression: a mini-review. Med Sci Monit. 2009;15(2):RA32–40.
Li L, Zhao F, Lu J, Li T, Yang H, Wu C, et al. Notch-1 signaling promotes the malignant features of human breast cancer through NF-kappaB activation. PLoS ONE. 2014;9(4):e95912. doi:10.1371/journal.pone.0095912.
Wang Z, Zhang Y, Li Y, Banerjee S, Liao J, Sarkar FH. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol Cancer Ther. 2006;5(3):483–93. doi:10.1158/1535-7163.MCT-05-0299.
Inoue J, Gohda J, Akiyama T, Semba K. NF-kappaB activation in development and progression of cancer. Cancer Sci. 2007;98(3):268–74. doi:10.1111/j.1349-7006.2007.00389.x.
Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446(5):475–82. doi:10.1007/s00428-005-1264-9.
Yamamoto M, Taguchi Y, Ito-Kureha T, Semba K, Yamaguchi N, Inoue J. NF-kappaB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat Commun. 2013;4:2299. doi:10.1038/ncomms3299.
Zhang X, Chen T, Zhang J, Mao Q, Li S, Xiong W, et al. Notch1 promotes glioma cell migration and invasion by stimulating beta-catenin and NF-kappaB signaling via AKT activation. Cancer Sci. 2012;103(2):181–90. doi:10.1111/j.1349-7006.2011.02154.x.
Li L, Zhang J, Xiong N, Li S, Chen Y, Yang H, et al. Notch-1 signaling activates NF-kappaB in human breast carcinoma MDA-MB-231 cells via PP2A-dependent AKT pathway. Med Oncol. 2016;33(4):33. doi:10.1007/s12032-016-0747-7.
Hales EC, Orr SM, Larson Gedman A, Taub JW, Matherly LH. Notch1 receptor regulates AKT protein activation loop (Thr308) dephosphorylation through modulation of the PP2A phosphatase in phosphatase and tensin homolog (PTEN)-null T-cell acute lymphoblastic leukemia cells. J Biol Chem. 2013;288(31):22836–48. doi:10.1074/jbc.M113.451625.
Zhang W, Grivennikov SI. Top Notch cancer stem cells by paracrine NF-kappaB signaling in breast cancer. Breast Cancer Res: BCR. 2013;15(5):316. doi:10.1186/bcr3565.
Zhao F, Li L, Guan L, Yang H, Wu C, Liu Y. Roles for GP IIb/IIIa and alphavbeta3 integrins in MDA-MB-231 cell invasion and shear flow-induced cancer cell mechanotransduction. Cancer Lett. 2014;344(1):62–73. doi:10.1016/j.canlet.2013.10.019.
Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285(5430):1028–32.
Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697–715. doi:10.1146/annurev.cellbio.12.1.697.
Deford P, Brown K, Richards RL, King A, Newburn K, Westover K, et al. MAGP2 controls Notch via interactions with RGD binding integrins: Identification of a novel ECM-integrin-Notch signaling axis. Exp Cell Res. 2016;341(1):84–91. doi:10.1016/j.yexcr.2016.01.011.
Hodkinson PS, Elliott PA, Lad Y, McHugh BJ, MacKinnon AC, Haslett C, et al. Mammalian NOTCH-1 activates beta1 integrins via the small GTPase R-Ras. J Biol Chem. 2007;282(39):28991–9001. doi:10.1074/jbc.M703601200.
Hodkinson PS, Elliott PA, Lad Y, McHugh BJ, MacKinnon AC, Haslett C, et al. Mammalian NOTCH-1 activates beta1 integrins via the small GTPase R-Ras. J Biol Chem. 2007;282(39):28991–9001. doi:10.1074/jbc.M703601200.
Kim KH, Chen CC, Alpini G, Lau LF. CCN1 induces hepatic ductular reaction through integrin alphavbeta(5)-mediated activation of NF-kappaB. J Clin Invest. 2015;125(5):1886–900. doi:10.1172/JCI79327.
Haque I, De A, Majumder M, Mehta S, McGregor D, Banerjee SK, et al. The matricellular protein CCN1/Cyr61 is a critical regulator of Sonic Hedgehog in pancreatic carcinogenesis. J Biol Chem. 2012;287(46):38569–79. doi:10.1074/jbc.M112.389064.
Legate KR, Montanez E, Kudlacek O, Fassler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006;7(1):20–31. doi:10.1038/nrm1789.
Hsu EC, Kulp SK, Huang HL, Tu HJ, Chao MW, Tseng YC, et al. Integrin-linked kinase as a novel molecular switch of the IL-6-NF-kappaB signaling loop in breast cancer. Carcinogenesis. 2016;37(4):430–42. doi:10.1093/carcin/bgw020.
Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging Notch signaling in bone cells. Cancer Cell. 2011;19(2):192–205. doi:10.1016/j.ccr.2010.12.022.
Hsu EC, Kulp SK, Huang HL, Tu HJ, Salunke SB, Sullivan NJ, et al. Function of integrin-linked kinase in modulating the stemness of IL-6-abundant breast cancer cells by regulating gamma-secretase-mediated Notch1 activation in caveolae. Neoplasia. 2015;17(6):497–508. doi:10.1016/j.neo.2015.06.001.
Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117(12):3988–4002. doi:10.1172/JCI32533.
Tahir SA, Yang G, Goltsov A, Song KD, Ren C, Wang J, et al. Caveolin-1-LRP6 signaling module stimulates aerobic glycolysis in prostate cancer. Cancer Res. 2013;73(6):1900–11. doi:10.1158/0008-5472.CAN-12-3040.
Lee C, Lee C, Lee S, Siu A, Ramos DM. The cytoplasmic extension of the integrin beta6 subunit regulates epithelial-to-mesenchymal transition. Anticancer Res. 2014;34(2):659–64.
Ding Y, Shen Y. Notch increased vitronection adhesion protects myeloma cells from drug induced apoptosis. Biochem Biophys Res Commun. 2015;467(4):717–22. doi:10.1016/j.bbrc.2015.10.076.
Masia A, Almazan-Moga A, Velasco P, Reventos J, Toran N, Sanchez de Toledo J, et al. Notch-mediated induction of N-cadherin and alpha9-integrin confers higher invasive phenotype on rhabdomyosarcoma cells. Br J Cancer. 2012;107(8):1374–83. doi:10.1038/bjc.2012.411.
Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. doi:10.1038/nrg2843.
Sun DW, Zhang HD, Mao L, Mao CF, Chen W, Cui M, et al. Luteolin inhibits breast cancer development and progression in vitro and in vivo by suppressing Notch signaling and regulating MiRNAs. Cell Physiol Biochem. 2015;37(5):1693–711. doi:10.1159/000438535.
Wang Z, Li Y, Kong D, Ahmad A, Banerjee S, Sarkar FH. Cross-talk between miRNA and Notch signaling pathways in tumor development and progression. Cancer Lett. 2010;292(2):141–8. doi:10.1016/j.canlet.2009.11.012.
Mansour MR, Sanda T, Lawton LN, Li X, Kreslavsky T, Novina CD, et al. The TAL1 complex targets the FBXW7 tumor suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. J Exp Med. 2013;210(8):1545–57. doi:10.1084/jem.20122516.
Dorhoi A, Iannaccone M, Farinacci M, Fae KC, Schreiber J, Moura-Alves P, et al. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J Clin Invest. 2013;123(11):4836–48. doi:10.1172/JCI67604.
Barbarulo A, Grazioli P, Campese AF, Bellavia D, Di Mario G, Pelullo M, et al. Notch3 and canonical NF-kappaB signaling pathways cooperatively regulate Foxp3 transcription. J Immunol. 2011;186(11):6199–206. doi:10.4049/jimmunol.1002136.
Kumar V, Palermo R, Talora C, Campese AF, Checquolo S, Bellavia D, et al. Notch and NF-kB signaling pathways regulate miR-223/FBXW7 axis in T-cell acute lymphoblastic leukemia. Leukemia. 2014;28(12):2324–35. doi:10.1038/leu.2014.133.
Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci: CMLS. 2009;66(5):773–87. doi:10.1007/s00018-008-8465-8.
Brabletz S, Bajdak K, Meidhof S, Burk U, Niedermann G, Firat E, et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011;30(4):770–82. doi:10.1038/emboj.2010.349.
Yang Y, Ahn YH, Gibbons DL, Zang Y, Lin W, Thilaganathan N, et al. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J Clin Invest. 2011;121(4):1373–85. doi:10.1172/JCI42579.
Ohashi S, Natsuizaka M, Naganuma S, Kagawa S, Kimura S, Itoh H, et al. A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer Res. 2011;71(21):6836–47. doi:10.1158/0008-5472.CAN-11-0846.
Shimono Y, Mukohyama J, Nakamura S, Minami H. MicroRNA regulation of human breast cancer stem cells. J Clin Med. 2015;. doi:10.3390/jcm5010002.
Xu YF, Hannafon BN, Ding WQ. microRNA regulation of human pancreatic cancer stem cells. Stem Cell Investig. 2017;4:5. doi:10.21037/sci.2017.01.01.
Chen J, Zhang H, Chen Y, Qiao G, Jiang W, Ni P, et al. miR-598 inhibits metastasis in colorectal cancer by suppressing JAG1/Notch2 pathway stimulating EMT. Exp Cell Res. 2017;352(1):104–12. doi:10.1016/j.yexcr.2017.01.022.
Wang XW, Xi XQ, Wu J, Wan YY, Hui HX, Cao XF. MicroRNA-206 attenuates tumor proliferation and migration involving the downregulation of NOTCH3 in colorectal cancer. Oncol Rep. 2015;33(3):1402–10. doi:10.3892/or.2015.3731.
Rizzo P, Miao H, D’Souza G, Osipo C, Song LL, Yun J, et al. Cross-talk between Notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008;68(13):5226–35. doi:10.1158/0008-5472.CAN-07-5744.
Clementz AG, Rogowski A, Pandya K, Miele L, Osipo C. NOTCH-1 and NOTCH-4 are novel gene targets of PEA3 in breast cancer: novel therapeutic implications. Breast Cancer Res: BCR. 2011;13(3):R63. doi:10.1186/bcr2900.
Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C, et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014;4(10):1154–67. doi:10.1158/2159-8290.CD-13-0830.
Samon JB, Castillo-Martin M, Hadler M, Ambesi-Impiobato A, Paietta E, Racevskis J, et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther. 2012;11(7):1565–75. doi:10.1158/1535-7163.MCT-11-0938.
Bhagat TD, Zou Y, Huang S, Park J, Palmer MB, Hu C, et al. Notch pathway is activated via genetic and epigenetic alterations and is a therapeutic target in clear cell renal cancer. J Biol Chem. 2017;292(3):837–46. doi:10.1074/jbc.M116.745208.
Notch inhibitor shows modest efficacy. Cancer Discov. 2017;7(2):OF3. doi:10.1158/2159-8290.CD-NB2016-159.
Gavai AV, Quesnelle C, Norris D, Han WC, Gill P, Shan W, et al. Discovery of clinical candidate BMS-906024: a potent pan-Notch inhibitor for the treatment of leukemia and solid tumors. ACS Med Chem Lett. 2015;6(5):523–7. doi:10.1021/acsmedchemlett.5b00001.
Knoechel B, Bhatt A, Pan L, Pedamallu CS, Severson E, Gutierrez A, et al. Complete hematologic response of early T-cell progenitor acute lymphoblastic leukemia to the gamma-secretase inhibitor BMS-906024: genetic and epigenetic findings in an outlier case. Cold Spring Harb Mol Case Stud. 2015;1(1):a000539. doi:10.1101/mcs.a000539.
Acknowledgements
We would like to thank the National Natural Science Foundation of China (11772088, 31470906, 31700811, 11502049, 81671821, 31470959, 81471785), the Basic Research Program of Sichuan Science and Technology (2017JY0019, 2017JY0217), the China Postdoctoral Science Foundation (2016M592657), the Fundamental Research Funds for the Central Universities (ZYGX2016Z001, ZYGX2015J143) and the Postdoctoral Science Foundation of University of Electronic Science and Technology of China (Y0200623601737) for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Li, L., Tang, P., Li, S. et al. Notch signaling pathway networks in cancer metastasis: a new target for cancer therapy. Med Oncol 34, 180 (2017). https://doi.org/10.1007/s12032-017-1039-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-017-1039-6