
Abstract
Endoplasmic reticulum (ER)-mitochondria regions are specialized subdomains called also mitochondria-associated membranes (MAMs). MAMs allow regulation of lipid synthesis and represent hubs for ion and metabolite signaling. As these two organelles can module both the amplitude and the spatiotemporal patterns of calcium (Ca2+) signals, this particular interaction controls several Ca2+-dependent pathways well known for their contribution to tumorigenesis, such as metabolism, survival, sensitivity to cell death, and metastasis. Mitochondria-mediated apoptosis arises from mitochondrial Ca2+ overload, permeabilization of the mitochondrial outer membrane, and the release of mitochondrial apoptotic factors into the cytosol. Decreases in Ca2+ signaling at the ER-mitochondria interface are being studied in depth as failure of apoptotic-dependent cell death is one of the predominant characteristics of cancer cells. However, some recent papers that linked MAMs Ca2+ crosstalk-related upregulation to tumor onset and progression have aroused the interest of the scientific community.
In this review, we will describe how different MAMs-localized proteins modulate the effectiveness of Ca2+-dependent apoptotic stimuli by causing both increases and decreases in the ER-mitochondria interplay and, specifically, by modulating Ca2+ signaling.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Ca2+ is the third most abundant metal in nature, and it was adopted as a regulator in the early evolutionary stages in prokaryotes (Cai et al. 2015). Ca2+ ions play a crucial role in countless biological processes, and one of their most important contributions is undoubtedly represented by Ca2+ signaling, a complex network of extra- and intracellular messenger systems that mediates a wide range of pathways (Rimessi et al. 2020). The characterization of the complex network involving Ca2+ signaling has been in progress for approximately 140 years since the first experiments examining the contraction of isolated rat hearts (Ringer 1883). Since then, extensive progress has been made in understanding the numerous molecular pathways involved, although many aspects are still being debated and still need to be defined. Evolutionarily, cells have developed systems to constantly maintain Ca2+ concentrations at very low background levels to avoid the precipitation of phosphate salts, making this ion the logical choice for the exchange of signals (Carafoli and Krebs 2016). The crucial role of Ca2+ in cell biology results from the ability of cells to shape Ca2+ signals in the dimensions of space, time, and amplitude (Alonso et al. 2009).
Ca2+ enters cells through an assortment of Ca2+-permeable channels that respond to different stimuli or acts as a second messenger, e.g., in the phosphoinositol signaling pathway, in which inositol trisphosphate (IP3) binds to Ca2+ channels on the endoplasmic reticulum (ER), transporting Ca2+ into the cytoplasm. Once in the cell, the effects of Ca2+ can be mediated by direct binding to its effectors, such as the phosphatase calcineurin, or indirectly by activating the ubiquitous Ca2+-binding protein calmodulin, leading to the regulation of target molecules such as the Ca2+/calmodulin-dependent kinases CaMKII and CaMKIV (Kerkhofs et al. 2017). Temporally and spatially defined Ca2+ changes in the cytoplasm or in well-defined organelles represent a highly versatile intracellular signal capable of regulating many different processes, including depolarization, hormonal secretion, contraction of smooth and striated muscles, and cellular replication and activation of cytoplasmic, mitochondrial, and nuclear enzymes (Giorgi et al. 2018a).
Proteins that participate in Ca2+ signaling are mostly ubiquitous, but their distribution is highly tissue-specific (Berridge et al. 2003). Cells that need rapid Ca2+ signals, such as myocytes, express many voltage-activated calcium channels to allow quick Ca2+ entry through the plasma membrane, which then, via ryanodine receptors (RyRs) on the sarcoplasmic reticulum, triggers further calcium release. However, nonexcitable cells display calcium oscillations that last for tens of seconds and preferentially use the phosphoinositol signaling pathway to control gene expression and metabolism (Cui et al. 2017).
Therefore, a lack of Ca2+ ions can lead to various issues, and excess Ca2+ ions have harmful effects. Indeed, a sustained rise in intracellular Ca2+ is considered the initial step of irreversible cellular injury, mediated by the activation of the intracellular self-destructive lysosomal enzymes responsible for breakdown of subcellular organelle membranes and increases in oxidative stress and for the hyperactivation of phospholipases and endonucleases, which, through DNA damage, participate in apoptosis (Danese et al. 2017). Intracellular Ca2+ signals are controlled by Ca2+ influx through the plasma membrane (PM) and Ca2+ release from intracellular stores, mainly the ER and Golgi. Intracellular Ca2+ stores are constantly refilled while cytosolic Ca2+ is extruded from the cell by the plasma membrane Ca2+ ATPase (PMCA) pump, to maintain the optimal cytosolic Ca2+ concentration (Marchi et al. 2018).
In the cell, one of the organelles in which changes in [Ca2+] are particularly important is the mitochondrion (Giorgi et al. 2018b), which decodes Ca2+ signals in very sensitive and specific inputs that regulate metabolism, energy production, autophagy, and apoptosis (Giorgi et al. 2018a).
Membrane juxtaposition of both the mitochondria and the ER leads to the highly specialized MAMs compartment, which can be defined as areas of close organelle apposition but that are biochemically distinct from pure mitochondria and pure ER (Morciano et al. 2018). These contact sites are part of abundant heterotypic contacts, which, especially in recent years, have been well characterized and which include the ER-plasma membrane, ER-Golgi, lipid droplets–peroxisomes, mitochondria-lipid droplets, mitochondria–vacuoles/endosomes/lysosomes, ER-lipid droplets, mitochondria-plasma membrane, mitochondria–peroxisomes, ER-lipid droplets, and mitochondrial inner and outer membranes (Eisenberg-Bord et al. 2016).
To witness the strong tethering between the ER and mitochondria, an isolated MAM fraction contains membrane fragments of the outer mitochondrial membrane, the ER, and some plasma membrane proteins (Poston et al. 2013). Tomography analysis has revealed the morphology of these ER-mitochondria-connecting tethers (Csordas et al. 2006). The maintenance of this delicate structural juxtaposition results strategic for the regulation of a huge number of biological processes, essentially through Ca2+ exchange. Poston et al. reported that there are around 1,000 molecular components of the MAMs fraction (Poston et al. 2013) and their study led to an elucidation of the multiple roles played by this particular subcellular compartment. In particular, MAMs co-regulate and influence Ca2+ signaling/dynamics, synthesis/transport of lipids and lipid intermediates, autophagy, apoptosis, and energy metabolism.
Noteworthy is the fact that MAM structures are sensitive to physiological cell conditions and this reflects in a transient and highly variable MAM composition. The length of ER-mitochondria tethers is a determining factor, critical for an efficient Ca2+ transfer, and an ER-mitochondria physical distance modulation is a condition found in different pathophysiological situations. About that, these two organelles’ interplay is also involved in mitochondrial shape and size, and MAM-regulated mitochondrial fusion/fission process undoubtedly covers a crucial role in governing mitochondrial dynamics. Dynamin-related protein 1 (Drp1) is responsible for mitochondrial fission; following its activation, Drp1 translocates from the cytosol to the mitochondria and oligomerizes and constricts this organelle until its division is achieved. Focusing on mitochondrial fusion, mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) are responsible for the outer membrane fusion, while optic atrophy 1 (Opa1) mediates mitochondrial inner membrane fusion (Ponte et al. 2020).
MAMs are enriched in channels involved in calcium transfer, allowing perfect and synergistic signaling between the ER and mitochondria. Moreover, MAMs target many proteins with oncogenic/oncosuppressive functions that modulate cell signaling pathways involved in physiopathological processes (Danese et al. 2017).
As Ca2+ signaling-governed processes (such as energy production, metabolism, autophagy, and apoptosis) are dysregulated in cancer cells and play a key role in Ca2+ transfer and signaling in MAMs, the perturbation of these Ca2+ transport systems at the ER and the mitochondria in relation to tumor onset and progression has become a very hot topic, especially in recent times. In fact, the recent characterization of the many oncogenes and tumor suppressors residing at the MAMs has led many research groups to elucidate how these proteins mediate their functions by altering ER-mitochondrial Ca2+ transfer, thereby promoting or preventing cancer cell survival. Increases or decreases in calcium exchange through the MAMs interface can either exert protumorigenic effects (such as promoting metastatic transformations) or antitumorigenic effects (such as restoring sensitivity to apoptosis) in a cancer type- and cancer state-specific manner (Kerkhofs et al. 2018).
The aim of this review is to clarify how the perturbation of Ca2+ signaling at the ER-mitochondria interface can play a double-sided role in tumor pathology and progression. Modulation of calcium signaling at the MAMs, highly dynamic signaling hubs, could therefore represent a good therapeutic strategy in the future.
2 MAM-Localized Ca2+ Signaling Modulators in Cancer: Channels and Receptors
Ca2+ signaling represents an important tool that regulates many physiological cellular events from proliferation to cell death. Given the pivotal role it plays in such events, it is understandable why, over the past decades, remodeling of its shape has been demonstrated to be involved in the onset of many pathological conditions, such as tumor progression (Monteith et al. 2012; Prevarskaya et al. 2014; Marchi et al. 2020). Proteins involved in the maintenance of Ca2+ homeostasis consist of pumps, exchangers, and channels and have been described as part of the Ca2+ signaling “toolkit” (Berridge et al. 2003).
In resting conditions, the free cytosolic Ca2+ concentration is much lower than that in most extracellular fluids, and an ion concentration gradient is generated. Thus, when Ca2+-permeable ion channels in the plasma membrane are open, Ca2+ flux into the cell increases (Carafoli 2002). However, as already mentioned, Ca2+ signaling can be generated by both external and internal cellular sources.
In the cell, the main ion reservoir from which Ca2+ can be transferred is the endoplasmic reticulum. On the one hand, the ER is the primary cell Ca2+ store; on the other hand, the main cellular Ca2+ signaling translators are the mitochondria.
Indeed, depletion of luminal ER Ca2+ levels is followed by a rapid increase in ion mitochondrial concentration. To ensure this interaction is effective, the ER and the mitochondria are juxtaposed on the MAMs at a short distance of approximately 10–25 nm (Csordas et al. 2006; Rizzuto et al. 1998; Marchi et al. 2014) in the smooth ER and at approximately 50 nm in the rough ER (Wang et al. 2015; Giacomello and Pellegrini 2016).
2.1 ER Side
Many ER-resident proteins involved in Ca2+ transfer have been found at the MAMs: the sarco-/endoplasmic reticulum Ca2+ ATPase (SERCA) and inositol 1,4,5-trisphosphate receptors (IP3R), among others. SERCAs are members of the P-type ATPase superfamily of primary active transporters (a large family of membrane-embedded pumps (Wang et al. 2015)) and can maintain the correct cytosolic and reticular Ca2+ concentrations.
The 110 kDa SERCA protein has 10 helix intramembrane domains involved in the interaction with two Ca2+ ions transferred to the ER lumen at the expense of adenosine triphosphate (ATP). The Ca2+ flux is coupled to the exchange of two to three protons moved to the cytoplasm (Palmgren and Nissen 2011). In addition to transmembrane domains, SERCA has three cytoplasmic regions: the nucleotide-binding domain (N), designed for ATP binding; the phosphorylation (P) domain, which hosts the amino acid residue phosphorylated by ATP; and the actuator (A) domain at the N-terminus, which controls enzyme dephosphorylation. During ATP hydrolysis, conformational changes in the protein domains occur, and as consequence, the intermembrane domains warp, enabling Ca2+ transport (Toyoshima et al. 2000; Moller et al. 2010).
To date, at least 12 isoforms of SERCA (SERCA1a-b, SERCA2a-d, SERCA3a-f) have been identified in vertebrates (Lipskaia et al. 2014), each characterized by tissue and developmental specificity. This diversity is because SERCAs are encoded by three different genes located on three chromosomes (ATP2A1, ATP2A2, and ATP2A3), each generating alternative splicing variants that differ mainly in the C-terminus of the protein.
The diversities in the coding sequencing of these proteins do not affect the protein tertiary structures, which are highly conserved among all isoforms, but instead lead to differences in Ca2+ affinity. Among all these proteins, ubiquitous SERCA2b is the isoform with the highest Ca2+ affinity and plays a crucial role in the regulation of ER Ca2+ uptake and Ca2+ homeostasis (Vandecaetsbeek et al. 2009). All SERCA isoforms are present along the entire ER membrane and are not particularly enriched in MAMs.
SERCA activity can be modulated by many proteins. Among them, the recently identified ER-luminal protein disulfide isomerase thioredoxin-related transmembrane protein 1 (TMX1) displays palmitoylation-dependent MAMs localization. TMX1 can directly interact with SERCA2b (Gutierrez and Simmen 2018; Lynes et al. 2012) and inhibit its activity, reducing Ca2+ transfer.
If SERCA activity is lowered by TMX1, its activity is enhanced by the redox active form of the redox-sensitive selenoprotein N (SEPN1) (Gutierrez and Simmen 2018). MAMs result particularly enriched in redox regulatory proteins, and TMX1 and SEPN1 are among them (Krols et al. 2016; Marino et al. 2015).
Calnexin is a chaperone protein that localizes at the ER-mitochondrial contact sites in a palmitoylation-dependent manner (Lynes et al. 2012). The primary function of this protein is to interact with misfolded proteins to improve the folding efficiency of ER proteins (Lamriben et al. 2016). Upon palmitoylation, calnexin moves to the MAMs, where it interacts with SERCA2b, increasing Ca2+ transfer from the cytosol to the ER (Lynes et al. 2013). Interestingly, the modulation of SERCA2b activity by calnexin is counteracted by TMX1 in a way that may suggest competition for the same binding site (Krols et al. 2016; Raturi et al. 2016).
IP3Rs are large-conductance nonselective cation channels that together with the RyRs, which is mainly expressed in sarcoplasmic reticulum, are major structures through which Ca2+ exits the ER (Ashby and Tepikin 2001).
IP3R channels are homo- or heterotetramers composed of four subunits of approximately 300 kDa each. The molecular structure of the IP3R monomer, determined by cryogenic electron microscopy, consists of three structural domains: an N-terminal ligand-binding domain, containing both the IP3-binding core and the suppressor region, a central modulatory domain, and a Ca2+ channel region at the C-terminus containing six intramembrane helices. The C-tails interact directly with the N-terminal domains of the other subunits (Fan et al. 2015).
In vertebrates, there are three different isoforms of IP3R (IP3R1, IP3R2, and IP3R3) encoded by three genes (ITPPR1, ITPR2, and ITPR3, in humans). Despite the high homology in the amino acid sequences (60–80%), these isoforms have a different expression pattern, with IP3R1 mainly expressed in neuronal cells, IP3R2 in muscle and liver cells, and ubiquitous IP3R3 in most cultured cells (Mikoshiba 2007; Foskett et al. 2007). In addition, the different isoforms show differences in ligand-binding sensitivity and regulation by Ca2+ and ATP (Newton et al. 1994; Miyakawa et al. 1999; Tu et al. 2005; Khan et al. 2006; Betzenhauser et al. 2008; Wagner 2nd et al. 2008; Vervloessem et al. 2015).
IP3Rs are enriched at MAMs levels, where they also exert a structural role, being in close proximity with the mitochondrial voltage-dependent anion channel 1 (VDAC1) and by interacting with the chaperone glucose-regulated protein GRP75 which acts as a tether between the two proteins and organelles (Bernard-Marissal et al. 2018). It has also been recently highlighted that IP3R isoforms differently regulate ER-mitochondrial contacts and local calcium transfer, proving that IP3Rs structural role in MAM compartment is crucial (Bartok et al. 2019).
The activity of IP3R receptors is regulated primarily by inositol trisphosphate (IP3), released at the plasma membrane after the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) by phospholipase C (PLC).
However, IP3Rs can also be modulated by ATP, post-translational modification (Mak and Foskett 2015; Bansaghi et al. 2014; Yule et al. 2010; Prole and Taylor 2016; Ivanova et al. 2014; Ramos-Franco et al. 1998), and Ca2+ ions, which act both from the luminal ER side, increasing the sensitivity to its ligand, and from the cytoplasmatic sides from which Ca2+ plays a dual role as an activator at low concentrations and an inhibitor if its concentration is higher than 300 nM (Table 1).
As noted earlier, there is a juxtaposition between the two MAM-forming organelles, and Ca2+ release from the ER is followed by uptake at the mitochondrial interface.
2.2 Mitochondrial Side
After being released from the ER, Ca2+ ions can first cross the outer mitochondrial membrane through VDAC and, once in the mitochondrial intramembrane space, enter the matrix through the mitochondrial Ca2+ uniporter (MCU).
VDAC is a 30-kDa protein existing in all eukaryotic cells in three different isoforms: VDAC1 and VDAC2 are expressed in most mammals, and VDAC3 is the isoform with the lowest expression (De Pinto et al. 2010; Huang et al. 2014; Maldonado et al. 2013). VDAC is the most abundant outer mitochondrial membrane protein, and due to its permeability not only to anions but also to respiratory substrates, ATP, reactive oxygen species (ROS), and cytochrome C can be considered master regulators of mitochondrial bioenergetics (Shoshan-Barmatz et al. 2010; Weisthal et al. 2014). The permeability of this channel is highly impacted by its two conformational states, opened and closed, since in the closed state, the channel is permeable only to small ions but not to anionic metabolites (Shoshan-Barmatz et al. 2010; Gincel et al. 2000; Schein et al. 1976). The switch between the opened and closed states is regulated by many factors, including Bcl2 family members (Tsujimoto and Shimizu 2000), Ca2+ ions (Bathori et al. 2006), and voltage. Indeed, high mitochondrial voltages induce VDAC to close (Gincel et al. 2000) in a N-terminus-mediated manner (Abu-Hamad et al. 2009).
Among VDAC channels, the most frequently expressed and consequently studied isoform is VDAC1 (Messina et al. 2012), which has been shown to be targeted to the MAMs (Hajnoczky et al. 2002; Shoshan-Barmatz and Gincel 2003; Colombini 2012) and to regulate the Ca2+ flux through the mitochondria outer membrane (Rapizzi et al. 2002). If regulation of mitochondrial Ca2+ signaling is not a unique feature of VDAC1, the ability to transmit proapoptotic stimuli to the mitochondria seems to be an exclusive characteristic of this isoform (De Stefani et al. 2012).
To reach the mitochondrial matrix and regulate all the previously mentioned processes, Ca2+ entering the outer mitochondrial membrane has to permeate the inner mitochondrial membrane that, unlike the outer membrane, is not permeable to ions. The accumulation of Ca2+ inside the mitochondrial matrix follows an electrogenic gradient and is driven by the low Ca2+ affinity uniporter complex MCU. Due to the low Ca2+ affinity of this MCU complex, the rapid mitochondrial ion accumulation is difficult to explain without considering the presence of close contacts between the ER and the mitochondria, which create microdomains with a high Ca2+ concentration (Rizzuto et al. 1998).
MCU is a complex of approximately 480 kDa composed of the channel-forming subunits MCUa and MCUb, organized mainly in pentamers. MCUa and MCUb have opposite effects on Ca2+ ion transfer (allowing and inhibiting permeation, respectively), and their relative quantities in the complex regulate the Ca2+ transport capability of MCU itself. In addition to the channel-forming subunits, mitochondrial calcium uptake 1 and 2 (MICU1 and MICU2) and the essential MICU regulator (EMRE) are part of the uniporter complex and play a pivotal role in regulating the integrity of the complex itself (De Stefani et al. 2015; Oxenoid et al. 2016; Raffaello et al. 2013; Sancak et al. 2013). MCU complexes were enriched in MAMs, positioned more to the mitochondrial periphery, indicating high accessibility to cytoplasm-derived Ca2+ inputs (Marchi et al. 2017).
Among the mitochondrial Ca2+ uptake family of regulator proteins MICU1 and MICU2, the best characterized is MICU1, which functions as a gatekeeper that can sense the Ca2+ levels of the intermembrane space. Indeed, at low concentrations, the gate is closed, but as soon as the Ca2+ levels pass the [Ca2+] threshold of 700 nM for MICU1-MICU2 heterodimers and 300 nM for MICU1 homodimers, the Ca2+-binding EF hands of MICU1 bind the ion and undergo a conformational change that opens the channel (Csordas et al. 2013; Mallilankaraman et al. 2012a; Perocchi et al. 2010; Petrungaro et al. 2015; Park et al. 2020) (Table 1).
3 Decrease in ER-Mitochondria Ca2+ Crosstalk
3.1 Dysfunctional ER-Ca2+ Release
As described in the introductory section, in recent years, increasing evidence has shown that organelles communicate with each other through Ca2+ signaling. In particular, at the MAMs level, interorganellar Ca2+ signaling is profoundly spatiotemporally regulated. Interestingly, in the tumor setting, an alteration of Ca2+ signaling has been shown to affect malignant transformation and tumor progression through the control of cell death programs and metabolism (Rimessi et al. 2020; Monteith et al. 2007).
In this context, the ER not only plays a decisive role in Ca2+ signaling but also guarantees a control system for correct protein folding and stress sensing. Alterations in ER homeostasis, including substantial Ca2+ depletion, are associated with the pathophysiology of many diseases, including cancer (Mekahli et al. 2011).
The normal Ca2+ exchange between the ER and the mitochondria requires adequate filling of the ER Ca2+ stores. Thus, decreasing the ER Ca2+ levels will compromise ER-mitochondrial Ca2+ transfer. As a consequence, changes in the ER Ca2+ store content affect the Ca2+ efflux from the ER to the mitochondria and ultimately cell survival (Ivanova et al. 2017).
The maintenance of physiological low levels of mitochondrial Ca2+ uptake by IP3R is crucial to preserve cellular bioenergetics in normal and cancer cells by enabling the dehydrogenase activation of the tricarboxylic acid (TCA) cycle, strong ATP production and metabolic intermediates for the generation of building blocks, allowing the cells to enter the cell cycle and proliferate. In breast cancer cells but not in normal cells, Ca2+ release suppression mediated by the inhibition of IP3R activity caused cell death through a deregulated autophagic mechanism (Singh et al. 2017a) and mitotic disruption, as reported by Cárdenas C. et al. (2016).
Regarding type 3 IP3R, the depletion of IP3R3 or its pharmacological blocking increased the level of the autophagic marker microtubule-associated protein 1A/1B-light chain 3 (LC3)-II through the upregulation of autophagic protein 5 (Atg5) and ROS generation, leading to the blockage of tumor growth in a mouse model of breast cancer (Singh et al. 2017a). This finding is correlated with the high expression of IP3R3 in human malignant tissues and high concentrations of metabolites in the serum of patients (Singh et al. 2017b).
Moreover, it has been reported that the inhibition of IP3R with caffeine, a nonspecific inhibitor of these receptors, leads to a decreased migration of glioblastoma cells and a substantially increased mean survival in a mouse glioblastoma xenograft model (Kang et al. 2010). In the Caco-2 colon cancer cell line, IP3R3 silencing, or nonspecific pharmacological inhibition by 2-APB in gastric cancer cells, induced apoptosis, while overexpression protected cells from staurosporine-induced apoptotic death (Shibao et al. 2010).
Interestingly, various MAM-located oncosuppressors and oncogenes have been reported to interact with IP3Rs, including the oncogene protein kinase B (PKB), also known as Akt, promyelocytic leukemia protein (PML), BRCA1 associated protein 1 (BAP1), phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and B-cell lymphoma 2 (Bcl-2) family proteins, modifying the Ca2+ release patterns and cell fate (Bononi et al. 2017; Akl and Bultynck 2013; Missiroli et al. 2017; Kuchay et al. 2017; Giorgi et al. 2010). Although the aforementioned proteins are all present at the ER-mitochondria interface, only PTEN and PML are particularly enriched on MAMs (Missiroli et al. 2016; Bononi et al. 2013).
Akt, as well as protein kinase C (PKC) isozymes (Pinton et al. 2004), is a key player in regulating multiple signaling pathways through calcium signaling tuning, such as cell metabolism, cell proliferation, and survival (Szado et al. 2008). Notably, in human breast cancers, the phosphoinositide 3-kinase (PI3K)/Akt/mTOR pathway is frequently dysregulated (Gonzalez-Angulo et al. 2011; Stemke-Hale et al. 2008).
On the ER side, IP3R Akt-mediated phosphorylation results in a decreased magnitude of Ca2+ release and, as a result, reduced mitochondrial Ca2+ uptake. Moreover, this decrease in Ca2+ flux protected glioblastoma cell lines from the effects of apoptotic stimuli (Szado et al. 2008).
In 2012, our group demonstrated that Akt specifically phosphorylates type 3 IP3R, leading to diminished mitochondrial Ca2+ influx and, consequently, protecting cells from apoptosis (Marchi et al. 2012).
PML tumor suppressor protein has been implicated in diverse cellular processes ranging from tumor suppression to defense against virus infection (Bernardi and Pandolfi 2007; Everett and Chelbi-Alix 2007; Hsu and Kao 2018; Pinton et al. 2011). An extranuclear fraction of PML has been demonstrated to be targeted to the MAMs in a p53-dependent manner (Missiroli et al. 2016) and to form a multicomplex with type 3 IP3R, the serine threonine kinase Akt and protein phosphatase 2A (PP2A) (Giorgi et al. 2010).
It has been shown that PML regulates the phosphorylation of IP3R by controlling the activity of Akt through the recruitment of the PP2A phosphatase at the MAMs interface. Hence, PML can coordinate Ca2+ mobilization into the mitochondria, which then triggers the cell death program. Conversely, in the absence of PML, PP2A does not assemble with IP3R and Akt, resulting in a higher activation of Akt (phospho-Akt). Once activated, Akt can hyperphosphorylate IP3R, thereby suppressing ER Ca2+ release to the mitochondria (Giorgi et al. 2011).
BAP1 is a member of the ubiquitin C-terminal hydrolase (UCH) subfamily of deubiquitylating enzymes and has tumor suppressor activity, which has been mainly correlated with its nuclear localization (Lee et al. 2014; Ismail et al. 2014). When BAP1 localizes to the ER, it binds, deubiquitylates, and stabilizes the activity of the IP3R3 channel, modulating Ca2+ release from the ER to the cytosol and then to the mitochondria, promoting apoptosis. In BAP1+/− carriers, the reduced level of BAP1 resulted in a diminished IP3R3 quote with a subsequent Ca2+ transfer decrease from the ER to the mitochondria. This event caused a reduced propensity of BAP1+/− cells to undergo apoptosis following DNA damage induced by asbestos or UV light (Bononi et al. 2017).
PTEN is another Ca2+-related tumor suppressor that has been shown to be mutated or suppressed in many tumors (Salmena et al. 2008). Bononi et al. demonstrated that a fraction of cellular PTEN is localized at the MAMs, where it interacts with IP3R3, antagonizing its Akt-mediated phosphorylation and enhancing Ca2+ transfer from the ER to mitochondria. In this way, it reestablishes cellular sensitivity to Ca2+-mediated proapoptotic stimuli. Conversely, PTEN knockdown reduced the Ca2+ release from the ER and decreased mitochondrial Ca2+ transients, thus preventing cell death activation (Bononi et al. 2013). Moreover, a novel role for PTEN has been proposed; it can compete with F-box and leucine-rich repeat protein 2 (FBXL2), an E3-ubiquitin ligase F-box protein, for binding to IP3R3 to prevent its degradation. It has been demonstrated that FBXL2 degradation of IP3R3 is enhanced in cancer cells in which PTEN expression is lowered, thereby resulting in the inhibition of apoptosis (Kuchay et al. 2017).
The Bcl-2 family of anti- and proapoptotic proteins is predominantly localized to the mitochondria, ER, and MAMs, and their activities strongly reflect their intracellular localization (Morciano et al. 2018). Bcl-2 is a proto-oncogene known for its involvement in inhibition of apoptosis through its interaction with the proapoptotic proteins BCL2 associated X protein (Bax) and Bcl-2 homologous antagonist/killer (Bak) (Rimessi et al. 2020). Indeed, at the ER, Bcl-2 prevents excessive Ca2+ flux by directly targeting all three IP3R receptor isoforms, which would lead to mitochondrial Ca2+ overload and opening of the permeability transition pore (mPTP) (Chen et al. 2015; Bonora et al. 2017). Dysregulation of Bcl-2 expression has been highlighted in various cancers, including hematopoietic, lung, breast, and prostate tumors (Morciano et al. 2018).
Bcl-XL is another antiapoptotic member of the same family that is frequently overexpressed in many tumors, such as multiple myeloma, melanoma, glioblastoma, prostate cancer, colorectal cancer, non-small cell lung cancer, and pancreatic cancers (Trisciuoglio et al. 2017; Scherr et al. 2016; Zhang et al. 2014; Yoshimine et al. 2013). This protein is localized at the MAMs (Monaco et al. 2015), where it directly binds the IP3R channels, regulating IP3R-related Ca2+ release. Bcl-XL caused a strong sensitization of IP3R, promoting prosurvival Ca2+ oscillations (White et al. 2005).
Among the antiapoptotic proteins of the Bcl-2 family, myeloid cell leukemia 1 (Mcl-1) also lowers the calcium ER store content by stimulating IP3Rs outside of the MAMs, thereby increasing Ca2+ leakage from the ER, resulting in a decline in the basal ER Ca2+ levels (Eckenrode et al. 2010). In the presence of low [IP3], in Mcl-1-expressing cells, store depletion becomes more prominent, indicating that the sensitivity of IP3-dependent Ca2+ release is enhanced by Mcl-1. Mcl-1-mediated IP3R sensitization also contributes to low-level IP3R-mediated Ca2+ signaling from the ER to the mitochondria and thereby stimulates mitochondrial bioenergetics (Bittremieux et al. 2016).
At the MAMs, oncogenic H-Ras also affects Ca2+ transfer to the mitochondria to promote evasion from the apoptotic cascade (Rimessi et al. 2014). In colorectal cancer cells, oncogenic K-Ras modified the expression of IP3Rs, weakening the Ca2+ release from the ER to allow cells to escape Ca2+-mediated apoptosis (Pierro et al. 2014). Indeed, Ras-driven mitochondrial dysfunction causes metabolic and redox changes that favor tumorigenesis (Hu et al. 2012). Hence, proper maintenance of IP3R3 protein levels is crucial for preventing oncogenesis by strengthening tumor-suppressive ER-mitochondrial Ca2+ transfer.
Furthermore, MAMs are a molecular platform for the regulation of many oxidoreductases. In this context, endoplasmic reticulum oxidoreductin 1-α (ERO1-α) activity is broadly investigated for its enrichment at ER-mitochondria contact sites (Anelli et al. 2012) and its high expression in different tumor types (Kakihana et al. 2012). This oxidase impacts ER-Ca2+ storage and IP3-dependent fluxes. During ER stress, ERO1-α oxidizes type 1 IP3R, promoting the release of Ca2+ from the ER (Anelli et al. 2012). Furthermore, endoplasmic reticulum resident protein 44 (ERp44) (an ER luminal chaperone protein) binds to IP3R1 and inhibits its channel activity under reducing conditions, resulting in the blockade of Ca2+ transfer to the mitochondria (Higo et al. 2005). Oxidation of IP3R1 by ERO1-α causes the dissociation of ERp44, thus leading to the activation of Ca2+ release via IP3R1 (Li et al. 2009). ERO1-α silencing has been demonstrated to profoundly affect mitochondrial Ca2+ uptake, likely modifying MCU activity. Thus, ERO1-α links redox and Ca2+ homeostasis in MAMs (Anelli et al. 2012).
Recently, the oncogenic transcription factor signal transducer and activator of transcription 3 (STAT3), which mediates the signaling of cytokines, growth factors, and oncogenes (Yu et al. 2014), has been shown to localize only to MAMs (Su et al. 2020). At this location, it modulates ER-mitochondria Ca2+ release by interacting with the IP3R3 channel and promoting its degradation, resulting in greater cellular resistance to apoptotic stimuli (Avalle et al. 2019). In breast cancer cell lines, silencing STAT3 enhances the ER Ca2+ release and sensitivity to apoptosis following oxidative stress, correlating with increased IP3R3 levels. This evidence suggests that STAT3-mediated IP3R3 downregulation in the ER crucially contributes to its antiapoptotic functions via Ca2+ flux modulation.
Together with the IP3R receptors, RyRs and SERCA are the major Ca2+ players in the ER (Berridge 2012). In general, RyRs regulate melanocyte and T cell proliferation (Hakamata et al. 1994; Kang et al. 2000) and astrocyte migration (Matyash et al. 2002). Ryanodine receptor type 2 (RyR2), a member of the RyR family, controls the Ca2+ release from the sarcoplasmic reticulum into the cytosol (Ding et al. 2017). Different studies have confirmed the association of RyR2 with several cancer types, including melanoma (Carpi et al. 2018), breast cancer (Lu et al. 2017), lymphoma (McCarthy et al. 2003), and prostate cancer (Mariot et al. 2000). Recently, it has been reported that RyR2 is downregulated in thyroid carcinoma tissues and that low expression levels of RyR2 are closely associated with poor prognosis in thyroid carcinoma patients (Xu et al. 2019).
Over the past years, the tumor suppressor p53 has been shown to be altered in many human cancer tissues, including colon, breast, lung, brain, bladder, pancreatic, stomach, and esophageal cancer (Vogelstein et al. 2000). Some of p53 fraction is located at the MAMs, where it directly binds to the SERCA pump, changing its oxidative state and thus leading to an increased Ca2+ load, followed by an enhanced flux to the mitochondria. Consequently, during apoptotic stimulation, more Ca2+ can be released from the ER into the mitochondria, enhancing mitochondrial Ca2+ overload, opening of the mitochondrial mPTP, release of caspase cofactors, and ultimately induction of the intrinsic apoptosis pathway (Morciano et al. 2018). Dysregulation of p53-dependent Ca2+ homeostasis led to reduced ER Ca2+ release, resulting in a low responsiveness to apoptotic stimulation (Giorgi et al. 2015).
We must also note the phosphofurin acid cluster sorting 2 protein (PACS-2) and PKR-like ER kinase (PERK). PACS-2 is a multifunctional protein involved in retrograde ER-Golgi trafficking of multiple proteins (Youker et al. 2009). Although it is unclear whether a direct interaction of PACS-2 at the MAMs occurs, it was demonstrated that depletion of PACS-2 reduces mitochondrial-ER contact sites and mediates apoptosis (Simmen et al. 2005). PACS-2 was also demonstrated to be a fundamental player in rapamycin complex 2 (mTORC2)-dependent regulation of MAMs integrity (Betz et al. 2013). PERK is a protein kinase that, together with inositol-requiring enzyme 1 (IRE1) and transcription factor 6 (ATF6), acts as an ER stress sensor from the ER membrane, controlling UPR functioning. The function of this protein in the MAMs is independent of its role as an ER stress sensor and transcriptional regulator of redox homeostasis. Indeed, PERK maintains, through its cytoplasmic domains, the juxtaposition of the ER and the mitochondria, acting as a structural tether and permitting the transmission of ROS-mediated signals (Verfaillie et al. 2012).
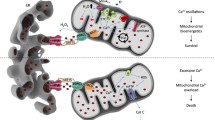
In conclusion, changes in the ER Ca2+-store content would perturb Ca2+ transfer from the ER to the mitochondria and ultimately influence cell death or survival. A reduction in intracellular store Ca2+ release is certainly the main mechanism adopted by cancer cells to escape mitochondria-mediated apoptosis (Fig. 1).

Downregulation of MAMs Ca2+ crosstalk in cancer: graphical representation of the calcium signaling regulators involved in a cancer-related decreased Ca2+ crosstalk state. See text for further details. Ca2+, calcium; ER, endoplasmic reticulum
3.2 Perturbed Mitochondrial Ca2+ Uptake
Cancer-derived modifications in cellular physiology could be related to impairment of the Ca2+ signaling network, which is frequently associated with the dysregulation of several Ca2+ channels and pumps (Prevarskaya et al. 2014; Hanahan and Weinberg 2000).
In addition to limiting the excessive release of Ca2+ from the ER, cancer cells can effectively prevent mitochondrial Ca2+ overload by limiting mitochondrial Ca2+ uptake.
Among the proteins responsible for limitation of mitochondrial calcium influx are Bcl-2 and Bcl-XL, the antiapoptotic Bcl-2-family proteins discussed in the previous paragraph; Bcl-2 and Bcl-XL are partially localized at the mitochondrial outer membrane and, similar to other antiapoptotic proteins, are frequently upregulated in cancer; these proteins can regulate mitochondrial Ca2+ uptake through VDAC1 (Shoshan-Barmatz et al. 2010).
Considering that VDAC1 is involved in death and cell survival, it is not surprising that this channel could be a target for Bcl-2 family proteins (De Stefani et al. 2012). These proteins target the N-terminal region of VDAC1 (Abu-Hamad et al. 2009; Arbel and Shoshan-Barmatz 2010), and it has been demonstrated that only the Bcl-XL BH4 domain is essential to bind VDAC1 and inhibit cell death (Monaco et al. 2015). Several studies demonstrated that the interaction between Bcl-XL and VDAC1 suppresses proapoptotic Ca2+ uptake, preventing the dissipation of the mitochondrial potential and the release of cytochrome c and apoptosis-inducing factor (AIF) through the outer membrane.
Indeed, studies concerning mitochondrial Ca2+ uptake that compare Bcl-XL-overexpressing versus Bcl-XL-deficient cells have demonstrated that this protein may be involved in MAMs microdomain reorganization and results in an alteration of the capacity of mitochondrial Ca2+ uptake, proving that Bcl-XL inhibits VDAC1 (Monaco et al. 2015; Bittremieux et al. 2016; Shimizu et al. 2000; Li et al. 2008).
Nevertheless, VDAC1 in hepatocarcinoma tissues can be downregulated by the small noncoding RNA miR-7, influencing tumor proliferation and metastasis (Chaudhuri et al. 2016a; Bargaje et al. 2012). Chaudhuri et al. showed that in human neuroblastoma cells and in mouse primary cortical neurons, miR-7 can reduce VDAC1 expression, with consequent inhibition of mitochondrial Ca2+ uptake, membrane depolarization, mitochondrial fragmentation, cytochrome c release, and ROS production, promoting cancer cell survival (Chaudhuri et al. 2016a).
MCU allows calcium ion permeation into the mitochondrial matrix, and its overexpression leads to an increase in mitochondrial Ca2+ entry and ROS production, influencing the migration, invasion, and size of different tumor types (Yu et al. 2017; Tang et al. 2015; Wang et al. 2007). However, a reduction in MCU expression decreases mitochondrial Ca2+ uptake, the opening of the mPTP and the release of proapoptotic factors, thus having a protective effect on apoptosis (Marchi et al. 2019b; Sebag et al. 2018; Oropeza-Almazan et al. 2017; Yuan et al. 2017; Liao et al. 2015; Qiu et al. 2013; Penston and Wormsley 1986).
Marchi et al. showed that, through MCU downregulation, the miR-25 MCU-targeting microRNA could perturb Ca2+ homeostasis, reducing the concentration of mitochondrial Ca2+ levels in HeLa cells. However, high levels of miR-25 have been observed both in prostate and colon cancer. The miR-25-dependent reduction in mitochondrial Ca2+ uptake correlates with resistance to proapoptotic stimuli and can be reversed by anti-miR-25 overexpression. Treatment with anti-miR-25 can restore the MCU expression levels and reverse the pathophysiology, thus suggesting a novel therapeutic target for prostate and colon cancer (Marchi et al. 2013).
One gene that is frequently deleted in many human cancers, principally in those caused by environmental carcinogens, is fragile histidine triad (FHIT). Consequently, its product, the Fhit protein, is absent or reduced in most cancers (Huebner and Croce 2003). The Fhit protein is localized in the mitochondria and the cytosol and acts as a tumor suppressor, increasing susceptibility to apoptosis (Siprashvili et al. 1997). Reintroduction of Fhit to the highly carcinogen-susceptible Fhit−/− mouse model reduced tumor sizes by activating apoptotic cell death (Zanesi et al. 2005). The Fhit protein generates ROS and enhances mitochondrial Ca2+ uptake by increasing mitochondrial Ca2+ hotspots. Therefore, Fhit acts as a tumor suppressor by modulating MCU opening and enhancing the susceptibility of cells to apoptosis, thus potentiating the effect of apoptotic agents (Rimessi et al. 2009).
Transient receptor potential cation channel subfamily C member 3 (TRPC3) belongs to a group of nonselective cation channels that are involved in different cellular mechanisms. TRPC3 channels can influence the mitochondrial membrane potential following their up- and downregulation. The activation of Ca2+-sensitive downstream pathways occurs through the influx of calcium from transient receptor potential channels (TRP channels), which act as apoptotic regulators (Wang et al. 2019; Takahashi et al. 2018; Raphael et al. 2014; Monet et al. 2010). However, Shengjie Feng et al. have shown that a fraction of the TRPC3 protein is localized to the mitochondria and mediates mitochondrial Ca2+ uptake when the cytosolic calcium concentration is elevated. Since, as we previously noted, mitochondrial membrane potential seems to be affected by TRPC3 channels and because mitochondrial Ca2+ uptake is not abolished when MCU expression is downregulated (De Stefani et al. 2011), TRPC3 might be another channel that allows the entry of calcium into the mitochondria, in addition to MCU (Kirichok et al. 2004). In particular, resistance to apoptosis and the proliferation of some tumors could be related to its downregulation, which results in reduced mitochondrial calcium uptake (Feng et al. 2013).
Fetal and adult testis-expressed 1 protein (FATE1) is a 21-kDa protein that belongs to the cancer-testis antigen proteins that are mainly expressed in the testis under physiological conditions and are upregulated in different cancer types (Dong et al. 2003; Whitehurst 2014; Simpson et al. 2005). This molecule, being a member of the mitochondrial fission factor (Miff) protein family, shares some structural similarities with Mff (Gandre-Babbe and van der Bliek 2008). The oncoprotein FATE1, which is located on the mitochondrial outer membrane preferentially in the MAMs compartment, is implicated in the regulation of Ca2+-dependent apoptosis in cancer cells, acting as an anti-tether agent through the modulation of the distance between the ER and the mitochondria (Doghman-Bouguerra et al. 2016), being a direct connection between its increased expression and MAMs morphology in adrenocortical carcinoma (AAC) patients with a poor prognosis (Doghman-Bouguerra et al. 2016). Overexpression of FATE1 in adenoid cystic carcinoma (ACC) was related to a decrease in mitochondrial Ca2+ uptake that confers resistance to proapoptotic stimuli and chemotherapeutic drugs (Doghman-Bouguerra et al. 2016).
In most human cancer types, including head and neck squamous cell carcinoma (HNSCC), high levels of enhancer of zeste homolog 2 (EZH2) have been detected. EZH2 is the enzymatic subunit of the PRC2 complex (polycomb repressive complex 2), which methylates lysine 9 and lysine 27 of histone H3, and is fundamental for transcriptional repression (Kim and Roberts 2016; Schuettengruber et al. 2007; Boyer et al. 2006). EZH2 acts as an oncogene, and its high expression levels are associated with tumor cell proliferation and migration (Zhou et al. 2015a; Ning et al. 2015). Furthermore, it has been shown that inhibition of EZH2 in HNSCC cells in vitro and in vivo induces loss of mitochondrial membrane potential (ΔΨm) with consequent activation of cell death pathways. Inhibition of EZH2 involves accumulation of Ca2+ into the mitochondria, induced by inactivation of MICU1 (Zhou et al. 2015b; Cosentino and Garcia-Saez 2014) (Fig. 1).
4 Upregulation of ER-Mitochondria Ca2+ Crosstalk
4.1 New Insights into Ca2+ Signaling Perturbation in the MAMs
The numerous molecular pathways described thus far all involve a decreased uptake of Ca2+ to the mitochondria, resulting from decreased ER release or mitochondrial defects. Historically, reports that have assessed the remodeling of MAMs Ca2+ signaling associated with tumorigenesis, invasion, and metastasis all led to the conclusion that cancer cells undergo minor mitochondria-dependent apoptosis because of decreases in the Ca2+ release from the ER. Recently, the characterization of new MAM-localized proteins and the finding of new mechanisms of action led the scientific community to consider that even an upregulation of Ca2+ signaling at the MAMs level could be harmful and drive tumor onset and progression. In the following paragraphs, we will describe how this condition, hitherto described as the cause of apoptotic cell death, can lead to the onset and development of tumor diseases.
4.2 Increased ER-Ca2+ Release
The endoplasmic reticulum is an organelle that contains a network of tubules and flattened sacs and is mainly known for its major role in the production, processing, and transport of proteins and lipids. The ER also represents the major intracellular store of Ca2+, an ion that is necessary on its lumen for second-messenger-induced Ca2+ release, the control of capacitative Ca2+ influx, and intra-ER chaperone activities such as polypeptide translocation, protein folding, and ER-associated degradation (Buck et al. 2007). In normal tissue cells, a sustained Ca2+ flux from the ER to the mitochondria can enhance the sensitivity of mitochondria to apoptotic stimuli; however, in some cases, an increase in Ca2+ ion leakage from the ER to the MAMs can promote tumor formation, especially in specific tissues and organs. For ER-mitochondria interorganellar Ca2+ signaling and, in particular, increased ER Ca2+ release, the recent revelation of the mechanisms by which IP3R3 upregulation drives oncogenesis via ER-mitochondrial Ca2+ crosstalk is particularly important. This statement is particularly strong because until last year, IP3R3 was well characterized as a Ca2+-related proapoptotic protein. In fact, the tumor suppressors BAP1 and PTEN have a stabilizing effect on IP3R3 in the ER, promoting susceptibility to cell death (Bononi et al. 2017; Kuchay et al. 2017), and in contrast, the oncogene K-RasG13D downregulates IP3R3, preventing the apoptotic death of cancer cells (Pierro et al. 2014). Three recent works by Guerra et al. (2019), Rezuchova et al. (2019), and Ueasilamongkol et al. (2020), for the first time, have deviated from the idea that IP3Rs only have an anti-oncogenic potential by driving proapoptotic Ca2+ signals to mitochondria but attributed an oncogenic potential to ER-mitochondria Ca2+ crosstalk. In an analysis of tumor tissues, the IP3R3-protein levels were elevated in hepatocellular carcinoma biopsies compared to healthy liver biopsies (Guerra et al. 2019), in clear cell renal cell carcinoma kidney biopsies compared to healthy regions (Rezuchova et al. 2019) and in cholangiocarcinoma cancer biopsies and cancer cell lines compared to normal tissues and normal cholangiocyte cell models (Ueasilamongkol et al. 2020). In all cases, only type 3 IP3Rs were found to be overexpressed in tumor tissues, with no changes or slight downregulation of type 1 and type 2. In particular, IP3R3 seems to be completely absent in normal human hepatocytes but is clearly present in biopsies from individuals with hepatitis B virus, hepatitis C virus (HCV), non-alcoholic fatty liver disease (NAFLD), and alcoholic liver disease (ALD), which are the four most common predisposing factors to the development of hepatocellular carcinoma (Guerra et al. 2019). This increase was more pronounced in the late stages of hepatocellular carcinoma.
Notably, in cholangiocarcinoma cells, most IP3R3 is localized to the MAMs, while in normal cholangiocytes, it resides in the ER subapical pole. In these cells, MAM localization promotes basal respiration by increasing mitochondrial Ca2+ signaling, and thus, depletion of this channel in these cells is deleterious for nuclear and mitochondrial functionality (Ueasilamongkol et al. 2020). In HepG2 cells, IP3R3 upregulation promotes cell death, but its chronic overexpression can increase the resistance of these cells to cell death inducers, enhancing malignant cell survival (Guerra et al. 2019).
The common key in all these cases is the extreme adaptation ability that drives oncogenesis and malignant cell transformation. These cancer cells became addicted to high IP3R3 levels at the MAM compartment for their survival, to maintain sustained cell metabolism and to obtain malignant features such as increased motility, migration, and invasion.
We want to include in this section the already mentioned ERO1-α, an extensively studied protein due to its ability to regulate many processes. ERO1-α is particularly enriched at the ER-mitochondria interface, controlling ER redox homeostasis and oxidative folding and regulating Ca2+ efflux from the ER and, consequently, mitochondrial Ca2+ accumulation (Anelli et al. 2012). ERO1-α is highly expressed in different tumor types and is associated with a poor prognosis in breast cancer (Kutomi et al. 2013). In fact, the expression of ERO1-α in triple-negative breast cancer cells is correlated with that of programmed cell death-ligand 1 (PD-L1), both at the protein and mRNA levels, via hypoxia-inducible factor 1-α (HIF-1α). Depletion of ERO1-α led to a significant reduction in PD-L1-mediated T-cell apoptosis, suggesting that ERO1-α has a key role in tumor-mediated immunosuppression (Tanaka et al. 2017).
Another MAMs Ca2+- and tumor-related protein that acts at the ER level is the receptor chaperone stress-activated chaperone sigma-1 receptor (Sig1R), which senses ER Ca2+ concentrations and regulates cell survival. This protein could be considered “borderline” in this section considering its mechanism of action; in fact, Sig1R is an ER-localized protein that favors the efflux of calcium ions from the endoplasmic reticulum and has been described as being overexpressed in breast cancer, especially in cancer cells with metastatic potential (Gueguinou et al. 2017). ER chaperones are important in maintaining proper intracellular Ca2+ levels, protein folding, and the unfolded protein response (UPR) under ER stress conditions (Bartoszewska and Collawn 2020).
Two MAM-localized chaperones that belong to the heat shock 70 kDa (HSP70) protein family are of considerable importance in Ca2+ signaling: chaperone glucose-regulated protein GRP75 and glucose-regulated protein 78 (GRP78, also known as immunoglobulin heavy-chain-binding protein BiP) (Brocchieri et al. 2008; Wadhwa et al. 2002).
GRP75 ensures the juxtaposition between IP3R and VDAC1 in the mitochondrial outer membrane (Szabadkai et al. 2006). Its localization is mainly mitochondrial, but it is also present at low levels in the cytoplasm, nucleus, ER, and Golgi apparatus (Ran et al. 2000; Wadhwa et al. 1995), where it exerts many different functions from the import of unfolded proteins into the mitochondrial matrix to modulation of exocytosis and endocytosis (Flachbartova and Kovacech 2013; Voos and Rottgers 2002; Schneider et al. 1996; Kronidou et al. 1994; Scherer et al. 1992). Sig1Rs are particularly enriched at the MAMs and in normal tissues form a complex with GRP78, another MAM-localized chaperone. GRP78 can bind to misfolded proteins and to unassembled complexes and modulates ER-associated degradation (ERAD), which regulates the UPR (Pfaffenbach and Lee 2011; Wang et al. 2009; Little et al. 1994). Its molecular structure displays two domains: the substrate-binding domain (SBD), involved in binding unfolded peptides, and the nucleotide-binding domain (NBD), which binds ATP to be hydrolyzed to obtain energy to prevent unfolded protein aggregation at the N-terminus (Luo et al. 2006; Lindquist and Craig 1988). GRP78, like almost all other chaperones, is useful for storing ER Ca2+ as a high-capacity Ca2+-binding protein under physiological conditions (Hendershot 2004).
Szabadkai et al. highlighted the mechanism by which Sig1R, dissociating from BiP, binds IP3R3 following the activation of IP3Rs. This event leads to IP3R3 stabilization at the MAMs and to an enhancement of IP3R3-mediated Ca2+ fluxes to the mitochondria (Szabadkai et al. 2006). Although BiP is an excellent target to consider for neuroprotective therapeutic strategies (Enogieru et al. 2019), it also influences how tumor cells survive, proliferate, and develop chemoresistance. During chronic ER stress conditions that involve prolonged ER Ca2+ depletion, Sig1R localization changes from the MAMs to the peripheral ER, reducing cellular damage and thus preventing cell death. Another mechanism of Ca2+ homeostasis perturbation implemented by Sig1R that has direct consequences on cell invasiveness in breast cancer has been described by Gueguinou et al. (2017). Sig1R favors the migration of cancer cells by forming a functional molecular platform with the calcium-activated K+ channels SK3 and ORAI calcium release-activated calcium modulator 1 (Orai1) (Gueguinou et al. 2017) (Fig. 2).

Upregulation of MAMs Ca2+ crosstalk in cancer: graphical representation of the calcium signaling regulators involved in a cancer-related increased Ca2+ crosstalk state. See text for further details. Ca2+, calcium; ER, endoplasmic reticulum
4.3 Increased Mitochondrial Ca2+ Uptake
Before the identification of the molecular players forming the MCU complex, the role of mitochondrial Ca2+ in cancer progression was simply confined to receiving Ca2+ from the ER, thereby regulating the apoptotic response. Low ER Ca2+ release results in reduced mitochondrial [Ca2+], mPTP inhibition, and resistance to chemotherapeutic-induced cell death. Consistent with this view, many oncogenic factors act at the MAMs to limit ER-mitochondria Ca2+ transfer (see the “Downregulation of ER-mitochondria calcium crosstalk” section). However, many mitochondrial Ca2+ channels that are responsible for favoring Ca2+ accumulation, such as VDACs, are overexpressed, rather than reduced, in cancer (Mazure 2017). These observations suggest that an increased intrinsic capacity of the mitochondrial compartment to accumulate Ca2+ could contribute to sustained malignant progression, although, at least theoretically, it predisposes cells to Ca2+-induced cell death. The oncogenic mechanisms regulated by mitochondrial Ca2+ mainly rely on the association between Ca2+ and the formation of mitogenic ROS, as well as pure stimulation of mitochondrial metabolism. Ca2+ accumulation activates four mitochondrial dehydrogenases, which in turn stimulate the respiratory chain and hence ATP production (Denton 2009). Thus, as a consequence of increased metabolic activity, ROS are generated inside the matrix, but they fail to trigger cell death, probably due to the superior antioxidant defense that often distinguishes the malignant phenotype (Gorrini et al. 2013).
The correlation between augmented mitochondrial Ca2+ entry, ROS production, and cancer growth appears evident for tumors overexpressing the uniporter complex pore-forming subunit MCU. Indeed, increased levels of MCU have been reported in different tumors, including breast and hepatocellular carcinomas (Vultur et al. 2018). In breast cancer, MCU-dependent mitochondrial Ca2+ entry is associated with ROS overproduction and higher metastatic potential through a mechanism that involves the downstream activation of HIF1-α transcriptional activity (Tosatto et al. 2016). Consistent with these observations, upregulation of MCU in triple-negative breast cancer cells promoted metastasis in an in vivo mouse model by enhancing glycolysis, a series of neoplastic events that is counteracted by the tumor-suppressor activity of miRNA-340 (Yu et al. 2017). Moreover, receptor-interacting protein kinase 1 (RIPK1) binds MCU to promote Ca2+ entry and colorectal cancer progression through stimulation of mitochondrial bioenergetics (Zeng et al. 2018). In hepatocellular carcinomas, the Ca2+-ROS axis orchestrated by MCU resulted in activation of metalloproteinase-2 (MMP2) (Ren et al. 2017), a zinc-dependent endopeptidase associated with extracellular matrix degradation and metastasis (Shay et al. 2015).
The link between Ca2+ and ROS overproduction is also relevant for the cancer-related functions of MICU1, the principal member of the MCU complex that regulates the gating of the channel (Kamer and Mootha 2015). Our group recently showed that MICU1 downregulation, as a result of higher AKT activity, could sustain cancer progression through Ca2+-dependent ROS generation (Marchi et al. 2019a). Indeed, loss of MICU1 disinhibits MCU, leading to Ca2+ permeation under resting (nonstimulated) conditions and increased mitochondrial ROS levels (Csordas et al. 2013), which could ultimately result in cell death (Mallilankaraman et al. 2012a; Liu et al. 2016). This finding implies that malignant cells showing low MICU1 levels predispose concomitant mechanisms to minimize the detrimental effects induced by ROS. Consistent with this view, MICU1 depletion in normal hepatocytes triggered extensive cell death, but upon pharmacological inhibition of mPTP opening, the loss of MICU1 conferred a strong proliferative advantage (Antony et al. 2016). Moreover, a combination of high mitochondrial Ca2+ entry through genetic manipulation of the MCU complex and mPTP closure exacerbated the tumorigenic potential of different cancer cells (Marchi et al. 2019b). Taken together, these observations suggest that variations in the composition of the MCU complex are a key event that cooperates with other oncogenic pathways to favor cancer growth.
Further evidence that supports this scenario derives from the protumorigenic role of MCU regulator 1 (MCUR1), which has been described as a matrix-located, positive regulator of the uniporter complex (Mallilankaraman et al. 2012b). In hepatocellular carcinomas, MCUR1 was strongly upregulated, and ROS production was augmented, leading to ROS-dependent degradation of p53 and consequent resistance to apoptosis (Ren et al. 2018). Notably, the cancer cell detoxification capacity was also increased due to activation of nuclear factor erythroid 2-related factor 2 (NRF2) (Jin et al. 2019), a master gene in the orchestration of the cellular antioxidant response (Cuadrado et al. 2019). Thus, MCUR1 can regulate two cancer hallmarks at once: Ca2+-mediated metastatic potential and resistance to apoptosis. However, the expression of MCUR1 correlates with the permeability transition and reduced cell survival (Chaudhuri et al. 2016b), indicating that MCUR1 oncogenic activities might be solely due to the concomitant inhibition of the functions of the mPTP through a superior mechanism. Nevertheless, it has been proposed that MCUR1 could act as a complex IV assembly factor rather than as an MCU interactor (Paupe et al. 2015). In this context, variations in mitochondrial Ca2+ uptake and ROS levels are side products of respiratory chain defects; therefore, the active role of Ca2+ in MCUR1-mediated oncogenesis should be completely reevaluated.
Overall, these observations indicate that increased mitochondrial Ca2+ uptake acts with other oncogenic mechanisms (e.g., mPTP inhibition or activation of antioxidant systems) to sustain cancer growth and dissemination. The protumorigenic role of mitochondrial Ca2+ signaling involves other pathways in addition to ROS production and excess malignant cell bioenergetics, including the MCU-dependent control of cytosolic Ca2+ through store-operated Ca2+ entry (SOCE). The activity of the MCU complex sustains cytosolic Ca2+ fluxes through SOCE, which in turn regulates cytoskeletal dynamics and cellular migration (Prudent et al. 2016). Moreover, recent findings suggest that spontaneous mitochondrial Ca2+ oscillations through the MCU complex are essential for mitotic entry and cell cycle progression (Koval et al. 2019; Zhao et al. 2019), thus revealing another mechanism that could account for the aberrant proliferation of cancer cells with an altered composition of the MCU complex (Fig. 2).
5 Conclusions
The importance of the multiple and complex signaling pathways generated by the displacement of Ca2+ ions and, specifically, the Ca2+-dependent communication between structurally and functionally interconnected intracellular organelles has been increasingly highlighted and described, especially in recent years. Evidence of this phenomenon is the dramatic effects on cell health that derive from perturbation of the MAMs morphology and modification of the ER-mitochondria tethering distance. Moreover, alterations in the MAMs protein pool and functionality have been connected with several pathological conditions, including diabetes, neurodegeneration, infection, and antiviral response and cancer (Pinton 2018). Tumor cells, in fact, could modify the systems that maintain cellular Ca2+ homeostasis to promote their survival and metastasis. The crucial role of the regulation of spatiotemporal Ca2+ signaling in the MAMs in cancer is confirmed by evidence that different oncogenes and tumor suppressors reside at the ER-mitochondria interface.
As shown previously, both an increase and a decrease of calcium ion exchange between these two organelles can, in a nonexclusive way, lead to the promotion or suppression of tumor behaviors in many tissues. This phenomenon is an indication of how the equilibrium that rules calcium homeostasis in this subcellular compartment is delicate, complex, and intimate. Specifically, although Ca2+ oscillations are essential at MAMs to feed mitochondrial metabolism, a persistent increase in mitochondrial [Ca2+] can lead to cell death. In this scenario, by limiting mitochondrial calcium uptake, many cancer cells develop resistance to death. On the other hand, it was also highlighted that an increased mitochondrial ability to accumulate Ca2+ supports malignant progression, by boosting mitochondrial metabolism and sustaining mitogenic ROS production. Thus, depending on the tumor context, MAM-localized Ca2+ signaling can exert different functions, also according to the different oncogenic paths involved.
Several questions have yet to be answered, many aspects remain to be clarified, and molecular pathways must be described to reach a good understanding of the complex mechanisms that stem from calcium signaling at the MAMs, knowledge that will be very useful in the development of novel therapeutic strategies for several tumors.
References
Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, Ben-Romano R, Friedman O, Shoshan-Barmatz V (2009) The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci 122(Pt 11):1906–1916. https://doi.org/10.1242/jcs.040188
Akl H, Bultynck G (2013) Altered Ca(2+) signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim Biophys Acta 1835(2):180–193. https://doi.org/10.1016/j.bbcan.2012.12.001
Alonso MT, Manjarres IM, Garcia-Sancho J (2009) Modulation of calcium signalling by intracellular organelles seen with targeted aequorins. Acta Physiol (Oxf) 195(1):37–49. https://doi.org/10.1111/j.1748-1716.2008.01920.x
Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A, Pinton P, Ripamonti M, Rizzuto R, Sitia R (2012) Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid Redox Signal 16(10):1077–1087. https://doi.org/10.1089/ars.2011.4004
Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, Rubin E, Csordas G, Seifert EL, Hoek JB, Hajnoczky G (2016) MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat Commun 7:10955. https://doi.org/10.1038/ncomms10955
Arbel N, Shoshan-Barmatz V (2010) Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J Biol Chem 285(9):6053–6062. https://doi.org/10.1074/jbc.M109.082990
Ashby MC, Tepikin AV (2001) ER calcium and the functions of intracellular organelles. Semin Cell Dev Biol 12(1):11–17. https://doi.org/10.1006/scdb.2000.0212
Avalle L, Camporeale A, Morciano G, Caroccia N, Ghetti E, Orecchia V, Viavattene D, Giorgi C, Pinton P, Poli V (2019) STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca(2+) fluxes and apoptotic responses. Cell Death Differ 26(5):932–942. https://doi.org/10.1038/s41418-018-0171-y
Bansaghi S, Golenar T, Madesh M, Csordas G, RamachandraRao S, Sharma K, Yule DI, Joseph SK, Hajnoczky G (2014) Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J Biol Chem 289(12):8170–8181. https://doi.org/10.1074/jbc.M113.504159
Bargaje R, Gupta S, Sarkeshik A, Park R, Xu T, Sarkar M, Halimani M, Roy SS, Yates J, Pillai B (2012) Identification of novel targets for miR-29a using miRNA proteomics. PLoS One 7(8):e43243. https://doi.org/10.1371/journal.pone.0043243
Bartok A, Weaver D, Golenar T, Nichtova Z, Katona M, Bansaghi S, Alzayady KJ, Thomas VK, Ando H, Mikoshiba K, Joseph SK, Yule DI, Csordas G, Hajnoczky G (2019) IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat Commun 10(1):3726. https://doi.org/10.1038/s41467-019-11646-3
Bartoszewska S, Collawn JF (2020) Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell Mol Biol Lett 25:18. https://doi.org/10.1186/s11658-020-00212-1
Bathori G, Csordas G, Garcia-Perez C, Davies E, Hajnoczky G (2006) Ca2+−dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J Biol Chem 281(25):17347–17358. https://doi.org/10.1074/jbc.M600906200
Bernardi R, Pandolfi PP (2007) Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8(12):1006–1016. https://doi.org/10.1038/nrm2277
Bernard-Marissal N, Chrast R, Schneider BL (2018) Endoplasmic reticulum and mitochondria in diseases of motor and sensory neurons: a broken relationship? Cell Death Dis 9(3):333. https://doi.org/10.1038/s41419-017-0125-1
Berridge MJ (2012) Calcium signalling remodelling and disease. Biochem Soc Trans 40(2):297–309. https://doi.org/10.1042/BST20110766
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4(7):517–529. https://doi.org/10.1038/nrm1155
Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN (2013) Feature article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A 110(31):12526–12534. https://doi.org/10.1073/pnas.1302455110
Betzenhauser MJ, Wagner LE 2nd, Iwai M, Michikawa T, Mikoshiba K, Yule DI (2008) ATP modulation of Ca2+ release by type-2 and type-3 inositol (1, 4, 5)-triphosphate receptors. Differing ATP sensitivities and molecular determinants of action. J Biol Chem 283(31):21579–21587. https://doi.org/10.1074/jbc.M801680200
Bittremieux M, Parys JB, Pinton P, Bultynck G (2016) ER functions of oncogenes and tumor suppressors: modulators of intracellular Ca(2+) signaling. Biochim Biophys Acta 1863(6 Pt B):1364–1378. https://doi.org/10.1016/j.bbamcr.2016.01.002
Bittremieux M, La Rovere RM, Akl H, Martines C, Welkenhuyzen K, Dubron K, Baes M, Janssens A, Vandenberghe P, Laurenti L, Rietdorf K, Morciano G, Pinton P, Mikoshiba K, Bootman MD, Efremov DG, De Smedt H, Parys JB, Bultynck G (2019) Constitutive IP3 signaling underlies the sensitivity of B-cell cancers to the Bcl-2/IP3 receptor disruptor BIRD-2. Cell Death Differ 26(3):531–547. https://doi.org/10.1038/s41418-018-0142-3
Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, Pandolfi PP, Pinton P (2013) Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ 20(12):1631–1643. https://doi.org/10.1038/cdd.2013.77
Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji M, Pellegrini L, Signorato V, Olivetto F, Pastorino S, Nasu M, Napolitano A, Gaudino G, Morris P, Sakamoto G, Ferris LK, Danese A, Raimondi A, Tacchetti C, Kuchay S, Pass HI, Affar EB, Yang H, Pinton P, Carbone M (2017) BAP1 regulates IP3R3-mediated Ca(2+) flux to mitochondria suppressing cell transformation. Nature 546(7659):549–553. https://doi.org/10.1038/nature22798
Bonora M, Morganti C, Morciano G, Pedriali G, Lebiedzinska-Arciszewska M, Aquila G, Giorgi C, Rizzo P, Campo G, Ferrari R, Kroemer G, Wieckowski MR, Galluzzi L, Pinton P (2017) Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep 18(7):1077–1089. https://doi.org/10.15252/embr.201643602
Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R (2006) Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441(7091):349–353. https://doi.org/10.1038/nature04733
Brocchieri L, Conway de Macario E, Macario AJ (2008) hsp70 genes in the human genome: conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC Evol Biol 8:19. https://doi.org/10.1186/1471-2148-8-19
Buck TM, Wright CM, Brodsky JL (2007) The activities and function of molecular chaperones in the endoplasmic reticulum. Semin Cell Dev Biol 18(6):751–761. https://doi.org/10.1016/j.semcdb.2007.09.001
Cai X, Wang X, Patel S, Clapham DE (2015) Insights into the early evolution of animal calcium signaling machinery: a unicellular point of view. Cell Calcium 57(3):166–173. https://doi.org/10.1016/j.ceca.2014.11.007
Campbell KJ, Dhayade S, Ferrari N, Sims AH, Johnson E, Mason SM, Dickson A, Ryan KM, Kalna G, Edwards J, Tait SWG, Blyth K (2018) MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis 9(2):19. https://doi.org/10.1038/s41419-017-0035-2
Carafoli E (2002) Calcium signaling: a tale for all seasons. Proc Natl Acad Sci U S A 99(3):1115–1122. https://doi.org/10.1073/pnas.032427999
Carafoli E, Krebs J (2016) Why calcium? How calcium became the best communicator. J Biol Chem 291(40):20849–20857. https://doi.org/10.1074/jbc.R116.735894
Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, Urra F, Smith N, Molgo J, Diehl JA, Ridky TW, Foskett JK (2016) Selective vulnerability of cancer cells by inhibition of Ca(2+) transfer from endoplasmic reticulum to mitochondria. Cell Rep 14(10):2313–2324. https://doi.org/10.1016/j.celrep.2016.02.030
Carpi S, Polini B, Poli G, Alcantara Barata G, Fogli S, Romanini A, Tuccinardi T, Guella G, Frontini FP, Nieri P, Di Giuseppe G (2018) Anticancer activity of Euplotin C, isolated from the marine ciliate Euplotes crassus, against human melanoma cells. Mar Drugs 16(5). https://doi.org/10.3390/md16050166
Chaudhuri AD, Choi DC, Kabaria S, Tran A, Junn E (2016a) MicroRNA-7 regulates the function of mitochondrial permeability transition pore by targeting VDAC1 expression. J Biol Chem 291(12):6483–6493. https://doi.org/10.1074/jbc.M115.691352
Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE (2016b) Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc Natl Acad Sci U S A 113(13):E1872–E1880. https://doi.org/10.1073/pnas.1602264113
Chen Q, Xu H, Xu A, Ross T, Bowler E, Hu Y, Lesnefsky EJ (2015) Inhibition of Bcl-2 sensitizes mitochondrial permeability transition pore (MPTP) opening in ischemia-damaged mitochondria. PLoS One 10(3):e0118834. https://doi.org/10.1371/journal.pone.0118834
Chen G, Park D, Magis AT, Behera M, Ramalingam SS, Owonikoko TK, Sica GL, Ye K, Zhang C, Chen Z, Curran WJ, Deng X (2019) Mcl-1 interacts with Akt to promote lung cancer progression. Cancer Res 79(24):6126–6138. https://doi.org/10.1158/0008-5472.CAN-19-0950
Colombini M (2012) VDAC structure, selectivity, and dynamics. Biochim Biophys Acta 1818(6):1457–1465. https://doi.org/10.1016/j.bbamem.2011.12.026
Cosentino K, Garcia-Saez AJ (2014) Mitochondrial alterations in apoptosis. Chem Phys Lipids 181:62–75. https://doi.org/10.1016/j.chemphyslip.2014.04.001
Crottes D, Guizouarn H, Martin P, Borgese F, Soriani O (2013) The sigma-1 receptor: a regulator of cancer cell electrical plasticity? Front Physiol 4:175. https://doi.org/10.3389/fphys.2013.00175
Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174(7):915–921. https://doi.org/10.1083/jcb.200604016
Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente PS, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnoczky G (2013) MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab 17(6):976–987. https://doi.org/10.1016/j.cmet.2013.04.020
Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, Attucks OC, Franklin S, Levonen AL, Kensler TW, Dinkova-Kostova AT (2019) Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov 18(4):295–317. https://doi.org/10.1038/s41573-018-0008-x
Cui C, Merritt R, Fu L, Pan Z (2017) Targeting calcium signaling in cancer therapy. Acta Pharm Sin B 7(1):3–17. https://doi.org/10.1016/j.apsb.2016.11.001
Danese A, Patergnani S, Bonora M, Wieckowski MR, Previati M, Giorgi C, Pinton P (2017) Calcium regulates cell death in cancer: roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim Biophys Acta Bioenerg 1858(8):615–627. https://doi.org/10.1016/j.bbabio.2017.01.003
De Pinto V, Guarino F, Guarnera A, Messina A, Reina S, Tomasello FM, Palermo V, Mazzoni C (2010) Characterization of human VDAC isoforms: a peculiar function for VDAC3? Biochim Biophys Acta 1797(6–7):1268–1275. https://doi.org/10.1016/j.bbabio.2010.01.031
De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476(7360):336–340. https://doi.org/10.1038/nature10230
De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, Rizzuto R (2012) VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ 19(2):267–273. https://doi.org/10.1038/cdd.2011.92
De Stefani D, Patron M, Rizzuto R (2015) Structure and function of the mitochondrial calcium uniporter complex. Biochim Biophys Acta 1853(9):2006–2011. https://doi.org/10.1016/j.bbamcr.2015.04.008
Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787(11):1309–1316. https://doi.org/10.1016/j.bbabio.2009.01.005
Ding Z, Yuan J, Liang Y, Wu J, Gong H, Ye Y, Jiang G, Yin P, Li Y, Zhang G, Yang C, Guo J, Chen Z, Wang X, Weng L, Zou Y (2017) Ryanodine receptor type 2 plays a role in the development of cardiac fibrosis under mechanical stretch through TGFbeta-1. Int Heart J 58(6):957–961. https://doi.org/10.1536/ihj.16-572
Doghman-Bouguerra M, Granatiero V, Sbiera S, Sbiera I, Lacas-Gervais S, Brau F, Fassnacht M, Rizzuto R, Lalli E (2016) FATE1 antagonizes calcium- and drug-induced apoptosis by uncoupling ER and mitochondria. EMBO Rep 17(9):1264–1280. https://doi.org/10.15252/embr.201541504
Dong XY, Su YR, Qian XP, Yang XA, Pang XW, Wu HY, Chen WF (2003) Identification of two novel CT antigens and their capacity to elicit antibody response in hepatocellular carcinoma patients. Br J Cancer 89(2):291–297. https://doi.org/10.1038/sj.bjc.6601062
Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C (2010) Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem 285(18):13678–13684. https://doi.org/10.1074/jbc.M109.096040
Eisenberg-Bord M, Shai N, Schuldiner M, Bohnert M (2016) A tether is a tether is a tether: tethering at membrane contact sites. Dev Cell 39(4):395–409. https://doi.org/10.1016/j.devcel.2016.10.022
Enogieru AB, Omoruyi SI, Hiss DC, Ekpo OE (2019) GRP78/BIP/HSPA5 as a therapeutic target in models of Parkinson’s disease: a mini review. Adv Pharm Sci 2019:2706783. https://doi.org/10.1155/2019/2706783
Everett RD, Chelbi-Alix MK (2007) PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89(6–7):819–830. https://doi.org/10.1016/j.biochi.2007.01.004
Fan G, Baker ML, Wang Z, Baker MR, Sinyagovskiy PA, Chiu W, Ludtke SJ, Serysheva II (2015) Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 527(7578):336–341. https://doi.org/10.1038/nature15249
Feng S, Li H, Tai Y, Huang J, Su Y, Abramowitz J, Zhu MX, Birnbaumer L, Wang Y (2013) Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc Natl Acad Sci U S A 110(27):11011–11016. https://doi.org/10.1073/pnas.1309531110
Feng YX, Jin DX, Sokol ES, Reinhardt F, Miller DH, Gupta PB (2017) Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat Commun 8(1):1079. https://doi.org/10.1038/s41467-017-01052-y
Flachbartova Z, Kovacech B (2013) Mortalin – a multipotent chaperone regulating cellular processes ranging from viral infection to neurodegeneration. Acta Virol 57(1):3–15. https://doi.org/10.4149/av_2013_01_3
Foskett JK, White C, Cheung KH, Mak DO (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87(2):593–658. https://doi.org/10.1152/physrev.00035.2006
Frenzel A, Grespi F, Chmelewskij W, Villunger A (2009) Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis 14(4):584–596. https://doi.org/10.1007/s10495-008-0300-z
Gandre-Babbe S, van der Bliek AM (2008) The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19(6):2402–2412. https://doi.org/10.1091/mbc.E07-12-1287
Giacomello M, Pellegrini L (2016) The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ 23(9):1417–1427. https://doi.org/10.1038/cdd.2016.52
Gincel D, Silberberg SD, Shoshan-Barmatz V (2000) Modulation of the voltage-dependent anion channel (VDAC) by glutamate. J Bioenerg Biomembr 32(6):571–583. https://doi.org/10.1023/a:1005670527340
Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J, Bernardi R, Rizzuto R, Tacchetti C, Pinton P, Pandolfi PP (2010) PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330(6008):1247–1251. https://doi.org/10.1126/science.1189157
Giorgi C, Wieckowski MR, Pandolfi PP, Pinton P (2011) Mitochondria associated membranes (MAMs) as critical hubs for apoptosis. Commun Integr Biol 4(3):334–335. https://doi.org/10.4161/cib.4.3.15021
Giorgi C, Bonora M, Missiroli S, Poletti F, Ramirez FG, Morciano G, Morganti C, Pandolfi PP, Mammano F, Pinton P (2015) Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget 6(3):1435–1445. https://doi.org/10.18632/oncotarget.2935
Giorgi C, Danese A, Missiroli S, Patergnani S, Pinton P (2018a) Calcium dynamics as a machine for decoding signals. Trends Cell Biol 28(4):258–273. https://doi.org/10.1016/j.tcb.2018.01.002
Giorgi C, Marchi S, Pinton P (2018b) The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol 19(11):713–730. https://doi.org/10.1038/s41580-018-0052-8
Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K, Sahin A, Liu S, Barrera JA, Burgues O, Lluch AM, Chen H, Hortobagyi GN, Mills GB, Meric-Bernstam F (2011) PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther 10(6):1093–1101. https://doi.org/10.1158/1535-7163.MCT-10-1089
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12(12):931–947. https://doi.org/10.1038/nrd4002
Gueguinou M, Crottes D, Chantome A, Rapetti-Mauss R, Potier-Cartereau M, Clarysse L, Girault A, Fourbon Y, Jezequel P, Guerin-Charbonnel C, Fromont G, Martin P, Pellissier B, Schiappa R, Chamorey E, Mignen O, Uguen A, Borgese F, Vandier C, Soriani O (2017) The SigmaR1 chaperone drives breast and colorectal cancer cell migration by tuning SK3-dependent Ca(2+) homeostasis. Oncogene 36(25):3640–3647. https://doi.org/10.1038/onc.2016.501
Guerra MT, Florentino RM, Franca A, Lima Filho AC, Dos Santos ML, Fonseca RC, Lemos FO, Fonseca MC, Kruglov E, Mennone A, Njei B, Gibson J, Guan F, Cheng YC, Ananthanarayanan M, Gu J, Jiang J, Zhao H, Lima CX, Vidigal PT, Oliveira AG, Nathanson MH, Leite MF (2019) Expression of the type 3 InsP3 receptor is a final common event in the development of hepatocellular carcinoma. Gut 68(9):1676–1687. https://doi.org/10.1136/gutjnl-2018-317811
Gutierrez T, Simmen T (2018) Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium 70:64–75. https://doi.org/10.1016/j.ceca.2017.05.015
Hajnoczky G, Csordas G, Yi M (2002) Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium 32(5–6):363–377. https://doi.org/10.1016/s0143416002001872
Hakamata Y, Nishimura S, Nakai J, Nakashima Y, Kita T, Imoto K (1994) Involvement of the brain type of ryanodine receptor in T-cell proliferation. FEBS Lett 352(2):206–210. https://doi.org/10.1016/0014-5793(94)00955-4
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70. https://doi.org/10.1016/s0092-8674(00)81683-9
Hendershot LM (2004) The ER function BiP is a master regulator of ER function. Mt Sinai J Med 71(5):289–297
Higo T, Hattori M, Nakamura T, Natsume T, Michikawa T, Mikoshiba K (2005) Subtype-specific and ER luminal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 120(1):85–98. https://doi.org/10.1016/j.cell.2004.11.048
Hsu KS, Kao HY (2018) Correction to: PML: regulation and multifaceted function beyond tumor suppression. Cell Biosci 8:18. https://doi.org/10.1186/s13578-018-0213-7
Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, Ogasawara M, Trachootham D, Feng L, Pelicano H, Chiao PJ, Keating MJ, Garcia-Manero G, Huang P (2012) K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res 22(2):399–412. https://doi.org/10.1038/cr.2011.145
Huang H, Shah K, Bradbury NA, Li C, White C (2014) Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis 5:e1482. https://doi.org/10.1038/cddis.2014.419
Huebner K, Croce CM (2003) Cancer and the FRA3B/FHIT fragile locus: it’s a HIT. Br J Cancer 88(10):1501–1506. https://doi.org/10.1038/sj.bjc.6600937
Ismail IH, Davidson R, Gagne JP, Xu ZZ, Poirier GG, Hendzel MJ (2014) Germline mutations in BAP1 impair its function in DNA double-strand break repair. Cancer Res 74(16):4282–4294. https://doi.org/10.1158/0008-5472.CAN-13-3109
Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G (2014) Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta 1843(10):2164–2183. https://doi.org/10.1016/j.bbamcr.2014.03.007
Ivanova H, Kerkhofs M, La Rovere RM, Bultynck G (2017) Endoplasmic reticulum-mitochondrial Ca(2+) fluxes underlying cancer cell survival. Front Oncol 7:70. https://doi.org/10.3389/fonc.2017.00070
Jin M, Wang J, Ji X, Cao H, Zhu J, Chen Y, Yang J, Zhao Z, Ren T, Xing J (2019) MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J Exp Clin Cancer Res 38(1):136. https://doi.org/10.1186/s13046-019-1135-x
Kakihana T, Nagata K, Sitia R (2012) Peroxides and peroxidases in the endoplasmic reticulum: integrating redox homeostasis and oxidative folding. Antioxid Redox Signal 16(8):763–771. https://doi.org/10.1089/ars.2011.4238
Kamer KJ, Mootha VK (2015) The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol 16(9):545–553. https://doi.org/10.1038/nrm4039
Kang HY, Kim NS, Lee CO, Lee JY, Kang WH (2000) Expression and function of ryanodine receptors in human melanocytes. J Cell Physiol 185(2):200–206. https://doi.org/10.1002/1097-4652(200011)185:2<200::AID-JCP4>3.0.CO;2-6
Kang SS, Han KS, Ku BM, Lee YK, Hong J, Shin HY, Almonte AG, Woo DH, Brat DJ, Hwang EM, Yoo SH, Chung CK, Park SH, Paek SH, Roh EJ, Lee SJ, Park JY, Traynelis SF, Lee CJ (2010) Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res 70(3):1173–1183. https://doi.org/10.1158/0008-5472.CAN-09-2886
Kerkhofs M, Giorgi C, Marchi S, Seitaj B, Parys JB, Pinton P, Bultynck G, Bittremieux M (2017) Alterations in Ca(2+) signalling via ER-mitochondria contact site remodelling in cancer. Adv Exp Med Biol 997:225–254. https://doi.org/10.1007/978-981-10-4567-7_17
Kerkhofs M, Bittremieux M, Morciano G, Giorgi C, Pinton P, Parys JB, Bultynck G (2018) Emerging molecular mechanisms in chemotherapy: Ca(2+) signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis 9(3):334. https://doi.org/10.1038/s41419-017-0179-0
Khan MT, Wagner L 2nd, Yule DI, Bhanumathy C, Joseph SK (2006) Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem 281(6):3731–3737. https://doi.org/10.1074/jbc.M509262200
Kim KH, Roberts CW (2016) Targeting EZH2 in cancer. Nat Med 22(2):128–134. https://doi.org/10.1038/nm.4036
Kirichok Y, Krapivinsky G, Clapham DE (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427(6972):360–364. https://doi.org/10.1038/nature02246
Kiss DL, Baez W, Huebner K, Bundschuh R, Schoenberg DR (2017) Impact of FHIT loss on the translation of cancer-associated mRNAs. Mol Cancer 16(1):179. https://doi.org/10.1186/s12943-017-0749-x
Koval OM, Nguyen EK, Santhana V, Fidler TP, Sebag SC, Rasmussen TP, Mittauer DJ, Strack S, Goswami PC, Abel ED, Grumbach IM (2019) Loss of MCU prevents mitochondrial fusion in G1-S phase and blocks cell cycle progression and proliferation. Sci Signal 12(579). https://doi.org/10.1126/scisignal.aav1439
Krols M, Bultynck G, Janssens S (2016) ER-mitochondria contact sites: a new regulator of cellular calcium flux comes into play. J Cell Biol 214(4):367–370. https://doi.org/10.1083/jcb.201607124
Kronidou NG, Oppliger W, Bolliger L, Hannavy K, Glick BS, Schatz G, Horst M (1994) Dynamic interaction between Isp45 and mitochondrial hsp70 in the protein import system of the yeast mitochondrial inner membrane. Proc Natl Acad Sci U S A 91(26):12818–12822. https://doi.org/10.1073/pnas.91.26.12818
Kuchay S, Giorgi C, Simoneschi D, Pagan J, Missiroli S, Saraf A, Florens L, Washburn MP, Collazo-Lorduy A, Castillo-Martin M, Cordon-Cardo C, Sebti SM, Pinton P, Pagano M (2017) PTEN counteracts FBXL2 to promote IP3R3- and Ca(2+)-mediated apoptosis limiting tumour growth. Nature 546(7659):554–558. https://doi.org/10.1038/nature22965
Kutomi G, Tamura Y, Tanaka T, Kajiwara T, Kukita K, Ohmura T, Shima H, Takamaru T, Satomi F, Suzuki Y, Torigoe T, Sato N, Hirata K (2013) Human endoplasmic reticulum oxidoreductin 1-alpha is a novel predictor for poor prognosis of breast cancer. Cancer Sci 104(8):1091–1096. https://doi.org/10.1111/cas.12177
Kveiborg M, Thomas G (2018) PACS-2 functions in colorectal cancer. Aging (Albany NY) 10(6):1190–1191. https://doi.org/10.18632/aging.101489
Lamriben L, Graham JB, Adams BM, Hebert DN (2016) N-glycan-based ER molecular chaperone and protein quality control system: the Calnexin binding cycle. Traffic 17(4):308–326. https://doi.org/10.1111/tra.12358
Lee HS, Lee SA, Hur SK, Seo JW, Kwon J (2014) Stabilization and targeting of INO80 to replication forks by BAP1 during normal DNA synthesis. Nat Commun 5:5128. https://doi.org/10.1038/ncomms6128
Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, Bossy-Wetzel E, Bennett MV, Pypaert M, Hickman JA, Smith PJ, Hardwick JM, Jonas EA (2008) Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A 105(6):2169–2174. https://doi.org/10.1073/pnas.0711647105
Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR, Tabas I (2009) Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J Cell Biol 186(6):783–792. https://doi.org/10.1083/jcb.200904060
Liao Y, Hao Y, Chen H, He Q, Yuan Z, Cheng J (2015) Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death. Protein Cell 6(6):434–442. https://doi.org/10.1007/s13238-015-0144-6
Lindquist S, Craig EA (1988) The heat-shock proteins. Annu Rev Genet 22:631–677. https://doi.org/10.1146/annurev.ge.22.120188.003215
Lipskaia L, Keuylian Z, Blirando K, Mougenot N, Jacquet A, Rouxel C, Sghairi H, Elaib Z, Blaise R, Adnot S, Hajjar RJ, Chemaly ER, Limon I, Bobe R (2014) Expression of sarco (endo) plasmic reticulum calcium ATPase (SERCA) system in normal mouse cardiovascular tissues, heart failure and atherosclerosis. Biochim Biophys Acta 1843(11):2705–2718. https://doi.org/10.1016/j.bbamcr.2014.08.002
Little E, Ramakrishnan M, Roy B, Gazit G, Lee AS (1994) The glucose-regulated proteins (GRP78 and GRP94): functions, gene regulation, and applications. Crit Rev Eukaryot Gene Expr 4(1):1–18. https://doi.org/10.1615/critreveukargeneexpr.v4.i1.10
Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, Yu ZX, Springer DA, Halsey C, Liu C, Murphy E, Finkel T (2016) MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep 16(6):1561–1573. https://doi.org/10.1016/j.celrep.2016.07.011
Lu H, Chen I, Shimoda LA, Park Y, Zhang C, Tran L, Zhang H, Semenza GL (2017) Chemotherapy-induced Ca(2+) release stimulates breast cancer stem cell enrichment. Cell Rep 18(8):1946–1957. https://doi.org/10.1016/j.celrep.2017.02.001
Luo S, Mao C, Lee B, Lee AS (2006) GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol 26(15):5688–5697. https://doi.org/10.1128/MCB.00779-06
Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L, Berthiaume LG, Simmen T (2012) Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J 31(2):457–470. https://doi.org/10.1038/emboj.2011.384
Lynes EM, Raturi A, Shenkman M, Ortiz Sandoval C, Yap MC, Wu J, Janowicz A, Myhill N, Benson MD, Campbell RE, Berthiaume LG, Lederkremer GZ, Simmen T (2013) Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. J Cell Sci 126(Pt 17):3893–3903. https://doi.org/10.1242/jcs.125856
Mak DO, Foskett JK (2015) Inositol 1,4,5-trisphosphate receptors in the endoplasmic reticulum: a single-channel point of view. Cell Calcium 58(1):67–78. https://doi.org/10.1016/j.ceca.2014.12.008
Maldonado EN, Sheldon KL, DeHart DN, Patnaik J, Manevich Y, Townsend DM, Bezrukov SM, Rostovtseva TK, Lemasters JJ (2013) Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J Biol Chem 288(17):11920–11929. https://doi.org/10.1074/jbc.M112.433847
Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M (2012a) MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151(3):630–644. https://doi.org/10.1016/j.cell.2012.10.011
Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, Madesh M (2012b) MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14(12):1336–1343. https://doi.org/10.1038/ncb2622
Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, Pinton P (2012) Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis 3:e304. https://doi.org/10.1038/cddis.2012.45
Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corra F, Giorgi C, De Marchi E, Poletti F, Gafa R, Lanza G, Negrini M, Rizzuto R, Pinton P (2013) Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol 23(1):58–63. https://doi.org/10.1016/j.cub.2012.11.026
Marchi S, Patergnani S, Pinton P (2014) The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta 1837(4):461–469. https://doi.org/10.1016/j.bbabio.2013.10.015
Marchi S, Bittremieux M, Missiroli S, Morganti C, Patergnani S, Sbano L, Rimessi A, Kerkhofs M, Parys JB, Bultynck G, Giorgi C, Pinton P (2017) Endoplasmic reticulum-mitochondria communication through Ca(2+) signaling: the importance of mitochondria-associated membranes (MAMs). Adv Exp Med Biol 997:49–67. https://doi.org/10.1007/978-981-10-4567-7_4
Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, Giorgi C, Pinton P (2018) Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69:62–72. https://doi.org/10.1016/j.ceca.2017.05.003
Marchi S, Corricelli M, Branchini A, Vitto VAM, Missiroli S, Morciano G, Perrone M, Ferrarese M, Giorgi C, Pinotti M, Galluzzi L, Kroemer G, Pinton P (2019a) Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca(2+) levels and tumor growth. EMBO J 38(2). https://doi.org/10.15252/embj.201899435
Marchi S, Vitto VAM, Patergnani S, Pinton P (2019b) High mitochondrial Ca(2+) content increases cancer cell proliferation upon inhibition of mitochondrial permeability transition pore (mPTP). Cell Cycle 18(8):914–916. https://doi.org/10.1080/15384101.2019.1598729
Marchi S, Giorgi C, Galluzzi L, Pinton P (2020) Ca(2+) fluxes and cancer. Mol Cell 78(6):1055–1069. https://doi.org/10.1016/j.molcel.2020.04.017
Marino M, Stoilova T, Giorgi C, Bachi A, Cattaneo A, Auricchio A, Pinton P, Zito E (2015) SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal muscle pathology, counteracts hyperoxidation by means of redox-regulating SERCA2 pump activity. Hum Mol Genet 24(7):1843–1855. https://doi.org/10.1093/hmg/ddu602
Mariot P, Prevarskaya N, Roudbaraki MM, Le Bourhis X, Van Coppenolle F, Vanoverberghe K, Skryma R (2000) Evidence of functional ryanodine receptor involved in apoptosis of prostate cancer (LNCaP) cells. Prostate 43(3):205–214. https://doi.org/10.1002/(sici)1097-0045(20000515)43:3<205::aid-pros6>3.0.co;2-m
Matyash M, Matyash V, Nolte C, Sorrentino V, Kettenmann H (2002) Requirement of functional ryanodine receptor type 3 for astrocyte migration. FASEB J 16(1):84–86. https://doi.org/10.1096/fj.01-0380fje
Mazure NM (2017) VDAC in cancer. Biochim Biophys Acta Bioenerg 1858(8):665–673. https://doi.org/10.1016/j.bbabio.2017.03.002
McCarthy TV, Datar S, Mackrill JJ (2003) Activation of ryanodine receptor/Ca2+ release channels downregulates CD38 in the Namalwa B lymphoma. FEBS Lett 554(1–2):133–137. https://doi.org/10.1016/s0014-5793(03)01122-0
Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L (2011) Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol 3(6). https://doi.org/10.1101/cshperspect.a004317
Messina A, Reina S, Guarino F, De Pinto V (2012) VDAC isoforms in mammals. Biochim Biophys Acta 1818(6):1466–1476. https://doi.org/10.1016/j.bbamem.2011.10.005
Mikoshiba K (2007) The IP3 receptor/Ca2+ channel and its cellular function. Biochem Soc Symp 74:9–22. https://doi.org/10.1042/BSS0740009
Missiroli S, Bonora M, Patergnani S, Poletti F, Perrone M, Gafa R, Magri E, Raimondi A, Lanza G, Tacchetti C, Kroemer G, Pandolfi PP, Pinton P, Giorgi C (2016) PML at mitochondria-associated membranes is critical for the repression of autophagy and cancer development. Cell Rep 16(9):2415–2427. https://doi.org/10.1016/j.celrep.2016.07.082
Missiroli S, Danese A, Iannitti T, Patergnani S, Perrone M, Previati M, Giorgi C, Pinton P (2017) Endoplasmic reticulum-mitochondria Ca(2+) crosstalk in the control of the tumor cell fate. Biochim Biophys Acta Mol Cell Res 1864(6):858–864. https://doi.org/10.1016/j.bbamcr.2016.12.024
Miyakawa T, Maeda A, Yamazawa T, Hirose K, Kurosaki T, Iino M (1999) Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J 18(5):1303–1308. https://doi.org/10.1093/emboj/18.5.1303
Moller JV, Olesen C, Winther AM, Nissen P (2010) The sarcoplasmic Ca2+-ATPase: design of a perfect chemi-osmotic pump. Q Rev Biophys 43(4):501–566. https://doi.org/10.1017/S003358351000017X
Monaco G, Decrock E, Arbel N, van Vliet AR, La Rovere RM, De Smedt H, Parys JB, Agostinis P, Leybaert L, Shoshan-Barmatz V, Bultynck G (2015) The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J Biol Chem 290(14):9150–9161. https://doi.org/10.1074/jbc.M114.622514
Monet M, Lehen'kyi V, Gackiere F, Firlej V, Vandenberghe M, Roudbaraki M, Gkika D, Pourtier A, Bidaux G, Slomianny C, Delcourt P, Rassendren F, Bergerat JP, Ceraline J, Cabon F, Humez S, Prevarskaya N (2010) Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res 70(3):1225–1235. https://doi.org/10.1158/0008-5472.CAN-09-2205
Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ (2007) Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer 7(7):519–530. https://doi.org/10.1038/nrc2171
Monteith GR, Davis FM, Roberts-Thomson SJ (2012) Calcium channels and pumps in cancer: changes and consequences. J Biol Chem 287(38):31666–31673. https://doi.org/10.1074/jbc.R112.343061
Morciano G, Marchi S, Morganti C, Sbano L, Bittremieux M, Kerkhofs M, Corricelli M, Danese A, Karkucinska-Wieckowska A, Wieckowski MR, Bultynck G, Giorgi C, Pinton P (2018) Role of mitochondria-associated ER membranes in calcium regulation in Cancer-specific settings. Neoplasia 20(5):510–523. https://doi.org/10.1016/j.neo.2018.03.005
Munoz-Maldonado C, Zimmer Y, Medova M (2019) A comparative analysis of individual RAS mutations in cancer biology. Front Oncol 9:1088. https://doi.org/10.3389/fonc.2019.01088
Newton CL, Mignery GA, Sudhof TC (1994) Co-expression in vertebrate tissues and cell lines of multiple inositol 1,4,5-trisphosphate (InsP3) receptors with distinct affinities for InsP3. J Biol Chem 269(46):28613–28619
Ning X, Shi Z, Liu X, Zhang A, Han L, Jiang K, Kang C, Zhang Q (2015) DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Lett 359(2):198–205. https://doi.org/10.1016/j.canlet.2015.01.005
Niu Z, Wang M, Zhou L, Yao L, Liao Q, Zhao Y (2015) Elevated GRP78 expression is associated with poor prognosis in patients with pancreatic cancer. Sci Rep 5:16067. https://doi.org/10.1038/srep16067
Oropeza-Almazan Y, Vazquez-Garza E, Chapoy-Villanueva H, Torre-Amione G, Garcia-Rivas G (2017) Small interfering RNA targeting mitochondrial calcium uniporter improves cardiomyocyte cell viability in hypoxia/reoxygenation injury by reducing calcium overload. Oxid Med Cell Longev 2017:5750897. https://doi.org/10.1155/2017/5750897
Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, Cong Y, Mootha VK, Chou JJ (2016) Architecture of the mitochondrial calcium uniporter. Nature 533(7602):269–273. https://doi.org/10.1038/nature17656
Palmgren MG, Nissen P (2011) P-type ATPases. Annu Rev Biophys 40:243–266. https://doi.org/10.1146/annurev.biophys.093008.131331
Park J, Lee Y, Park T, Kang JY, Mun SA, Jin M, Yang J, Eom SH (2020) Structure of the MICU1-MICU2 heterodimer provides insights into the gatekeeping threshold shift. IUCrJ 7(Pt 2):355–365. https://doi.org/10.1107/S2052252520001840
Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA (2015) CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab 21(1):109–116. https://doi.org/10.1016/j.cmet.2014.12.004
Penston J, Wormsley KG (1986) H2-receptor antagonists and gastric cancer. Med Toxicol 1(3):163–168. https://doi.org/10.1007/bf03259835
Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467(7313):291–296. https://doi.org/10.1038/nature09358
Petrungaro C, Zimmermann KM, Kuttner V, Fischer M, Dengjel J, Bogeski I, Riemer J (2015) The Ca(2+)-dependent release of the Mia40-induced MICU1-MICU2 Dimer from MCU regulates mitochondrial Ca(2+) uptake. Cell Metab 22(4):721–733. https://doi.org/10.1016/j.cmet.2015.08.019
Pfaffenbach KT, Lee AS (2011) The critical role of GRP78 in physiologic and pathologic stress. Curr Opin Cell Biol 23(2):150–156. https://doi.org/10.1016/j.ceb.2010.09.007
Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL (2014) Oncogenic K-Ras suppresses IP(3)-dependent Ca(2)(+) release through remodelling of the isoform composition of IP(3)Rs and ER luminal Ca(2)(+) levels in colorectal cancer cell lines. J Cell Sci 127(Pt 7):1607–1619. https://doi.org/10.1242/jcs.141408
Pinton P (2018) Mitochondria-associated membranes (MAMs) and pathologies. Cell Death Dis 9(4):413. https://doi.org/10.1038/s41419-018-0424-1
Pinton P, Leo S, Wieckowski MR, Di Benedetto G, Rizzuto R (2004) Long-term modulation of mitochondrial Ca2+ signals by protein kinase C isozymes. J Cell Biol 165(2):223–232. https://doi.org/10.1083/jcb.200311061
Pinton P, Giorgi C, Pandolfi PP (2011) The role of PML in the control of apoptotic cell fate: a new key player at ER-mitochondria sites. Cell Death Differ 18(9):1450–1456. https://doi.org/10.1038/cdd.2011.31
Ponte S, Carvalho L, Gagliardi M, Campos I, Oliveira PJ, Jacinto A (2020) Drp1-mediated mitochondrial fission regulates calcium and F-actin dynamics during wound healing. Biol Open 9(5). https://doi.org/10.1242/bio.048629
Poston CN, Krishnan SC, Bazemore-Walker CR (2013) In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J Proteomics 79:219–230. https://doi.org/10.1016/j.jprot.2012.12.018
Prevarskaya N, Ouadid-Ahidouch H, Skryma R, Shuba Y (2014) Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos Trans R Soc Lond B Biol Sci 369(1638):20130097. https://doi.org/10.1098/rstb.2013.0097
Prole DL, Taylor CW (2016) Inositol 1,4,5-trisphosphate receptors and their protein partners as signalling hubs. J Physiol 594(11):2849–2866. https://doi.org/10.1113/JP271139
Prudent J, Popgeorgiev N, Gadet R, Deygas M, Rimokh R, Gillet G (2016) Mitochondrial Ca(2+) uptake controls actin cytoskeleton dynamics during cell migration. Sci Rep 6:36570. https://doi.org/10.1038/srep36570
Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, Hardingham GE (2013) Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4:2034. https://doi.org/10.1038/ncomms3034
Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I, Rizzuto R (2013) The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32(17):2362–2376. https://doi.org/10.1038/emboj.2013.157
Rai K, Pilarski R, Cebulla CM, Abdel-Rahman MH (2016) Comprehensive review of BAP1 tumor predisposition syndrome with report of two new cases. Clin Genet 89(3):285–294. https://doi.org/10.1111/cge.12630
Ramos-Franco J, Fill M, Mignery GA (1998) Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biophys J 75(2):834–839. https://doi.org/10.1016/S0006-3495(98)77572-1
Ran Q, Wadhwa R, Kawai R, Kaul SC, Sifers RN, Bick RJ, Smith JR, Pereira-Smith OM (2000) Extramitochondrial localization of mortalin/mthsp70/PBP74/GRP75. Biochem Biophys Res Commun 275(1):174–179. https://doi.org/10.1006/bbrc.2000.3237
Raphael M, Lehen'kyi V, Vandenberghe M, Beck B, Khalimonchyk S, Vanden Abeele F, Farsetti L, Germain E, Bokhobza A, Mihalache A, Gosset P, Romanin C, Clezardin P, Skryma R, Prevarskaya N (2014) TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc Natl Acad Sci U S A 111(37):E3870–E3879. https://doi.org/10.1073/pnas.1413409111
Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE, Rizzuto R (2002) Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol 159(4):613–624. https://doi.org/10.1083/jcb.200205091
Raturi A, Gutierrez T, Ortiz-Sandoval C, Ruangkittisakul A, Herrera-Cruz MS, Rockley JP, Gesson K, Ourdev D, Lou PH, Lucchinetti E, Tahbaz N, Zaugg M, Baksh S, Ballanyi K, Simmen T (2016) TMX1 determines cancer cell metabolism as a thiol-based modulator of ER-mitochondria Ca2+ flux. J Cell Biol 214(4):433–444. https://doi.org/10.1083/jcb.201512077
Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, Guo X, Ji L, Huang Q, Zhang H, Yang H, Xing J (2017) MCU-dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 36(42):5897–5909. https://doi.org/10.1038/onc.2017.167
Ren T, Wang J, Zhang H, Yuan P, Zhu J, Wu Y, Huang Q, Guo X, Zhang J, Ji L, Li J, Zhang H, Yang H, Xing J (2018) MCUR1-mediated mitochondrial calcium signaling facilitates cell survival of hepatocellular carcinoma via reactive oxygen species-dependent P53 degradation. Antioxid Redox Signal 28(12):1120–1136. https://doi.org/10.1089/ars.2017.6990
Revathidevi S, Munirajan AK (2019) Akt in cancer: mediator and more. Semin Cancer Biol 59:80–91. https://doi.org/10.1016/j.semcancer.2019.06.002
Rezuchova I, Hudecova S, Soltysova A, Matuskova M, Durinikova E, Chovancova B, Zuzcak M, Cihova M, Burikova M, Penesova A, Lencesova L, Breza J, Krizanova O (2019) Type 3 inositol 1,4,5-trisphosphate receptor has antiapoptotic and proliferative role in cancer cells. Cell Death Dis 10(3):186. https://doi.org/10.1038/s41419-019-1433-4
Rimessi A, Marchi S, Fotino C, Romagnoli A, Huebner K, Croce CM, Pinton P, Rizzuto R (2009) Intramitochondrial calcium regulation by the FHIT gene product sensitizes to apoptosis. Proc Natl Acad Sci U S A 106(31):12753–12758. https://doi.org/10.1073/pnas.0906484106
Rimessi A, Marchi S, Patergnani S, Pinton P (2014) H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene 33(18):2329–2340. https://doi.org/10.1038/onc.2013.192
Rimessi A, Pedriali G, Vezzani B, Tarocco A, Marchi S, Wieckowski MR, Giorgi C, Pinton P (2020) Interorganellar calcium signaling in the regulation of cell metabolism: a cancer perspective. Semin Cell Dev Biol 98:167–180. https://doi.org/10.1016/j.semcdb.2019.05.015
Ringer S (1883) A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J Physiol 4(1):29–42.3. https://doi.org/10.1113/jphysiol.1883.sp000120
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280(5370):1763–1766. https://doi.org/10.1126/science.280.5370.1763
Salmena L, Carracedo A, Pandolfi PP (2008) Tenets of PTEN tumor suppression. Cell 133(3):403–414. https://doi.org/10.1016/j.cell.2008.04.013
Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342(6164):1379–1382. https://doi.org/10.1126/science.1242993
Schein SJ, Colombini M, Finkelstein A (1976) Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J Membr Biol 30(2):99–120. https://doi.org/10.1007/bf01869662
Scherer PE, Manning-Krieg UC, Jeno P, Schatz G, Horst M (1992) Identification of a 45-kDa protein at the protein import site of the yeast mitochondrial inner membrane. Proc Natl Acad Sci U S A 89(24):11930–11934. https://doi.org/10.1073/pnas.89.24.11930
Scherr AL, Gdynia G, Salou M, Radhakrishnan P, Duglova K, Heller A, Keim S, Kautz N, Jassowicz A, Elssner C, He YW, Jaeger D, Heikenwalder M, Schneider M, Weber A, Roth W, Schulze-Bergkamen H, Koehler BC (2016) Bcl-xL is an oncogenic driver in colorectal cancer. Cell Death Dis 7(8):e2342. https://doi.org/10.1038/cddis.2016.233
Schneider HC, Westermann B, Neupert W, Brunner M (1996) The nucleotide exchange factor MGE exerts a key function in the ATP-dependent cycle of mt-Hsp70-Tim44 interaction driving mitochondrial protein import. EMBO J 15(21):5796–5803
Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G (2007) Genome regulation by polycomb and trithorax proteins. Cell 128(4):735–745. https://doi.org/10.1016/j.cell.2007.02.009
Sebag SC, Koval OM, Paschke JD, Winters CJ, Comellas AP, Grumbach IM (2018) Inhibition of the mitochondrial calcium uniporter prevents IL-13 and allergen-mediated airway epithelial apoptosis and loss of barrier function. Exp Cell Res 362(2):400–411. https://doi.org/10.1016/j.yexcr.2017.12.003
Shay G, Lynch CC, Fingleton B (2015) Moving targets: emerging roles for MMPs in cancer progression and metastasis. Matrix Biol 44-46:200–206. https://doi.org/10.1016/j.matbio.2015.01.019
Shibao K, Fiedler MJ, Nagata J, Minagawa N, Hirata K, Nakayama Y, Iwakiri Y, Nathanson MH, Yamaguchi K (2010) The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 48(6):315–323. https://doi.org/10.1016/j.ceca.2010.09.005
Shimizu S, Konishi A, Kodama T, Tsujimoto Y (2000) BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A 97(7):3100–3105. https://doi.org/10.1073/pnas.97.7.3100
Shoshan-Barmatz V, Gincel D (2003) The voltage-dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem Biophys 39(3):279–292. https://doi.org/10.1385/CBB:39:3:279
Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N (2010) VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med 31(3):227–285. https://doi.org/10.1016/j.mam.2010.03.002
Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G (2005) PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 24(4):717–729. https://doi.org/10.1038/sj.emboj.7600559
Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ (2005) Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 5(8):615–625. https://doi.org/10.1038/nrc1669
Singh A, Chagtoo M, Tiwari S, George N, Chakravarti B, Khan S, Lakshmi S, Godbole MM (2017a) Inhibition of inositol 1, 4, 5-trisphosphate receptor induce breast cancer cell death through deregulated autophagy and cellular bioenergetics. J Cell Biochem 118(8):2333–2346. https://doi.org/10.1002/jcb.25891
Singh A, Sharma RK, Chagtoo M, Agarwal G, George N, Sinha N, Godbole MM (2017b) 1H NMR metabolomics reveals Association of High Expression of inositol 1, 4, 5 trisphosphate receptor and metabolites in breast cancer patients. PLoS One 12(1):e0169330. https://doi.org/10.1371/journal.pone.0169330
Siprashvili Z, Sozzi G, Barnes LD, McCue P, Robinson AK, Eryomin V, Sard L, Tagliabue E, Greco A, Fusetti L, Schwartz G, Pierotti MA, Croce CM, Huebner K (1997) Replacement of Fhit in cancer cells suppresses tumorigenicity. Proc Natl Acad Sci U S A 94(25):13771–13776. https://doi.org/10.1073/pnas.94.25.13771
Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, van de Vijver MJ, Valero V, Gray JW, Bernards R, Mills GB, Hennessy BT (2008) An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 68(15):6084–6091. https://doi.org/10.1158/0008-5472.CAN-07-6854
Su Y, Huang X, Huang Z, Huang T, Xu Y, Yi C (2020) STAT3 localizes in mitochondria-associated ER membranes instead of in mitochondria. Front Cell Dev Biol 8:274. https://doi.org/10.3389/fcell.2020.00274
Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175(6):901–911. https://doi.org/10.1083/jcb.200608073
Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Soderberg O, Bootman MD, Roderick HL (2008) Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A 105(7):2427–2432. https://doi.org/10.1073/pnas.0711324105
Takahashi N, Chen HY, Harris IS, Stover DG, Selfors LM, Bronson RT, Deraedt T, Cichowski K, Welm AL, Mori Y, Mills GB, Brugge JS (2018) Cancer cells co-opt the neuronal redox-sensing channel TRPA1 to promote oxidative-stress tolerance. Cancer Cell 33(6):985–1003. e1007. https://doi.org/10.1016/j.ccell.2018.05.001
Takei N, Yoneda A, Sakai-Sawada K, Kosaka M, Minomi K, Tamura Y (2017) Hypoxia-inducible ERO1alpha promotes cancer progression through modulation of integrin-beta1 modification and signalling in HCT116 colorectal cancer cells. Sci Rep 7(1):9389. https://doi.org/10.1038/s41598-017-09976-7
Tanaka T, Kutomi G, Kajiwara T, Kukita K, Kochin V, Kanaseki T, Tsukahara T, Hirohashi Y, Torigoe T, Okamoto Y, Hirata K, Sato N, Tamura Y (2017) Cancer-associated oxidoreductase ERO1-alpha promotes immune escape through up-regulation of PD-L1 in human breast cancer. Oncotarget 8(15):24706–24718. https://doi.org/10.18632/oncotarget.14960
Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, Cai G, Meng X, Zou F (2015) Mitochondrial Ca(2)(+) uniporter is critical for store-operated Ca(2)(+) entry-dependent breast cancer cell migration. Biochem Biophys Res Commun 458(1):186–193. https://doi.org/10.1016/j.bbrc.2015.01.092
Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, Duchen MR, Rosato A, Bogeski I, Szabadkai G, Rizzuto R, Mammucari C (2016) The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol Med 8(5):569–585. https://doi.org/10.15252/emmm.201606255
Toyoshima C, Nakasako M, Nomura H, Ogawa H (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 a resolution. Nature 405(6787):647–655. https://doi.org/10.1038/35015017
Trisciuoglio D, Tupone MG, Desideri M, Di Martile M, Gabellini C, Buglioni S, Pallocca M, Alessandrini G, D'Aguanno S, Del Bufalo D (2017) BCL-XL overexpression promotes tumor progression-associated properties. Cell Death Dis 8(12):3216. https://doi.org/10.1038/s41419-017-0055-y
Tsujimoto Y, Shimizu S (2000) VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ 7(12):1174–1181. https://doi.org/10.1038/sj.cdd.4400780
Tu H, Wang Z, Bezprozvanny I (2005) Modulation of mammalian inositol 1,4,5-trisphosphate receptor isoforms by calcium: a role of calcium sensor region. Biophys J 88(2):1056–1069. https://doi.org/10.1529/biophysj.104.049601
Ueasilamongkol P, Khamphaya T, Guerra MT, Rodrigues MA, Gomes DA, Kong Y, Wei W, Jain D, Trampert DC, Ananthanarayanan M, Banales JM, Roberts LR, Farshidfar F, Nathanson MH, Weerachayaphorn J (2020) Type 3 inositol 1,4,5-trisphosphate receptor is increased and enhances malignant properties in cholangiocarcinoma. Hepatology 71(2):583–599. https://doi.org/10.1002/hep.30839
Vandecaetsbeek I, Trekels M, De Maeyer M, Ceulemans H, Lescrinier E, Raeymaekers L, Wuytack F, Vangheluwe P (2009) Structural basis for the high Ca2+ affinity of the ubiquitous SERCA2b Ca2+ pump. Proc Natl Acad Sci U S A 106(44):18533–18538. https://doi.org/10.1073/pnas.0906797106
Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19(11):1880–1891. https://doi.org/10.1038/cdd.2012.74
Vervloessem T, Yule DI, Bultynck G, Parys JB (2015) The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca(2)(+)-release channel. Biochim Biophys Acta 1853(9):1992–2005. https://doi.org/10.1016/j.bbamcr.2014.12.006
Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408(6810):307–310. https://doi.org/10.1038/35042675
Voos W, Rottgers K (2002) Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim Biophys Acta 1592(1):51–62. https://doi.org/10.1016/s0167-4889(02)00264-1
Vultur A, Gibhardt CS, Stanisz H, Bogeski I (2018) The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflugers Arch 470(8):1149–1163. https://doi.org/10.1007/s00424-018-2162-8
Wadhwa R, Pereira-Smith OM, Reddel RR, Sugimoto Y, Mitsui Y, Kaul SC (1995) Correlation between complementation group for immortality and the cellular distribution of mortalin. Exp Cell Res 216(1):101–106. https://doi.org/10.1006/excr.1995.1013
Wadhwa R, Taira K, Kaul SC (2002) An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: what, when, and where? Cell Stress Chaperones 7(3):309–316. https://doi.org/10.1379/1466-1268(2002)007<0309:ahfcmm>2.0.co;2
Wagner LE 2nd, Joseph SK, Yule DI (2008) Regulation of single inositol 1,4,5-trisphosphate receptor channel activity by protein kinase A phosphorylation. J Physiol 586(15):3577–3596. https://doi.org/10.1113/jphysiol.2008.152314
Wang X, Perez E, Liu R, Yan LJ, Mallet RT, Yang SH (2007) Pyruvate protects mitochondria from oxidative stress in human neuroblastoma SK-N-SH cells. Brain Res 1132(1):1–9. https://doi.org/10.1016/j.brainres.2006.11.032
Wang M, Wey S, Zhang Y, Ye R, Lee AS (2009) Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal 11(9):2307–2316. https://doi.org/10.1089/ARS.2009.2485
Wang PT, Garcin PO, Fu M, Masoudi M, St-Pierre P, Pante N, Nabi IR (2015) Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J Cell Sci 128(15):2759–2765. https://doi.org/10.1242/jcs.171132
Wang Y, Qi YX, Qi Z, Tsang SY (2019) TRPC3 regulates the proliferation and apoptosis resistance of triple negative breast cancer cells through the TRPC3/RASA4/MAPK pathway. Cancers (Basel) 11(4). https://doi.org/10.3390/cancers11040558
Weisthal S, Keinan N, Ben-Hail D, Arif T, Shoshan-Barmatz V (2014) Ca(2+)-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim Biophys Acta 1843(10):2270–2281. https://doi.org/10.1016/j.bbamcr.2014.03.021
White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK (2005) The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol 7(10):1021–1028. https://doi.org/10.1038/ncb1302
Whitehurst AW (2014) Cause and consequence of cancer/testis antigen activation in cancer. Annu Rev Pharmacol Toxicol 54:251–272. https://doi.org/10.1146/annurev-pharmtox-011112-140326
Xu N, Zhang D, Chen J, He G, Gao L (2019) Low expression of ryanodine receptor 2 is associated with poor prognosis in thyroid carcinoma. Oncol Lett 18(4):3605–3612. https://doi.org/10.3892/ol.2019.10732
Yoshimine S, Kikuchi E, Kosaka T, Mikami S, Miyajima A, Okada Y, Oya M (2013) Prognostic significance of Bcl-xL expression and efficacy of Bcl-xL targeting therapy in urothelial carcinoma. Br J Cancer 108(11):2312–2320. https://doi.org/10.1038/bjc.2013.216
Youker RT, Shinde U, Day R, Thomas G (2009) At the crossroads of homoeostasis and disease: roles of the PACS proteins in membrane traffic and apoptosis. Biochem J 421(1):1–15. https://doi.org/10.1042/BJ20081016
Yu H, Lee H, Herrmann A, Buettner R, Jove R (2014) Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14(11):736–746. https://doi.org/10.1038/nrc3818
Yu C, Wang Y, Peng J, Shen Q, Chen M, Tang W, Li X, Cai C, Wang B, Cai S, Meng X, Zou F (2017) Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget 8(48):83831–83844. https://doi.org/10.18632/oncotarget.19747
Yuan Z, Cao A, Liu H, Guo H, Zang Y, Wang Y, Wang Y, Wang H, Yin P, Peng W (2017) Calcium uptake via mitochondrial uniporter contributes to palmitic acid-induced apoptosis in mouse podocytes. J Cell Biochem 118(9):2809–2818. https://doi.org/10.1002/jcb.25930
Yule DI, Betzenhauser MJ, Joseph SK (2010) Linking structure to function: recent lessons from inositol 1,4,5-trisphosphate receptor mutagenesis. Cell Calcium 47(6):469–479. https://doi.org/10.1016/j.ceca.2010.04.005
Zanesi N, Pekarsky Y, Croce CM (2005) A mouse model of the fragile gene FHIT: from carcinogenesis to gene therapy and cancer prevention. Mutat Res 591(1–2):103–109. https://doi.org/10.1016/j.mrfmmm.2005.05.016
Zeng F, Chen X, Cui W, Wen W, Lu F, Sun X, Ma D, Yuan Y, Li Z, Hou N, Zhao H, Bi X, Zhao J, Zhou J, Zhang Y, Xiao RP, Cai J, Zhang X (2018) RIPK1 binds MCU to mediate induction of mitochondrial Ca(2+) uptake and promotes colorectal oncogenesis. Cancer Res 78(11):2876–2885. https://doi.org/10.1158/0008-5472.CAN-17-3082
Zhang T, Zhao C, Luo L, Zhao H, Cheng J, Xu F (2012) The expression of Mcl-1 in human cervical cancer and its clinical significance. Med Oncol 29(3):1985–1991. https://doi.org/10.1007/s12032-011-0005-y
Zhang K, Jiao K, Xing Z, Zhang L, Yang J, Xie X, Yang L (2014) Bcl-xL overexpression and its association with the progress of tongue carcinoma. Int J Clin Exp Pathol 7(11):7360–7377
Zhao H, Li T, Wang K, Zhao F, Chen J, Xu G, Zhao J, Li T, Chen L, Li L, Xia Q, Zhou T, Li HY, Li AL, Finkel T, Zhang XM, Pan X (2019) AMPK-mediated activation of MCU stimulates mitochondrial Ca(2+) entry to promote mitotic progression. Nat Cell Biol 21(4):476–486. https://doi.org/10.1038/s41556-019-0296-3
Zhou X, Ren Y, Zhang J, Zhang C, Zhang K, Han L, Kong L, Wei J, Chen L, Yang J, Wang Q, Zhang J, Yang Y, Jiang T, Li M, Kang C (2015a) HOTAIR is a therapeutic target in glioblastoma. Oncotarget 6(10):8353–8365. https://doi.org/10.18632/oncotarget.3229
Zhou X, Ren Y, Kong L, Cai G, Sun S, Song W, Wang Y, Jin R, Qi L, Mei M, Wang X, Kang C, Li M, Zhang L (2015b) Targeting EZH2 regulates tumor growth and apoptosis through modulating mitochondria dependent cell-death pathway in HNSCC. Oncotarget 6(32):33720–33732. https://doi.org/10.18632/oncotarget.5606
Acknowledgments
PP is grateful to Camilla degli Scrovegni for continuous support. The Signal Transduction Laboratory is supported by the Italian Association for Cancer Research (AIRC: IG-23670 to P.P. and IG-19803 to C.G.), A-ROSE, Telethon (GGP11139B to P.P), Progetti di Rilevante Interesse Nazionale (PRIN2017E5L5P3 to P.P and PRIN20177E9EPY to C.G.), the Italian Ministry of Health (GR-2013-02356747 to C.G.), the European Research Council (ERC, 853057- InflaPML to C.G.), and local funds from the University of Ferrara to PP and C.G. SM is supported by the Italian Ministry of Health (GR-2016-02364602) and local funds from Marche Polytechnic University (Ancona, Italy). MRW was supported by the Internal Project of The Children’s Memorial Health Institute (No S141/2014).
The authors declare no conflict of interests. All authors read and approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Danese, A. et al. (2020). Cancer-Related Increases and Decreases in Calcium Signaling at the Endoplasmic Reticulum-Mitochondria Interface (MAMs). In: Pedersen, S.H.F., Barber, D.L. (eds) Organelles in Disease. Reviews of Physiology, Biochemistry and Pharmacology, vol 185. Springer, Cham. https://doi.org/10.1007/112_2020_43
Download citation
DOI: https://doi.org/10.1007/112_2020_43
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-22594-9
Online ISBN: 978-3-031-22595-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)