
Abstract
Glycophobia in structural biology is strongly associated with the unpredictable, heterogeneous nature of protein glycosylation and the complex, flexible structures of the glycoprotein glycans. Moreover, glycoproteins cannot be produced by conventional bacterial expression systems. Nuclear magnetic resonance (NMR) spectroscopy assisted by other analytical and preparative techniques can now successfully address these issues. Recombinant glycoproteins can be expressed with stable isotope labeling using a variety of eukaryotic production vehicles. Glycoforms of glycoproteins can be remodeled by genetic engineering of the production vehicles as well as in vitro enzymatic reactions. Stable-isotope-assisted NMR techniques have provided detailed information regarding conformational dynamics and interactions of the carbohydrate chains in solution, giving insights into the functional mechanisms of glycoprotein glycans.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Advancements in biomolecular NMR spectroscopy have been driven by the constant development of methods for the expression of isotope-labeled biomacromolecules and novel hardware, exemplified by higher-field-strength magnets along with sophisticated measurement techniques. This progress has enabled high-throughput three-dimensional (3D)-structure determination of proteins, quantitative characterization of conformational dynamics of biomolecules on a broad range of timescales [1, 2], depiction of huge protein complexes beyond 1 MDa [3], and observation of protein behaviors in intracellular environments [4]. Advances in biomolecular NMR spectroscopy have also provided technical basis for high-throughput analyses of protein–ligand interactions using chemical libraries [5, 6]. To date, proteins subjected to NMR-based structural analysis have been conventionally produced using bacterial protein expression systems or cell-free expression systems [7]. Although more than half of proteins in nature are estimated to be modified with carbohydrate moieties [8], these recombinant proteins are not glycosylated by the aforementioned expression systems.
Carbohydrate chains have a significant impact on physicochemical and biofunctional properties of the carrier proteins [9]. Thermal stability and solubility of proteins are governed by the carbohydrate chains, which can also affect protein conformations, and thereby control their biological functions. Moreover, glycans directly involved in interactions with other proteins, which are collectively termed lectins, mediate a variety of biological processes involved in cell–cell communications, viral and bacterial infections, immune responses, and protein-fate determination. Therefore, without knowledge of the glycan structures, one can barely understand the functional mechanisms of glycoproteins in the biological context. However, despite their biological importance, structural biology studies of glycoproteins have been falling behind. Indeed, atomic coordinates of glycoproteins represent only 4% of the total entries in the Protein Data Bank, and most of these provide structural information on only one or two glycan residues (Fig. 15.1).

Current statistics regarding Protein Data Bank entries on N-linked glycoprotein structures (data were extracted on May 17, 2017). a Number and percentage of X-ray crystallography and NMR structures of N-linked glycoproteins. b Size distribution of N-glycans among glycoprotein structures revealed by X-ray crystallography (top) and NMR spectroscopy (bottom)
Besides the traditional application for the determination of the covalent structures of oligosaccharides [10,11,12], NMR spectroscopy can play versatile roles in characterizing dynamic conformations and interactions of oligosaccharides. In this chapter, we will outline the major obstacles in studying glycoproteins via NMR spectroscopy and the current state-of-the-art methodology for NMR-based structural glycobiology.
2 Enigmatic Aspects of Carbohydrate Structures
Needless to say, any structural biology studies presuppose the covalent structures of the target biomolecules, e.g., the amino acid sequences of proteins. An obvious example can be seen in structural genomics, in which accumulating genome sequence data have prompted comprehensive structural determination of the encoded proteins. From this perspective, one of the biggest problems in structural biology of glycoproteins is that the covalent structures of glycans are not directly encoded in the genome. Although the necessary sequence for eukaryotic N-glycosylation, i.e., Asn-X-Ser/Thr(/Cys) in which X is not proline, is widely known, this is insufficient [9]. Furthermore, no consensus motif has been established for other glycosylation sites, despite a certain tendency of amino acid sequences in O-glycosylation sites [9]. Hence, glycosylation sites of a protein with a known amino acid sequence remain unpredictable. Moreover, it is much more difficult to deduce the sequence of carbohydrate moiety at each glycosylation site. Indeed, the carbohydrate moieties displayed at one site of protein often have a high degree of sequence divergence and heterogeneity, depending not only on the protein structure but also on physiological and pathological conditions [9]. In the case of immunoglobulin G (IgG), which is a major serum glycoprotein, its glycosylation depends on aging, pregnancy, delivery, and onset of connective tissue disease typified by rheumatoid arthritis [13,14,15,16,17]. Of course, recombinant glycoproteins produced in different production vehicles exhibit different glycoforms and even cell culture conditions affect the glycosylation profiles [18, 19].
The intrinsic complexity of oligosaccharide structures brings further difficulty in structural analysis of glycoproteins. The building blocks of sugar chains, i.e., monosaccharides, are structurally simple with little variation in their functional groups in comparison with amino acids [9]. However, unlike polypeptide chains and nucleic acids, sugar residues are connected through multiple types of glycosidic linkages, potentially giving rise to a wide variety of isomeric and branching structures. Indeed, the number of all possible isomers of a tetrasaccharide composed of four different monosaccharides was calculated to be 34,560, whereas only 24 different tetrapeptides are made from four different amino acid residues [20]. Glycosidic linkage isomerism is critical to glycoprotein functions, as best exemplified by infections with influenza viruses, which bind host cells by recognizing specific sialyl linkage isomers [21]. Paradoxically, the structural simplicity (or similarity) of sugar residues precludes structural analyses: Isomeric structures such as glucose, mannose, and galactose cannot be distinguished by conventional mass spectrometry (MS), whereas the little diversity of the functional groups causes a chemical shift degeneracy in NMR spectroscopy.
Another difficulty for structural analysis of glycoproteins is associated with the motional freedom of glycosidic linkages in the carbohydrate moieties, giving rise to conformational fluctuations. This flexible property of glycans, along with their structural heterogeneity, hampers crystallization of glycoproteins, and, even if it succeeds, interpretation of their electron densities is unavoidably ambiguous [22]. Moreover, a tricky problem is that mutational deglycosylation can cause unexpected or even undesirable modifications of the proteins, such as sulfation and nicks [23, 24].
All these characteristics of the carbohydrate chains make structural analysis of glycoproteins (and even oligosaccharides) a challenging task. Namely, one who attempts to perform structural glycobiology must conduct a series of analyses, from the determination of carbohydrate sequences to characterization of the conformational dynamics of target glycoproteins including their glycans. NMR spectroscopy plays a central role in addressing these issues because it can, in principle, characterize dynamic structures of heterogeneous biomacromolecules in solution. However, it should be noted that a variety of preparative and analytical techniques that complement NMR approach are indispensable for conducting structural analysis of glycoproteins. In the following sections, we will describe how one can perform structural glycobiology by NMR spectroscopy aided by these complementing methods.
3 Expression of Isotope-Labeled Glycoproteins
As glycoproteins cannot be produced by conventional protein expression systems such as Escherichia coli, appropriate eukaryotic vehicles have to be chosen for the production of NMR quantities of glycoproteins with stable isotope labeling. Typical choices would be cell lines derived from mammals. Several groups developed metabolic isotope labeling of recombinant proteins using mammalian cells, including Chinese hamster ovary (CHO) cells, mouse hybridomas, and adenovirus vector-based mammalian cells [25,26,27,28,29]. In particular, uniform and amino acid-selective 13C and/or 15N labeling techniques were established using IgG as a model glycoprotein, yielding several NMR applications [27, 29]. The composition of the serum-free medium used for the production of isotopically labeled IgG is shown in Table 15.1. Amino acid-selective labeling is accomplished by replacing specific amino acid(s) with their isotope-labeled counterpart(s), although this often suffers from metabolic isotope scrambling and dilution. To suppress the undesirable metabolic processes, metabolic inhibitors such as β-chloro-l-alanine are often added to the medium [30].
For uniform 13C/15N labeling of a glycoprotein, it is necessary to replace all the metabolic precursors with the isotope-labeled ones, which are commercially very expensive. A more practical approach is to use an isotope-labeled amino acid mixture derived from algae with the modification of the medium composition (Table 15.1). Using this protocol, uniformly 13C/15N-labeled IgG glycoproteins were produced through mammalian cell culture. Figure 15.2A shows a 1H–15N HSQC spectrum of 13C/15N-labeled human IgG-Fc obtained from CHO cells [31].

1H–15N HSQC spectra of isotope-labeled human IgG1-Fc are shown obtained from a CHO cells, b transgenic tobacco plants, and c silkworms. d Superposition of 1H–15N HSQC spectra of the uniformly 15N/13C-labeled IgG1-Fc expressed by transgenic silkworm (red) and CHO cells (black). e Mapping of the amino acid residues with observable chemical shift changes [(0.2δ 2N + δ 2H )1/2 > 0.1 ppm and >0.2 ppm in pink and red, respectively] on the crystal structure [PDB code: 3AVE] [57]. The N-glycans are represented by gray sphere. The molecular graphics were generated with PyMOL [98]. In (a) backbone assignments are annotated by the resonance peaks with one-letter amino acid codes and the sequence numbers. Side-chain resonances corresponding to NH2 amides are connected by horizontal lines. Adapted from Refs. [31, 34, 35] with the modification
Isotope labeling of the carbohydrate moieties can be simply achieved by using 13C-labeled glucose as a metabolic precursor. 13C- and/or 15N-labeled glucosamine (but not N-acetylglucosamine) can be used for metabolic labeling of GlcNAc residues in the carbohydrate moieties of glycoproteins [32, 33].
In addition to mammalian cells, metabolic isotope labeling techniques have been developed for NMR applications in various eukaryotic expression systems including yeast, Dictyostelium discoideum, insects, and plants [34,35,36,37,38,39]. As an insect cell-based production vehicle, Sf9 cells infected with recombinant baculoviruses are widely used for the expression of membrane glycoproteins for NMR as well as crystallographic analyses [36, 40]. Recently, silkworms have been demonstrated as promising production vehicles for isotope-labeled recombinant glycoproteins [35]. In this approach, baculovirus-infected fifth instar larvae were reared with an artificial diet containing a protein mixture derived from Candida utilis grown in a culture medium containing 15N-labeled ammonium sulfate as a nitrogen source. 15N-enrichment level of recombinant human IgG achieved in this system was approximately 80%.
Plant expression systems are now attracting attention from pharmaceutical interests. Transgenic tobacco (Nicotiana benthamiana) was tested for the expression of 15N-labeled IgG [34]. Four-week-old seedlings of transgenic tobacco were placed in a drip hydroponic system and cultivated for 49 days with isotopically labeled Murashige and Skoog medium containing potassium nitrate (K15NO3) and ammonium nitrate (15NH 154 NO3) as the major nitrogen sources. The degree of 15N-enrichment of recombinant human IgG thus produced was in the approximately 50–60% range.
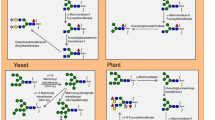
Figure 15.2 compares the 1H–15N HSQC spectra of recombinant human IgG1-Fc expressed by transgenic tobacco and silkworm as well as CHO cells [31, 34, 35]. Although the spectra are similar, significant chemical shift differences were observed for the amino acid residues in close proximity to N-glycans. The plant-derived IgG1-Fc exhibits α1,3-linked fucose and β1,2-linked xylose residues in the common conserved Man3GlcNAc2 core, whereas the insect cell-derived glycoprotein expresses paucimannose-type glycans as the major components (Fig. 15.3). Because these glycoforms are not expressed in mammalian vehicles, the observed spectral differences could be attributed to microenvironmental alterations caused by differential glycosylation.

N-glycan processing pathways as the basis of glycoform-engineering strategies. Schematic drawing of the N-glycan processing pathway in the ER common to mammalian cells, insect, yeast, and plant, and the distinct pathways in the Golgi complex along with a prokaryotic N-glycan pathway exemplified by that in C. jejuni. Adapted from Ref. [94] with the modification
Yeast can also be employed for the expression of isotope-labeled recombinant glycoproteins [37, 38, 41]. For example, Pichia pastoris strain GS115 was used for producing 15N-labeled human chorionic gonadotropin and human follicle-stimulating hormone [37]. From cultures of P. pastoris using 15NH4Cl/glucose–glycerol–methanol as nitrogen and carbon sources, human chorionic gonadotropin was obtained with an 15N enrichment of 70%, with modification by the yeast-strain-specific high-mannose-type oligosaccharides ranging from Man8GlcNAc2 to Man11GlcNAc2. Kamiya et al. [38, 41] described a method for the overexpression of uniformly 13C-labeled decasaccharides M8B and undecasaccharide M9 using genetically engineered Saccharomyces cerevisiae cells grown in medium containing 13C-labeled glucose.
Deuteration is effective to suppress dipolar relaxation for dealing with larger biomacromolecules [42, 43]. However, one major problem in eukaryotic expression systems is difficulty in metabolic deuteration using heavy water because the cells cannot survive in such a milieu. Recently, using deuterated yeast extract, Grzesiek and coworkers successfully established a methodology for deuteration of recombinant proteins expressed in insect cells with a deuteration degree of >60% without any decrease in protein yield [44]. This line of study will open up new avenues for NMR analyses of larger glycoproteins expressed by eukaryotic expression systems.
Although E. coli lacks a protein glycosylation pathway, protein N-glycosylation has been found in a variety of bacteria including Campylobacter jejuni, which expresses proteins modified with the N-linked heptasaccharide, GalNAcα1-4GalNAcα1-4(Glcβ1-3)GalNAcα1-4GalNAcα1-4GalNAcα1-3Bac [45] (Fig. 15.3). A 13C/15N-labeled recombinant protein produced in E. coli was successfully modified in vitro with this oligosaccharide by using enzymes from C. jejuni [46].
4 Glycosylation Profiling
Because the glycoform of a given glycoprotein is generally unpredictable, experimental glycosylation profiling is necessary for identifying its covalent structure. High-performance liquid chromatography (HPLC) mapping is a useful technique for the quantitative profiling of protein glycosylation. In particular, Takahashi and coworkers have developed 3D HPLC mapping using pyridyl-2-amination of N-linked oligosaccharides chemically or enzymatically cleaved from glycoproteins [47, 48] (Fig. 15.4). The fluorescence-labeled oligosaccharides are subjected to three different HPLC columns, i.e., diethylaminoethyl (DEAE), octadecylsilica (ODS), and amide, in a sequential manner, enabling identification of their structures based on their elution positions on the columns and reference to a database. A strength of this method is the ability to discriminate isomeric oligosaccharides by their differential elution times, primarily on the ODS column. Figure 15.4a illustrates a typical elution profile on an ODS column of the pyridylamino derivatives of N-glycans from human IgG1-Fc, which exhibit microheterogeneities resulting from the presence or absence of non-reducing terminal galactose residues [49].

Scheme of N-glycosylation profiling based on the 3D HPLC map (left) and elution profiles on an ODS column of pyridylamino derivatives of the N-linked oligosaccharides released from the human IgG1-Fc glycoforms (right). a Non-treated Fc, b Fc(G2), c Fc(G0), and d Fc(M3). Key F fucose; G galactose; GN N-acetylglucosamine; M mannose. Partially adapted from Ref. [49] with the modification
MS also offers useful tools for rapid and sensitive profiling of glycosylation of glycoproteins. MS analyses enable site-specific glycosylation profiling as well as determination of entire molecular glycoforms of multiple glycosylated proteins including glycan combinations [50,51,52,53]. Although discrimination of isomeric oligosaccharides is not possible by conventional MS measurement, attempts have been made to achieve it by analyzing the fragmentation patterns observed in multistage tandem mass (MSn) spectra [54, 55].
5 Remodeling of Glycoprotein Glycoforms
Heterogeneous glycosylation hampers unambiguous structural studies of glycoproteins, making structural biologists glycophobics. To overcome this problem, various techniques have been applied for remodeling glycoprotein glycoforms to reduce glycan heterogeneity. Glycoform remodeling is also useful for systematic analyses of the structure–function relationships of glycoproteins, addressing the functional properties of individual glycoforms.
Glycosidase treatment has been traditionally used for trimming of glycoprotein glycans. Glycoproteins with a uniform glycan structure can also be prepared by in vitro enzymatic attachment of non-reducing terminal sugar residue(s) [49, 56]. Figure 15.4b, c shows elution profiles of N-glycans derived from digalactosyl (G2) and galactosyl (G0) glycoforms of human IgG1-Fc prepared by enzymatic galactosylation and degalactosylation, respectively. The IgG1-Fc glycans could be further trimmed into the trimannosyl core FucGlcNAc2Man3 by N-acetylhexosaminidase treatment (Fig. 15.4d) and subsequently into the Fucα1 → 6GlcNAc disaccharide (FGN) with endo D treatment. The glycan could be completely eliminated by PNGase F treatment. Figure 15.5 compares the 1H–15N HSQC spectra of a series of Fc glycoforms (G2, G0, M3, FGN, and deglycosylation), displaying progressive spectral changes of the polypeptide backbone of Fc upon stepwise trimming of the glycans [49]. The results indicated that removal of the cleavage at the GlcNAcβ1-4GlcNAc linkage of the Fc glycans conformationally affected areas proximal to the hinge region, including the Fcγ receptor-binding sites, thereby impairing receptor binding. Therefore, the carbohydrate moieties of Fc play crucial roles in maintaining the structural integrity of the functional sites of IgG.

a 1H–15N HSQC spectral changes upon trimming of the carbohydrate chains of uniformly 13C/15N-labeled human IgG1-Fc. The spectra of the different glycoforms of Fc with different colors were superimposed. Black Fc(G2); blue Fc(G0); green Fc(M3); orange Fc(FGN); and red deglycosylated Fc. b The carbohydrate sequence attached to Asn297 of human IgG1-Fc. The arrows indicate the cleavage sites of the glycosidases used, and the resultant glycoforms are also shown. Adapted from Ref. [49] with the permission
Another approach for controlling glycosylation is genetic engineering of glycoprotein-producing cells by knocking out genes responsible for specific processes during glycan formation. For example, CHO cells lacking FUT8, an enzyme catalyzing the transfer of fucose from GDP-fucose to GlcNAc in an α-1,6 linkage, produce a completely non-fucosylated glycoform of IgG [57], which has a higher affinity for Fcγ receptor III on natural killer cells and consequently exerts an increased magnitude of antibody-dependent cell-mediated cytotoxicity (ADCC) [58,59,60]. Figure 15.6 compares the 1H–15N HSQC spectra of the fucosylated and non-fucosylated forms of IgG-Fc labeled with 15N at the amide groups of tyrosine residues, showing chemical shift perturbation and line broadening of the peak originating from Tyr296, which is proximal to the fucose residue (vide infra) [57].

a 1H–15N HSQC spectral comparison between the fucosylated (red) and Fuc non-fucosylated (black) glycoforms of 15N-Tyr-labeled human IgG1-Fc. Tyr296 of non-fucosylated Fc exhibits a significant broadening of the HSQC peak. b Mapping on the crystal structure of fucosylated IgG-Fc of the amino acid residues showing the chemical shift difference between fucosylated and non-fucosylated IgG-Fc. The chemical shift differences are quantified for each residue according to the equation (0.2δ 2N + δ 2H )1/2, where δN and δH represent the differences in nitrogen and proton chemical shifts between fucosylated and non-fucosylated IgG-Fc. The amino acid residues showing and not showing observable chemical shift differences (>0.1 ppm) are colored red and green on the crystal structure [PDB code: 3AVE] [57], respectively. The N-glycans are represented by stick models. The molecular graphics were generated with PyMOL [98]. Adapted from Ref. [57] with the permission
Genetic engineering of S. cerevisiae by deletion of genes involved in N-glycan processing has enabled overexpression of homogeneous high-mannose-type oligosaccharides M9 and M8B with 13C labeling [38, 41] (Fig. 15.7). Several attempts have been made to produce recombinant proteins with human-like glycosylation in yeast [61,62,63]. Currently, sialylated biantennary N-glycans can be expressed by engineered P. pastoris, in which four genes responsible for yeast-specific glycosylation were knocked out and 14 heterologous genes involved in the biosynthesis of human-type glycosylation were introduced [62]. Furthermore, IgG modified with complex-type N-glycans could be expressed in silkworm by co-expression of human β1,2-N-acetylglucosaminyltransferase II and β1,4-galactosyltransferase [64]. This genetically engineered production vehicle may be used for the expression of isotope-labeled recombinant glycoproteins for NMR studies.

NMR spectra of the high-mannose-type undecasaccharide M9. a 1H–13C HSQC spectra of M9 (black) uniformly labeled with 13C compared with that of M8B (red). b 1H–13C HSQC spectra of M9, metabolically 13C-labeled with d-[1-13C]glucose (black), d-[2-13C]glucose (blue), d-[3-13C]glucose (green), d-[4-13C]glucose (magenta), d-[5-13C]glucose (orange), or d-[6-13C]glucose (cyan). The six spectra were superposed, and the 13C-labeled positions in the glucose isotopomers used as metabolic precursors are shown with circles in the same colors as the corresponding spectra. c 1H–13C HSQC spectra of M9, modified with a lanthanide chelating tag with Tm3+ (red) and La3+ (blue). Chemical shift differences induced by PCS are indicated by arrows. Adapted from the Refs. [38, 41, 69] with the permission
6 Spectral Observations and Assignments
Carbohydrate NMR spectral analyses are often hampered by peak overlapping, particularly when dealing with liberated oligosaccharides or solvent-exposed glycans. A straightforward approach for solving this problem is to employ a higher magnetic field so as to increase spectral resolution [65, 66]. Line narrowing of oligosaccharide CH peaks is expected in a higher magnetic field, because in systems where dipole–dipole interaction is a dominant relaxation mechanism, T 2 longevity of the molecules with an appropriate motional correlation time increases in the higher field [67, 68]. Paramagnetic lanthanide probes can also be used for improving chemical shift dispersion. Figure 15.7c illustrates an HSQC spectrum of high-mannose-type undecasaccharide M9 with a lanthanide chelating tag covalently attached at its reducing terminus with La3+ and Tm3+ [69]. The overlapping peaks originating from the three non-reducing terminal mannose residues exhibit different pseudo-contact shifts (PCSs), dissolving the chemical shift degeneracy because their spatial arrangements are different with respect to the reducing terminus. PCS thus provides conformational information of the oligosaccharides (vide infra).
Although scalar connectivity-based techniques are applicable for spectral assignments of glycoprotein polypeptide chains, sequential assignments of the glycan remain partially dependent on nuclear Overhauser effect (NOE) connectivities. Therefore, selective isotope labeling of the carbohydrate moieties facilitates spectral assignments. Metabolic labeling using appropriate isotope-labeled metabolic precursors enables residue-specific and/or position-specific 13C incorporation [28, 29]. Figure 15.7b shows superposition of six 1H-13C HSQC spectra of M9 labeled with 13C at different positions using six different glucose isotopomers as metabolic precursors [41]. Peak classification into the sugar residue types can be made based on differential protonation levels during metabolic labeling using perdeuterated 13C-glucose as a metabolic precursor [38, 70]. In vitro enzymatic attachment of an isotope-labeled sugar onto the non-reducing end of the glycan is also useful for selective observation of NMR signals originating from the terminal sugar residues [33, 71, 72]. Yamaguchi et al. [73] proposed that 13C-detected 13C–13C NOE spectroscopy (NOESY) experiments are useful for spectral assignments of large glycoproteins because the observed peaks can be easily classified into monosaccharide types with specific linkage configurations by inspecting the intraresidue 13C–13C connectivity patterns (Fig. 15.8).

Two-dimensional 13C–13C NOESY patterns expected for the sugar residues in a biantennary complex-type oligosaccharide (left) and the oligosaccharide region of the two-dimensional 13C–13C NOESY spectrum of 13C-labeled mouse IgG2b–Fc (right). Adapted from Ref. [73] with the modification
Although carbohydrate hydroxyl groups are functionally crucial, as exemplified by hydrogen bonding, direct observation of their 1H NMR signals is extremely difficult in aqueous solution. An indirect approach utilizes the H/D isotope effect on the 13C chemical shift at 13C–OH groups to estimate exchange rate for OH with the solvent in a 50/50 H2O/D2O solution at steady state [74, 75]. This technique is applicable for the estimation of the NH exchange rate of the acetamide group of N-acetylglucosamine residues of a glycoprotein with appropriate 13C labeling (Fig. 15.9) [27]. Freedberg and coworkers used a 1H–13C HSQC–total correlated spectroscopy (TOCSY)-based technique to measure OH exchange rates in a 50/50 H2O/D2O solution, enabling quantitative characterization of transient hydrogen bonds involving the hydroxyl groups [76].

Schematic view of deuterium–hydrogen exchange of the amide group of GlcNAc residue on the line shape of the carbonyl 13C resonance due to deuterium isotope shift and (left) and 400 MHz 2D H(C)CO spectra of IgG-Fc showing the peaks from the GlcNAc acetamide groups. The Fc sample was dissolved in a sodium phosphate buffer H2O:D2O = 1:1. The nomenclature of oligosaccharide residues is represented in Fig. 15.5b. Adapted from Ref. [27] with the permission
7 NMR Analyses of Dynamic Conformations and Interactions of Oligosaccharides
In the case of other biomolecules, conformations of oligosaccharides are primarily characterized based on NOE data complemented with those of J coupling and residual dipolar coupling [77,78,79]. NOE data are also used for probing carbohydrate–protein interactions. Figure 15.10a shows intramolecular NOE connectivities observed between the core fucose of biantennary complex-type glycan and the aromatic ring of its spatially proximal tyrosine residue (Tyr296) in human IgG1-Fc. Core fucosylation precludes optimum binding of IgG1 to Fcγ receptor IIIa, which is mediated by the Tyr296 side chain, and thereby compromises ADCC activity [58]. This is, at least partially, attributed to steric masking of the functionally important tyrosine by the fucose. In the absence of the core fucose, Tyr296 gains motional freedom as exemplified by exchange line broadening of the 1H–15N HSQC peak originating from this tyrosine [57].

Parts of a 2D HSQC–NOESY spectrum of human IgG1-Fc (metabolically labeled with [13C6]glucose) exhibiting intramolecular NOE connectivities between Tyr296 and the core fucose residue and b 15N-edited NOESY spectrum of the 15N-labeled Fc-derived heptapeptide carrying Man3GlcNAc2 bound to the carbohydrate-binding domain of Fbs1 exhibiting intermolecular NOE peak (boxed) between Tyr279 Hε (Fbs1) and Asn3 Hδ (glycopeptide). Adapted from Refs. [27, 69] with the permission
Glycoprotein recognition by lectins can be investigated by observing intermolecular NOEs using isotope-labeled oligosaccharides or glycopeptides [80, 81]. Figure 15.10B shows an 15N-edited NOESY spectrum indicating intermolecular NOE connectivities between the carbohydrate-binding domain of Fbs1, a cytosolic lectin, as well as the substrate recognition part of a multisubunit ubiquitin ligase and its cognate glycopeptide cleaved from metabolically 13C/15N-labeled IgG [80]. In this specific case, the lectin contacts not only the innermost part of the glycan but also the side chain of the glycosylated asparagine. A glycan–polypeptide junction of glycoprotein is usually shielded from the solvent by spatially surrounding amino acid residues but can be exposed in its misfolded state and targeted for ubiquitin-mediated proteasomal degradation. Fbs1 captures the junction and protects it against attack by the cytosolic deglycosylation enzyme during ubiquitination of glycoprotein substrates. Because carbohydrate–lectin interactions are typically weak, with dissociation constants in the submillimolar range, transferred NOE and saturation transfer difference experiments are effective to characterize such weak interactions [82,83,84].
NOE-based conformational analysis of carbohydrate chains is hampered by low proton density, in comparison with that of protein. Moreover, conformational dynamics of carbohydrate chains hamper quantitative interpretation of NOE data. Recently, paramagnetism-assisted NMR approaches have been applied to conformational analysis of oligosaccharides [85,86,87]. In particular, PCS data, obtained employing paramagnetic lanthanide probes, are used as source of long distance (as far as 40 Å) information independent of the correlation time of the heterogeneous internal motion of oligosaccharides. Paramagnetic relaxation enhancements have also been used to characterize conformation and metal binding of oligosaccharides and their clusters [88,89,90].
Because oligosaccharides are extremely flexible molecules, NMR provides information averaged over their dynamic conformational ensembles. To address this issue, NMR data are treated as population-weighted averages of the possible conformers and reproduced by a population-optimized combination of selected low-energy conformers. In a recently proposed approach, NMR data are used for the validation of conformational ensembles derived from molecular dynamics (MD) simulation of flexible oligosaccharides that exhibit shallow and broad energy minima in their conformational space [69, 91,92,93]. In this approach, experimentally observed PCS data are compared with those back-calculated from the MD-derived conformational ensembles (Fig. 15.11). NMR validation showed that conformational spaces of the triantennary high-mannose-type oligosaccharides could be sufficiently explored by replica-exchange MD simulation with a total simulation time of several microseconds [69]. In conjunction with crystallographic data of lectin-bound oligosaccharides, carbohydrate recognition by lectins is characterized by conformational selection as well as induced-fit mechanisms [87, 93].

Conformational ensemble of M9 obtained from replica-exchange MD calculation validated by the PCS data (left) and the correlation between experimentally observed PCS values with Tm3+ and theoretical PCS data back-calculated from the ensemble model (right). Adapted from Ref. [69] with the permission
Oligosaccharides accommodated in a protein can be less mobile than liberated ones. As a typical example, the pair of biantennary oligosaccharides packed within the quaternary structure of IgG-Fc is visualized for the most part in the crystalline state [57, 58]. However, the terminal galactose residues exhibit different dynamic properties, which have been characterized on the basis of relaxation data [33, 71]. Therefore, the glycans attached to proteins can exhibit heterogeneous properties in terms of conformational dynamics.
8 Perspectives
NMR spectroscopy in conjunction with computational approaches has now successfully characterized dynamic conformational ensembles of oligosaccharides and glycoprotein glycans. This prompts us to improve functionality of carbohydrate chains by designing and optimizing their conformational spaces. To realize this glycoengineering, it is obviously necessary to create glycoproteins with tailored and even artificial glycoforms.
A hybrid approach combining cell engineering and chemoenzymatic techniques has the ability to deal with glycoform heterogeneity of glycoproteins in NMR structural glycobiology [94]. This line of technical development will realize isotope-labeled glycoproteins with tailored glycoforms. By installing glycosylation systems of C. jejuni and S. cerevisiae into E. coli, bacterial expression of recombinant glycoproteins having the core structure of eukaryotic glycans, i.e., Man3GlcNAc2, has been achieved [95]. This glycan can be substituted with synthetic donor oligosaccharides by in vitro enzymatic transglycosylation catalyzed by endo-β-N-acetylglucosaminidases, leaving the innermost GlcNAc residue as acceptor site [96].
Furthermore, synthetic approaches have been successfully developed to prepare glycoproteins involving chemical ligation of recombinant non-glycosylated peptides and chemically synthesized glycopeptides [97]. These technical advancements will open up new possibilities to create glycoproteins with artificial glycoforms. NMR spectroscopy along with computational approaches will play critical roles in detailed characterization of dynamic conformations and interactions of those neo-glycoproteins for better understanding of the functional mechanisms of carbohydrate chains.
References
Boehr, D.D., Nussinov, R., Wright, P.E.: The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 (2009)
Kovermann, M., Rogne, P., Wolf-Watz, M.: Protein dynamics and function from solution state NMR spectroscopy. Q. Rev. Biophys. 49, e6 (2016)
Rosenzweig, R., Kay, L.E.: Bringing dynamic molecular machines into focus by methyl-TROSY NMR. Annu. Rev. Biochem. 83, 291–315 (2014)
Freedberg, D.I., Selenko, P.: Live cell NMR. Annu. Rev. Biophys. 43, 171–192 (2014)
Skinner, A.L., Laurence, J.S.: High-field solution NMR spectroscopy as a tool for assessing protein interactions with small molecule ligands. J. Pharm. Sci. US 97, 4670–4695 (2008)
Dias, D.M., Ciulli, A.: NMR approaches in structure-based lead discovery: recent developments and new frontiers for targeting multi-protein complexes. Prog. Biophys. Mol. Biol. 116, 101–112 (2014)
Carlson, E.D., Gan, R., Hodgman, C.E., Jewett, M.C.: Cell-free protein synthesis: applications come of age. Biotechnol. Adv. 30, 1185–1194 (2012)
Apweiler, R., Hermjakob, H., Sharon, N.: On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8 (1999)
Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E.: Essentials of Glycobiology, 2nd edn. Cold Spring Harbor Laboratory Press, New York (2009)
Bubb, W.A.: NMR spectroscopy in the study of carbohydrates: characterizing the structural complexity. Concepts Magn. Resonance Part A 19A, 1–19 (2003)
Prestegard, J.H., Koerner, T.A.W., Demou, P.C., Yu, R.K.: Complete analysis of oligosaccharide primary structure using two-dimensional high-field proton NMR. J. Am. Chem. Soc. 104, 4993–4995 (1982)
Vliegenthart, J.F.: High resolution 1H-NMR spectroscopy of carbohydrate structures. Adv. Exp. Med. Biol. 125, 77–91 (1980)
Albrecht, S., Unwin, L., Muniyappa, M., Rudd, P.M.: Glycosylation as a marker for inflammatory arthritis. Cancer Biomark. 14, 17–28 (2014)
Adamczyk, B., Albrecht, S., Stockmann, H., Ghoneim, I.M., Al-Eknah, M., Al-Busadah, K.A., Karlsson, N.G., Carrington, S.D., Rudd, P.M.: Pregnancy-associated changes of IgG and serum N-glycosylation in Camel (Camelus dromedarius). J. Proteome Res. 15, 3255–3265 (2016)
Parekh, R.B., Dwek, R.A., Sutton, B.J., Fernandes, D.L., Leung, A., Stanworth, D., Rademacher, T.W., Mizuochi, T., Taniguchi, T., Matsuta, K., et al.: Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 316, 452–457 (1985)
Yamada, E., Tsukamoto, Y., Sasaki, R., Yagyu, K., Takahashi, N.: Structural changes of immunoglobulin G oligosaccharides with age in healthy human serum. Glycoconj. J. 14, 401–405 (1997)
Kibe, T., Fujimoto, S., Ishida, C., Togari, H., Wada, Y., Okada, S., Nakagawa, H., Tsukamoto, Y., Takahashi, N.: Glycosylation and placental transport of immunoglobulin G. J. Clin. Biochem. Nutr. 21, 57–63 (1996)
Park, S.S., Park, J., Ko, J., Chen, L., Meriage, D., Crouse-Zeineddini, J., Wong, W., Kerwin, B.A.: Biochemical assessment of erythropoietin products from Asia versus US Epoetin alfa manufactured by Amgen. J. Pharm. Sci. 98, 1688–1699 (2009)
Zeck, A., Pohlentz, G., Schlothauer, T., Peter-Katalinic, J., Regula, J.T.: Cell type-specific and site directed N-glycosylation pattern of FcγRIIIa. J. Proteome Res. 10, 3031–3039 (2011)
Lindhorst, T.K.: Essentials of Carbohydrate Chemistry and Biochemistry, 3rd Completely Revised and Enlarged Edition edn. Wiley, London (2007)
de Graaf, M., Fouchier, R.A.: Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J. 33, 823–841 (2014)
Wormald, M.R., Petrescu, A.J., Pao, Y.L., Glithero, A., Elliott, T., Dwek, R.A.: Conformational studies of oligosaccharides and glycopeptides: complementarity of NMR, X-ray crystallography, and molecular modelling. Chem. Rev. 102, 371–386 (2002)
Masuda, K., Yamaguchi, Y., Takahashi, N., Jefferis, R., Kato, K.: Mutational deglycosylation of the Fc portion of immunoglobulin G causes O-sulfation of tyrosine adjacently preceding the originally glycosylated site. FEBS Lett. 584, 3474–3479 (2010)
Shibata-Koyama, M., Iida, S., Okazaki, A., Mori, K., Kitajima-Miyama, K., Saitou, S., Kakita, S., Kanda, Y., Shitara, K., Kato, K., Satoh, M.: The N-linked oligosaccharide at FcγRIIIa Asn-45: an inhibitory element for high FcγRIIIa binding affinity to IgG glycoforms lacking core fucosylation. Glycobiology 19, 126–134 (2009)
Yamaguchi, Y., Kato, K.: Dynamics and interactions of glycoconjugates probed by stable-isotope-assisted NMR spectroscopy. Methods Enzymol. 478, 305–322 (2010)
Sastry, M., Xu, L., Georgiev, I.S., Bewley, C.A., Nabel, G.J., Kwong, P.D.: Mammalian production of an isotopically enriched outer domain of the HIV-1 gp120 glycoprotein for NMR spectroscopy. J. Biomol. NMR 50, 197–207 (2011)
Kato, K., Yamaguchi, Y., Arata, Y.: Stable-isotope-assisted NMR approaches to glycoproteins using immunoglobulin G as a model system. Prog. Nucl. Magn. Reson. Spectrosc. 56, 346–359 (2010)
Yamaguchi, Y., Yagi, H., Kato, K.: Stable isotope labeling of glycoproteins for NMR study. In: NMR in Glycoscience and Glycotechnology, pp. 194–207. RSC Publishing, Cambridge (2017)
Kato, K., Yamaguchi, Y.: Glycoproteins and antibodies: solution NMR studies. In: Encyclopedia of Magnetic Resonance, vol. 3, pp. 1779–1790. Wiley, Chichester (2012)
Kato, K., Matsunaga, C., Igarashi, T., Kim, H., Odaka, A., Shimada, I., Arata, Y.: Complete assignment of the methionyl carbonyl carbon resonances in switch variant anti-dansyl antibodies labeled with [1-13C]methionine. Biochemistry 30, 270–278 (1991)
Yagi, H., Zhang, Y., Yagi-Utsumi, M., Yamaguchi, T., Iida, S., Yamaguchi, Y., Kato, K.: Backbone 1H, 13C, and 15N resonance assignments of the Fc fragment of human immunoglobulin G glycoprotein. Biomol. NMR Assign. 9, 257–260 (2015)
Kato, K., Yamaguchi, Y.: Structural analyses of glycoconjugates by NMR. In: Experimental Glycoscience, pp. 45–50. Springer, Tokyo (2008)
Yamaguchi, Y., Kato, K., Shindo, M., Aoki, S., Furusho, K., Koga, K., Takahashi, N., Arata, Y., Shimada, I.: Dynamics of the carbohydrate chains attached to the Fc portion of immunoglobulin G as studied by NMR spectroscopy assisted by selective 13C labeling of the glycans. J. Biomol. NMR 12, 385–394 (1998)
Yagi, H., Fukuzawa, N., Tasaka, Y., Matsuo, K., Zhang, Y., Yamaguchi, T., Kondo, S., Nakazawa, S., Hashii, N., Kawasaki, N., Matsumura, T., Kato, K.: NMR-based structural validation of therapeutic antibody produced in Nicotiana benthamiana. Plant Cell Rep. 34, 959–968 (2015)
Yagi, H., Nakamura, M., Yokoyama, J., Zhang, Y., Yamaguchi, T., Kondo, S., Kobayashi, J., Kato, T., Park, E.Y., Nakazawa, S., Hashii, N., Kawasaki, N., Kato, K.: Stable isotope labeling of glycoprotein expressed in silkworms using immunoglobulin G as a test molecule. J. Biomol. NMR 62, 157–167 (2015)
Walton, W.J., Kasprzak, A.J., Hare, J.T., Logan, T.M.: An economic approach to isotopic enrichment of glycoproteins expressed from Sf9 insect cells. J. Biomol. NMR 36, 225–233 (2006)
Blanchard, V., Gadkari, R.A., George, A.V., Roy, S., Gerwig, G.J., Leeflang, B.R., Dighe, R.R., Boelens, R., Kamerling, J.P.: High-level expression of biologically active glycoprotein hormones in Pichia pastoris strains–selection of strain GS115, and not X-33, for the production of biologically active N-glycosylated 15N-labeled phCG. Glycoconj. J. 25, 245–257 (2008)
Kamiya, Y., Yamamoto, S., Chiba, Y., Jigami, Y., Kato, K.: Overexpression of a homogeneous oligosaccharide with 13C labeling by genetically engineered yeast strain. J. Biomol. NMR 50, 397–401 (2011)
Cubeddu, L., Moss, C.X., Swarbrick, J.D., Gooley, A.A., Williams, K.L., Curmi, P.M., Slade, M.B., Mabbutt, B.C.: Dictyostelium discoideum as expression host: isotopic labeling of a recombinant glycoprotein for NMR studies. Protein Exp. Purif. 19, 335–342 (2000)
Nettleship, J.E., Assenberg, R., Diprose, J.M., Rahman-Huq, N., Owens, R.J.: Recent advances in the production of proteins in insect and mammalian cells for structural biology. J. Struct. Biol. 172, 55–65 (2010)
Kamiya, Y., Yanagi, K., Kitajima, T., Yamaguchi, T., Chiba, Y., Kato, K.: Application of metabolic 13C labeling in conjunction with high-field nuclear magnetic resonance spectroscopy for comparative conformational analysis of high mannose-type oligosaccharides. Biomolecules 3, 108–123 (2013)
Sattler, M., Fesik, S.W.: Use of deuterium labeling in NMR: overcoming a sizeable problem. Structure 4, 1245–1249 (1996)
Kainosho, M., Guntert, P.: SAIL–stereo-array isotope labeling. Q. Rev. Biophys. 42, 247–300 (2009)
Opitz, C., Isogai, S., Grzesiek, S.: An economic approach to efficient isotope labeling in insect cells using homemade 15N-, 13C- and 2H-labeled yeast extracts. J. Biomol. NMR 62, 373–385 (2015)
Guarino, C., DeLisa, M.P.: A prokaryote-based cell-free translation system that efficiently synthesizes glycoproteins. Glycobiology 22, 596–601 (2012)
Slynko, V., Schubert, M., Numao, S., Kowarik, M., Aebi, M., Allain, F.H.: NMR structure determination of a segmentally labeled glycoprotein using in vitro glycosylation. J. Am. Chem. Soc. 131, 1274–1281 (2009)
Takahashi, N., Nakagawa, H., Fujikawa, K., Kawamura, Y., Tomiya, N.: Three-dimensional elution mapping of pyridylaminated N-linked neutral and sialyl oligosaccharides. Anal. Biochem. 226, 139–146 (1995)
Takahashi, N., Kato, K.: GALAXY(Glycoanalysis by the Three Axes of MS and Chromatography): a Web Application that Assists Structural Analyses of N-glycans. Trends Glycosci. Glycotech. 15, 235–251 (2003)
Yamaguchi, Y., Nishimura, M., Nagano, M., Yagi, H., Sasakawa, H., Uchida, K., Shitara, K., Kato, K.: Glycoform-dependent conformational alteration of the Fc region of human immunoglobulin G1 as revealed by NMR spectroscopy. Biochim. Biophys. Acta 1760, 693–700 (2006)
Yagi, H., Kuo, C.W., Obayashi, T., Ninagawa, S., Khoo, K.H., Kato, K.: Direct mapping of additional modifications on phosphorylated O-glycans of alpha-dystroglycan by mass spectrometry analysis in conjunction with knocking out of causative genes for dystroglycanopathy. Mol. Cell. Proteomics 15, 3424–3434 (2016)
Masuda, K., Yamaguchi, Y., Kato, K., Takahashi, N., Shimada, I., Arata, Y.: Pairing of oligosaccharides in the Fc region of immunoglobulin G. FEBS Lett. 473, 349–357 (2000)
Thaysen-Andersen, M., Packer, N.H.: Advances in LC-MS/MS-based glycoproteomics: getting closer to system-wide site-specific mapping of the N- and O-glycoproteome. Biochim. Biophys. Acta 1844, 1437–1452 (2014)
Terral, G., Beck, A., Cianferani, S.: Insights from native mass spectrometry and ion mobility-mass spectrometry for antibody and antibody-based product characterization. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1032, 79–90 (2016)
Ito, H., Kameyama, A., Sato, T., Narimatsu, H.: Preparation of a glycan library using a variety of glycosyltransferases. Methods Mol. Biol. 534, 283–291 (2009)
Kato, K., Yamaguchi, Y., Takahashi, N., Nishimura, M., Iwamoto, S., Sekiya, S., Tanaka, K.: Discrimination of isomeric fragment ions observed in tandem mass spectra of biantennary oligosaccharides by use of selective isotope labeling. J. Mass Spectrom. Soc. Jpn. 52, 284–288 (2004)
Tanji, H., Ohto, U., Shibata, T., Miyake, K., Shimizu, T.: Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 339, 1426–1429 (2013)
Matsumiya, S., Yamaguchi, Y., Saito, J., Nagano, M., Sasakawa, H., Otaki, S., Satoh, M., Shitara, K., Kato, K.: Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J. Mol. Biol. 368, 767–779 (2007)
Mizushima, T., Yagi, H., Takemoto, E., Shibata-Koyama, M., Isoda, Y., Iida, S., Masuda, K., Satoh, M., Kato, K.: Structural basis for improved efficacy of therapeutic antibodies upon defucosylation of their Fc glycans. Genes Cells 16, 1071–1080 (2011)
Shields, R.L., Lai, J., Keck, R., O’Connell, L.Y., Hong, K., Meng, Y.G., Weikert, S.H., Presta, L.G.: Lack of fucose on human IgG1N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J. Biol. Chem. 277, 26733–26740 (2002)
Shinkawa, T., Nakamura, K., Yamane, N., Shoji-Hosaka, E., Kanda, Y., Sakurada, M., Uchida, K., Anazawa, H., Satoh, M., Yamasaki, M., Hanai, N., Shitara, K.: The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 278, 3466–3473 (2003)
Chiba, Y., Akeboshi, H.: Glycan engineering and production of ‘humanized’ glycoprotein in yeast cells. Biol. Pharm. Bull. 32, 786–795 (2009)
Hamilton, S.R., Davidson, R.C., Sethuraman, N., Nett, J.H., Jiang, Y., Rios, S., Bobrowicz, P., Stadheim, T.A., Li, H., Choi, B.K., Hopkins, D., Wischnewski, H., Roser, J., Mitchell, T., Strawbridge, R.R., Hoopes, J., Wildt, S., Gerngross, T.U.: Humanization of yeast to produce complex terminally sialylated glycoproteins. Science 313, 1441–1443 (2006)
Irani, Z.A., Kerkhoven, E.J., Shojaosadati, S.A., Nielsen, J.: Genome-scale metabolic model of Pichia pastoris with native and humanized glycosylation of recombinant proteins. Biotechnol. Bioeng. 113, 961–969 (2016)
Kato, T., Kako, N., Kikuta, K., Miyazaki, T., Kondo, S., Yagi, H., Kato, K., Park, E.Y.: N-glycan modification of a recombinant protein via coexpression of human glycosyltransferases in silkworm pupae. Sci. Rep. 7, 1409 (2017)
Kato, K., Sasakawa, H., Kamiya, Y., Utsumi, M., Nakano, M., Takahashi, N., Yamaguchi, Y.: 920 MHz ultra-high field NMR approaches to structural glycobiology. Biochim. Biophys. Acta 1780, 619–625 (2008)
Blundell, C.D., Reed, M.A., Overduin, M., Almond, A.: NMR spectra of oligosaccharides at ultra-high field (900 MHz) have better resolution than expected due to favourable molecular tumbling. Carbohydr. Res. 341, 1985–1991 (2006)
Canet, D., Robert, J.B.: Behaviour of the NMR relaxation parameters at high fields. In: NMR at very high field, vol. 25, pp 45–89. Springer, Berlin (1991)
Meersmann, T., Schwager, M., Varma, V., Bodenhausen, G.: Little-known advantages of very high fields in NMR. J. Magn. Reson. Ser. A 119, 275–279 (1996)
Yamaguchi, T., Sakae, Y., Zhang, Y., Yamamoto, S., Okamoto, Y., Kato, K.: Exploration of conformational spaces of high-mannose-type oligosaccharides by an NMR-validated simulation. Angew. Chem. Int. Ed. Engl. 53, 10941–10944 (2014)
Yamaguchi, Y., Takizawa, T., Kato, K., Arata, Y., Shimada, I.: 1H and 13C NMR assignments for the glycans in glycoproteins by using 2H/13C-labeled glucose as a metabolic precursor. J. Biomol. NMR 18, 357–360 (2000)
Barb, A.W., Prestegard, J.H.: NMR analysis demonstrates immunoglobulin G N-glycans are accessible and dynamic. Nat. Chem. Biol. 7, 147–153 (2011)
Gilhespy-Muskett, A.M., Partridge, J., Jefferis, R., Homans, S.W.: A novel 13C isotopic labelling strategy for probing the structure and dynamics of glycan chains in situ on glycoproteins. Glycobiology 4, 485–489 (1994)
Yamaguchi, Y., Walchli, M., Nagano, M., Kato, K.: A C-13-detection NMR approach for large glycoproteins. Carbohyd. Res. 344, 535–538 (2009)
Hanashima, S., Kato, K., Yamaguchi, Y.: 13C-NMR quantification of proton exchange at LewisX hydroxyl groups in water. Chem. Commun. (Camb.) 47, 10800–10802 (2011)
Pfeffer, P.E., Valentine, K.M., Parrish, F.W.: Deuterium-induced differential isotope shift C-13 NMR. 1. Resonance re-assignments of monosaccharides and disaccharides. J. Am. Chem. Soc. 101, 1265–1274 (1979)
Battistel, M.D., Azurmendi, H.F., Freedberg, D.I.: Glycan OH exchange rate determination in aqueous solution: seeking evidence for transient hydrogen bonds. J. Phys. Chem. B 121, 683–695 (2017)
Hadad, M., J,, Zhang, W., Turney, T., Sernau, L., Wang, X., Woods, R.J., Incandela, A., Surjancev, I., Wang, A., Yoon, M.Y., Coscia, A., Euell, C., Meredith, R., Carmichael, I., Serianni, A.: NMR Spin-couplings in saccharides: relationships between structure, conformation and the magnitudes of JHH, JCH and JCC values. In: NMR in Glycoscience and Glycotechnology, pp. 20–100. RSC Publishing, Cambridge (2017)
Prestegard, J.H., Eletsky, A.: Paramagnetic, RDC and RCSA constraints in the structural analysis of glycans. In: NMR in Glycoscience and Glycotechnology, pp. 123–149. RSC Publishing, Cambridge (2017)
Schubert, M.: Insights into carbohydrate recognition by 3D structure determination of protein–carbohydrate complexes using NMR. In: NMR in Glycoscience and Glycotechnology, pp. 101–122. RSC Publishing, Cambridge. (2017)
Yamaguchi, Y., Hirao, T., Sakata, E., Kamiya, Y., Kurimoto, E., Yoshida, Y., Suzuki, T., Tanaka, K., Kato, K.: Fbs1 protects the malfolded glycoproteins from the attack of peptide: N-glycanase. Biochem. Biophys. Res. Commun. 362, 712–716 (2007)
Nestor, G., Anderson, T., Oscarson, S., Gronenborn, A.M.: Exploiting uniformly 13C-labeled carbohydrates for probing carbohydrate-protein interactions by nmr spectroscopy. J. Am. Chem. Soc. 139, 6210–6216 (2017)
Yamaguchi, Y., Kato, K.: Analyses of sugar–protein interactions by NMR. In: Experimental Glycoscience, pp. 121–123. Springer, Tokyo (2008)
Mayer, M., Meyer, B.: Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 123, 6108–6117 (2001)
Jimenes-Barbero, J., Peters, T.: TR-NOE experiments to study carbohydrate-protein interactions. In: NMR Spectroscopy of Glycoconjugates, pp. 289–309. Wiley-VCH, Weinheim (2006)
Canales, A., Mallagaray, A., Pérez-Castells, J., Boos, I., Unverzagt, C., André, S., Gabius, H.-J., Cañada, F.J., Jiménez-Barbero, J.: Breaking pseudo-symmetry in multiantennary complex N-glycans using lanthanide-binding tags and NMR pseudo-contact shifts. Angew. Chem. Int. Ed. 52, 13789–13793 (2013)
Kato, K., Yamaguchi, T.: Paramagnetic NMR probes for characterization of the dynamic conformations and interactions of oligosaccharides. Glycoconj. J. 32, 505–513 (2015)
Kato, K., Yagi, H., Yamaguchi, T.: NMR characterization of dynamic conformations and interactions of oligosaccharides. In: Modern Magnetic Resonance. Springer, Berlin (2017, in press)
Yamaguchi, T., Kamiya, Y., Choo, Y.M., Yamamoto, S., Kato, K.: Terminal spin labeling of a high-mannose-type oligosaccharide for quantitative NMR analysis of its dynamic conformation. Chem. Lett. 42, 544–546 (2013)
Yan, G., Yamaguchi, T., Suzuki, T., Yanaka, S., Sato, S., Fujita, M., Kato, K.: Hyper-assembly of self-assembled glycoclusters mediated by specific carbohydrate–carbohydrate interactions. Chem. Asian J. 12, 968–972 (2017)
Demarco, M.L., Woods, R.J., Prestegard, J.H., Tian, F.: Presentation of membrane-anchored glycosphingolipids determined from molecular dynamics simulations and NMR paramagnetic relaxation rate enhancement. J. Am. Chem. Soc. 132, 1334–1338 (2010)
Zhang, Y., Yamamoto, S., Yamaguchi, T., Kato, K.: Application of paramagnetic NMR-validated molecular dynamics simulation to the analysis of a conformational ensemble of a branched oligosaccharide. Molecules 17, 6658 (2012)
Yamamoto, S., Zhang, Y., Yamaguchi, T., Kameda, T., Kato, K.: Lanthanide-assisted NMR evaluation of a dynamic ensemble of oligosaccharide conformations. Chem. Commun. 48, 4752–4754 (2012)
Suzuki, T., Kajino, M., Yanaka, S., Zhu, T., Yagi, H., Satoh, T., Yamaguchi, T., Kato, K.: Conformational analysis of a high-mannose-type oligosaccharide displaying glucosyl determinant recognised by molecular chaperones using NMR-validated molecular dynamics simulation. ChemBioChem 18, 396–401 (2017)
Kamiya, Y., Satoh, T., Kato, K.: Recent advances in glycoprotein production for structural biology: toward tailored design of glycoforms. Curr. Opin. Struct. Biol. 26, 44–53 (2014)
Valderrama-Rincon, J.D., Fisher, A.C., Merritt, J.H., Fan, Y.Y., Reading, C.A., Chhiba, K., Heiss, C., Azadi, P., Aebi, M., DeLisa, M.P.: An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat. Chem. Biol. 8, 434–436 (2012)
Schwarz, F., Huang, W., Li, C., Schulz, B.L., Lizak, C., Palumbo, A., Numao, S., Neri, D., Aebi, M., Wang, L.X.: A combined method for producing homogeneous glycoproteins with eukaryotic N-glycosylation. Nat. Chem. Biol. 6, 264–266 (2010)
Kajihara, Y., Tanabe, Y., Sasaoka, S., Okamoto, R.: Homogeneous human complex-type oligosaccharides in correctly folded intact glycoproteins: evaluation of oligosaccharide influence on protein folding, stability, and conformational properties. Chemistry 18, 5944–5953 (2012)
DeLano, W.L.: The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos (2002)
Acknowledgements
We thank Mr. Masaki Kato (Structural Glycobiology Team, RIKEN) for statistic analysis of PDB data and Drs. Takumi Yamaguchi (School of Material Science, Japan Advanced Institute of Science and Technology) and Maho Yagi-Utsumi (Institute for Molecular Science, National Institutes of Natural Sciences) for useful discussion. This work was partly supported by the Nanotechnology Platform Program (Molecule and Material Synthesis) of MEXT, MEXT/JSPS Grants in Aid for Scientific Research (JP25102008, JP15K07935 and JP17H05893), the Japan Agency for Medical Research and Development, and the Cooperative Research Program of Institute for Protein Research, Osaka University, NMRCR-16-05.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kato, K., Yanaka, S., Yagi, H. (2018). Technical Basis for Nuclear Magnetic Resonance Approach for Glycoproteins. In: The Nuclear Magnetic Resonance Society of Japan (eds) Experimental Approaches of NMR Spectroscopy. Springer, Singapore. https://doi.org/10.1007/978-981-10-5966-7_15
Download citation
DOI: https://doi.org/10.1007/978-981-10-5966-7_15
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-5965-0
Online ISBN: 978-981-10-5966-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)