
Abstract
Physical activity has long-lasting beneficial effects by inducing metabolic adaptation, retarding biological ageing and reducing the risk of various age-related disorders and lifestyle-associated diseases such as type 2 diabetes mellitus, cancer, cardiovascular disorders and various types of inflammation, thereby extending healthy lifespan. Recent studies revealed that epigenetic mechanisms such as DNA methylation, histone modifications and microRNA expression are involved in exercise-induced adaptive responses. In this chapter, we first describe the processes of DNA methylation, histone modifications and microRNA, and then overview the effect of exercise on these epigenetic regulatory mechanisms. Finally, we discuss the relevance of epigenetics to ageing.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
5.1 Introduction: Genetics and Epigenetics
Gene expression is determined by the nucleotide sequences encoded in the genome and components of the transcriptional machinery. Expression changes during the development and differentiation of cells that have the same genome, starting from a single fertilized egg, and eventually giving rise to an individual consisting of many billions of differentiated and undifferentiated cells in a multi-cellular organism. Each tissue and organ consists of different sets of cells that express both cell-specific and common genes throughout life. Long-term cell-specific gene expression is determined by mechanisms including DNA methylation and histone modifications . Such mechanisms of gene expression are termed epigenetics.
Epigenetics describes a phenomenon in which a fixed pattern of gene expression in a cell or an organism is inherited from one generation to the next in cells or organisms, without changing the nucleotide sequence of the genome. However, this definition has been broadened to include the long-term stable control of cell-specific gene expression without changes in the nucleotide sequence. In addition, the recent development of molecular biology and technologies that allow the detection and quantification of minor RNAs identified the expression of microRNA (miRNA) as an important epigenetic mechanism that modulates gene expression. First, we briefly overview the processes of DNA methylation, histone modifications and miRNA expression, describe the effects of exercise on the epigenetic changes and finally discuss the relevance of epigenetics to ageing.
5.2 DNA Methylation
Genomic DNA methylation is the classical epigenetic modification and has a well-established mechanism of inheritance between generations. It involves the addition of a methyl group to a cytosine base at CpG dinucleotide sequences, although it can also occur in cytosine bases at other sequences. CpG islands are CpG-rich sequences that are often located in the promoter region of genes and are usually hypomethylated compared with CpG sequences downstream of the islands known as CpG shores. The hypermethylation of CpG islands is associated with transcriptional repression. Most CpG sequences in regions other than promoters are also methylated. Methylation is catalysed by a family of DNA methyltransferases, which use the substrate S-adenosylmethionine as the methyl donor. There are two categories of DNA methyltransferases: one that transmits the methylation pattern to the next generation during DNA replication (DNMT1, maintenance DNA methyltransferase) and another that introduces a methyl group to previously un-methylated cytosines to modulate gene expression and other chromatin functions (DNMT3a and DNMT3b, de novo DNA methyltransferases). DNA methylation at promoter regions makes the chromatin conformation more condensed, thereby suppressing transcription, whereas modifications inside the gene enhance transcription. DNA methylation influences histone modifications, and the two processes exhibit cross-talk. In addition, some miRNAs modulate DNA methyltransferases, thereby affect the status of DNA methylation [1].
The mechanism by which methyl groups are removed from methylated DNA has long been unclear, but it is now thought to be initiated by the oxidation of 5′-methylcytosine into 5-hyroxymethylcytosine. This oxidation is catalysed by the ten-eleven translocation (TET) family enzymes. Oxidised 5′-methylcytosine can be depleted passively during DNA replication or reverted to cytosine actively by base excision repair reactions that are catalysed by thymine DNA glycosylase [2]. Therefore, the methylation and demethylation of DNA can be modulated dynamically by methyltransferases and demethylation reactions.
5.3 Histone Modifications
Histones are highly basic proteins that are associated with nuclear DNA to form nucleosome complexes. These complexes exhibit a compact conformation of beads on a string, which packs DNA in the confined space in the nucleus and thereby represses the expression of most genes. The nucleosome consists of two of each histones, H2A, H2B, H3, and H4 to form an octamer of histones. Another histone, H1, is located between nucleosome particles, and is called the linker histone. A DNA sequence of ~150 nucleotides wraps around the histone octamer. Histones are modified by acetylation (at Lys [K] residues), methylation (K and Arg [R] residues), phosphorylation (Ser [S], Thr [T] and Tyr [Y] residues), ubiquitination (K), deimination (R), sumoylation (K) and carbonylation (unidentified residues). These modifications occur predominantly at the N-terminal regions of histones, which protrude from the globular domains and are therefore named histone tails. The modification of histones alters the conformation of chromatin to influence DNA transcription, replication and repair. The acetylation of K residues is one of the most frequent histone modifications. It neutralizes the positive charge of histones to relax the structure of the chromatin by reducing the electrostatic interaction with negatively charged DNAs, thereby activating transcription.
Histone acetylation is catalysed by histone acetyl transferases (HATs) using acetyl CoA as the substrate. There are many HAT isozymes, including p300 and CBP (cyclic AMP response element binding protein [CREB] binding protein). Acetylated histones are deacetylated by the catalytic action of histone deacetylases (HDACs). HDACs have attracted much attention because deacetylation is likely to play a key role in the dynamics of acetylation. HDACs are categorized as class I to IV. Class II HDACs are characterised by their localisation in both the nucleus and cytoplasm and trafficking between the two compartments, depending on the situation. Class III enzymes, also called silent information regulators (SIRTs), are nicotinamide adenine dinucleotide (NAD)-dependent enzymes whose activity is influenced by the energy status of the cell, as well as metabolic scenarios such as calorie restriction and DNA repair in which the amount of the coenzyme NAD can change significantly. The HATs and HDACs involved in histone dynamics are also called lysine acetyl transferases (KATs) and lysine deacetylase (KDACs), respectively, because some enzymes catalyse the acetylation and deacetylation of proteins in general, not just histones. For example, many regulatory proteins such as transcription factors and mitochondrial proteins involved in energy metabolism undergo reversible acetylation, which modulates their activity.
Methylation, another frequent histone modification, activates or inactivates the function of chromatin depending on which residues are modified [3]. For example, H3K4 tri-methylation (H3K4me3) at the promoter region activates gene expression [4], whereas H3K27 methylation at the same region suppresses transcription. The methylation is catalysed by histone methyltransferases (HMTs), and can occur as the mono-, di- and tri-methylation of K residues and the mono- and di-methylation of R residues at the ε-amino group. Unlike acetylation, methylation does not change the charge of the histones. The function of chromatin can be induced by the steric effects of methyl group(s) interacting with proteins such as transcription factors. Because of the interaction between histone methyltransferases with SET (Suppressor of variegation 3–9, Enhancer of zeste and Trithorax) domains and DNA methyltransferases, histone methylation influences DNA methylation and vice versa [5].
Other important modifications that can change the architecture of chromatin include the phosphorylation at S, T and Y residues, which can influence the transcription of genes on which the modified chromatin is localised. For example, the phosphorylation of histone H3 S10 enhances acetylation at K14 and suppresses acetylation at K9. The mono-ubiquitination at lysine residues in H2A and H2B also modulates transcription [6].
5.4 MicroRNA
miRNA is a recently identified epigenetic mechanism of gene regulation. miRNAs are non-coding RNAs ~22 nucleotides long, which are derived from the transcription of non-coding DNA. They bind to mRNA at either 3′- or 5′-terminal untranslated regions, decreasing the stability of the mRNA or repressing translation to modulate the amount of protein synthesised. Some miRNAs up-regulate gene expression by increasing the efficiency of translation [7]. miRNA precursors (primary miRNA, pri-miRNA) are transcribed from the miRNA coding regions of genes by RNA polymerase II and are processed to functional mature miRNAs by two endonucleases: Drosha in the nucleus and Dicer in the cytoplasm. The final products then form complexes with proteins that regulate translation of specific mRNAs with complementary sequences to the miRNA [8]. The export of intermediate products from the nucleus to the cytoplasm is dependent on the nuclear export receptor Exportin 5. There are more than 2500 different miRNAs in humans, the number being increasing. A single miRNA can modulate multiple mRNAs, and a single mRNA can be influenced by multiple miRNAs. The expression of more than 60 % of human genes are thought to be modified by miRNAs [9].
In addition to their intracellular roles in the regulation of gene expression, miRNAs are exported into the circulation as microvesicles (exosomes) wrapped in membranes or in complexes with proteins [10]. Circulating miRNAs can play roles in cell-cell communication, and are often used as biomarkers of physiological and pathological conditions. The intercellular transfer of functional miRNAs is mediated by exosomes [11].
5.5 Effects of Exercise on Epigenetics
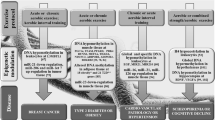
Regular physical activity exerts long-lasting beneficial effects by inducing metabolic adaptations, thereby reducing the risk of various age-related disorders and extending a healthy life span. These adaptive changes are thought to be induced by altered gene expression. Recent studies revealed that epigenetic mechanisms such as DNA methylation, histone modifications and microRNA expression are involved in this adaptation. Here, we review selected studies as examples of the complex epigenetic regulation that is induced by exercise.
5.5.1 DNA Methylation
Acute and regular exercise influences DNA methylation in skeletal muscle and other tissues by inducing the expression of genes that promote health. Barres et al. performed skeletal muscle biopsies in healthy sedentary individuals, and showed that an acute bout of aerobic exercise decreases the methylation of global DNA and the promoters of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) and pyruvate dehydrogenase kinase, isozyme 4 (PDK4) genes [12]. In addition, the mRNA expression of these genes was elevated markedly after exercise. Such exercise-induced changes were not seen in other genes such as myocyte enhancer factor 2A (MEF2a), myogenic differentiation 1 (MYOD1), citrate synthase (CS) and glyceraldehyde 3-phosphate dehydrogenase; a house-keeping protein (GAPDH). These results were reproduced in a model system where mouse soleus muscle was stimulated by ex vivo contraction, which decreased DNA methylation and increased mRNA expression; these changes were not dependent on factors external to the muscle itself. Therefore, the authors provided evidence that promoter hypomethylation is an early mechanism of the exercise-induced activation of responsive genes in skeletal muscle. In another model system using rat myotube cultures, the authors reported that ionomycin, which activates Ca2+ release, or the AMP activated protein kinase (AMPK) activator 5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR), similarly induced gene expression without promoter hypomethylation, suggesting that DNA methylation does not exclusively regulate exercise-induced gene activation.
Adipose tissue is important not only as an endocrine organ that produces adipokines and other factors with systemic effects but also as has a central role in preventing obesity and type 2 diabetes [13]. Rönn et al. [14] examined the effects of 6 months of exercise intervention on the genome-wide DNA methylation at CpG sites in subcutaneous adipose tissue biopsies in previously sedentary but healthy males. They found that genes associated with obesity and type 2 diabetes exhibited differential DNA methylation patterns before and after the exercise intervention. They studied three genes (RALBP1, HDAC4 and NCOR2) in detail, and showed that DNA methylation was increased in the exercised group. The mRNA expression of these genes was decreased significantly after exercise, as expected from the changes in the methylation levels. Ral-binding protein 1 (RALBP1) is an effector protein of the small GTPases RalA and RalB, which play roles in the pathogenesis of metabolic syndrome. Therefore, the authors assessed whether gene expression was suppressed by exercise. Data revealed that DNA methylation is responsible for the suppression of transcription using a luciferase reporter gene construct linked to a methylated human gene promoter in vitro. HDAC4, a histone deacetylase, suppresses GLUT4 transcription in adipocytes; therefore, reducing the transcription of HDAC4 by increasing DNA methylation likely elevates GLUT4 levels and subsequently glucose uptake. NCOR (Nuclear receptor corepressor) is a transcriptional corepressor that binds to and suppresses the enzyme activity of HDAC4. The authors used an in vitro model of 3T3 L1 adipocytes to reveal that silencing HDAC4 or NCOR2 increases lipogenesis by reducing the suppression of GLUT4 and increasing adipocyte glucose uptake.
Exercise has beneficial effects on brain functions such as promoting neurogenesis, learning, memory and improving emotions. Gomez-Pinilla et al. [15] studied the effects of 1 week of voluntary exercise on the methylation of brain-derived neurotrophic factor (BDNF) gene, as well as histone modifications in the hippocampus of young rats. BDNF is the most abundant neurotrophin in mammalian brains, and it promotes the growth, maintenance, function and protection of neurons. Exercise up-regulates BDNF expression in the hippocampus [16]. The transcription of promoter IV of BDNF is suppressed by methyl-CpG-binding protein (MeCP2) when the DNA is methylated in sedentary animals, and is activated by exercise. Therefore, exercise up-regulates BDNF mRNA. The authors found that exercise stimulated DNA demethylation of promoter IV, which is one of multiple promoters and is subjected to epigenetic regulation; therefore, the demethylation led to the dissociation of MeCP2 from the site occupying the promoter. Neuronal depolarisation induces the calcium-calmodulin-dependent protein kinase II (CaMKII) -dependent phosphorylation of MeCP2; phospho-MeCP2 is then dissociated from its binding site on the promoter, which allows transcription to start. As well as decreasing promoter methylation, exercise increased the acetylation of histone H3 in the nucleosomes that were associated with the BDNF promoter region (see also “Histone modifications” below). Exercise reduced HDAC5 mRNA and protein levels significantly, possibly by increasing H3 acetylation. Phospho-cyclic AMP-response-element binding protein (CREB) recruits CREB-binding protein, which has HAT-promoting activity. This also contributes to increased H3 acetylation. Therefore, voluntary exercise changes DNA methylation and histone acetylation at the promoter or BDNF gene, which up-regulates the expression of BDNF in the hippocampus to promote neural function.
5.5.2 Histone Modifications
McGee et al. [17] studied the effect of a single bout of cycling on global histone modifications in the skeletal muscle of young males. There was no significant change in histone H3 acetylation at K9 and K14 immediately after 1 h of exercise, which was reportedly associated with the initiation of transcription. In contrast, the acetylation of H3K36, which is associated with elongation, was increased. The authors suggested that exercise remodels chromatin to increase the transcription of genes related to energy and other metabolism in skeletal muscle and also increase transcript elongation [18, 19]. The increased acetylation of H3K36 was caused by the transport of class IIa HDACs from the nucleus by exercise. McGee et al. showed that the levels of HDAC4 and HDAC5 in the nucleus were decreased significantly after exercise, even though the total amount of enzymes in the cell was unchanged [17]. The exercise-induced up-regulation of AMP-activated protein kinase (AMPK) and CaMKII was apparently responsible for the increased HDAC phosphorylation that caused their nuclear export, thereby reducing the deacetylation (i.e., increasing the acetylation) of histone H3. Therefore, phosphorylation-dependent nuclear export plays a role in exercise-induced chromatin remodelling. The role of CaMKII activation in H3 hyperacetylation at the MEF2 binding site in the promoter region of GLUT4 was also suggested to be the mechanism by which GLUT4 expression is increased in skeletal muscle after intermittent swimming exercise in rats [20].
Much attention has been paid to the beneficial effects of exercise in the ageing brain. However, studies in young brains are limited, particularly regarding the epigenetic effects on gene expression. Able and Rissman studied the effects of 1 week of voluntary wheel running in young (46 days old) mice and found significantly increased H3 global acetylation in the hippocampus and cerebellum compared with sedentary animals [21]. This is consistent with previous findings where exercise increased hippocampal H3 acetylation in the chromatin at the BDNF gene promoter in adult rats [15]. Several HDAC mRNAs, including HDAC5, were decreased by exercise in both regions, consistent with the hypothesis that increased H3 acetylation could lead to increased expression of the BDNF gene [21]. The expression of DNMTs (DNMT1, DNMT3a and 3b) in the hippocampus was down-regulated by exercise. The suppression of DNMT gene activity might contribute to increased BDNF gene expression by increasing DNA methylation . A strong negative correlation was found between BDNF and HDAC1 expression, supporting the hypothesis that histone acetylation up-regulates the expression of BDNF. Therefore, it is possible that exercise promotes brain function by stimulating epigenetic modifications and up-regulating the expression of the genes that are required for neural function in the developing brain in young animals, such as BDNF and synapsin, which is involved in synaptic vesicle trafficking. These findings highlight the importance of physical activity for stimulating brain function in children. Moreover, it is important to note that running distance correlated positively with BDNF expression in both the hippocampus and cerebellum, further emphasizing the importance of physical activity.
The extent of histone acetylation that influences gene expression is dependent on HAT and HDAC activity, as well as the availability of HAT substrate (acetyl CoA). In view of reports demonstrating that histone acetylation in the hippocampus is a significant epigenetic change that is induced by exercise, Elsner et al. [22] studied the effects of a single session of forced treadmill exercise and chronic regular exercise for 2 weeks on the activity of these enzymes in the hippocampus of young rats. A single session of treadmill running suppressed the HDAC activity remarkably compared with sedentary control animals. However, chronic exercise had no significant effect on HDAC activity. HAT activity, which was studied using histone H3 and H4 peptides as substrates, was increased significantly toward H4, but not H3, by a single exercise session, whereas chronic exercise had no effect. These findings are consistent with the observations of other investigators such as Gomez-Pinilla et al. who reported that voluntary exercise reduced HDAC5 mRNA levels in the rat hippocampus, as discussed above. Similarly, McGee and Hargreaves demonstrated that the amount of HDAC4 and HDAC5 proteins was down-regulated in human skeletal muscle after exercise [15, 23].
Histone phosphorylation is generally less well studied than acetylation and methylation. Nevertheless, the phosphorylation of S, T, and Y residues plays an important role in modulating chromatin activity [24]. Chandramohan et al. reported that the acquisition of the behavioural immobility response by forced swimming exercise as a form of psychological stress induced a transient increase in the number of immuno-positive neurons in the dentate gyrus granular cell layer. This was associated with the phosphorylation of histone H3 S10 and the acetylation of H3K14 in the promoter region of the c-Fos gene, which led to the induction of c-Fos expression [25]. The authors showed that chromatin modifications altered the transcription of genes associated with neural functions by modulating the signal transduction pathways involving N-methyl-D-aspartate (NMDA) receptors and extracellular signal-regulated kinases (ERK), which are associated with learning and memory. The same authors investigated the effects of voluntary exercise on a running wheel for 4 weeks before exposure to stressful conditions on the above parameters [26]. Voluntary exercise increased the number of neurons that were positive for the expression of phospho-acetyl histone H3 and c-Fos in the dentate gyrus of rats exposed to stress compared with control sedentary animals.
Skeletal muscle is composed of mainly fast or slow fibres that express different amounts of myosin heavy chain (MHC). The soleus, a typical slow-type muscle, expresses predominantly type I MHC, while the plantaris, a typical fast-type muscle, expresses primarily type IIb and IIx MHC. MHC expression in slow-type fibres undergoes a shift to that typical of fast-type fibres under muscle unloading. Pandorf et al. [27] assessed whether the histone modifications in chromatin at the MHC gene differ among muscles (soleus and plantaris) or fibre types in rats. They also examined the effect of muscle unloading on possible chromatin remodeling. The association between the modification and the expression of the specific type of MHC was assessed using chromatin immunoprecipitation with antibodies specific for each modification (i.e., diacetylation at H3K9 and 14 and trimethylation at H3K4 [H3K4me3]) followed by PCR to quantify the amount of DNA precipitated. Data revealed high levels of expression of type I MHC in the soleus and type IIb and IIx MHC in the plantaris. Consistent with this differing pattern of gene expression, H3 acetylation was high at the type I MHC gene and low at the type IIb MHC and IIx MHC genes in the soleus. The opposite was true in the plantaris. A similar modification pattern was observed at H3K4me3. Animals were subjected to hind limb suspension to unload the legs for 7 days as an inactivity model, which shifts the muscle fibre type from slow to fast in the soleus. This resulted in a shift in the MHC gene expression toward that of the fast type. Therefore, the authors demonstrated that H3 acetylation and H3K4me3 are modulated dynamically in parallel with the shift in fibre type from slow to fast.
5.5.3 miRNA (Micro RNA) Expression
miRNAs play a different role in the epigenetic regulation of gene expression, as compared to DNA methylation and histone modifications, in that it does not involve a direct modification of chromatin. Instead, miRNAs regulate the amount of protein expressed in cells post-transcriptionally by modulating mRNA stability and the efficiency of translation. Nevertheless, they are, of late, becoming increasingly important in the regulation of cell- and tissue-specific gene expression, as well as potential biomarkers for physiological and pathological conditions. In particular, some miRNAs are transported in the blood and, therefore, can be quantified by blood sampling and RT-PCR. As such, there are an increasing number of publications discussing the roles of miRNAs in health and disease, including exercise. Although many are correlative, studies assessing the molecular mechanisms behind the regulation of miRNA expression and consequences of increases or decreases in their expression on the amount of target proteins have emerged.
miRNAs have been studied extensively in skeletal muscle [28]. Many specific miRNAs are enriched in muscle, such as miR-1 and miR-133, which play physiological and pathological roles in myogenesis, muscle growth, differentiation and disease. They are known collectively as myomiRs.
Russell et al. used thigh skeletal muscle biopsies to study the effect of an acute bout of exercise and short-term (10 days) endurance training using cycling on the expression of miRNAs and components involved in miRNA biogenesis; specifically, the two nucleases involved in the processing of precursor miRNAs and exportin 5, which exports the processed products from the nucleus to the cytoplasm [29]. They found a significant increase in miR-1 and miR-133 and a decrease in miR-9, miR-23 and miR-31 after the exercise. The down-regulation of miR-23 would up-regulate the synthesis of PGC-1α, its target protein, which regulates mitochondrial biogenesis. Therefore, miR-23 might be involved in the adaptive response to endurance exercise. miR-1 levels remained high and miR-31 remained low after training, suggesting that exercise had long-lasting effects on adaptation. The predicted target proteins of the miRNAs affected by the exercise were searched using sequence matches with the mRNAs of target proteins using bioinformatics. HDAC4 was a predicted target of miR-1, -133, -9 and -23, whereas nuclear respiratory factor 1 (NRF1) might be a target of miR-9, -23 and -31. Negative correlations were found between miR-9 and HDAC4, miR-31 and HDAC4, and miR-31 and NRF1 protein. These findings prompted the authors to assess the potential relationship between the miRNA levels and protein expression in myotube cultures. Although luciferase reporter assays using the HDAC4 and NRF1 genes revealed reduced luciferase activity in cells co-transfected with miR-31, as expected, the expression of HDAC4 and NRF1 was not affected. The authors hypothesized that this negative result might be due to the non-physiological conditions in the cell culture system, and suggested that different effects might be observed in human skeletal muscle.
Circulating miRNAs have been used as biomarkers for various physiological and pathological conditions. Physical activity, both acute exercise and endurance training, changes the levels of circulating miRNAs. Nielsen et al. [30] assessed alterations in the miRNA levels in the plasma of exercised humans to assess whether the response to different stimuli could be used as a signature for exercise interventions. They excluded samples that showed signs of haemolysis because red blood cells are an abundant source of miRNAs. The miRNA expression pattern changed immediately after cycling for 60 min (the down-regulation of eight miRNAs, including miR-106a and miR-221, and the upregulation of species including miR-338-3p, miR-330-3p). These findings suggest that circulating miRNAs are adjusted rapidly in response to different exercise stimuli, suggesting that these different patterns could be used to monitor exercise. Interestingly, some miRNAs increased 1 h after exercise, such as miR-143 and miR-145, which were enriched in the liver. Therefore, tissues other than muscle secrete miRNAs in response to exercise, which might affect the function of other organs. Although the liver could be thought of as a tissue that is less responsive to exercise, as compared with skeletal or cardiac muscle, it is important to study tissues such as the brain and liver, which might respond to physical activity to exert systemic beneficial effects. Consistent with this, Radak et al. [31] reported that the oxidative modification of proteins in rat brains was reduced and cognitive function was improved by regular swimming exercise. In addition, Nakamoto et al. [32] showed that regular exercise in rats using treadmill running reduced the oxidative modification of nuclear and mitochondrial DNA and up-regulated the expression of the repair enzyme OGG1 in the liver. Therefore, the systemic effects of exercise have been reported. Nevertheless, it should be noted that the circulating miRNAs might be derived passively, for example from damaged muscle due to exercise; however, not all muscle-enriched miRNAs were detected in the circulation [30]. Therefore, miRNA secretion is likely to be a selective process. As such, although the physiological consequences of miRNA secretion are not yet fully elucidated, the patterns of miRNAs in the blood might be a good biomarker of different physiological and pathological conditions without the need for tissue isolation.
5.6 Relevance to Ageing
Ageing can be defined as the gradual loss of homeostasis, which leads to decline in physiological function and increased susceptibility to diseases over time. In terms of longevity, the contribution of genetics is estimated to be 25–30 %, and the rest is likely to be due to environmental and lifestyle factors, as well as probability or chance, perhaps with the exception of long-lived cohorts such as centenarians aside. Epigenetic alterations might also contribute to longevity, reduce the risk of ageing -related diseases and also maintain a good quality of life in old age.
A report by Fraga et al. [33] suggests that epigenetic modifications might play a role in human ageing. They demonstrated that there were far more differences in the patterns of DNA methylation and histone acetylation in circulating lymphocytes in older (50 years of age) genetically identical monozygotic twins compared with younger (3 years of age) twins. Importantly, consistent with the epigenetic changes, the differences in gene expression between the older pairs were much greater than were those in young pairs. These findings suggest that an identical genome in early life could undergo different epigenetic modifications throughout life. The relatively small contribution of genetics towards longevity determination might be partly due to variable epigenetic modifications throughout life, which might lead to different disease susceptibility. However, it is important to note that a possible shift in the cell population (e.g. a shift to more memory T cells and fewer naive T cells during ageing) over time might have influenced this result [34].
Ageing is usually associated with reduced levels of global DNA methylation in CpG sequences, as well as the hypermethylation of some areas such as promoter regions. However, the physiological implication of changes in DNA methylation is generally unclear, although the age-related hypermethylation of the promoter regions of tumour suppressor genes increases the risk of carcinogenesis. Maegawa et al. [35] studied the widespread and tissue-specific changes in DNA methylation in mice with age. They found that 21 % of the promoter regions in the intestine exhibited increased methylation, whereas 13 % showed decreased methylation when animals were compared at 3 and 35 months of age. In the human colon, the proportion of autosomal genes which showed age-related hypermethylation was 10 %, while 1 % of genes showed hypomethylation when young (29–41 years) and old (61–72 years) individuals were compared. The authors concluded that the dysregulation of DNA methylation is a common feature of ageing in mammals. Apart of the aberrant methylation of protein-coding genes with age, ribosomal DNA clusters are also hypermethylated in the liver of old rats [36]. This hypermethylation might be associated with reduced gene transcription, which might lead to ageing-related changes in the expression of ribosomal RNA in old animals [37].
Frailty is an important issue for elderly individuals. Although it is not a disease and does not increase the mortality rate directly, it can cause ageing-related diseases. Bellizzi et al. found that worsening frailty status, as measured by loss of bodyweight, sarcopenia, muscle weakness, and reduced physical activity, was associated with decreased global DNA methylation in peripheral blood cells of individuals aged 65–105 over a 7-year-follow-up [38]. It was speculated that environmental factors such as diet and lifestyle might influence the methylation in various tissues, which could affect gene expression and thereby lead to local or systemic frailty.
Changes in the post-translational modification of histones occur with age, which might reduce gene expression. Kawakami et al. [39] reported that the acetylation of H3K9 was decreased and the phosphorylation of H3S10 was increased significantly in rat livers with age. Because these modifications suppress gene activity, these findings suggest that the age-related decline in chromatin functions might be due to such epigenetic changes.
Memory impairment is a common feature of ageing animals. The involvement of epigenetics is correlated with the ageing-related alterations in gene expression in the brain. Peleg et al. [40] studied histone acetylation in the hippocampus of young (3 months) and older (16 months) mice subjected to contextual fear conditioning. The older mice exhibited impaired associative learning, as detected by reduced freezing behaviour upon conditioning. The histone acetylation at H3K9 and K14 or H4K5, K8, K12 or K16 was similar in the two age groups of naïve mice. There was a transient increase in H3K9 and K14 and H4K5, K8 and K12 acetylation in young mice, whereas H4K12 acetylation was not up-regulated in the old mice. The increase in the other sites was similar in both groups of animals, suggesting that memory impairment in older animals correlated with defective learning-induced H4K12 acetylation. Data revealed that the hippocampal transcription of old mice remained almost unchanged in response to fear conditioning, whereas it increased in the young animals. They further used chromatin immunoprecipitation to demonstrate that the high level of gene expression induced by the conditioning correlated with increased H4K12 acetylation along the coding regions of genes, suggesting that transcription elongation was impaired in the old mice. Interestingly, the administration of HDAC inhibitors such as sodium butylate to older mice prior to the conditioning increased H4K12 acetylation significantly in the coding regions of learning-regulated genes. These findings suggest that the dysregulation of H4K12 is causally related to age-associated memory impairment. It is interesting to note that transgenic model mice with induced neuronal loss that are housed continuously in an environmentally enriched cage with wheels for voluntary running and other devices for physical activity exhibited increased histone H3 and H4 acetylation in the hippocampus at multiple sites, including H4K12 [41]. The model mice with neurodegenerative diseases that experienced environmental enrichment re-established access to long-term memories, exhibited dendrite sprouting and had an increased number of synapses, which could be mimicked by treatment with HDAC inhibitors that up-regulate histone acetylation. Therefore, physical exercise in an enriched environment might facilitate recovery from the impaired learning and memory that occurs with ageing by increasing histone acetylation.
Reports on the possible involvement of miRNAs in ageing are limited (see the special issue of Ageing Research Reviews 17: 1–98, 2014). Ageing-related decline in muscle function is a major concern for elderly individuals; therefore, they must maintain physical activity in daily life. Drummond et al. studied the expression of muscle-specific miRNAs and primary transcript pri-miRNAs in muscle biopsies taken from young (29 ± 2 years) and old (70 ± 2 year) males in response to leg extension exercises and the ingestion of leucine-enriched essential amino acid solution as an anabolic stimulus trying to correlate the stimulus with the expression of muscle specific miRNAs, upstream regulators (MyoD and myogenin) and downstream target proteins insulin-like growth factor-1 (IGF-1), HDAC4 and MEF2) that can be related to the promotion of the protein synthesis in the muscle [42]. The levels of pri-miRNA-1-1, -1-2, -133a-1 and 133a-2 were higher in older than younger males at baseline (before exercise). The expression of these miRNAs was reduced 6 h after exercise in young males compared with baseline. Mature miRNA-1 was down-regulated in response to the anabolic stimulus in only young individuals. The authors did not detect ageing-related differences in protein expression of IGF-1, HDAC4 and MEF2 at baseline, all of which are predicted or validated targets of miR-1. Studies in nematodes showed a decrease in miR-1 expression with advancing age [43]. Therefore, studies in worms and humans are contradictory. Drummond et al. [42] speculated that this discrepancy might be because nematodes lack satellite cells, which play an essential role in the muscle growth of mammals and have different types of muscle fibre. The failure of older subjects to down-regulate the expression of miR-1 following anabolic stimuli might be responsible for the reduced muscle protein synthesis observed in elderly subjects. Because they did not detect age-related changes in myogenic regulatory factors such as myogenin and MyoD, which are responsible for miR-1 expression, it is unclear whether transcription factors play a role in miRNA regulation in response to anabolic stimuli.
Age-associated cognitive decline is also a serious problem in older age individuals. The involvement of epigenetics , including miRNAs, in the brains of patients with neurodegenerative disease such as Alzheimer’s disease and Parkinson’s disease was suggested. Inukai et al. examined the expression of miRNAs in the brains of young (5 month-old) and old (24–25 month-old) mice, and discovered several novel miRNA candidates for predicted target proteins, including components of the insulin signal transduction pathways that are relevant to ageing [44]. Many miRNAs in the brain exhibit dynamic changes in expression by more than two-fold during ageing. The expression of most (80–95 %) of these miRNA was decreased with age, consistent with the findings in the human blood mononuclear cells of young and old individuals [45]. However, this contradicts a report showing the predominant up-regulation of miRNAs in mouse brains during ageing [46]. Therefore, studies on miRNAs in ageing are forming an exciting novel field, but further developments are needed.
One of the issues that attract the interest of researchers in gerontology is predicting the age of individuals and tissues. Recently, Horvath developed a method to predict the age of human cells and tissues using a large number of data sets assessing the methylation of CpG dinucleotides [47]. He found that the DNA methylation age is close to zero in embryonic and iPS cells and that it correlates with cell passage number. However, this differed from mitotic age because it tracks chronological age in non-proliferative tissues (for example, brain tissue). He stated that this model identified a highly heritable measure of ageing acceleration in studies of twins. This prediction was applicable to chimpanzee tissues as well. Although this prediction is interesting, it remains unclear whether it reflects physiological ageing accurately. These findings did not provide evidence of an association between premature ageing in progeria and accelerated DNA methylation age.
5.7 Concluding Remarks
In this chapter, we described advances in studies assessing the influence of exercise -induced epigenetic changes on gene expression and its relevance to the mechanisms of ageing . It is clear that DNA methylation , histone modifications and microRNA expression alter the phenotypes of many cell types, tissues and organs in response to physical activity or inactivity as well as in ageing. Nevertheless, it will take much more time for the entire picture to emerge regarding the epigenetic mechanisms that regulate gene expression during exercise, nutrition, ageing and different pathologies.
References
Fabbri M, Garzon R, Cimmino A et al (2007) MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA 104(40):15805–15810
Kohli RM, Zhang Y (2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502(7472):472–479
Lee MG, Villa R, Trojer P et al (2007) Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318(5849):447–450
Mikkelsen TS, Ku M, Jaffe DB et al (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448(7153):553–560
Cedar H, Bergman Y (2009) Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10(5):295–304
Zhang Y (2003) Transcriptional regulation by histone ubiquitination and deubiquitination. Genes Dev 17(22):2733–2740
Vasudevan S (2012) Functional validation of microRNA-target RNA interactions. Methods 58(2):126–134
Winter J, Jung S, Keller S et al (2009) Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11(3):228–234
Friedman RC, Farh KK, Burge CB et al (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19(1):92–105
Mitchell PS, Parkin RK, Kroh EM et al (2008) Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA 105(30):10513–10518
Valadi H, Ekstrom K, Bossios A et al (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9(6):654–659
Barres R, Yan J, Egan B et al (2012) Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab 15(3):405–411
Golbidi S, Laher I (2014) Exercise induced adipokine changes and the metabolic syndrome. J Diab Res 2014:726861
Rönn T, Volkov P, Davegardh C et al (2013) A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PLoS Genet 9(6):e1003572
Gomez-Pinilla F, Zhuang Y, Feng J et al (2011) Exercise impacts brain-derived neurotrophic factor plasticity by engaging mechanisms of epigenetic regulation. Eur J Neurosci 33(3):383–390
Neeper SA, Gomez-Pinilla F, Choi J et al (1995) Exercise and brain neurotrophins. Nature 373(6510):109
McGee SL, Fairlie E, Garnham AP et al (2009) Exercise-induced histone modifications in human skeletal muscle. J Physiol 587(Pt 24):5951–5958
Mahoney DJ, Parise G, Melov S et al (2005) Analysis of global mRNA expression in human skeletal muscle during recovery from endurance exercise. FASEB J 19(11):1498–1500
Holloszy JO (2008) Regulation by exercise of skeletal muscle content of mitochondria and GLUT4. J Physiol Pharmacol 59(Suppl 7):5–18
Smith JA, Kohn TA, Chetty AK et al (2008) CaMK activation during exercise is required for histone hyperacetylation and MEF2A binding at the MEF2 site on the Glut4 gene. Am J Physiol Endocrinol Metab 295(3):E698–E704
Abel JL, Rissman EF (2013) Running-induced epigenetic and gene expression changes in the adolescent brain. Int J Dev Neurosci 31(6):382–390
Elsner VR, Lovatel GA, Bertoldi K et al (2011) Effect of different exercise protocols on histone acetyltransferases and histone deacetylases activities in rat hippocampus. Neuroscience 192:580–587
McGee SL (1985) Hargreaves M (2011) Histone modifications and exercise adaptations. J Appl Physiol 110(1):258–263
Rossetto D, Avvakumov N, Cote J (2012) Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 7(10):1098–1108
Chandramohan Y, Droste SK, Arthur JS et al (2008) The forced swimming-induced behavioural immobility response involves histone H3 phospho-acetylation and c-Fos induction in dentate gyrus granule neurons via activation of the N-methyl-D-aspartate/extracellular signal-regulated kinase/mitogen- and stress-activated kinase signalling pathway. Eur J Neurosci 27(10):2701–2713
Collins A, Hill LE, Chandramohan Y et al (2009) Exercise improves cognitive responses to psychological stress through enhancement of epigenetic mechanisms and gene expression in the dentate gyrus. PLoS ONE 4(1):e4330
Pandorf CE, Haddad F, Wright C et al (2009) Differential epigenetic modifications of histones at the myosin heavy chain genes in fast and slow skeletal muscle fibers and in response to muscle unloading. Am J Physiol Cell Physiol 297(1):C6–C16
Sharma M, Juvvuna PK, Kukreti H et al (2014) Mega roles of microRNAs in regulation of skeletal muscle health and disease. Front Physiol 5:239
Russell AP, Lamon S, Boon H et al (2013) Regulation of miRNAs in human skeletal muscle following acute endurance exercise and short-term endurance training. J Physiol 591(Pt 18):4637–4653
Nielsen S, Akerstrom T, Rinnov A et al (2014) The miRNA plasma signature in response to acute aerobic exercise and endurance training. PLoS ONE 9(2):e87308
Radak Z, Kaneko T, Tahara S et al (2001) Regular exercise improves cognitive function and decreases oxidative damage in rat brain. Neurochem Int 38(1):17–23
Nakamoto H, Kaneko T, Tahara S et al (2007) Regular exercise reduces 8-oxodG in the nuclear and mitochondrial DNA and modulates the DNA repair activity in the liver of old rats. Exp Gerontol 42(4):287–295
Fraga MF, Ballestar E, Paz MF et al (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 102(30):10604–10609
Martin GM (2005) Epigenetic drift in aging identical twins. Proc Natl Acad Sci USA 102(30):10413–10414
Maegawa S, Hinkal G, Kim HS et al (2010) Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 20(3):332–340
Oakes CC, Smiraglia DJ, Plass C et al (2003) Aging results in hypermethylation of ribosomal DNA in sperm and liver of male rats. Proc Natl Acad Sci USA 100(4):1775–1780
Mori N, Mizuno D, Goto S (1978) Increase in the ratio of 18S RNA to 28S RNA in the cytoplasm of mouse tissues during aging. Mech Ageing Dev 8(4):285–297
Bellizzi D, D’Aquila P, Montesanto A et al (2012) Global DNA methylation in old subjects is correlated with frailty. Age 34(1):169–179
Kawakami K, Nakamura A, Ishigami A et al (2009) Age-related difference of site-specific histone modifications in rat liver. Biogerontology 10(4):415–421
Peleg S, Sananbenesi F, Zovoilis A et al (2010) Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328(5979):753–756
Fischer A, Sananbenesi F, Wang X et al (2007) Recovery of learning and memory is associated with chromatin remodelling. Nature 447(7141):178–182
Drummond MJ, McCarthy JJ, Fry CS et al (2008) Aging differentially affects human skeletal muscle microRNA expression at rest and after an anabolic stimulus of resistance exercise and essential amino acids. Am J Physiol Endocrinol Metab 295(6):E1333–E1340
Ibanez-Ventoso C, Yang M, Guo S et al (2006) Modulated microRNA expression during adult lifespan in Caenorhabditis elegans. Aging Cell 5(3):235–246
Inukai S, de Lencastre A, Turner M et al (2012) Novel microRNAs differentially expressed during aging in the mouse brain. PLoS ONE 7(7):e40028
Noren Hooten N, Abdelmohsen K, Gorospe M et al (2010) microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE 5(5):e10724
Li N, Bates DJ, An J et al (2011) Up-regulation of key microRNAs, and inverse down-regulation of their predicted oxidative phosphorylation target genes, during aging in mouse brain. Neurobiol Aging 32(5):944–955
Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol 14(10):R115
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Goto, S., Kawakami, K., Naito, H., Katamoto, S., Radak, Z. (2015). Epigenetic Modulation of Gene Expression by Exercise. In: Yu, B. (eds) Nutrition, Exercise and Epigenetics: Ageing Interventions. Healthy Ageing and Longevity, vol 2. Springer, Cham. https://doi.org/10.1007/978-3-319-14830-4_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-14830-4_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-14829-8
Online ISBN: 978-3-319-14830-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)