
Abstract
Inter-organellar contact sites establish microdomains for localised Ca2+-signalling events. One of these microdomains is established between the ER and the mitochondria. Importantly, the so-called mitochondria-associated ER membranes (MAMs) contain, besides structural proteins and proteins involved in lipid exchange, several Ca2+-transport systems, mediating efficient Ca2+ transfer from the ER to the mitochondria. These Ca2+ signals critically control several mitochondrial functions, thereby impacting cell metabolism, cell death and survival, proliferation and migration. Hence, the MAMs have emerged as critical signalling hubs in physiology, while their dysregulation is an important factor that drives or at least contributes to oncogenesis and tumour progression. In this book chapter, we will provide an overview of the role of the MAMs in cell function and how alterations in the MAM composition contribute to oncogenic features and behaviours.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Calcium signaling
- Ca2+-transport systems
- IP3 receptors
- Voltage-dependent anion channels
- Chaperones
- Cell death and survival
- Mitochondrial bioenergetics
- Autophagy
- Mitochondria-associated ER membranes (MAMs)
- Cancer
17.1 Introduction
Eukaryotic cells are faced with many challenges to sustain life (Chen and Silver 2012). To tackle these challenges, nature has come up with cellular compartmentalisation resulting in different organelles including the nucleus, endoplasmic reticulum (ER), mitochondria, peroxisomes and Golgi apparatus (Diekmann and Pereira-Leal 2013). The ability of eukaryotic cells to restrict processes to a subcellular localisation brings about numerous advantages (Chen and Silver 2012). However, to ensure the smooth orchestration of cellular processes, communication between the organelles is critical.
One way of inter-organellar communication is Ca2+ signalling. The fact that Ca2+ is not homogeneously distributed throughout the cell makes it possible to use Ca2+ as a messenger (Clapham 2007). Ca2+ signals typically arise from the ER and the lysosomes (Berridge et al. 2000; Berridge et al. 2003). These Ca2+ signals impact the other organelles via microdomains established by membrane contact sites (La Rovere et al. 2016; Raffaello et al. 2016). Important target organelles of ER-derived Ca2+ signals are the mitochondria, which help to maintain Ca2+ homeostasis in the cell (Rizzuto et al. 2012). Ca2+ uptake in the mitochondria is driven by the negative mitochondrial membrane potential (Rizzuto et al. 2000). The outer mitochondrial membrane (OMM) is freely permeable to Ca2+ due to the presence of porins, like the type 1 voltage-gated anion channel (VDAC1) (Gincel et al. 2001; Rapizzi et al. 2002). While Ca2+ easily reaches the intermembrane space, a more intriguing problem was the entry of Ca2+ into the mitochondrial matrix. Initial research pointed to the existence of a mitochondrial uniporter, albeit one with low affinity for Ca2+ (Carafoli 2012; Marchi and Pinton 2014). However, the low affinity of this uniporter could not easily be matched to observations that Ca2+ was dynamically and efficiently exchanged between the cytosol and the mitochondria, considering that cytosolic [Ca2+] is typically in the low-to-middle nanomolar range (Rizzuto et al. 1998; Csordás et al. 1999; Carafoli 2012).
The mechanism underlying this seemingly paradoxical observation is the presence of close contacts between ER and mitochondria, which favour quasi-synaptic Ca2+ transfer from the ER to the mitochondria (Rizzuto et al. 1998; Csordás et al. 1999). These sites can be isolated biochemically as mitochondria-associated ER membranes (MAMs). MAMs are parts of the ER membrane that are in close proximity to the mitochondrial membrane and are tethered to it (Szabadkai et al. 2006; van Vliet et al. 2014). At the MAMs, the distance between the ER and mitochondria is believed to be approximately 10 to 25 nm (Rizzuto et al. 1998; Csordás et al. 2006; Marchi et al. 2014b), allowing proteins situated on the ER membrane and OMM to interact and enabling efficient Ca2+-based communication between the ER and mitochondria (Decuypere et al. 2011; Rowland and Voeltz 2012; Marchi et al. 2014b). In this way, MAMs provide a microdomain in which the [Ca2+] is several folds higher than in the bulk cytosol (estimated to be >10 μM at the ER-mitochondrial interface) (Csordás et al. 2010), alleviating the problem of the paradoxical low affinity of the mitochondrial Ca2+ uniporter (Rizzuto et al. 1993, 1998). Apart from Ca2+ homeostasis, MAMs are implicated in several processes critical to cell function, e.g. lipid transport, ER stress, apoptosis, autophagy, inflammation and anti-viral response (Vance 2014; van Vliet et al. 2014).
Due to their involvement in these specific functions, MAMs contain a select protein population. The inositol 1,4,5-trisphosphate (IP3) receptor (IP3R) and VDAC1 channels are present in this sub-organellar domain (Várnai et al. 2005; Szabadkai et al. 2006), underlying the prominent role of the MAMs in ER-mitochondrial Ca2+ signalling. Furthermore, proteins indirectly involved in ER-mitochondrial Ca2+ flux can be found in and around the MAMs as well. These include glucose-regulated protein 75 (GRP75) (Szabadkai et al. 2006), mitofusin-2 (Mfn2) (de Brito and Scorrano 2008), phosphofurin acidic cluster sorting protein-2 (PACS-2) (Myhill et al. 2008), promyelocytic leukaemia protein (PML) (Pinton et al. 2011), sigma-1 receptor (Sig-1R) (Hayashi and Su 2007) and protein kinase RNA-like endoplasmic reticulum kinase (PERK) (Verfaillie et al. 2012), amongst others. Since Ca2+ signalling fulfils an important role in several cell physiological processes and considering its dysregulation in pathophysiological conditions, the expression of these proteins is often altered, and their functional activity is converted to promote tumour growth, proliferation, migration, apoptosis resistance and changes in cellular metabolism (Urra et al. 2016; Marchi and Pinton 2016). Furthermore, the MAMs harbour an increasing number of oncogenes and tumour suppressors that functionally impact ER-mitochondrial Ca2+ transfer and oncogenic features (Marchi et al. 2014a; Giorgi et al. 2015b; Bittremieux et al. 2016).
In this chapter, we will discuss (1) the MAM components, including the Ca2+-transport systems, chaperones and structural proteins that are present, (2) how MAM components impact ER-mitochondrial Ca2+ transfer and their structural organisation and (3) how alterations in the function of these MAM components drive oncogenesis and tumour progression. An overview of the MAM components to be discussed in this chapter can be found in Fig. 17.1.

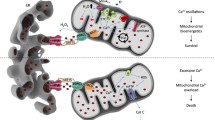
MAM components playing a role in ER-mitochondrial Ca 2+ signalling. The MAMs harbour a specific protein population consisting of Ca2+-transport proteins and chaperones, as well as of proteins that enable their structural organisation. The principal components of the ER-mitochondrial Ca2+ exchange at the MAMs are the IP3R and VDAC1, which are physically coupled by the chaperone protein GRP75. When Ca2+ is released from the ER by the IP3R, it freely permeates the OMM via VDAC1, to be transported to the mitochondrial matrix by the MCU, located in the IMM. The chaperone Sig-1R is able to modify IP3R-mediated Ca2+ signalling. Sig-1R is held inactive by binding to GRP78, but under ER stress binding to GRP78 is disrupted and Sig-1R interacts with the IP3R, stabilising the IP3R and enabling proficient Ca2+ signalling even under conditions of ER stress. The efficiency of Ca2+ exchange between ER and mitochondria is influenced by the presence and action of tethering proteins like PERK and Mfn2 and anti-tethering proteins like FATE1. The functional effect of tethering and anti-tethering proteins at the MAMs is indicated by arrows facing each other or arrows pointing in opposite directions, respectively. Besides its function as a tethering protein, PACS-2 also contributes to MAM organisation, while simultaneously having a role in the enrichment of the chaperone calnexin at the MAMs. Furthermore, calnexin is enriched at the MAMs by palmitoylation, a process that switches calnexin function from quality control/protein folding to ER Ca2+-signalling control by enhancing SERCA activity. The interaction between calnexin and SERCA2b appears to be counteracted by the thiol reductase TMX1 (not shown for clarity reasons), which inhibits SERCA2b activity (For more details, please see text)
17.2 MAM Components
17.2.1 The Ca2+-Signalling Machinery at the MAMs
17.2.1.1 IP3R
The IP3R is an intracellular Ca2+-release channel, present in the ER membrane (Ashby and Tepikin 2001; Choe and Ehrlich 2006). IP3Rs are opened by IP3 (Foskett et al. 2007; Parys and De Smedt 2012; Fedorenko et al. 2014), a second messenger released into the cytosol after phosphatidylinositol 4,5-bisphosphate cleavage by phospholipase C. IP3R activity is tightly controlled by cytosolic [Ca2+] in a biphasic manner (Iino 1990; Finch et al. 1991; Bezprozvanny et al. 1991; Parys et al. 1992). The Ca2+-flux properties of the IP3R are also regulated by other cellular factors, including ATP, regulatory proteins (Choe and Ehrlich 2006; Foskett et al. 2007; Parys and De Smedt 2012) and protein kinases and phosphatases (Vanderheyden et al. 2009). Structurally, the IP3R consists of three domains: an N-terminal ligand-binding domain, which is subdivided in a suppressor region and the IP3-binding core, a central modulatory domain and a pore-forming region in the C-terminal tail (Yoshikawa et al. 1999). The IP3R comes in three different isoforms (IP3R1, IP3R2 and IP3R3) which are encoded by different genes (ITPR1, ITPR2 and ITPR3) and display 60–80% homology at the level of the amino acid sequence (Mikoshiba 2007; Foskett et al. 2007). Sensitivity towards their ligand IP3 as well as regulation by Ca2+, ATP and phosphorylation appears to be isoform specific (Newton et al. 1994; Miyakawa et al. 1999; Tu et al. 2005; Khan et al. 2006; Betzenhauser et al. 2008; Wagner et al. 2008; Vervloessem et al. 2015).
A subset of IP3Rs is present at the MAMs, where it is responsible for ER-mitochondrial Ca2+ flux (Várnai et al. 2005; Mendes et al. 2005). By varying the spatio-temporal pattern of Ca2+ release from the ER, Ca2+ signalling can convey messages, which are differentially decoded at the subcellular level (Berridge et al. 2000, 2003). For example, constitutive low-level IP3R-mediated ER-mitochondrial Ca2+ transfer maintains mitochondrial bioenergetics through stimulation of mitochondrial respiration and ATP production (Cárdenas et al. 2010; Kaufman and Malhotra 2014), whereas excessive Ca2+ release from the ER triggers apoptotic cell death due to mitochondrial Ca2+ overload (Giorgi et al. 2012; Morciano et al. 2015). Hence, IP3Rs located at the MAMs play an important role in determining cell fate. Especially IP3R3 seems to be abundant at the MAMs, since it has been proposed that pro-apoptotic ER-mitochondrial Ca2+ transfers preferentially occur via IP3R3 (Blackshaw et al. 2000; Mendes et al. 2005). However, also the other IP3R isoforms have been implicated in Ca2+-mediated cell death (Gutstein and Marks 1997; Jayaraman and Marks 1997; Assefa et al. 2004; Li et al. 2009; Akl et al. 2013) and thus may reside in the MAMs in certain cell types or systems.
17.2.1.2 VDAC1
As described above, Ca2+ released by the IP3R is able to cross the OMM through VDAC1, a weakly anion-selective channel that is Ca2+ permeable and which is enriched at the MAMs (Hajnóczky et al. 2002; Shoshan-Barmatz and Gincel 2003; Colombini 2012). Apart from its role in mitochondrial Ca2+ transport, VDAC allows for substrates of the electron transport chain, like malate, succinate and nicotinamide adenine dinucleotide, to access the mitochondria (Shoshan-Barmatz et al. 2010). Moreover, VDAC’s channel properties permit ATP, produced by oxidative phosphorylation, as well as other mitochondrial products like reactive oxygen species (ROS) to diffuse into the cytosol (Shoshan-Barmatz et al. 2010). Additionally, VDAC1 oligomers have been implicated in the release of cytochrome c into the cytosol (Weisthal et al. 2014). As it were, VDAC functions as the channel that allows the mitochondria to communicate with their subcellular environment.
VDAC is able to switch between an open and a closed state in a voltage-dependent manner. While the channel is stable in the open state at low voltages, high voltages cause VDAC to switch to the closed state (Hodge and Colombini 1997; Gincel et al. 2000). Interestingly, the open state shows a weak selectivity towards anions, as opposed to the closed state, which blocks the passage of large anionic molecules, while it has been proposed to be selective for cations (Gincel et al., 2000; Schein et al., 1976; Shoshan-Barmatz et al., 2010). At the structural level, the N-terminus of the protein, which forms an α-helix, is important for its voltage-dependent gating (Abu-Hamad et al. 2009). Different mechanistic models have been proposed, albeit the exact mechanism has not been established yet (Shoshan-Barmatz et al. 2010), and this voltage-dependency has only been observed in vitro (Shoshan-Barmatz et al. 2010). Apart from voltage-dependent gating, the closed or open state of the channel is affected by modulators interacting with VDAC1. Examples include Bcl-XL (Vander Heiden et al. 2000, 2001), hexokinase (Azoulay-Zohar et al. 2004), tubulin (Rostovtseva et al. 2008), mitochondrial membrane lipids (Rostovtseva et al. 2006) and Ca2+ (Báthori et al. 2006).
In mammals, there are three known isoforms of VDAC: VDAC1, VDAC2 and VDAC3, with VDAC1 being the isoform that is expressed at the highest level and consequently has been studied most extensively (Messina et al. 2012). Recombinant expression of VDAC1 enhanced the Ca2+ transfer to the mitochondria (Rapizzi et al. 2002), yet this property seems not to be unique for VDAC1, as also the other isoforms display it (De Stefani et al. 2012). However, there is a unique role for VDAC1, but not for VDAC2 nor VDAC3, in conveying pro-apoptotic Ca2+ signals to the mitochondria (De Stefani et al. 2012).
17.2.2 The Chaperones
17.2.2.1 GRP75
GRP75 is a chaperone protein belonging to the heat shock 70 kDa (HSP70) protein family (Wadhwa et al. 2002a). GRP75 has been found at different subcellular localisations, e.g. the cytosol, the mitochondria, the ER and the Golgi apparatus (Wadhwa et al. 1995; Ran et al. 2000). Moreover, GRP75 is a pleiotropic protein. For example, GRP75 located in the mitochondrial matrix helps to import unfolded proteins into the matrix in an ATP-dependent manner in cooperation with Tim44 (Scherer et al. 1992; Kronidou et al. 1994; Schneider et al. 1996; Voos and Röttgers 2002). Furthermore, GRP75 is thought to play a role in endocytosis as well as exocytosis (Flachbartová and Kovacech 2013). At the MAMs, GRP75 plays an important role in Ca2+ signalling, as it forms a physical link between the IP3R and VDAC1 (Szabadkai et al. 2006; Betz et al. 2013; Rieusset et al. 2016), thereby increasing the efficiency of ER-mitochondrial Ca2+ signalling.
17.2.2.2 Sig-1R
Sig-1R is a chaperone, which was first mistakenly identified as an opioid receptor subtype (Su 1982; Hayashi and Su 2007; Tagashira et al. 2014). Sig-1R mainly resides in the ER, specifically at the MAMs, under resting conditions (Hayashi and Su 2007). There, Sig-1R is bound to an ER lumen chaperone, glucose-regulated protein 78 (GRP78, also known as BiP) (Hayashi and Su 2007). Upon ER stress, however, this association is broken, and Sig-1R gains its chaperone function, targeting its client proteins like the ER stress sensor inositol-requiring enzyme 1 (IRE1) and the IP3R, thereby regulating ER-mitochondrial Ca2+ signalling (cfr. infra) (Hayashi and Su 2007; Mori et al. 2013). Interestingly, Shioda et al. (2012) reported the existence of a truncated splice form of Sig-1R, which did not bind to the IP3R. Overexpression of this splice variant decreased mitochondrial Ca2+ uptake, while promoting IP3R degradation, as opposed to the non-truncated Sig-1R (Shioda et al. 2012). Furthermore, Sig-1R can undergo translocation to various subcellular localisations upon stimulation by agonists. These locations include the plasma membrane and the nuclear envelope (Su et al. 2010; Mavlyutov et al. 2015; Tsai et al. 2015; Chu and Ruoho 2016).
17.2.2.3 Calnexin
Calnexin is another ER chaperone that is enriched at the MAMs and this in a palmitoylation-dependent manner (Lynes et al. 2012). This chaperone interacts with glycoproteins that are monoglucosylated (Hebert et al. 1995). Functionally, calnexin improves efficiency of ER protein folding and helps retaining misfolded proteins in the ER (Lamriben et al. 2016). Furthermore, calnexin modifies Ca2+ signalling in the cell through its functional interaction with sarco/endoplasmic reticulum Ca2+-ATPase 2b (SERCA2b) (Roderick et al. 2000). When the cytosolic domain of calnexin is phosphorylated, the interaction inhibits SERCA2b activity, diminishing intracellular Ca2+ oscillations (Roderick et al. 2000). In further work, it was shown that calnexin interaction with SERCA was critically dependent on calnexin palmitoylation and was impaired upon ER stress induction (Lynes et al. 2013). The binding of calnexin to SERCA2b appeared to promote its activity, given the higher ER Ca2+-store content of cells overexpressing wild-type calnexin, but not a palmitoylation-deficient calnexin mutant (Lynes et al. 2013). Interestingly, the binding of calnexin to SERCA2b is influenced by the thioredoxin-related transmembrane protein (TMX1), which is also targeted to the MAMs through palmitoylation (Lynes et al. 2012), since knockout of TMX1 improved binding of calnexin to SERCA2b (Krols et al. 2016; Raturi et al. 2016). TMX1 and calnexin thus may target overlapping binding sites in SERCA2b. As such, the stimulatory effect of calnexin on SERCA activity may be partially related to its ability to reduce the binding of TMX1, which inhibits SERCA activity, to SERCA2b (Krols et al. 2016; Raturi et al. 2016).
17.2.3 Proteins Defining MAM Structure
17.2.3.1 Mfn2
Mfn2, a GTPase protein situated in the OMM, was first studied for its function as a mitochondrial fusion protein, together with its homologue Mfn1 (Ranieri et al. 2013). Interestingly, in 2008 it was found that Mfn2 is also located on the ER membrane and enriched at the MAMs, where it acts as a tether that links mitochondria to the ER and supports efficient Ca2+ signalling between the two organelles (de Brito and Scorrano 2008). This tethering function of Mfn2 is realised by the interaction of Mfn2 in the ER membrane with Mfn1 or Mfn2 localised in the OMM. Recently, however, a discussion has risen about the role of Mfn2 as a mitochondrial tether in the MAMs. Ultrastructural analyses as well as functional, biochemical and genetic approaches showed that Mfn2 antagonised ER-mitochondrial tethering (Cosson et al. 2012; Filadi et al. 2015), since ablation of Mfn2 resulted in an increased inter-organellar proximity. It was proposed that Mfn2 functions as an anti-tether that maintains a correct, non-toxic distance between both the ER and the mitochondria (Filadi et al. 2015). However, very recently, the role of Mfn2 as a bona fide ER-mitochondrial tether has been confirmed in a series of experiments aiming to critically reappraise its function (Naon et al. 2016).
17.2.3.2 PACS-2
PACS-2 is an ER-associated protein involved in retrograde ER-Golgi trafficking of multiple proteins (Youker et al. 2009). This sorting protein was initially studied for its role in mitochondrial network and MAM organisation (Simmen et al. 2005). PACS-2 regulates communication between the ER and the mitochondria by controlling contact sites between the two organelles (Simmen et al. 2005). In this way, PACS-2 mediates apoptosis (cfr. infra) and ER homeostasis, while promoting transfer of lipids between the ER and mitochondria (Simmen et al. 2005). Interestingly, PACS-2 can also assist in calnexin enrichment at the MAMs in concert with the coat protein complex COPI (Myhill et al. 2008). Also mechanistic target of rapamycin complex 2 (mTORC2) can be found at the MAMs where it regulates MAM integrity via PACS-2 phosphorylation in a protein kinase B (PKB/Akt)-dependent way (Betz et al. 2013).
17.2.3.3 PERK
PERK, a kinase protein located in the ER membrane, functions as an ER stress sensor that controls the unfolded protein response (UPR), alongside IRE1 and activating transcription factor 6 (ATF6) (Sano and Reed 2013). The main effect of the UPR on the cell is the diminishment of mRNA translation to avoid further accumulation of improperly folded proteins, while at the same time, the transcription of chaperones is stimulated (Sano and Reed 2013). Furthermore, retrograde transport of unfolded proteins to the cytosol takes place, where they undergo ubiquitination and subsequent degradation (Sano and Reed 2013). PERK is mainly responsible for the halt of translation by virtue of phosphorylating eukaryotic translation initiation factor 2 (eIF2α), a protein needed for correct mRNA translation (Sano and Reed 2013). The phosphorylation disrupts recycling of eIF2α from its GTP-free form to its GTP-bound form (Sano and Reed 2013). Furthermore, eIF2α phosphorylation is responsible for the preferential translation of UPR-involved genes. In addition, PERK phosphorylates nuclear erythroid 2 p45-related factor 2 (Nrf2) inducing the expression of antioxidant genes to alleviate oxidative stress (Cullinan and Diehl 2004). When ER stress continues for a longer period, the cell will brace itself for apoptosis. In this process, PERK contributes by promoting the transcription of CCAAT-enhancer-binding protein homologous protein, a pro-apoptotic transcription factor (Oyadomari and Mori 2004; Sano and Reed 2013). Moreover, PERK was shown to be involved in autophagy regulation via ATF4-dependent transcription of autophagy-related (ATG) genes (Harding et al. 2000).
Additionally, PERK also serves as an ER-mitochondria tether at the MAMs, thereby facilitating the propagation of ROS signals between these two organelles (Verfaillie et al. 2012). Hence, PERK-knockout cells displayed significantly weaker ER-mitochondria contact sites, counteracting ROS-triggered apoptosis. This function of PERK to maintain ER-mitochondria juxtapositions was independent of its kinase activity but required its cytoplasmic domains (Verfaillie et al. 2012).
17.2.3.4 Foetal and Adult Testis Expressed 1 (FATE1)
FATE1 is a protein that belongs to the group of cancer-testis antigens (CTAs) (Dong et al. 2003), which is a heterogeneous group of proteins with limited expression in normal testis tissue. However, in several types of cancer, these CTAs are upregulated (Simpson et al. 2005; Whitehurst 2014). Recently, FATE1 was found to reside at the MAMs where it regulates ER-mitochondrial distance and ER-mitochondrial Ca2+ flux (cfr. infra) (Doghman-Bouguerra et al. 2016). Remarkably, silencing FATE1 led to an increased sensitivity towards paclitaxel, a chemotherapeutic drug, in non-small cell lung cancer cell lines (Whitehurst et al. 2007).
17.3 MAMs in Cancer
All types of cancer share certain stereotypical traits, called the hallmarks of cancer (Hanahan and Weinberg 2011; Giampazolias and Tait 2016). These features, acquired gradually during the development of tumours, include sustaining proliferative signalling, resisting cell death, activating invasion and metastasis, inducing angiogenesis and rewiring metabolism (Hanahan and Weinberg 2011; Giampazolias and Tait 2016). Importantly, MAMs and mitochondria play key roles in many cellular processes such as cell death, cell migration and energy production (Giampazolias and Tait 2016). Therefore, functioning of these cellular compartments is frequently altered and affected during acquisition of the hallmarks of cancer. In this section, we discuss the role of the proteins listed above in the various hallmarks of cancer.
17.3.1 Tumour Growth, Proliferation and Metastasis
17.3.1.1 The IP3R
Since Ca2+ signalling controls a plethora of cellular functions that relate to cancer hallmarks, IP3Rs have emerged as important regulators of tumour biology. A striking example of the importance of the receptor’s function is the observation that lack of IP3R-mediated Ca2+ signalling in thymocytes causes the development of malignancies in mice, resembling T-cell acute lymphoblastic leukaemia (Ouyang et al. 2014). Furthermore, there is a growing body of evidence that suggests IP3R-mediated Ca2+ release plays a role in cancer cell migration (Wei et al. 2009, 2012; Huang et al. 2016). Also, migrating fibroblasts displayed cytosolic Ca2+ flickers mediated in part by IP3R2 (Wei et al. 2009, 2012). Furthermore, IP3R3 is overexpressed in glioblastoma cells, whereas reducing its expression via siRNA attenuated migration via inhibition of cytosolic Ca2+ signalling (Kang et al. 2010). Recently, it was also found that overexpression of ER protein 44, which negatively regulates Ca2+ release, prevented migration of A549 cells by suppressing IP3R2-dependent Ca2+ release (Huang et al. 2016). Moreover, the IP3R was shown to play a role in MCF-7 cell growth, since growth inhibition occurred upon blockage of the IP3R (Szatkowski et al. 2010). Interestingly, 17-β-estradiol, which induces cell proliferation in MCF-7 cells, may do so by elevating IP3R3 levels (Szatkowski et al. 2010). In the same cell line, a molecular and functional coupling between IP3R3 and large-conductance Ca2+- and voltage-dependent K+ (BKCa) channels was responsible for ATP-induced proliferation in a cyclin-D1/cyclin-dependent kinase 4-dependent mechanism (Mound et al. 2013). Ablation of IP3R3 or BKCa resulted in attenuated proliferation (Mound et al. 2013). Interestingly, the IP3R is also implied in senescence (Wiel et al. 2014), protecting cells from tumour onset and progression (Ben-Porath and Weinberg 2004; Collado and Serrano 2010; Kang et al. 2011). It was shown that loss of IP3R2 allowed cells to avoid oncogene-induced senescence (Wiel et al. 2014). This was also the case for the mitochondrial Ca2+ uniporter. This points to mitochondrial Ca2+ accumulation playing an important role in senescence through lowering the mitochondrial membrane potential and ROS (Wiel et al. 2014).
17.3.1.2 VDAC1
The expression levels of VDAC1 are correlated with tumour growth in different types of cancer. Zhang et al. showed that a decrease in miRNA-320a allowed for a high VDAC1 expression in non-small cell lung cancer cells and that this was correlated with the initiation and progression of cancer (Zhang et al. 2016b). Furthermore, cervical cancer tissues positive for VDAC1 showed an increased tumour size and deep stromal invasion compared to tissues negative for VDAC1 (Wu et al., 2016a). In the same study, VDAC1 knockdown inhibited cell proliferation and migration (Wu et al. 2016a), which was also shown in human papilloma virus-related cervical cancers (Zhang et al. 2016a). This evidence suggests that VDAC1 promotes tumour survival and invasion. Interestingly, knockout of VDAC1 in MEF cells increased proliferation rates under hypoxic conditions through activation of the extracellular signal-regulated protein kinase (ERK) 1/2 pathway (Brahimi-Horn et al. 2015).
Moreover, VDAC1’s role in Ca2+ signalling has been linked to cell migration. Myeloid cell leukaemia sequence 1 (Mcl-1), an anti-apoptotic protein from the B-cell lymphoma 2 (Bcl-2) protein family, is able to bind VDAC1 with high affinity, thereby seemingly promoting mitochondrial Ca2+ uptake. Mcl-1 binding to VDAC1 promoted cell migration without affecting cell proliferation. The pro-migration effect of Mcl-1 could be antagonised by VDAC-based peptides that interfere with VDAC1/Mcl-1-complex formation (Huang et al. 2014). Also other anti-apoptotic Bcl-2 proteins, including Bcl-XL, inhibit VDAC1-mediated Ca2+ uptake in the mitochondria (Arbel et al. 2012; Monaco et al. 2015; Vervliet et al. 2016). The mechanism involved Bcl-XL’s Bcl-2 homology 4 (BH4) domain and VDAC1’s N-terminus (Monaco et al. 2015). The inhibitory impact of Bcl-2 proteins on VDAC1-mediated Ca2+ uptake in the mitochondria is consistent with the original papers that describe Bcl-2 proteins as negative regulators of VDAC1-mediated apoptosis (Shimizu et al. 1999, 2000).
17.3.1.3 The Chaperones
A study by Vilner et al. (1995) demonstrated that Sig-1R was overexpressed in a large range of cancer cell lines, both human and rodent. Later studies added that there might be a link between Sig-1R overexpression and metastasis. This was proposed by Aydar et al. since the highest expression levels were found in metastatic cell lines (Aydar et al. 2006). Sig-1R mRNA levels were also found to be higher in invasive breast cancer tissue derived from patients, compared to normal breast tissue (Wang et al. 2004); Sig-1R mRNA was overexpressed in colorectal cancer and colorectal cancer liver metastases (Skrzycki and Czeczot 2013), and overexpression of Sig-1R in hilar cholangiocarcinoma was linked to poor differentiation, lymph node metastasis and advanced disease stage (Xu et al. 2014).
Concerning the molecular mechanisms that are possibly involved, the ability of Sig-1R to interact with several ion channels seems to be important for various oncogenic features (Crottès et al. 2013). In K562 myeloid leukaemia cells, the link between Sig-1R and expression of the potassium ion channel human ether-à-go-g-related gene (hERG), which controls several processes like migration and adhesion (Pillozzi et al. 2007, 2011), was investigated (Crottès et al. 2011). It was observed that Sig-1R is important for hERG maturation by improving maturation efficiency and stabilisation of the α-subunit (Crottès et al. 2011). Other ion channels that interact with Sig-1R and that play a role in cancer include L-type voltage-gated Ca2+ channels, voltage-gated Na+ channels and Ca2+-activated K+ channels (for extensive review, see Crottès et al. 2013). However, the interaction of Sig-1R with these ion channels is not necessarily related to its role at the MAMs.
Not only by directly interacting with other proteins, but also indirectly, Sig-1R is able to modify properties of ion channels: Palmer et al. (2007) found that Sig-1R can bind cholesterol and stabilise lipid rafts via the insertion of cholesterol. In turn, the cholesterol level of lipid rafts can impact the signalling molecules present in these domains (Gniadecki 2004; Palmer et al. 2007), thereby altering the activity of ion channels nearby.
In addition, GRP75 overexpression is correlated with tumour growth and invasion (Kaul et al. 1998; Yi et al. 2008; Jin et al. 2016). Also, in K562 cells high expression levels of GRP75, as well as other chaperone proteins, coincided with resistance towards the proteasome inhibitor bortezomib (Kliková et al. 2015), and inhibition of GRP75 reduced cisplatin resistance in ovarian cancer (Yang et al. 2013).
Also calnexin, as a chaperone, may play a role in tumoural growth in response to growth factors. Lakkaraju and van der Goot (2013) found that in squamous carcinoma cells, caspase-8-mediated cleavage of calnexin occurs upon stimulation of the cells with epidermal growth factor. This yields a calnexin fragment that inhibits protein inhibitor of activated STAT3 (PIAS3), an inhibitor of signal transducer and activator of transcription 3 (STAT3), which functions as an oncogenic transcription factor (Lakkaraju and van der Goot 2013). This, in turn, promotes STAT3-dependent transcription and possibly tumour growth (Lakkaraju and van der Goot 2013).
17.3.1.4 Proteins Defining MAM Structure
The importance of Mfn2 for cell proliferation is suggested by findings in vascular smooth muscle cells: overexpression of Mfn2 in cultured vascular smooth muscle cells inhibited proliferation by blocking the mitogen-activated protein kinase (MAPK)/ERK signalling pathway (Chen et al. 2004). This mechanism was found to be independent of its role in mitochondrial fusion (Chen et al. 2004; Guo et al. 2007). In concert with observations of Mfn2 acting in an anti-proliferative way, Zhang et al. (2013) showed that Mfn2 expression was lower in gastric tumours than in normal mucosal tissue and that expression levels were negatively correlated with tumour size, while Wu et al. (2016b) observed that poor overall survival in hepatocellular carcinoma patients correlated with low Mfn2 expression levels. Also in primary breast cancer, a loss of Mfn2 was detected (Kannan et al. 2016). Strikingly, knockdown of TMX1 in HeLa and A375P melanoma cells generates a similar phenotype as low-level Mfn2 expression (Raturi et al. 2016). This includes increased SERCA activity and altered MAM structure (Raturi et al. 2016). In the case of TMX1, Raturi et al. propose that the stimulatory effect on tumour growth upon TMX1 knockdown is due to an elevated Ca2+ retention capacity at the ER combined with an increased ER-mitochondrial distance (Raturi et al. 2016). This, in turn, leads to reduced ER-mitochondrial Ca2+ flux and impairment of mitochondrial metabolism, possibly contributing to the Warburg effect (cfr. infra) (Raturi et al. 2016).
Interestingly, another study revealed that Mfn2 deficiency decreased proliferation by blocking autophagy in HeLa cells (Ding et al. 2015). Similarly, A549 human lung adenocarcinoma cells showed disturbed cell proliferation and invasion upon Mfn2 knockdown (Lou et al. 2015). A recent bioinformatics study conducted on the same cell line revealed that Mfn2 knockdown resulted in repression of genes implicated in cell-cycle progression as well as DNA replication and MAPK signalling pathway (Lou et al. 2016). These opposing data suggest that Mfn2’s role in cancer is highly context dependent. It may however also relate to a critical window for proper ER-mitochondria distance, in which too close apposition results in excessive apoptosis sensitivity, whereas too far apposition results in defective energetic and metabolic features, as discussed elsewhere (Naon and Scorrano 2014).
PERK, the ER stress sensor, also fulfils this double-edged function in relation to tumour growth. For example, PERK activity has been linked to cell-cycle arrest (Brewer and Diehl 2000; Hamanaka et al. 2005). Since PERK is involved in a general slowdown of the translation process, several proteins are impacted including the drivers of the cell cycle, which comprise the cyclins (Brewer and Diehl 2000; Hamanaka et al. 2005). In this case, cyclin D1’s expression is severely altered due to its short half-life (Brewer and Diehl 2000; Hamanaka et al. 2005). This brings about a redistribution of p21, which blocks cyclin-dependent kinase 2, resulting in cell-cycle arrest in the G1 phase (Brewer and Diehl 2000; Hamanaka et al. 2005).
In addition, PERK causes an increase in p53 through decreased E3 ubiquitin ligase human double minute 2 (Hdm2)-dependent removal, promoting apoptosis when a certain threshold is reached (Li et al. 2006; Zhang et al. 2006). Another feature of this p53 accumulation is the induction of p21 and subsequent cell-cycle arrest, as stated above (Ono et al. 1997). Furthermore, PERK is involved in the upregulation of p47, an N-terminal truncated analogue of p53, which mediates cell-cycle arrest in the G2 phase (Bourougaa et al. 2010). Conversely, PERK has been proposed to improve the degradation of p53 in a glycogen synthase kinase 3 β (GSK3β)-dependent manner (Qu et al. 2004; Pluquet et al. 2005).
Also, PERK and its downstream signalling axis have been implicated in metastasis of several cancers, e.g. cervix cancer, breast cancer and head and neck squamous cell carcinoma (Nagelkerke et al. 2013, 2015; Mujcic et al. 2013). Epithelia to mesenchymal transition, which is an indication of the level of invasiveness, is also correlated with PERK signalling, which is underpinned by observations in primary breast cancer, colon cancer, gastric cancer and lung cancer (Feng et al. 2014). Furthermore, the human epidermal growth factor receptor 2 (HER2)/Neu protein is able to induce PERK activity, which allows for redox homeostasis via Nrf2 (Bobrovnikova-Marjon et al. 2010). Subsequently, loss of PERK in HER2/Neu-dependent mammary adenocarcinoma was responsible for growth attenuation and decreased metastasis (Bobrovnikova-Marjon et al. 2010).
About the role of PACS-2 in tumour progression, not much is known. However, PACS-2 was shown to be a regulator of ADAM17, a metalloproteinase that is involved in epithelial development, growth and tumour progression (Dombernowsky et al. 2015). More specifically, loss of PACS-2 diminished ADAM17 levels at the cell surface due to increased degradation (Dombernowsky et al. 2015).
17.3.2 Apoptosis
Another major hallmark of cancer cells is their ability to evade apoptosis (Hanahan and Weinberg 2011). As briefly indicated before, Ca2+ signalling is able to regulate the apoptotic process. While Ca2+ oscillations were found to be pro-survival signals due to stimulation of critical enzymes of the tricarboxylic acid cycle (TCA), high-amplitude Ca2+ signals that last for a longer time can cause apoptosis through mitochondrial Ca2+ overload (Hajnóczky et al. 1995; Orrenius et al. 2003; Joseph and Hajnóczky 2007; Roderick and Cook 2008; Denton 2009). This, in turn, causes the opening of the mitochondrial permeability transition pore, mitochondrial swelling and eventually the release of pro-apoptotic factors like cytochrome c in the cytosol (Halestrap 2014; Morciano et al. 2015; Jonas et al. 2015). In order to prevent Ca2+-induced apoptosis and/or to promote Ca2+-dependent bioenergetics, cells may rewire their Ca2+-signalling toolkit (Capiod et al. 2007; Chen et al. 2013; Stewart et al. 2015).
17.3.2.1 The IP3R
The IP3R exerts a central role in ER-mitochondrial Ca2+ signalling, making it prone to the electrical rewiring of the cancer cell. IP3R expression levels are altered in various cancers, supporting the critical role of the IP3R in Ca2+ signalling from the ER. For instance, hormone-refractory prostate tumour cells showed increased levels of IP3R1 (Boutin et al. 2015). This is thought to increase Ca2+ leakage from the ER, so that less Ca2+ is available for the induction of apoptosis by mitochondrial Ca2+ overload (Boutin et al. 2015). Interestingly, bladder cancer cells evade cell death by doing the opposite: treatment with cisplatin diminished IP3R1 levels, provoking cisplatin resistance (Tsunoda et al. 2005). By lowering IP3R expression levels, cancer cells prevent the event of toxic mitochondrial Ca2+ overload (Prevarskaya et al. 2014). Furthermore, some diffuse large B-cell lymphomas (DLBCLs) express high levels of IP3R2 (Akl et al. 2013). The reason for this IP3R2 elevation remains elusive, but one hypothesis is that in metabolically stressed cancer cells, low levels of ATP, a positive regulator of the IP3R, are insufficient to provide the basal Ca2+ signalling needed to fuel mitochondrial bioenergetics (Akl et al. 2013; Akl et al. 2014). Hence, by upregulation of the IP3R2, which is the IP3R isoform most sensitive to IP3, these cancer cells are able to survive (Akl et al. 2013, 2014).
In cancer cells, the process of apoptosis is not only influenced by modifying the expression levels of the IP3R, but also by altering its Ca2+-release properties. For instance, phosphorylation of the IP3R dramatically changes its function (Vanderheyden et al. 2009). PKB/Akt is a serine-threonine kinase that phosphorylates the IP3R C terminally via a substrate motive that is conserved in all isoforms (Khan et al. 2006). While ER Ca2+ levels remain unaffected in HeLa cells overexpressing PKB/Akt, IP3R-dependent ER Ca2+ release was shown to be negatively affected (Szado et al. 2008; Marchi et al. 2008). Again, this mechanism may protect against mitochondrial Ca2+ overload and subsequent apoptosis (Marchi et al. 2008). Furthermore, it was shown that the protective effect of PKB/Akt overexpression was isoform specific: in COS7 cells, almost completely lacking IP3R1, PKB/Akt activation led to a decreased IP3-induced Ca2+ release and conferred a protective effect against apoptosis (Marchi et al. 2012). However, in SH-SY 5Y cells, lacking IP3R3, ER Ca2+ release was not modified, while expressing the type 3 isoform in these cells restored the protective effect (Marchi et al. 2012). This suggests that the anti-apoptotic effect of PKB/Akt is mediated in an IP3R3-dependent way (Marchi et al. 2012). The effect of PKB/Akt-mediated phosphorylation is thought to be directly counteracted by phosphatase and tensin homolog (PTEN), which also localises at the MAMs and dephosphorylates the IP3R, thereby increasing again the IP3R-mediated Ca2+ release (Bononi et al. 2013).
A recent study showed that extra-nuclear PML contributes to protection against Ca2+-mediated apoptotic cell death via interaction with the IP3R (Giorgi et al. 2010). PML located at the MAMs physically interacts with the IP3R3. In cells expressing ER-targeted PML, apoptotic stimuli induced a higher cytosolic and mitochondrial Ca2+ response (Giorgi et al. 2010). Furthermore, it was revealed that PML-expressing cells displayed lower levels of phosphorylated IP3R3 and phosphorylated, active PKB/Akt and higher levels of the phosphatase 2A (PP2A) compared to PML-negative cells (Giorgi et al. 2010). PML stimulates pro-apoptotic Ca2+ signalling at the MAMs by recruiting PP2A to IP3R3-PKB/Akt complexes (Giorgi et al. 2010), resulting in a suppressed PKB/Akt-mediated IP3R3 phosphorylation, since PP2A negatively regulates the activity of PKB/Akt at the ER (Pinton et al. 2011; Bittremieux et al. 2016).
IP3R function can also be stimulated by phosphorylation (Gomez et al. 2016). During reperfusion injury of the heart, a fraction of the protein kinase GSK3β is localised at the sarco/endoplasmic reticulum (SR/ER) and the MAMs. There, it interacts with the IP3R Ca2+-channelling complex, regulating its protein composition and modulating Ca2+ transfer between the SR/ER and mitochondria. During hypoxia reoxygenation, GSK3β activity is augmented, resulting in increased IP3R phosphorylation and IP3R hyperactivity. Consequently, increased IP3R-mediated SR/ER-mitochondria Ca2+ transfer leads to cardiomyocyte cell death. Therefore, inhibition of GSK3β may protect the heart from lethal reperfusion injury by cellular Ca2+ overload (Gomez et al. 2016).
The critical role of IP3R3 underlying cellular apoptosis sensitivity has also been confirmed independently in two isogenic cell lines, one of which expresses oncogenic K-Ras and one in which this oncogenic allele was deleted via homologous recombination (Pierro et al. 2014). The presence of oncogenic K-Ras caused a lowering in the ER Ca2+-store content, thereby decreasing the likelihood of pro-apoptotic Ca2+ transfer and thus decreasing apoptotic sensitivity (Pierro et al. 2014). This was due to the increased expression level of IP3R1 relative to that of IP3R3 in the cell line expressing oncogenic K-Ras, augmenting basal Ca2+ leak via IP3R1 and suppressing pro-apoptotic Ca2+ transfer into the mitochondria via the IP3R3 (Pierro et al. 2014).
Apart from phosphorylation and expression regulation, IP3R-mediated Ca2+ release can be modulated directly by the binding of an increasing number of oncogenes and tumour suppressors (Akl and Bultynck 2013; Bittremieux et al. 2016). Notably, several members of the Bcl-2-protein family are known to interact with the IP3R. First, there is the anti-apoptotic protein Bcl-2 itself, whereas its canonical function comprises the sequestration of pro-apoptotic Bcl-2-protein family members like Bik and Bid via its BH3 domain (Youle and Strasser 2008; Czabotar et al. 2014) evidence was found that Bcl-2 interacts via its BH4 domain with 20 amino acids in the central, modulatory part of the IP3R (Rong et al. 2008, 2009; Monaco et al. 2012). This interaction protects cells against Ca2+-mediated apoptotic cell death (Hanson et al. 2008; Rong et al. 2008). The importance of Bcl-2’s complex formation with the IP3R became clear in Bcl-2-dependent chronic lymphocytic leukaemia and in DLBCL cell lines (Zhong et al. 2011; Akl et al. 2013). Some subtypes of DLBCL display high levels of IP3R2, the most sensitive isoform with respect to IP3 (Akl et al. 2013). Therefore, Bcl-2 overexpression is needed in these cells to avoid Ca2+-induced apoptosis triggered by the high expression levels of IP3R2, making these cells balancing on the edge of apoptosis, a state that was coined “primed-for-death at the ER” (Akl et al. 2013; Akl et al. 2014). Following this concept, TAT-IDP, a peptide mimicking the IP3R-binding site for Bcl-2, and its derivative BIRD-2, induced apoptosis by disrupting IP3R/Bcl-2 interaction and eliciting spontaneous toxic Ca2+ signalling (Zhong et al. 2011; Akl et al. 2013; Akl et al. 2015; Lavik et al. 2015; Greenberg et al. 2015).
Apart from Bcl-2, the closely related anti-apoptotic Bcl-XL protein is also able to interact with the IP3R, albeit not via its BH4 domain (White et al. 2005; Monaco et al. 2012). Bcl-XL, through its hydrophobic cleft, binds the IP3R at its C-terminal region (Eckenrode et al. 2010) by targeting two BH3-domain-like sequences (Yang et al. 2016). The binding between Bcl-XL and the IP3R results in a reduction in ER [Ca2+], increased Ca2+ oscillations in the cytosol and protection against apoptosis by sensitising the IP3R to low basal levels of IP3 (Li et al. 2007).
Bcl-XL does not only alter Ca2+ signalling in the cell via direct modulation of the IP3R, but also via a nuclear factor of activated T-cells (NFAT)-dependent pathway, which modifies IP3R expression levels (Li et al. 2007). Furthermore, Mcl-1, another Bcl-2-protein family member, was shown to interact directly with the IP3R via the last transmembrane domain in its C-terminal tail (Eckenrode et al. 2010). Like Bcl-XL, Mcl-1 was found to sensitise the IP3R, thereby increasing the frequency of Ca2+ oscillations in the cell and the number of oscillating cells (Eckenrode et al. 2010). Figure 17.2 displays the proteins that regulate ER-mitochondrial Ca2+ signalling via the IP3R and VDAC1. For reasons of clarity, a distinction was made between the regulation of pro-survival and pro-apoptotic Ca2+ signalling.

Regulation of pro-survival and pro-apoptotic Ca 2+ signalling at the MAMs. Arrow-headed lines indicate a stimulatory interaction, while bar-headed lines indicate an inhibitory interaction. (a) Regulation of pro-survival Ca2+ signalling at the MAMs. Bcl-2, Bcl-XL and Mcl-1 increase pro-survival Ca2+ oscillations and stimulate cell metabolism by interacting with the C-terminus of the IP3R, which results in a sensitisation of the channel to basal IP3 levels. Mcl-1 also enhances cell survival through binding to VDAC1, thereby increasing its activity and thus mitochondrial Ca2+ uptake. While these proteins directly impact the Ca2+-flux properties of the IP3R or VDAC1, Sig-1R indirectly promotes cell survival. Under conditions of ER stress, Sig-1R becomes active and stabilises the IP3R, ensuring the transmission of pro-survival Ca2+ signalling into the mitochondria. (b) Regulation of pro-apoptotic Ca2+ signalling at the MAMs. Bcl-2 and Bcl-XL do not only support pro-survival Ca2+ signalling, but also inhibit pro-apoptotic Ca2+ signalling. Binding of Bcl-2 via its BH4 domain to the central region of the IP3R diminishes the Ca2+ flux through the IP3R, while Bcl-XL inhibits VDAC1 via its BH4 domain that targets the N-terminus of VDAC1. Ca2+ release from the ER is also decreased by PKB/Akt, which inhibits IP3R function by phosphorylation. The phosphatase PTEN counteracts the function of PKB/Akt at the MAMs by dephosphorylating the IP3R. PML indirectly influences the phosphorylation state of the IP3R by recruiting the phosphatase PP2A, which negatively regulates PKB/Akt activity in the MAMs and counteracts PKB/Akt-mediated phosphorylation of IP3R in the MAMs. Thus, PML alleviates the suppression of IP3R-mediated Ca2+ flux from the ER to the mitochondria imposed by PKB/Akt (For more details, please see text)
Finally, inhibition of Bcl-2-family members has emerged as an attractive anticancer strategy, particularly by preventing the complex formation between the anti- and pro-apoptotic Bcl-2-family members (Davids and Letai 2012). However, the function of anti-apoptotic Bcl-2-family members appears to be involved in ER-mitochondrial contact sites as well. Indeed, recent studies showed that targeting the hydrophobic cleft using BH3 mimetics like ABT-737, a non-selective Bcl-2/Bcl-XL inhibitor, enhance anticancer treatments by increasing ER-mitochondria contact sites and stimulating ER-mitochondrial Ca2+ transfer (Fan et al. 2015; Xie et al. 2016). As such, ABT-737 could restore the sensitivity of cisplatin-resistant ovarian cancer cells to cisplatin treatment. This correlated with the ability of cisplatin to induce mitochondrial Ca2+ overload, an important feature of the successful induction of cell death by anticancer treatments (Bittremieux and Bultynck 2015; Bonora et al. 2015; Fan et al. 2015; Giorgi et al. 2015a; Xie et al. 2016).
17.3.2.2 VDAC1
As VDAC1 is the gateway for Ca2+ entry in the mitochondria, this protein’s function may also be influenced in cancer cells to ensure their survival. Bcl-XL was found to bind to and block VDAC1 with its BH4 domain, thereby inhibiting Ca2+-mediated apoptosis (Monaco et al. 2015). Intriguingly, cytosolic Ca2+ levels impact VDAC1 expression levels (Weisthal et al. 2014). An increase in cytosolic [Ca2+], elicited by, for example, H2O2, induces a rise in VDAC1 expression, which at the same time correlates with the ability to form oligomers in the OMM, through which the pro-apoptotic protein cytochrome c is released from the mitochondria (Weisthal et al. 2014).
VDAC1 does not only contribute to Ca2+-mediated apoptotic cell death, but also influences apoptosis occurring independently of Ca2+. For instance, in human glioma cells subjected to hypoxia, VDAC1 has been implicated in the activation of mitophagy (Qiao et al. 2016). In these cells, the mitochondrial deacetylase sirtuin-3 (Sirt3) stimulates the association between VDAC1 and parkin, an E3 ubiquitin ligase, stimulating mitophagy (Qiao et al. 2016; Bernardini et al. 2017). Consequently, knockdown of Sirt3 inhibited mitophagy, rendering the cells prone to apoptotic cell death (Qiao et al. 2016). Thus, VDAC1 in concert with Sirt3 plays a role in protecting cancer cells through mitophagy. Interestingly, parkin also seems to play a role in the regulation of mitochondrial homeostasis and energy metabolism (Calì et al. 2013). Overexpression of parkin in HeLa and SH-SY5Y neuroblastoma cells increased physical as well as functional interactions between the ER and the mitochondria, whereas parkin silencing caused mitochondrial fragmentation and compromised mitochondrial Ca2+ transients due to reduced ER-mitochondria tethering (Calì et al. 2013).
Furthermore, VDAC1 provides a link between apoptosis and differentiation of cancer cells. In glioblastoma tumour cells, in which VDAC1 expression was silenced, a shift from pro-apoptotic proteins linked to cell proliferation, including avian myelocytomatosis virus oncogene cellular homolog (c-Myc) and nuclear factor κB (NF-κB), to pro-apoptotic proteins regulating cell differentiation, including p53, was observed (Arif et al. 2016). This led to differentiation of the glioblastoma cells into astrocyte- and neuron-like cells. Additionally, several studies showed that hexokinase (HK), the enzyme catalysing the first step of glycolysis (Wilson 2003), bound to VDAC1, confers protection from apoptosis in HEK and HeLa cells (Bryson et al. 2002). This protection is proposed to result from the inhibition of the interaction between VDAC1 and the pro-apoptotic Bcl-2 family member Bax by HK (Bryson et al. 2002). Interestingly, for HK to execute its anti-apoptotic effect, its binding to VDAC1 is needed (Arzoine et al. 2009; Abu-Hamad et al. 2009). Furthermore, evidence also suggests that the binding of HK to VDAC1 reduced mitochondrial ROS generation (da-Silva et al. 2004; Sun et al. 2008). Since ROS production is often elevated in cancer cells (Liou and Storz 2010; Panieri and Santoro 2016), this may be another mechanism by which HK protects tumour cells against cell death, as both the HK-I and -II isoforms conveyed protection towards apoptosis-inducing oxidants through their association with VDAC1 (Bryson et al. 2002; Ahmad et al. 2002).
17.3.2.3 The Chaperones
Also the chaperone proteins at the MAMs are able to modify the cell’s apoptotic pathways. Similar to VDAC1, Sig-1R acts in a Ca2+-dependent and Ca2+-independent way. Apoptotic Ca2+ signalling is impacted by Sig-1R’s ability to bind to the IP3R. Knockdown of Sig-1R resulted in increased degradation of the IP3R3 via the proteasome, suggesting that it has a stabilising function (Hayashi and Su 2007). Furthermore, in conditions of physiologically normal ER [Ca2+], Sig-1R is in a resting state, bound to GRP78 (Hayashi and Su 2007). However, under conditions of depletion of the ER Ca2+ stores, the Sig-1R/GRP78 complex is disrupted, and Sig-1R obtains its chaperone activity (Hayashi and Su 2007). It targets the IP3R to ensure that IP3-mediated pro-survival Ca2+ signalling to the mitochondria occurs properly. Interestingly, while during short periods of ER Ca2+ depletion, Sig-1R remains localised at the MAMs it is redistributed throughout the ER upon longer ER Ca2+ depletion, again as a pro-survival mechanism under continued ER stress, which is often present in cancer cells (Hayashi and Su 2007). By stabilising the IP3R, it sustains pro-survival Ca2+ signalling during ER stress. Alternatively, it was reported that Sig-1R supports cell survival during ER stress via another client protein: IRE1 (Mori et al. 2013). Activation of IRE1 triggers its endonuclease activity, needed for splicing X-box binding protein-1 (XBP1) mRNA. This transcription factor then promotes the transcription of various ER chaperones (Yoshida et al. 2001). It is thought that Sig-1R’s stabilisation of IRE1 contributes to prolonged signalling along the IRE1-XBP1 axis, thereby supporting cell survival under conditions of augmented and prolonged ER stress (Mori et al. 2013).
GRP75, on the other hand, impacts apoptosis in cancer cells primarily through its alteration of MAPK/ERK signalling and influence on p53. In medullary thyroid carcinoma cells, apoptosis and inhibition of cell growth were caused by a depletion of GRP75 (Starenki et al. 2015). Investigations into the pathways involved revealed that a temporary activation of MAPK/ERK signalling was responsible for the growth arrest, while apoptosis was induced through mitochondrial dysfunction. This consisted of loss of the mitochondrial membrane potential, lowered oxygen consumption and an elevation of ROS levels. Furthermore, it was shown that these mitochondrial effects were linked to a decrease in Bcl-2 expression (Starenki et al. 2015). Similar results in different cancer cell lines confirmed GRP75 as a negative regulator of MAPK/ERK signalling (Wu et al. 2013). An additional contribution to the anti-apoptotic function of GRP75 is its capacity to bind the tumour suppressor p53 (Wadhwa et al. 1998; Kaul et al. 2001; Wadhwa et al. 2002b). The interaction prevents nuclear translocation of p53, abrogating its function as a transcription factor (Wadhwa et al. 2002b). Moreover, keeping p53 in the cytoplasm speeds up its proteasomal degradation (Kaul et al. 2005). Strikingly, applying the HSP90 inhibitor 17-AAG, which blocks other heat shock proteins but not GRP75, stimulated GRP75 expression and reinforced its binding to p53, weakening the effect of the HSP90 inhibitor in hepatocellular carcinoma (Guo et al. 2014).
Also calnexin has been implicated in the regulation of apoptotic cell death. However, it seems that calnexin may play an anti-apoptotic as well as a pro-apoptotic role dependently on the circumstances. Caspase-3 and Caspase-7 have both been shown to cleave calnexin in vitro, while overexpression of the cleavage product partially inhibited apoptosis (Takizawa et al. 2004). Indirectly, calnexin also fulfils a pro-apoptotic role: its cytosolic tail is able to recruit caspase-8, which is responsible for the cleavage of Bap31 (Breckenridge et al. 2002; Delom et al. 2007). Bap31’s cleaved form stimulates Ca2+ release from the ER and apoptosis ensues (Breckenridge et al. 2003). In the MCF-7 breast cancer cell line, which is resistant to tunicamycin-induced cell death, calnexin is able to sensitise the cells to tunicamycin, independently of its chaperone function (Delom et al. 2007).
17.3.2.4 Proteins Defining MAM Structure
In MCF-7 cells, Mfn2 mediated apoptosis via the phosphoinositide 3-kinase (PI3K)/PKB/Akt signalling pathway (Ma et al. 2015). These results mimic the findings in vascular smooth muscle cells. There, Mfn2 was shown to trigger mitochondrial apoptosis by inhibiting the GTPase Ras, resulting in decreased PKB/Akt signalling along the Ras-PI3K-PKB/Akt axis (Guo et al. 2007). However, it was not established whether lowered PKB/Akt signalling was linked to an increased Ca2+ signalling through reduced phosphorylation of the IP3R. In this regard, it is interesting to note that Wang et al. (2015) found that overexpression of Mfn2 induced Ca2+-dependent apoptosis in hepatocellular carcinoma.
Additionally, several studies have linked PACS-2 with the apoptotic process, both via the intrinsic pathway as through the extrinsic one. For example, PACS-2 has been shown to interact with the Bcl-2-family protein Bid (Simmen et al. 2005): upon the addition of apoptotic stimuli, PACS-2 is responsible for the translocation of Bid to the mitochondria, where cytochrome c release and caspase activation ensue (Simmen et al. 2005). Furthermore, PACS-2 is involved in tumour necrosis factor-related apoptosis inducing ligand (TRAIL)-triggered apoptosis, more specifically in lysosomal permeabilisation (Werneburg et al. 2012). PACS-2 recruits Bim and Bax, two other members of the Bcl-2 family, to the lysosomal membrane to bring about cathepsin B release and subsequent apoptosis (Werneburg et al. 2012). On this note, it is interesting that cellular inhibitor of apoptosis protein-1 and -2 (cIAP-1/cIAP-2) repress expression levels of PACS-2 by promoting its ubiquitinylation (Guicciardi et al. 2014). In this way, these cIAPs confer resistance to TRAIL-induced apoptosis in hepatobiliary cancer cell lines (Guicciardi et al. 2014). Curiously, PKB/Akt-mediated phosphorylation of Ser437 serves as a switch to shift from PACS-2’s trafficking function to its function as a promoter of apoptosis (Aslan et al. 2009). When phosphorylated, PACS-2 is bound to the 14-3-3 scaffold protein, which inhibits its role in apoptosis (Aslan et al. 2009), in a similar way as the Bcl-2-protein family Bad (Zha et al. 1996). Interestingly, phosphorylation of PACS-2 does not merely serve to repress apoptosis, but is also required for polycystin-2 localisation to the ER (Aslan et al. 2009).
Apart from engaging Bcl-2-protein family members, PACS-2 also influences the regulation of p53. Sirt1 deacetylates p53, but upon DNA damage, PACS-2 is shuttled to the nucleus, where it interacts with Sirt1, preventing the deacetylation of p53 and inducing p21-dependent cell-cycle arrest (Atkins et al. 2014). Contrasting with its pro-apoptotic function, however, a recent study has found that PACS-2 was necessary for NF-κB-dependent Bcl-XL induction in response to DNA damage (Barroso-González et al. 2016).
Lastly, PERK has been shown to play a role in the survival of c-Myc-dependent cancer cells via its involvement in autophagy (Hart et al. 2012). As an oncogene, c-Myc regulates ribosome expression and biogenesis, increasing protein synthesis (van Riggelen et al. 2010). This increased synthesis load is accompanied by elevated PERK activity, which promotes autophagy as a survival mechanism through the upregulation of unc-51-like kinase 1 and ATG5 (Hart et al. 2012). This is underpinned by the observation that upon loss of PERK in these cancer cells, the balance tips from survival to apoptosis due to decreased autophagy (Hart et al. 2012).
Furthermore, PERK seems to play an important role in tumour cell survival under hypoxic conditions. Hypoxia was shown to trigger PERK signalling in xenograft models, while a dominant-negative PERK or eIF2α was linked with an increase in apoptotic cells in hypoxic regions of the tumour (Bi et al. 2005). Delving into the molecular mechanisms responsible for this protection against hypoxia, an increase in ATG5 expression was found, indicating that the induction of autophagy provides the protection for tumour cells (Kouroku et al. 2007; Rouschop et al. 2010).
Recently, the CTA FATE1 has been discovered at the MAMs, where it controls ER-mitochondrial distance (Doghman-Bouguerra et al. 2016). In fact, FATE1 functions as an anti-tether: it diminishes ER-mitochondrial contact sites and decreases mitochondrial Ca2+ uptake. Hence, it regulates the sensitivity towards pro-apoptotic stimuli that elicit apoptosis via Ca2+ signalling (Doghman-Bouguerra et al. 2016). Another anti-apoptotic function of FATE1 is its role in the prevention of accumulation of Bik, a pro-apoptotic Bcl-2-protein family member (Maxfield et al. 2015). The underlying molecular mechanism consists of the recruitment of the E3 ligase RNF183 by FATE1 and subsequent stimulation of Bik degradation (Maxfield et al. 2015), allowing cells to survive even in the presence of apoptotic stimuli.
17.3.3 Cellular Energetics and Biochemical Pathways
17.3.3.1 The IP3R
Ca2+ plays an important role in controlling mitochondrial bioenergetics, since it stimulates ATP production and mitochondrial respiration as the α-ketoglutarate, isocitrate and pyruvate dehydrogenases are Ca2+-dependent rate-limiting enzymes of the TCA (Cárdenas et al. 2010; Kaufman and Malhotra 2014). These dehydrogenases are inhibited in the absence of constitutive low-level Ca2+ transfer from ER to mitochondria, which turns on AMP-activated kinase (AMPK). This results in an increase in basal autophagic flux that is independent of mTOR (Cárdenas et al. 2010; Cárdenas and Foskett 2012). The pro-survival low-level IP3R-mediated Ca2+ signalling can be modified by the activity of several anti-apoptotic Bcl-2-family proteins, which are often upregulated in cancer (Bittremieux et al. 2016). Bcl-2, Bcl-XL as well as Mcl-1 have been reported to interact with IP3Rs (White et al. 2005; Li et al. 2007; Eckenrode et al. 2010). The anti-apoptotic proteins target the C-terminal region of the IP3R (a.a. 2570-2749) (White et al. 2005), resulting in a sensitisation of the IP3Rs to basal IP3 levels, thereby enhancing IP3R-dependent Ca2+ oscillations and stimulating mitochondrial bioenergetics. Bcl-XL is also present in the MAMs, targeting and stimulating IP3Rs and driving mitochondrial metabolism (Williams et al. 2016). Bcl-XL is recruited to the MAMs during non-apoptotic ER stress induction, augmenting mitochondrial bioenergetics through interaction with IP3Rs. IP3R sensitisation by Bcl-XL occurs via its hydrophobic cleft, which binds two BH3-like domains in the C-terminus of IP3Rs, although the BH4 domain of Bcl-XL also contributes by targeting the central, modulatory domain of the IP3R (Yang et al. 2016; Williams et al. 2016). Therefore, BH3 mimetic drugs that target Bcl-XL may also antagonise Bcl-XL’s ability to sensitise IP3Rs and thus may suppress Ca2+-driven mitochondrial metabolism. This is very important, since cancer cells are particularly addicted to these basal Ca2+-signalling events to sustain adequate TCA cycling (Cárdenas et al. 2016; Bultynck 2016). This process provides mitochondrial substrates, like nucleosides, that are essential for proper cell-cycle progression and cell division. In the absence of these Ca2+ fluxes, non-tumorigenic cells tune down their cell cycle, while tumorigenic cells progress through the cell cycle irrespective of their energetic state, resulting in a mitotic catastrophe (Cárdenas et al. 2016; Bultynck 2016).
As described above, the phosphorylation state of the IP3R has a determining role in its Ca2+-release properties, and PML is able to alter this phosphorylation state. Recently, it has been discovered that MAM-localised PML inhibits autophagy via the control of ER-mitochondrial Ca2+ signalling (Missiroli et al. 2016). By stimulating ER-mitochondrial Ca2+ flux, the cell’s metabolism is being stimulated (Cárdenas et al. 2010; Kaufman and Malhotra 2014). p53 is acting as a molecular bridge to keep PML at its place in the MAMs (Missiroli et al. 2016). Upon PML loss, however, metabolic stimulation via Ca2+ is not present anymore, and this turns on AMPK signalling and subsequent autophagy (Missiroli et al. 2016). Additionally, also mTORC2 may control metabolism via IP3R3 phosphorylation, in a similar way it contributes to MAM integrity via PACS-2 phosphorylation (Betz et al. 2013). Activation of PKB/Akt in an mTORC2-dependent manner reduces IP3R-mediated Ca2+ signalling and hence might impact the rate of oxidative phosphorylation (Betz et al. 2013).
17.3.3.2 VDAC1
As a gateway connecting the mitochondria to their environment, VDAC1 is not only a regulator of apoptosis: its localisation at the OMM of the mitochondrion also allows for regulation of cellular bioenergetics. For a start, it has been reported that the Ca2+-flux properties of VDAC1 are stimulated by Mcl-1, which binds with high affinity to the anion channel in the OMM (Huang et al. 2014). This results in an increased mitochondrial Ca2+ uptake, thereby promoting ATP production and stimulating cell survival, as described above (Huang et al. 2014).
Furthermore, VDAC1 is thought to play a major role in constituting the Warburg effect through its interaction with HK (Bustamante and Pedersen 1977; Azoulay-Zohar et al. 2004; Pedersen 2008). A common characteristic of cancer cells is that they show high levels of glycolysis, even though they are oxygenated (Vander Heiden et al. 2009; Liberti and Locasale 2016). This aerobic glycolysis is commonly referred to as the Warburg effect (Vander Heiden et al. 2009; Liberti and Locasale 2016). It is proposed that, by binding VDAC1, ATP produced by oxidative phosphorylation is readily accessible for HK-I and -II to fuel the conversion of glucose to glucose-6-phosphate (G-6-P) (Pedersen 2008). This provides a functional coupling of glycolysis with the TCA. Interestingly, evidence suggests that HK is less sensitive to product inhibition by G-6-P through its interaction with VDAC1 (Bustamante and Pedersen 1977; Azoulay-Zohar et al. 2004). Another clue to VDAC1’s role in the Warburg effect is that especially the VDAC1-bound isoforms, HK-I and HK-II were found to show higher expression levels in several types of cancer, e.g. lymphoma, prostate and breast cancer (Pedersen 2008). Recently, mTORC2 was found to impact the binding of HK-II to VDAC1, again through PKB/Akt-dependent phosphorylation of HK-II, which stabilises its binding to VDAC1 (Betz et al. 2013).
The Warburg effect also comprises the suppression of oxidative phosphorylation in the mitochondria (Zheng 2012; Lu et al. 2015). A long unidentified player in this suppression is dimeric tubulin (Rostovtseva et al. 2008). Dimeric tubulin at concentrations in the nanomolar range was shown to reversibly block VDAC reconstituted into planar phospholipid membranes (Rostovtseva et al. 2008). Furthermore, this block of VDAC was demonstrated to decrease oxygen consumption in isolated mitochondria (Rostovtseva et al. 2008). In addition to this, it was shown that in HepG2 cells, an increase in dimeric tubulin resulted in mitochondrial depolarisation, while a decrease in dimeric tubulin was associated with mitochondrial hyperpolarisation (Maldonado et al. 2010). These results suggest that mitochondrial metabolism in cancer cells is attenuated by the tubulin-mediated blockage of VDAC (Maldonado et al. 2010). It is noteworthy that the effect of free tubulin on cellular metabolism was not found in primary hepatocytes: microtubule depolymerisation decreased the mitochondrial membrane potential, as in cancer cells, but inducing polymerisation did not increase it (Maldonado et al. 2010). This leaves the question whether the observations concerning free tubulin blocking VDAC are specific for cancer cells (Maldonado et al. 2010; Rostovtseva and Bezrukov 2012).
Apart from a shift towards aerobic glycolysis, other biochemical pathways like cholesterologenesis may be altered in cancer cells. Also in this process, a link to VDAC1 can be found: the channel is part of the polyprotein complex called the transduceosome, which is responsible for import of cholesterol into the mitochondria (McEnery et al. 1992; Liu et al. 2006; Rone et al. 2012). In this complex, VDAC1 is proposed to interact with the translocator protein, anchoring the transduceosome to the OMM and facilitating the binding and import of the steroidogenic acute regulatory protein (Hauet et al. 2005). It is proposed that HK bound to VDAC1 may influence the amount of cholesterol synthesis and its import in mitochondria in cancer cells (Campbell and Chan 2007), while at the same time, the channel properties of VDAC1 itself may be influenced by the augmented levels of cholesterol in the OMM (Pastorino and Hoek 2008).
17.4 Conclusions
The MAMs and their various components, including Ca2+-transport systems, chaperones and structural components, establish an important Ca2+-signalling domain between the ER, the main intracellular Ca2+-storage organelle and the mitochondria, the main organelle controlling cell death and survival processes, including cellular bioenergetics and autophagy, apoptosis sensitivity, growth and proliferation. Dysregulation of these processes is a hallmark of cancer. Hence, alterations and perturbations in the structural organisation and functional properties of the MAMs have emerged as an important nexus that underlies oncogenesis, tumour growth and metastasis and responses to chemotherapy. Moreover, several oncogenes and tumour suppressors are localised at the MAMs. Thus, changes in MAMs can drive oncogenesis, while cancer cells at a later stage could remodel MAMs to favour tumour growth, proliferation and metastatic behaviour.
References
Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, Ben-Romano R, Friedman O, Shoshan-Barmatz V (2009) The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci 122:1906–1916
Ahmad A, Ahmad S, Schneider BK, Allen CB, Chang LY, White CW (2002) Elevated expression of hexokinase II protects human lung epithelial-like A549 cells against oxidative injury. Am J Physiol Lung Cell Mol Physiol 283:573–584
Akl H, Bultynck G (2013) Altered Ca2+ signaling in cancer cells: Proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim Biophys Acta 1835:180–193
Akl H, Monaco G, La Rovere R, Welkenhuyzen K, Kiviluoto S, Vervliet T, Molgó J, Distelhorst CW, Missiaen L, Mikoshiba K et al (2013) IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis 4:e632
Akl H, Vervloessem T, Kiviluoto S, Bittremieux M, Parys JB, De Smedt H, Bultynck G (2014) A dual role for the anti-apoptotic Bcl-2 protein in cancer: mitochondria versus endoplasmic reticulum. Biochim Biophys Acta 1843:2240–2252
Akl H, La Rovere RM, Janssens A, Vandenberghe P, Parys JB, Bultynck G (2015) HA14-1 potentiates apoptosis in B-cell cancer cells sensitive to a peptide disrupting IP3 receptor/Bcl-2 complexes. Int J Dev Biol 59:391–398
Arbel N, Ben-Hail D, Shoshan-Barmatz V (2012) Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J Biol Chem 287:23152–23161
Arif T, Krelin Y, Shoshan-Barmatz V (2016) Reducing VDAC1 expression induces a non-apoptotic role for pro-apoptotic proteins in cancer cell differentiation. Biochim Biophys Acta 1857:1228–1242
Arzoine L, Zilberberg N, Ben-Romano R, Shoshan-Barmatz V (2009) Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J Biol Chem 284:3946–3955
Ashby MC, Tepikin AV (2001) ER calcium and the functions of intracellular organelles. Semin Cell Dev Biol 12:11–17
Aslan JE, You H, Williamson DM, Endig J, Youker RT, Thomas L, Shu H, Du Y, Milewski RL, Brush MH et al (2009) Akt and 14-3-3 control a PACS-2 homeostatic switch that integrates membrane traffic with TRAIL-induced apoptosis. Mol Cell 34:497–509
Assefa Z, Bultynck G, Szlufcik K, Nadif Kasri N, Vermassen E, Goris J, Missiaen L, Callewaert G, Parys JB, De Smedt H (2004) Caspase-3-induced truncation of type 1 inositol trisphosphate receptor accelerates apoptotic cell death and induces inositol trisphosphate-independent calcium release during apoptosis. J Biol Chem 279:43227–43236
Atkins KM, Thomas LL, Barroso-González J, Thomas L, Auclair S, Yin J, Kang H, Chung JH, Dikeakos JD, Thomas G (2014) The multifunctional sorting protein PACS-2 regulates SIRT1-mediated deacetylation of p53 to modulate p21-dependent cell-cycle arrest. Cell Rep 8:1545–1557
Aydar E, Onganer P, Perrett R, Djamgoz MB, Palmer CP (2006) The expression and functional characterization of sigma (sigma) 1 receptors in breast cancer cell lines. Cancer Lett 242:245–257
Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V (2004) In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J 377:347–355
Barroso-González J, Auclair S, Luan S, Thomas L, Atkins KM, Aslan JE, Thomas LL, Zhao J, Zhao Y, Thomas G (2016) PACS-2 mediates the ATM and NF-κB-dependent induction of anti-apoptotic Bcl-xL in response to DNA damage. Cell Death Differ 23:1448–1457
Báthori G, Csordás G, Garcia-Perez C, Davies E, Hajnóczky G (2006) Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J Biol Chem 281:17347–17358
Ben-Porath I, Weinberg RA (2004) When cells get stressed: an integrative view of cellular senescence. J Clin Invest 113:8–13
Bernardini JP, Lazarou M, Dewson G (2017) Parkin and mitophagy in cancer. Oncogene 36(10):1315–1327
Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1:11–21
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4:517–529
Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN (2013) Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA 110:12526–12534
Betzenhauser MJ, Wagner LE, Iwai M, Michikawa T, Mikoshiba K, Yule DI (2008) ATP modulation of Ca2+ release by type-2 and type-3 inositol (1, 4, 5)-triphosphate receptors. Differing ATP sensitivities and molecular determinants of action. J Biol Chem 283:21579–21587
Bezprozvanny I, Watras J, Ehrlich BE (1991) Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351:751–754
Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J et al (2005) ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 24:3470–3481
Bittremieux M, Bultynck G (2015) p53 and Ca2+ signaling from the endoplasmic reticulum: partners in anti-cancer therapies. Oncoscience 2:233–238
Bittremieux M, Parys JB, Pinton P, Bultynck G (2016) ER functions of oncogenes and tumor suppressors: Modulators of intracellular Ca2+ signaling. Biochim Biophys Acta 1863:1364–1378
Blackshaw S, Sawa A, Sharp AH, Ross CA, Snyder SH, Khan AA (2000) Type 3 inositol 1,4,5-trisphosphate receptor modulates cell death. FASEB J Off Publ Fed Am Soc Exp Biol 14:1375–1379
Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, Cavener D, Diehl JA (2010) PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 29:3881–3895
Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, Pandolfi PP, Pinton P (2013) Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ 20:1631–1643
Bonora M, Giorgi C, Pinton P (2015) Novel frontiers in calcium signaling: A possible target for chemotherapy. Pharmacol Res 99:82–85
Bourougaa K, Naski N, Boularan C, Mlynarczyk C, Candeias M, Marullo S, Fåhraeus R (2010) Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol Cell 38:78–88
Boutin B, Tajeddine N, Monaco G, Molgo J, Vertommen D, Rider M, Parys JB, Bultynck G, Gailly P (2015) Endoplasmic reticulum Ca2+ content decrease by PKA-dependent hyperphosphorylation of type 1 IP3 receptor contributes to prostate cancer cell resistance to androgen deprivation. Cell Calcium 57:312–320
Brahimi-Horn MC, Giuliano S, Saland E, Lacas-Gervais S, Sheiko T, Pelletier J, Bourget I, Bost F, Féral C, Boulter E et al (2015) Knockout of Vdac1 activates hypoxia-inducible factor through reactive oxygen species generation and induces tumor growth by promoting metabolic reprogramming and inflammation. Cancer Metab 3:8
Breckenridge DG, Nguyen M, Kuppig S, Reth M, Shore GC (2002) The procaspase-8 isoform, procaspase-8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc Natl Acad Sci USA 99:4331–4336
Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC (2003) Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 160:1115–1127
Brewer JW, Diehl JA (2000) PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci USA 97:12625–12630
Bryson JM, Coy PE, Gottlob K, Hay N, Robey RB (2002) Increased hexokinase activity, of either ectopic or endogenous origin, protects renal epithelial cells against acute oxidant-induced cell death. J Biol Chem 277:11392–11400
Bultynck G (2016) Onco-IP3Rs feed cancerous cravings for mitochondrial Ca2+. Trends Biochem Sci 41:390–393
Bustamante E, Pedersen PL (1977) High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc Natl Acad Sci USA 74:3735–3739
Calì T, Ottolini D, Negro A, Brini M (2013) Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca2+transfer to sustain cell bioenergetics. Biochim Biophys Acta 1832:495–508
Campbell AM, Chan SHP (2007) The voltage dependent anion channel affects mitochondrial cholesterol distribution and function. Arch Biochem Biophys 466:203–210
Capiod T, Shuba Y, Skryma R, Prevarskaya N (2007) Calcium signalling and cancer cell growth. Subcell Biochem 45:405–427
Carafoli E (2012) The interplay of mitochondria with calcium: an historical appraisal. Cell Calcium 52:1–8
Cárdenas C, Foskett JK (2012) Mitochondrial Ca2+ signals in autophagy. Cell Calcium 52:44–51
Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung KH, Yang J, Parker I et al (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142:270–283
Cárdenas C, Müller M, McNeal A, Lovy A, Jaňa F, Bustos G, Urra F, Smith N, Molgó J, Diehl JA, Ridky TW, Foskett JK (2016) Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep 14:2313–2324
Chen AH, Silver PA (2012) Designing biological compartmentalization. Trends Cell Biol 22:662–670
Chen K-H, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao R-P et al (2004) Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol 6:872–883
Chen Y-F, Chen Y-T, Chiu W-T, Shen M-R (2013) Remodeling of calcium signaling in tumor progression. J Biomed Sci 20:23
Choe C-U, Ehrlich BE (2006) The inositol 1,4,5-trisphosphate receptor (IP3R) and its regulators: sometimes good and sometimes bad teamwork. Sci STKE 2006:re15
Chu UB, Ruoho AE (2016) Biochemical pharmacology of the sigma-1 receptor. Mol Pharmacol 89:142–153
Clapham DE (2007) Calcium signaling. Cell 131:1047–1058
Collado M, Serrano M (2010) Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10:51–57
Colombini M (2012) VDAC structure, selectivity, and dynamics. Biochim Biophys Acta 1818:1457–1465
Cosson P, Marchetti A, Ravazzola M, Orci L (2012) Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PloS One 7:e46293
Crottès D, Martial S, Rapetti-Mauss R, Pisani DF, Loriol C, Pellissier B, Martin P, Chevet E, Borgese F, Soriani O (2011) Sig1R protein regulates hERG channel expression through a post-translational mechanism in leukemic cells. J Biol Chem 286:27947–27958
Crottès D, Guizouarn H, Martin P, Borgese F, Soriani O (2013) The sigma-1 receptor: a regulator of cancer cell electrical plasticity? Membr Physiol Membr Biophys 4:175
Csordás G, Thomas AP, Hajnóczky G (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J 18:96–108
Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella C, Hajnóczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174:915–921
Csordás G, Várnai P, Golenár T, Roy S, Purkins G, Schneider T, Balla T, Hajnóczky G (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39:121–132
Cullinan SB, Diehl JA (2004) PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem 279:20108–20117
Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15:49–63
da Silva WS, Gómez-Puyou A, de Gómez-Puyou MT, Moreno-Sanchez R, De Felice F, de Meis L, Oliveira MF, Galina A (2004) Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J Biol Chem 279:39846–39855
Davids MS, Letai A (2012) Targeting the B-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol 30:3127–3135
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610
De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, Rizzuto R (2012) VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ 19:267–273
Decuypere J-P, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB (2011) The IP3 receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta 1813:1003–1013
Delom F, Emadali A, Cocolakis E, Lebrun JJ, Nantel A, Chevet E (2007) Calnexin-dependent regulation of tunicamycin-induced apoptosis in breast carcinoma MCF-7 cells. Cell Death Differ 14:586–596
Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787:1309–1316
Diekmann Y, Pereira-Leal JB (2013) Evolution of intracellular compartmentalization. Biochem J 449:319–331
Ding Y, Gao H, Zhao L, Wang X, Zheng M (2015) Mitofusin 2-deficiency suppresses cell proliferation through disturbance of autophagy. PLoS One 10:e0121328
Doghman-Bouguerra M, Granatiero V, Sbiera S, Sbiera I, Lacas-Gervais S, Brau S, Fassnacht M, Rizzuto R, Lalli E (2016) FATE1 antagonizes calcium- and drug-induced apoptosis by uncoupling ER and mitochondria. EMBO Rep 17:1264–1280
Dombernowsky SL, Samsøe-Petersen J, Petersen CH, Instrell R, Hedegaard AM, Thomas L, Atkins KM, Auclair S, Albrechtsen R, Mygind KJ et al (2015) The sorting protein PACS-2 promotes ErbB signalling by regulating recycling of the metalloproteinase ADAM17. Nat Commun 6:7518
Dong X-Y, Su Y-R, Qian X-P, Yang X-A, Pang X-W, Wu H-Y, Chen W-F (2003) Identification of two novel CT antigens and their capacity to elicit antibody response in hepatocellular carcinoma patients. Br J Cancer 89:291–297
Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C (2010) Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem 285:13678–13684
Fan Z, Yu H, Cui N, Kong X, Liu X, Chang Y, Wu Y, Sun L, Wang G (2015) ABT737 enhances cholangiocarcinoma sensitivity to cisplatin through regulation of mitochondrial dynamics. Exp Cell Res 335:68–81
Fedorenko OA, Popugaeva E, Enomoto M, Stathopulos PB, Ikura M, Bezprozvanny I (2014) Intracellular calcium channels: inositol-1,4,5-trisphosphate receptors. Eur J Pharmacol 739:39–48
Feng Y-X, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TH, Jin DX, Reinhardt F, Ploegh HL, Wang Q, Gupta PB (2014) Epithelial-to-mesenchymal transition activates PERK-eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discov 4:702–715
Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2015) Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci USA 112:2174–2181
Finch EA, Turner TJ, Goldin SM (1991) Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science 252:443–446
Flachbartová Z, Kovacech B (2013) Mortalin - a multipotent chaperone regulating cellular processes ranging from viral infection to neurodegeneration. Acta Virol 57:3–15
Foskett JK, White C, Cheung K-H, Mak D-OD (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87:593–658
Giampazolias E, Tait SWG (2016) Mitochondria and the hallmarks of cancer. FEBS J 283:803–814
Gincel D, Silberberg SD, Shoshan-Barmatz V (2000) Modulation of the voltage-dependent anion channel (VDAC) by glutamate. J Bioenerg Biomembr 32:571–583
Gincel D, Zaid H, Shoshan-Barmatz V (2001) Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J 358:147–155
Giorgi C, Ito K, Lin H-K, Santangelo C, Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J, Bernardi R, Rizzuto R et al (2010) PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330:1247–1251
Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A, Suski JM et al (2012) Mitochondrial Ca2+ and apoptosis. Cell Calcium 52:36–43
Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, Galindo Ramirez F, Rizzuto R, Di Virgilio F, Zito E et al (2015a) p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci USA 112:1779–1784
Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P (2015b) Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal 22:995–1019
Gniadecki R (2004) Depletion of membrane cholesterol causes ligand-independent activation of Fas and apoptosis. Biochem Biophys Res Commun 320:165–169
Gomez L, Thiebaut P-A, Paillard M, Ducreux S, Abrial M, Crola Da Silva C, Durand A, Alam MR, Van Coppenolle F, Sheu S-S et al (2016) The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ 23:313–322
Greenberg EF, McColl KS, Zhong F, Wildey G, Dowlati A, Distelhorst CW (2015) Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1,4,5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis 6:e2034
Guicciardi ME, Werneburg NW, Bronk SF, Franke A, Yagita H, Thomas G, Gores GJ (2014) Cellular inhibitor of apoptosis (cIAP)-mediated ubiquitination of phosphofurin acidic cluster sorting protein 2 (PACS-2) negatively regulates tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) cytotoxicity. PloS One 9:e92124
Guo X, Chen K-H, Guo Y, Liao H, Tang J, Xiao R-P (2007) Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ Res 101:1113–1122
Guo W, Yan L, Yang L, Liu X, E Q, Gao P, Ye X, Liu W, Zuo J (2014) Targeting GRP75 improves HSP90 inhibitor efficacy by enhancing p53-mediated apoptosis in hepatocellular carcinoma. PloS One 9:e85766
Gutstein DE, Marks AR (1997) Role of inositol 1,4,5-trisphosphate receptors in regulating apoptotic signaling and heart failure. Heart Vessels Suppl 12:53–57
Hajnóczky G, Robb-Gaspers LD, Seitz MB, Thomas AP (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82:415–424
Hajnóczky G, Csordás G, Yi M (2002) Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium 32:363–377
Halestrap AP (2014) The C ring of the F1Fo ATP synthase forms the mitochondrial permeability transition pore: a critical appraisal. Front Oncol 4:234
Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA (2005) PERK and GCN2 contribute to eIF2α phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell 16:5493–5501
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hanson CJ, Bootman MD, Distelhorst CW, Wojcikiewicz RJ, Roderick HL (2008) Bcl-2 suppresses Ca2+ release through inositol 1,4,5-trisphosphate receptors and inhibits Ca2+ uptake by mitochondria without affecting ER calcium store content. Cell Calcium 44:324–338
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5:897–904
Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M (2012) ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 122:4621–4634
Hauet T, Yao Z-X, Bose HS, Wall CT, Han Z, Li W, Hales DB, Miller WL, Culty M, Papadopoulos V (2005) Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into leydig cell mitochondria. Mol Endocrinol Baltim Md 19:540–554
Hayashi T, Su T-P (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131:596–610
Hebert DN, Foellmer B, Helenius A (1995) Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81:425–433
Hodge T, Colombini M (1997) Regulation of metabolite flux through voltage-gating of VDAC channels. J Membr Biol 157:271–279
Huang H, Shah K, Bradbury NA, Li C, White C (2014) Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis 5:e1482
Huang X, Jin M, Chen Y-X, Wang J, Zhai K, Chang Y, Yuan Q, Yao K-T, Ji G (2016) ERP44 inhibits human lung cancer cell migration mainly via IP3R2. Aging 8:1276–1286
Iino M (1990) Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. J Gen Physiol 95:1103–1122
Jayaraman T, Marks AR (1997) T cells deficient in inositol 1,4,5-trisphosphate receptor are resistant to apoptosis. Mol Cell Biol 17:3005–3012
Jin H, Ji M, Chen L, Liu Q, Che S, Xu M, Lin Z (2016) The clinicopathological significance of Mortalin overexpression in invasive ductal carcinoma of breast. J Exp Clin Cancer Res CR 35:42
Jonas EA, Porter GA, Beutner G, Mnatsakanyan N, Alavian KN (2015) Cell death disguised: the mitochondrial permeability transition pore as the c-subunit of the F1FO ATP synthase. Pharmacol Res 99:382–392
Joseph SK, Hajnóczky G (2007) IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis Int J Program Cell Death 12:951–968
Kang SS, Han K-S, Ku BM, Lee YK, Hong J, Shin HY, Almonte AG, Woo DH, Brat DJ, Hwang EM et al (2010) Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res 70:1173–1183
Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A et al (2011) Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479:547–551
Kannan A, Wells RB, Sivakumar S, Komatsu S, Singh KP, Samten B, Philley JV, Sauter ER, Ikebe M, Idell S et al (2016) Mitochondrial reprogramming regulates breast cancer progression. Clin Cancer Res 22:3348–3360
Kaufman RJ, Malhotra JD (2014) Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim Biophys Acta 1843:2233–2239
Kaul SC, Duncan EL, Englezou A, Takano S, Reddel RR, Mitsui Y, Wadhwa R (1998) Malignant transformation of NIH3T3 cells by overexpression of mot-2 protein. Oncogene 17:907–911
Kaul SC, Reddel RR, Mitsui Y, Wadhwa R (2001) An N-terminal region of mot-2 binds to p53 in vitro. Neoplasia N Y N 3:110–114
Kaul SC, Aida S, Yaguchi T, Kaur K, Wadhwa R (2005) Activation of wild type p53 function by its mortalin-binding, cytoplasmically localizing carboxyl terminus peptides. J Biol Chem 280:39373–39379
Khan MT, Wagner L, Yule DI, Bhanumathy C, Joseph SK (2006) Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem 281:3731–3737
Kliková K, Štefaniková A, Pilchová I, Hatok J, Chudý P, Chudej J, Dobrota D, Račay P (2015) Differential impact of bortezomib on HL-60 and K562 cells. Gen Physiol Biophys 34:33–42
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T (2007) ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 14:230–239
Krols M, Bultynck G, Janssens S (2016) ER-Mitochondria contact sites: a new regulator of cellular calcium flux comes into play. J Cell Biol 214:367–370
Kronidou NG, Oppliger W, Bolliger L, Hannavy K, Glick BS, Schatz G, Horst M (1994) Dynamic interaction between Isp45 and mitochondrial hsp70 in the protein import system of the yeast mitochondrial inner membrane. Proc Natl Acad Sci USA 91:12818–12822
La Rovere RM, Roest G, Bultynck G, Parys JB (2016) Intracellular Ca2+ signaling and Ca2+microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 60:74–87
Lakkaraju AKK, van der Goot FG (2013) Calnexin controls the STAT3-mediated transcriptional response to EGF. Mol Cell 51:386–396
Lamriben L, Graham JB, Adams BM, Hebert DN (2016) N-Glycan-based ER molecular chaperone and protein quality control system: the Calnexin binding cycle. Traffic Cph Den 17:308–326
Lavik AR, Zhong F, Chang M-J, Greenberg E, Choudhary Y, Smith MR, McColl KS, Pink J, Reu FJ, Matsuyama S et al (2015) A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget 6:27388–27402
Li J, Lee B, Lee AS (2006) Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem 281:7260–7270
Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C (2007) Apoptosis regulation by Bcl-xL modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci USA 104:12565–12570
Li G, Mongillo M, Chin K-T, Harding H, Ron D, Marks AR, Tabas I (2009) Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J Cell Biol 186:783–792
Liberti MV, Locasale JW (2016) The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41:211–218
Liou G-Y, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44:479–496
Liu J, Rone MB, Papadopoulos V (2006) Protein-protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. J Biol Chem 281:38879–38893
Lou Y, Li R, Liu J, Zhang Y, Zhang X, Jin B, Liu Y, Wang Z, Zhong H, Wen S et al (2015) Mitofusin-2 over-expresses and leads to dysregulation of cell cycle and cell invasion in lung adenocarcinoma. Med Oncol 32:132
Lou Y, Zhang Y, Li R, Gu P, Xiong L, Zhong H, Zhang W, Han B (2016) Transcriptional profiling revealed the anti-proliferative effect of MFN2 deficiency and identified risk factors in lung adenocarcinoma. Tumor Biol 37:8643–8655
Lu J, Tan M, Cai Q (2015) The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett 356:156–164
Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L, Berthiaume LG, Simmen T (2012) Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J 31:457–470
Lynes EM, Raturi A, Shenkman M, Ortiz Sandoval C, Yap MC, Wu J, Janowicz A, Myhill N, Benson MD, Campbell RE et al (2013) Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. J Cell Sci 126:3893–3903
Ma LI, Chang Y, Yu L, He W, Liu Y (2015) Pro-apoptotic and anti-proliferative effects of mitofusin-2 via PI3K/Akt signaling in breast cancer cells. Oncol Lett 10:3816–3822
Maldonado EN, Patnaik J, Mullins MR, Lemasters JJ (2010) Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res 70:10192–10201
Marchi S, Pinton P (2014) The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol 592:829–839
Marchi S, Pinton P (2016) Alterations of calcium homeostasis in cancer cells. Curr Opin Pharmacol 29:1–6
Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, Pinton P (2008) Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun 375:501–505
Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, Pinton P (2012) Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis 3:e304
Marchi S, Giorgi C, Oparka M, Duszynski J, Wieckowski MR, Pinton P (2014a) Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol Cell Oncol 1:956469
Marchi S, Patergnani S, Pinton P (2014b) The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta 1837:461–469
Mavlyutov TA, Epstein M, Guo L-W (2015) Subcellular localization of the sigma-1 receptor in retinal neurons - an electron microscopy study. Sci Rep 5:10689
Maxfield KE, Taus PJ, Corcoran K, Wooten J, Macion J, Zhou Y, Borromeo M, Kollipara RK, Yan J, Xie J et al (2015) Comprehensive functional characterization of cancer-testis antigens defines obligate participation in multiple hallmarks of cancer. Nat Commun 6:8840
McEnery MW, Snowman AM, Trifiletti RR, Snyder SH (1992) Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci USA 89:3170–3174
Mendes CCP, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, Gomez MV, Nathanson MH, Leite MF (2005) The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem 280:40892–40900
Messina A, Reina S, Guarino F, De Pinto V (2012) VDAC isoforms in mammals. Biochim Biophys Acta 1818:1466–1476
Mikoshiba K (2007) The IP3 receptor/Ca2+ channel and its cellular function. Biochem Soc Symp:9–22
Missiroli S, Bonora M, Patergnani S, Poletti F, Perrone M, Gafà R, Magri E, Raimondi A, Lanza G, Tacchetti C, Kroemer G, Pandolfi PP, Pinton P, Giorgi C (2016) PML at mitochondria-associated membranes is critical for the repression of autophagy and cancer development. Cell Rep 16:2415–2427
Miyakawa T, Maeda A, Yamazawa T, Hirose K, Kurosaki T (1999) Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J 18:1303–1308
Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, De Maeyer M, Missiaen L, Distelhorst CW, De Smedt H et al (2012) Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ 19:295–309
Monaco G, Decrock E, Arbel N, van Vliet AR, La Rovere RM, De Smedt H, Parys JB, Agostinis P, Leybaert L, Shoshan-Barmatz V et al (2015) The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J Biol Chem 290:9150–9161
Morciano G, Giorgi C, Bonora M, Punzetti S, Pavasini R, Wieckowski MR, Campo G, Pinton P (2015) Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J Mol Cell Cardiol 78:142–153
Mori T, Hayashi T, Hayashi E, Su T-P (2013) Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PloS One 8:e76941
Mound A, Rodat-Despoix L, Bougarn S, Ouadid-Ahidouch H, Matifat F (2013) Molecular interaction and functional coupling between type 3 inositol 1,4,5-trisphosphate receptor and BKCa channel stimulate breast cancer cell proliferation. Eur J Cancer Oxf Engl 1990 49:3738–3751
Mujcic H, Nagelkerke A, Rouschop KMA, Chung S, Chaudary N, Span PN, Clarke B, Milosevic M, Sykes J, Hill RP et al (2013) Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin Cancer Res 19:6126–6137
Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K, Cooper TJ, Thomas G, Simmen T (2008) The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell 19:2777–2788
Nagelkerke A, Bussink J, Mujcic H, Wouters BG, Lehmann S, Sweep FC, Span PN (2013) Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res BCR 15:R2
Nagelkerke A, Sweep FC, Stegeman H, Grénman R, Kaanders JH, Bussink J, Span PN (2015) Hypoxic regulation of the PERK/ATF4/LAMP3-arm of the unfolded protein response in head and neck squamous cell carcinoma. Head Neck 37:896–905
Naon D, Scorrano L (2014) At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim Biophys Acta 1843:2184–2194
Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernández-Alvarez MI et al (2016) Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci USA 113:11249–11254
Newton CL, Mignery GA, Südhof TC (1994) Co-expression in vertebrate tissues and cell lines of multiple inositol 1,4,5-trisphosphate (InsP3) receptors with distinct affinities for InsP3. J Biol Chem 269:28613–28619
Ono Y, Tamiya T, Ichikawa T, Matsumoto K, Furuta T, Ohmoto T, Akiyama K, Seki S, Ueki K, Louis DN (1997) Accumulation of wild-type p53 in astrocytomas is associated with increased p21 expression. Acta Neuropathol (Berl) 94:21–27
Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4:552–565
Ouyang K, Leandro Gomez-Amaro R, Stachura DL, Tang H, Peng X, Fang X, Traver D, Evans SM, Chen J (2014) Loss of IP3R-dependent Ca2+ signalling in thymocytes leads to aberrant development and acute lymphoblastic leukemia. Nat Commun 5:4814
Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11:381–389
Palmer CP, Mahen R, Schnell E, Djamgoz MB, Aydar E (2007) Sigma-1 receptors bind cholesterol and remodel lipid rafts in breast cancer cell lines. Cancer Res 67:11166–11175
Panieri E, Santoro MM (2016) ROS homeostasis and metabolism: a dangerous liaison in cancer cells. Cell Death Dis 7:e2253
Parys JB, De Smedt H (2012) Inositol 1,4,5-trisphosphate and its receptors. Adv Exp Med Biol 740:255–279
Parys JB, Sernett SW, DeLisle S, Snyder PM, Welsh MJ, Campbell KP (1992) Isolation, characterization, and localization of the inositol 1,4,5-trisphosphate receptor protein in Xenopus laevis oocytes. J Biol Chem 267:18776–18782
Pastorino JG, Hoek JB (2008) Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr 40:171–182
Pedersen PL (2008) Voltage dependent anion channels (VDACs): a brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J Bioenerg Biomembr 40:123–126
Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL (2014) Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodelling of the isoform composition of IP3Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J Cell Sci 127:1607–1619
Pillozzi S, Brizzi MF, Bernabei PA, Bartolozzi B, Caporale R, Basile V, Boddi V, Pegoraro L, Becchetti A, Arcangeli A (2007) VEGFR-1 (FLT-1), β1 integrin, and hERG K+ channel for a macromolecular signaling complex in acute myeloid leukemia: role in cell migration and clinical outcome. Blood 110:1238–1250
Pillozzi S, Masselli M, De Lorenzo E, Accordi B, Cilia E, Crociani O, Amedei A, Veltroni M, D’Amico M, Basso G, Becchetti A, Campana D, Arcangeli A (2011) Chemotherapy resistance in acute lymphoblastic leukemia requires hERG1 channels and is overcome by hERG1 blockers. Blood 117:902–914
Pinton P, Giorgi C, Pandolfi PP (2011) The role of PML in the control of apoptotic cell fate: a new key player at ER–mitochondria sites. Cell Death Differ 18:1450–1456
Pluquet O, Qu L-K, Baltzis D, Koromilas AE (2005) Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3β. Mol Cell Biol 25:9392–9405
Prevarskaya N, Ouadid-Ahidouch H, Skryma R, Shuba Y (2014) Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos Trans R Soc Lond B Biol Sci 369:20130097
Qiao A, Wang K, Yuan Y, Guan Y, Ren X, Li L, Chen X, Li F, Chen AF, Zhou J et al (2016) Sirt3-mediated mitophagy protects tumor cells against apoptosis under hypoxia. Oncotarget 7:43390–43400
Qu L, Huang S, Baltzis D, Rivas-Estilla AM, Pluquet O, Hatzoglou M, Koumenis C, Taya Y, Yoshimura A, Koromilas AE (2004) Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3β. Genes Dev 18:261–277
Raffaello A, Mammucari C, Gherardi G, Rizzuto R (2016) Calcium at the center of Cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci 41(12):1035–1049
Ran Q, Wadhwa R, Kawai R, Kaul SC, Sifers RN, Bick RJ, Smith JR, Pereira-Smith OM (2000) Extramitochondrial localization of mortalin/mthsp70/PBP74/GRP75. Biochem Biophys Res Commun 275:174–179
Ranieri M, Brajkovic S, Riboldi G, Ronchi D, Rizzo F, Bresolin N, Corti S, Comi GP (2013) Mitochondrial fusion proteins and human diseases. Neurol Res Int 2013:293893
Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE, Rizzuto R (2002) Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol 159:613–624
Raturi A, Gutiérrez T, Ortiz-Sandoval C, Ruangkittisakul A, Herrera-Cruz MS, Rockley JP, Gesson K, Ourdev D, Lou P-H, Lucchinetti E (2016) TMX1 determines cancer cell metabolism as a thiol-based modulator of ER-mitochondria Ca2+ flux. J Cell Biol 214:433–444
Rieusset J, Fauconnier J, Paillard M, Belaidi E, Tubbs E, Chauvin MA, Durand A, Bravard A, Teixeira G, Bartosch B (2016) Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 59:614–623
Rizzuto R, Brini M, Murgia M, Pozzan T (1993) Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262:744–747
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280:1763–1766
Rizzuto R, Bernardi P, Pozzan T (2000) Mitochondria as all-round players of the calcium game. J Physiol 529(Pt 1):37–47
Rizzuto R, De Stefani D, Raffaello A, Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Bio 13:566–578
Roderick HL, Cook SJ (2008) Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer 8:361–375
Roderick HL, Lechleiter JD, Camacho P (2000) Cytosolic phosphorylation of calnexin controls intracellular Ca2+oscillations via an interaction with SERCA2b. J Cell Biol 149:1235–1248
Rone MB, Midzak AS, Issop L, Rammouz G, Jagannathan S, Fan J, Ye X, Blonder J, Veenstra T, Papadopoulos V (2012) Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol Endocrinol Baltim Md 26:1868–1882
Rong Y-P, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, Matsuyama S, Herlitze S, Roderick HL, Bootman MD et al (2008) Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell 31:255–265
Rong Y-P, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, Mignery GA, Roderick HL, Bootman MD, Distelhorst CW (2009) The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci USA 106:14397–14402
Rostovtseva TK, Bezrukov SM (2012) VDAC inhibition by tubulin and its physiological implications. Biochim Biophys Acta 1818:1526–1535
Rostovtseva TK, Kazemi N, Weinrich M, Bezrukov SM (2006) Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J Biol Chem 281:37496–37506
Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, Sackett DL (2008) Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci USA 105:18746–18751
Rouschop KMA, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW et al (2010) The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 120:127–141
Rowland AA, Voeltz GK (2012) Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13:607–625
Sano R, Reed JC (2013) ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833:3460–3470
Schein SJ, Colombini M, Finkelstein A (1976) Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J Membr Biol 30:99–120
Scherer PE, Manning-Krieg UC, Jenö P, Schatz G, Horst M (1992) Identification of a 45-kDa protein at the protein import site of the yeast mitochondrial inner membrane. Proc Natl Acad Sci USA 89:11930–11934
Schneider HC, Westermann B, Neupert W, Brunner M (1996) The nucleotide exchange factor MGE exerts a key function in the ATP-dependent cycle of mt-Hsp70-Tim44 interaction driving mitochondrial protein import. EMBO J 15:5796–5803
Shimizu S, Narita M, Tsujimoto Y (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399:483–487
Shimizu S, Konishi A, Kodama T, Tsujimoto Y (2000) BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci USA 97:3100–3105
Shioda N, Ishikawa K, Tagashira H, Ishizuka T, Yawo H, Fukunaga K (2012) Expression of a truncated form of the endoplasmic reticulum chaperone protein, σ1 receptor, promotes mitochondrial energy depletion and apoptosis. J Biol Chem 287:23318–23331
Shoshan-Barmatz V, Gincel D (2003) The voltage-dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem Biophys 39:279–292
Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N (2010) VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med 31:227–285
Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung C-H, Crump CM, Thomas G (2005) PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 24:717–729
Simpson AJ, Caballero OL, Jungbluth A, Chen Y-T, Old LJ (2005) Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 5:615–625
Skrzycki M, Czeczot H (2013) Altered expression level of Sigma1 receptor gene in human colorectal cancer. J Recept Signal Transduct Res 33:313–318
Starenki D, Hong S-K, Lloyd RV, Park J-I (2015) Mortalin (GRP75/HSPA9) upregulation promotes survival and proliferation of medullary thyroid carcinoma cells. Oncogene 34:4624–4634
Stewart TA, Yapa KTDS, Monteith GR (2015) Altered calcium signaling in cancer cells. Biochim Biophys Acta 1848:2502–2511
Su TP (1982) Evidence for sigma opioid receptor: binding of [3H]SKF-10047 to etorphine-inaccessible sites in guinea-pig brain. J Pharmacol Exp Ther 223:284–290
Su T-P, Hayashi T, Maurice T, Buch S, Ruoho AE (2010) The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci 31:557–566
Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H (2008) Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol 28:1007–1017
Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175:901–911
Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Söderberg O et al (2008) Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci USA 105:2427–2432
Szatkowski C, Parys JB, Ouadid-Ahidouch H, Matifat F (2010) Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiol-induced breast cancer epithelial cell growth. Mol Cancer 9:156
Tagashira H, Bhuiyan MS, Shioda N, Fukunaga K (2014) Fluvoxamine rescues mitochondrial Ca2+ transport and ATP production through σ(1)-receptor in hypertrophic cardiomyocytes. Life Sci 95:89–100
Takizawa T, Tatematsu C, Watanabe K, Kato K, Nakanishi Y (2004) Cleavage of calnexin caused by apoptotic stimuli: implication for the regulation of apoptosis. J Biochem (Tokyo) 136:399–405
Tsai S-YA, Chuang J-Y, Tsai M-S, Wang X-F, Xi Z-X, Hung J-J, Chang W-C, Bonci A, Su T-P (2015) Sigma-1 receptor mediates cocaine-induced transcriptional regulation by recruiting chromatin-remodeling factors at the nuclear envelope. Proc Natl Acad Sci USA 112:6562–6570
Tsunoda T, Koga H, Yokomizo A, Tatsugami K, Eto M, Inokuchi J, Hirata A, Masuda K, Okumura K, Naito S (2005) Inositol 1,4,5-trisphosphate (IP3) receptor type1 (IP3R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene 24:1396–1402
Tu H, Wang Z, Bezprozvanny I (2005) Modulation of mammalian inositol 1,4,5-trisphosphate receptor isoforms by calcium: a role of calcium sensor region. Biophys J 88:1056–1069
Urra H, Dufey E, Avril T, Chevet E, Hetz C (2016) Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2:252–262
van Riggelen J, Yetil A, Felsher DW (2010) MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer 10:301–309
van Vliet AR, Verfaillie T, Agostinis P (2014) New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta 1843:2253–2262
Vance JE (2014) MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim Biophys Acta 1841:595–609
Vander Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB (2000) Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc Natl Acad Sci USA 97:4666–4671
Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M (2001) Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem 276:19414–19419
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Vanderheyden V, Devogelaere B, Missiaen L, De Smedt H, Bultynck G, Parys JB (2009) Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim Biophys Acta 1793:959–970
Várnai P, Balla A, Hunyady L, Balla T (2005) Targeted expression of the inositol 1,4,5-triphosphate receptor (IP3R) ligand-binding domain releases Ca2+ via endogenous IP3R channels. Proc Natl Acad Sci USA 102:7859–7864
Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19:1880–1891
Vervliet T, Parys JB, Bultynck G (2016) Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene 35:5079–5092
Vervloessem T, Yule DI, Bultynck G, Parys JB (2015) The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca2+-release channel. Biochim Biophys Acta 1853:1992–2005
Vilner BJ, John CS, Bowen WD (1995) Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res 55:408–413
Voos W, Röttgers K (2002) Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim Biophys Acta 1592:51–62
Wadhwa R, Pereira-Smith OM, Reddel RR, Sugimoto Y, Mitsui Y, Kaul SC (1995) Correlation between complementation group for immortality and the cellular distribution of mortalin. Exp Cell Res 216:101–106
Wadhwa R, Takano S, Robert M, Yoshida A, Nomura H, Reddel RR, Mitsui Y, Kaul SC (1998) Inactivation of tumor suppressor p53 by mot-2, a hsp70 family member. J Biol Chem 273:29586–29591
Wadhwa R, Taira K, Kaul SC (2002a) An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: what, when, and where? Cell Stress Chaperones 7:309–316
Wadhwa R, Yaguchi T, Hasan MK, Mitsui Y, Reddel RR, Kaul SC (2002b) Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein. Exp Cell Res 274:246–253
Wagner LE, Joseph SK, Yule DI (2008) Regulation of single inositol 1,4,5-trisphosphate receptor channel activity by protein kinase A phosphorylation. J Physiol 586:3577–3596
Wang B, Rouzier R, Albarracin CT, Sahin A, Wagner P, Yang Y, Smith TL, Meric-Bernstam F, Marcelo Aldaz C, Marcelo AC et al (2004) Expression of sigma 1 receptor in human breast cancer. Breast Cancer Res Treat 87:205–214
Wang W, Xie Q, Zhou X, Yao J, Zhu X, Huang P, Zhang L, Wei J, Xie H, Zhou L et al (2015) Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett 358:47–58
Wei C, Wang X, Chen M, Ouyang K, Song L-S, Cheng H (2009) Calcium flickers steer cell migration. Nature 457:901–905
Wei C, Wang X, Zheng M, Cheng H (2012) Calcium gradients underlying cell migration. Curr Opin Cell Biol 24:254–261
Weisthal S, Keinan N, Ben-Hail D, Arif T, Shoshan-Barmatz V (2014) Ca2+-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim Biophys Acta 1843:2270–2281
Werneburg NW, Bronk SF, Guicciardi ME, Thomas L, Dikeakos JD, Thomas G, Gores GJ (2012) Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) protein-induced lysosomal translocation of proapoptotic effectors is mediated by phosphofurin acidic cluster sorting protein-2 (PACS-2). J Biol Chem 287:24427–24437
White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK (2005) The endoplasmic reticulum gateway to apoptosis by Bcl-XL modulation of the InsP3R. Nat Cell Biol 7:1021–1028
Whitehurst AW (2014) Cause and consequence of cancer/testis antigen activation in cancer. Annu Rev Pharmacol Toxicol 54:251–272
Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG et al (2007) Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 446:815–819
Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calvé B, Augert A, Ferrand M, Prevarskaya N, Simonnet H, Vindrieux D et al (2014) Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun 5:3792
Williams A, Hayashi T, Wolozny D, Yin B, Su T-C, Betenbaugh MJ, Su T-P (2016) The non-apoptotic action of Bcl-xL: regulating Ca2+ signaling and bioenergetics at the ER-mitochondrion interface. J Bioenerg Biomembr 48:211–225
Wilson JE (2003) Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol 206:2049–2057
Wu P-K, Hong S-K, Veeranki S, Karkhanis M, Starenki D, Plaza JA, Park J-I (2013) A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol Cell Biol 33:4051–4067
Wu C-H, Lin Y-W, Wu T-F, Ko J-L, Wang P-H (2016a) Clinical implication of voltage-dependent anion channel 1 in uterine cervical cancer and its action on cervical cancer cells. Oncotarget 7:4210–4225
Wu Y, Zhou D, Xu X, Zhao X, Huang P, Zhou X, Song W, Guo H, Wang W, Zheng S (2016b) Clinical significance of mitofusin-2 and its signaling pathways in hepatocellular carcinoma. World J Surg Oncol 14:179
Xie Q, Su J, Jiao B, Shen L, Ma L, Qu X, Yu C, Jiang X, Xu Y, Sun L (2016) ABT737 reverses cisplatin resistance by regulating ER-mitochondria Ca2+ signal transduction in human ovarian cancer cells. Int J Oncol
Xu D, Yi W, Chen Y, Ma L, Wang J, Yu G (2014) Overexpression of Sig1R is closely associated with tumor progression and poor outcome in patients with hilar cholangiocarcinoma. Med Oncol Northwood Lond Engl 31:261
Yang L, Li H, Jiang Y, Zuo J, Liu W (2013) Inhibition of mortalin expression reverses cisplatin resistance and attenuates growth of ovarian cancer cells. Cancer Lett 336:213–221
Yang J, Vais H, Gu W, Foskett JK (2016) Biphasic regulation of InsP3 receptor gating by dual Ca2+ release channel BH3-like domains mediates Bcl-xL control of cell viability. Proc Natl Acad Sci USA 113:1953–1962
Yi X, Luk JM, Lee NP, Peng J, Leng X, Guan X-Y, Lau GK, Beretta L, Fan S-T (2008) Association of mortalin (HSPA9) with liver cancer metastasis and prediction for early tumor recurrence. Mol Cell Proteomics 7:315–325
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891
Yoshikawa F, Iwasaki H, Michikawa T, Furuichi T, Mikoshiba K (1999) Trypsinized cerebellar inositol 1,4,5-trisphosphate receptor. Structural and functional coupling of cleaved ligand binding and channel domains. J Biol Chem 274:316–327
Youker RT, Shinde U, Day R, Thomas G (2009) At the crossroads of homoeostasis and disease: roles of the PACS proteins in membrane traffic and apoptosis. Biochem J 421:1–15
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59
Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ (1996) serine phosphorylation of death agonist bad in response to survival factor results in binding to 14-3-3 not BCL-XL. Cell 87:619–628
Zhang F, Hamanaka RB, Bobrovnikova-Marjon E, Gordan JD, Dai M-S, Lu H, Simon MC, Diehl JA (2006) Ribosomal stress couples the unfolded protein response to p53-dependent cell cycle arrest. J Biol Chem 281:30036–30045
Zhang G-E, Jin H-L, Lin X-K, Chen C, Liu X-S, Zhang Q, Yu J-R (2013) Anti-tumor effects of Mfn2 in gastric cancer. Int J Mol Sci 14:13005–13021
Zhang C, Ding W, Liu Y, Hu Z, Zhu D, Wang X, Yu L, Wang L, Shen H, Zhang W et al (2016a) Proteomics-based identification of VDAC1 as a tumor promoter in cervical carcinoma. Oncotarget 7:52317–52328
Zhang G, Jiang G, Wang C, Zhong K, Zhang J, Xue Q, Li X, Jin H, Li B, Zhang G et al (2016b) Decreased expression of microRNA-320a promotes proliferation and invasion of non-small cell lung cancer cells by increasing VDAC1 expression. Oncotarget 7:49470–49480
Zheng J (2012) Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett 4:1151–1157
Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, Rong Y-P, Molitoris JK, Lam M, Ryder C et al (2011) Induction of Ca2+ –driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood 117:2924–2934
Acknowledgements
GB and JBP are supported by grants from the Research Foundation - Flanders (FWO) (grants G.0571.12N, G.0819.13N, G.0C91.14N, G.0927.15N and G.0A34.16N) and the Research Council - KU Leuven (OT14/101). MB and MK are holders of a Ph.D. Fellowship from the FWO. P.P. is grateful to Camilla degli Scrovegni for continuous support. P.P. is supported by Telethon (GGP15219/B), the Italian Association for Cancer Research (AIRC) (IG-14442), the Italian Cystic Fibrosis Research Foundation (19/2014), the Italian Ministry of Education, University and Research (COFIN no. 20129JLHSY_002, FIRB no. RBAP11FXBC_002 and Futuro in Ricerca no. RBFR10EGVP_001), local funds from the University of Ferrara and the Italian Ministry of Health. C.G is supported by AIRC (MFAG-13521), the Italian Ministry of Health, Cariplo and local funds from the University of Ferrara.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Kerkhofs, M. et al. (2017). Alterations in Ca2+ Signalling via ER-Mitochondria Contact Site Remodelling in Cancer. In: Tagaya, M., Simmen, T. (eds) Organelle Contact Sites. Advances in Experimental Medicine and Biology, vol 997. Springer, Singapore. https://doi.org/10.1007/978-981-10-4567-7_17
Download citation
DOI: https://doi.org/10.1007/978-981-10-4567-7_17
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4566-0
Online ISBN: 978-981-10-4567-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)