
Abstract
Similar to fludarabine and cladribine, clofarabine (2-chloro-2′-fluoro-2′-deoxyarabinosyladenine) is resistant to deamination by adenosine deaminase due to the presence of a halogen group at the two position of the base. However, unlike other 2′-deoxyadenosine analogs, clofarabine also has a halogen in the sugar that prevents cleavage of the glycosidic bond by purine nucleoside phosphorylase. The cytotoxic activity of clofarabine is due to both its inhibition of ribonucleotide reductase and its efficient incorporation in DNA, where it inhibits DNA synthesis. While some activity has been observed in lymphoid malignancies, clinical efficacy has primarily been observed in acute leukemias. The recommended dose of clofarabine for adult acute leukemia (40 mg/m2/day × 5 days) results in plasma levels of around 1 μM. The accumulation of clofarabine triphosphate in circulating leukemia cells is dose dependent, with a long half-life. This is particularly the case in responders, resulting in incremental increases in clofarabine triphosphate with every daily infusion of the drug. The actions of clofarabine triphosphate on ribonucleotide reductase and incorporation in the DNA repair patch suggest that a mechanism-based combination with arabinosylcytosine and DNA-damaging agents would be effective. Combination clinical trials have been conducted, while new trials are underway.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The natural nucleobases and nucleosides, such as adenine and adenosine, are important molecules in cells because they are components of DNA, RNA, and various intracellular nucleotides, such as cAMP, ATP, CTP, GTP, and UTP as well as their corresponding deoxynucleotides. Nucleotides are synthesized de novo in human cells, and nucleobases and nucleosides are usually only present in cells at low concentrations. However, if available, these compounds are salvaged from the environment and used for the synthesis of DNA, RNA, and nucleotide triphosphates, a process that involves many enzymes. Analogs of these compounds are a major class of drugs (referred to as antimetabolites) that are important in the treatment of viral infections and cancer. The FDA has approved over 25 nucleoside analogs for use in the treatment of viral infections [1] and 14 antimetabolites for the treatment of cancer [2].
Nucleobase and nucleoside analogs are structurally similar to the natural compounds and are used by many of the anabolic enzymes in a manner similar to that of the natural compounds, usually without interfering with the activity of the enzyme. For the most part, the analog and its metabolites must appear to the cell as the natural compound. However, because of the small structural difference in these molecules, one of its metabolites eventually interacts with an enzyme in a negative way, resulting in its inhibition, and the resulting disruption of nucleic acid synthesis or function results in the death of the cancer cell or virus. The structural modifications must be small so that the anabolic enzymes recognize them as substrates and convert them to a metabolite that eventually disrupts a vital enzyme in nucleotide metabolism. However, it is important to realize that small structural differences in nucleoside analogs can have a major impact on their mechanism of action and their clinical activity. As we illustrate in this chapter, clofarabine (abbreviated in the literature as CAFDA, chloro-fluoro-ara-A, and Cl-F-ara-A) or 9-β-D-[2-deoxy-2-fluoro-arabinofuranosyl]-2-chloroadenine (Fig. 16.1) is an excellent example of this principle. The drug is sold as Clolar in the USA and Evoltra in Europe. The only structural difference between clofarabine and cladribine is the replacement of a hydrogen atom at the 2′ position with a fluorine atom. However, this small difference endows clofarabine with biochemical activities that are sufficiently different from cladribine as to result in different clinical activities.

Structures of deoxyadenosine, cladribine, fludarabine, and clofarabine
The basis of selectivity with antiviral agents usually involves selective activation by an enzyme expressed by the virus or selective inhibition of an enzyme important to the synthesis of the viral genetic material. With anticancer analogs, the basis for selectivity is much less clear because the enzymes involved in activation and activity in cancer cells are identical to those found in normal cells. However, the antitumor selectivity of nucleoside analogs can be attributed to enhanced metabolism in cancer cells or inhibition of DNA synthesis or function. The goal of cancer drugs is to prevent replication of the cancer cell because it is this replication that leads to the death of the patient. Since most host cells in humans are not replicating and therefore are not sensitive to agents that inhibit DNA synthesis, inhibition of DNA synthesis in cancer cells affords modest selectivity. However, some tissues have cells with rapid turnover (bone marrow, gastrointestinal tract, and hair follicles), and toxicity in these tissues due to the inhibition of DNA synthesis limits the amount of drug that can be administered.
Because clofarabine is an analog of 2′-deoxyadenosine (dAdo), the metabolism of dAdo in human cells, summarized in Fig. 16.2, introduces the enzymes that are important in the activation and antitumor activity of clofarabine. Adenosine deaminase is expressed in the plasma; therefore, the primary metabolic route for dAdo that is injected into animals is deamination by adenosine deaminase to deoxyinosine, which is rapidly cleaved by purine nucleoside phosphorylase (PNP) to hypoxanthine. Hypoxanthine can be reused by hypoxanthine/guanine phosphoribosyl transferase (HGPRT) for the generation of purine nucleotides, although it is more likely to be converted by xanthine oxidase to xanthine and uric acid, which is eventually excreted. Adenine-containing nucleosides are extremely poor substrates for mammalian PNP [3]; therefore, little, if any, dAdo that is administered intravenously is cleaved to adenine in mammalian systems. However, dAdo is a good substrate for bacterial PNP [4] and would be readily cleaved in the gastrointestinal tract. A small amount of dAdo administered to animals is phosphorylated to dAMP, primarily by deoxycytidine kinase (dCyd kinase), which is further phosphorylated to dADP and dATP. dATP is the primary acid-soluble intracellular metabolite of dAdo, and it is used as a substrate for the synthesis of DNA. An important enzyme in the generation of deoxynucleotides for DNA synthesis is ribonucleotide reductase. The activity of this enzyme is controlled by the natural NTPs and dNTPs to maintain the proper balance of nucleotides for DNA synthesis. As can be seen in Fig. 16.2, dATP represses ribonucleotide reductase activity, whereas ATP stimulates its activity.

Schema depicting the metabolism of deoxyadenosine. Enzymes are boxed in rectangles. Abbreviations: NDP nucleoside diphosphate, dCyd 2′-deoxycytidine, dAdo 2′-deoxyadenosine, dIno 2′-deoxyinosine, Hx hypoxanthine, Xa xanthine, AD adenosine deaminase, PNP purine nucleoside phosphorylase, HGPRT hypoxanthine-guanine phosphoribosyl transferase, RNR ribonucleotide reductase, IMP inosine 5′-monophosphate, GMP guanosine 5′-monophosphate, AMP adenosine 5′-monophosphate
2 Rationale for the Synthesis of Clofarabine
Numerous enzymes are involved in the activation and activity of nucleoside analogs, which is a primary reason why drug discovery within this class of compounds is a very empirical process. It is difficult, if not impossible, to design a compound that will behave as needed with each of the enzymes that must be involved in its activity. Therefore, the rational drug discovery strategy with these molecules is to generate numerous analogs that are based on a thorough understanding of the biological activity of the many synthesized compounds and then evaluate them in appropriate model systems to identify the agents with the desired activities.
A thorough explanation of the rationale for the synthesis of clofarabine can be found in a review by Secrist et al. [5]. In brief, nucleosides with halogens at the two position of the adenine base are known to be very poor substrates for adenosine deaminase [6] and therefore are known to be stable in plasma. These compounds (2-halo-2′-deoxyadenosine analogs) potently inhibit tumor cell growth. In addition, 2′-flouro nucleosides have been synthesized and are known to have biological activity [7]. 2′-Halogens on nucleoside analogs are also resistant to cleavage by acid and purine nucleoside phosphorylases, another feature that would contribute to their in vivo stability. Therefore, in a search for new anticancer agents, numerous 2′-halo (F, Cl, or Br) and 2-halo (F, Cl, or Br) dAdo analogs have been synthesized and evaluated for biological activity in both in vitro and in vivo tumor models [8]. Clofarabine was the most potent compound in in vitro cell-killing assays and demonstrated excellent activity against numerous human tumor xenografts in mice [9, 10].
3 Transport
Human cells will salvage natural nucleosides, if available, for nucleic acid synthesis in place of de novo synthesis, and cells express numerous transporters to aid the entry of these compounds. Both equilibrative transporters (hENT1, hENT2, and hENT3) and concentrative transporters (hCNT1, hCNT2, and hCNT3) are expressed on most human cells [11]. Because of the abundance of these transporters, natural nucleosides in the cell culture will equilibrate across the cell membrane in just a few seconds. To be active, all nucleoside analogs (with the possible exceptions of deoxycoformycin and forodesine) must be able to cross cellular membranes to interact with their cellular targets. Most nucleoside analogs that are useful in the treatment of various diseases are recognized by these transporters; therefore, they also equilibrate across cell membranes very quickly. Precise transport studies have been conducted with clofarabine, and it has been shown that it is transported into cells by both human equilibrative and concentrative nucleoside transporters [12]. Cells producing hENT1 exhibited the highest uptake of clofarabine (0.14 pmole/μl), followed by hCNT2 (0.025 pmole/μl), whereas uptake in cells producing hENT2 or hCNT1 was not higher than that in cells that were nucleoside transport deficient. In oocytes expressing recombinant transporters, the efficiency of transport by hCNT3 (1.3) was much greater than that of hENT1, hENT2, or hCNT2 (0.11, 0.20, or 0.15). Although not entirely consistent, these results indicate that clofarabine can enter cells by all known transporters except hCNT1, which is selective for pyrimidine nucleosides. In addition, at high concentrations, diffusion of clofarabine directly through the membrane is also possible [12].
Comparative studies have shown [12] that cladribine had the highest efficiency of transport with hENT1, with a Vmax/Km value of 1.8 versus 0.7 for clofarabine and 0.8 for fludarabine (9-β-D-[arabinofuranosyl]-2-F-adenine). The uptake and flux of clofarabine, cladribine, and fludarabine were very similar in human renal proximal tubule cells, which express equilibrative hENT1, hENT2, and hCNT3 [13]. These results indicate that although there are some differences in transport, all of these compounds are readily transported across human membranes.
Some studies have evaluated the importance of ABCG2 to the activity of clofarabine. This ABC transporter is known as the breast cancer resistance protein and transports various metabolites, including nucleoside monophosphates, out of cells. Cells transfected with DNA constructs, resulting in overexpression of human or mouse ABCG2, had significantly reduced transport of both clofarabine and cladribine metabolites [14]. These results were correlated with decreased levels of cellular metabolites of cladribine in ABCG2-expressing cells, which were much less sensitive to clofarabine and cladribine. In these studies, the ABCG2 transporter transported both cladribine monophosphate and cladribine itself. The results of a study by Nagai et al. [15] supported these conclusions and showed that ABCG2 primarily transports clofarabine. Their results indicate that the metabolism of clofarabine and its cytotoxicity are strongly determined by the interplay between dCyd kinase and ABCG2, and they suggest that inhibition of ABCG2 will improve antitumor activity in tumors in which dCyd kinase levels are low. The role of ABC transporters in nucleoside analog metabolism and activity has recently been reviewed [16].
4 Metabolism
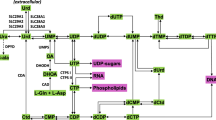
The metabolism of clofarabine is schematically presented in Fig. 16.3. Once inside the cell, clofarabine is converted to clofarabine triphosphate [17]. Xie and Plunkett [18] showed that at low concentrations, the primary intracellular metabolite was clofarabine monophosphate, which was approximately twice the level of clofarabine triphosphate. At high concentrations of clofarabine, the monophosphate and triphosphate levels were similar. Clofarabine diphosphate levels were very low, indicating that phosphorylation of the monophosphate of clofarabine is the rate-limiting step in its activation to the triphosphate. This characteristic is similar to that of cladribine [19] and indicates that the monophosphate kinase does not prefer molecules with substitutions at the two position of a purine nucleoside as large as a Cl atom. In the case of most other deoxynucleoside analogs used for the treatment of cancer, the nucleoside kinase is usually the rate-limiting enzyme in their activation, and the triphosphate usually accounts for more than 90% of the intracellular metabolites.

Schematic of clofarabine’s mechanism of action. Clofarabine is transported into cells by both diffusion and facilitated diffusion. It is monophosphorylated by deoxycytidine kinase (dCK) and then serially phosphorylated by other kinases to form clofarabine triphosphate, the active moiety. The triphosphate acts to terminate DNA chain elongation and inhibit repair by incorporating it into the DNA chain through competitive inhibition of DNA polymerases. It also inhibits ribonucleotide reductase, with a reduction of dNTP pools, and induces apoptosis through direct and indirect actions on mitochondria by releasing cytochrome c and other proapoptotic factors (Obtained with permission from Bonate et al. [73])
Clofarabine triphosphate accumulates at high levels in CEM cells [17, 18]: treatment of cells with low concentrations of clofarabine (1–3 μM for 2–4 h) resulted in concentrations of clofarabine triphosphate that were 50–100 μM, which were similar to intracellular dATP concentrations in untreated cells. The accumulation of clofarabine triphosphate was dose dependent up to 10 μM. Similar intracellular concentrations of clofarabine triphosphate were achieved in all phases of the cell cycle (G1, S, and G2-M) [18]. Clinical studies have indicated that plasma clofarabine levels of 1.5 μM are achieved at the maximally tolerated dose of clofarabine in patients with acute leukemias after a 1-h infusion, and this treatment resulted in a clofarabine triphosphate concentration of approximately 19 μM in blast cells [20].
When clofarabine was removed from the medium of K562 and CEM cell cultures, clofarabine triphosphate had a half-life in cells of 1–3 h [18, 21]. However, in circulating leukemic cells, more than 50% of the clofarabine triphosphate was still present 24 h after clofarabine transfusion [20]. The metabolism of clofarabine and cladribine has been compared in ex vivo studies with CLL and AML cells that were obtained from patients [22]. In these studies, the half-life of clofarabine triphosphate was longer than that in CEM cells but similar to that of cladribine triphosphate (7.3 vs. 4.3 h, respectively). The enzymes involved in the degradation of nucleoside triphosphates in cells have not been studied in great detail. Triphosphate metabolites of nucleoside analogs do not readily cross cell membranes and are therefore trapped in the cells in which they were created, whereas nucleosides themselves will freely distribute across the cell membrane. Therefore, the antitumor activity of these agents can be extended well beyond the time that the drug circulates in the plasma because the active metabolite is maintained in tumor cells long after the drug has disappeared from the plasma. The long retention time of clofarabine triphosphate contributes to its lasting antitumor activity, even though the plasma half-life is fairly short [20, 23].
Cytoplasmic 5′-nucleotidase cN-II is one of the seven 5′-nucleotidases found in mammals. This enzyme catalyzes the removal of the 5′-phosphate from purine 5′-monophosphates and is involved in the maintenance of cellular nucleotide pools. Therefore, it can also degrade analogs of purine nucleoside monophosphates and could theoretically interfere with the activation of clofarabine. In cell culture experiments with CEM and RL cells, inhibition of this enzyme did not affect the cytotoxicity of clofarabine and had only modest effects on the induction of apoptosis [24], indicating that it has only a modest effect on the metabolism of clofarabine when clofarabine is constantly present in the cell culture medium. The results of these studies suggest that the monophosphate pool maintained by the dCyd kinase/5′-nucleotidase activity ratio in the presence of excess extracellular clofarabine is sufficient to maintain cytotoxic clofarabine triphosphate levels in the cell. However, it is possible that such inhibitors would have a more dramatic effect on metabolism after the removal of clofarabine from the external environment, which happens in vivo. As clofarabine triphosphate is degraded to the clofarabine monophosphate, inhibition of this enzyme could prevent further degradation, resulting in re-phosphorylation by the monophosphate kinase and an increase in the intracellular half-life of clofarabine triphosphate.
As expected for a dAdo analog, clofarabine was incorporated into DNA, but not RNA, indicating that it is a DNA-directed drug [18]. At low concentrations, clofarabine is mostly incorporated into internal positions in the DNA (>80%), suggesting that DNA synthesis is not inhibited by the incorporation of clofarabine monophosphate into the growing DNA strand. However, at high concentrations (1 or 10 μM), much more clofarabine is found at terminal positions in DNA. At 10 μM, 65% of the clofarabine in DNA is found at terminal positions, which suggests that DNA synthesis is inhibited at the site of incorporation.
The primary enzyme involved in the activation of clofarabine in tumor cells is dCyd kinase [17, 18, 22]. Clofarabine is a very good substrate for this enzyme, with Km and Vmax values that are similar to those of dCyd [21] but much better than those of dAdo (the catalytic efficiency of dAdo is only 0.3% that of dCyd). Clofarabine was modestly better substrate for dCyd kinase than was cladribine. Its catalytic efficiency was three to four times that of cladribine, with a Vmax that was ten times greater. The X-ray crystal structure of dCyd kinase with clofarabine in the active site has been determined [25]. The results of these studies indicate that the conformation of the enzyme/clofarabine complex was similar to structures of the pyrimidine-bound complexes and that the interactions between the 2-Cl group and its surrounding hydrophobic residues contribute to the high catalytic efficiency of dCyd kinase with clofarabine.
Decreased dCyd kinase activity has been shown to result in resistance to clofarabine in various cell culture systems [22, 26–29]. Decreased histone acetylation [28], but not methylation of the dCyd kinase gene [26, 28, 29], was associated with decreased dCyd kinase expression. In HL-60 cells, the decreased activation of clofarabine was associated with decreased expression of dCyd kinase, deoxyguanosine kinase, and hENT1, hENT2, and hCNT3 genes [30]. Studies have shown that there are genetic variations in the expression of dCyd kinase activity in patient populations and suggest that these variations are responsible for the variable rates of activation of clofarabine and other nucleoside analogs [31].
Treatment of cells with clofarabine and other nucleoside analogs can enhance dCyd kinase activity in many, but not all, cell lines [32–34]. Others have shown that inhibition of DNA synthesis can enhance dCyd kinase activity [35, 36] by phosphorylating dCyd kinase at serine 74 [35, 37, 38]. The enhancement of dCyd kinase activity by clofarabine can be exploited to improve antitumor activity by combining clofarabine with other nucleoside analogs that are activated by dCyd kinase [33, 34, 39–41].
Clofarabine is also a good substrate for deoxyguanosine kinase [42], an enzyme that is expressed in mitochondria. The catalytic efficiency of clofarabine with this enzyme is similar to that of both deoxyguanosine and cladribine, although both the Km and Vmax values for the two deoxyadenosine analogs are 10–20 times greater than are those for deoxyguanosine. The contribution of deoxyguanosine kinase to the phosphorylation of clofarabine in cells is low due to the much higher expression of dCyd kinase activity in most cell types [43, 44], but it could be important in cells that express low amounts of dCyd kinase. Resistance to clofarabine in CEM cells was correlated with decreased dCyd kinase activity but not with decreased deoxyguanosine kinase [22, 27].
5 Mechanisms of Action
The mechanism of action of clofarabine resembles that of other dAdo analogs and is schematically presented in Fig. 16.3 . Below, we describe the primary actions of this nucleoside analog.
5.1 Inhibition of DNA Synthesis
As with other anticancer nucleoside analogs, the primary activity of clofarabine that is responsible for its antitumor activity is the inhibition of DNA synthesis [17, 18, 21, 45]. RNA and protein synthesis are inhibited by clofarabine only at high concentrations. Clofarabine triphosphate also inhibited yeast poly(A) polymerase activity [46], but it is unclear how much the inhibition of this enzyme contributes to the antitumor activity of clofarabine. DNA synthesis is immediately inhibited in cells treated with clofarabine, and a 3-h incubation with 0.3 μM clofarabine resulted in prolonged inhibition [45], which did not recover to more than 20% of control values 72 h after removal of the drug. The immediate and long-lasting inhibition of DNA synthesis in cells treated with clofarabine strongly suggests that the enzymes that are critical for DNA replication, such as DNA polymerase and ribonucleotide reductase, are the primary targets for clofarabine and that disruption of DNA function as a result of the incorporation of clofarabine into daughter strands is of secondary importance to the activity of clofarabine.
5.1.1 Inhibition of Ribonucleotide Reductase
Incubation of cells with clofarabine results in significant decreases in intracellular deoxynucleotide pools [17, 18, 45], suggesting that clofarabine triphosphate is a potent inhibitor of ribonucleotide reductase, a critical enzyme that is involved in the de novo synthesis of deoxynucleotides [47]. This hypothesis was confirmed in experiments in which clofarabine triphosphate potently inhibited ribonucleotide reductase in cell-free extracts [17], and incubation of cells with clofarabine dramatically decreased the conversion of radiolabeled purines and pyrimidines to their respective deoxynucleotide triphosphates [45]. Clofarabine had the most impact on dCTP and dATP pools, a modest impact on dGTP pools, and no effect on dTTP pools [17, 45]. The concentration of clofarabine triphosphate that is required to inhibit ADP reduction from ribonucleotide reductase isolated from K562 cells by 50% is 65 nM, which is similar to that seen with cladribine triphosphate [17].
The activity of ribonucleotide reductase in cells is tightly controlled by the natural nucleotides to ensure that the cell has all of the deoxynucleotides needed for DNA synthesis in the correct concentrations. It is known that dATP is a potent regulator of ribonucleotide reductase activity and inhibits the reduction of ADP, UDP, and CDP [47]. Therefore, the results described above suggest that clofarabine triphosphate interacts with ribonucleotide reductase in the allosteric binding site as an analog of dATP. However, in studies with purified human ribonucleotide reductase, Aye and Stubbe [48] showed that both clofarabine diphosphate and clofarabine triphosphate were potent inhibitors of ribonucleotide reductase activity. They showed that clofarabine triphosphate is a rapid reversible inhibitor, with a Ki of 40 nM, and that clofarabine diphosphate is a slow-binding, reversible inhibitor, with a Ki of 17 nM. Their results indicated that clofarabine triphosphate bound to the allosteric binding site (A site) on the α subunit. The A site controls the rate of the reaction, and when dATP is bound, the enzyme is inactive. Clofarabine diphosphate bound to the substrate-binding site (C site). Although clofarabine diphosphate levels are much lower than clofarabine triphosphate levels in cells [18], the relatively long half-life of the inhibited state means that clofarabine diphosphate could still significantly contribute to the inhibition of ribonucleotide reductase in cells. Finally, inhibition of enzyme activity by either compound was associated with protein oligomerization that was more kinetically stable than were dATP-induced hexamers [48–50].
5.1.2 Inhibition of DNA Polymerization
Clearly, inhibition of ribonucleotide reductase is sufficient to result in the inhibition of DNA synthesis; however, clofarabine triphosphate is also a good substrate and inhibitor of DNA polymerases α and ε, two important enzymes that are involved in the replication of chromosomal DNA [17, 45]. Clofarabine triphosphate was found to be a potent inhibitor of the incorporation of dATP by DNA polymerase α, with a Ki value of approximately 1 μM (Km for dATP was 4 μM), but it was a weak inhibitor of DNA polymerase β (involved in DNA repair) and γ (involved in mitochondrial DNA synthesis). A nucleoside triphosphate analog can interact with a DNA polymerase in one of three ways: it can interfere with DNA synthesis without being used as a substrate; it can be used as a substrate but not interfere with continued DNA synthesis; or it can be used as a substrate and cause disruption of subsequent DNA chain elongation. The results of studies in intact cells with radiolabeled clofarabine indicate that it is incorporated into DNA in internal and terminal positions [18], but these studies did not indicate whether DNA synthesis was inhibited due to the incorporation. In addition, inhibition of dATP incorporation by isolated polymerases does not indicate that DNA synthesis is actually inhibited because it is possible that clofarabine triphosphate inhibited dATP incorporation by acting as an alternative substrate without interfering with subsequent elongation.
The results of DNA-sequencing studies supported those of the radiolabeled studies, showing that clofarabine triphosphate is used by DNA polymerase α [17], with Km and Vmax values that are similar to those of dATP (as well as cladribine triphosphate). These studies also showed that the ability of polymerase α to add new nucleotides after the incorporation of clofarabine triphosphate was significantly less than that after the incorporation of dATP. The Km of adding dGTP after the incorporation of clofarabine triphosphate was 100-fold greater than that after the incorporation of dAMP. The Vmax for the incorporation of dGTP was similar, regardless of whether dAMP or clofarabine monophosphate was incorporated previously. Interestingly, the incorporation of cladribine had only a modest effect on the subsequent use of dGTP as a substrate (only a threefold difference to that seen after the incorporation of dAdo). The effect of clofarabine incorporation on subsequent chain elongation was similar to that of fludarabine, an agent known to interfere with DNA synthesis. The effect of clofarabine triphosphate on DNA polymerase ε activity was similar to that seen with DNA polymerase α, except that it more effectively inhibited chain elongation by DNA polymerase ε [45]. This enhanced inhibition of chain elongation resulted in significant inhibition of DNA synthesis by this enzyme at clofarabine triphosphate concentrations that were only 3% those of dATP.
Under conditions in which control reactions extended all of the labeled DNA primer to the end of the template strand, DNA polymerase α was able to extend the DNA chain with clofarabine triphosphate in place of dATP across a single thymidine residue in the template strand, but it was not able to extend past two consecutive thymidine residues [17]. This result was very similar to that seen with fludarabine triphosphate but was different from that seen with cladribine triphosphate. DNA polymerase α was able to extend the DNA chain with cladribine triphosphate across two consecutive thymidine residues in the template strand, but it could not extend beyond three consecutive thymidine residues. These results indicate that clofarabine caused significant chain termination by DNA polymerase α, which is consistent with the immediate and lasting inhibition of DNA synthesis seen in cells treated with clofarabine [18].
These results seem to be in conflict with the results that indicate that at low concentrations, more than 80% of the clofarabine incorporated into DNA is incorporated into internal positions [18], which suggests that elongation of DNA is not inhibited by the incorporation of clofarabine into DNA. The above results indicate that DNA polymerases can easily extend the DNA chain across single thymidine residues with clofarabine triphosphate but that chain extension is dramatically inhibited by the incorporation of two successive clofarabine nucleotides. Since it is likely that single incorporations are more prevalent in DNA than are multiple incorporations, there would be many more internal clofarabine molecules in DNA, even though the inhibition of DNA synthesis due to chain termination is the primary action of clofarabine that is responsible for its antitumor activity. In addition, chain termination caused by two successive clofarabine incorporations would have one internal and one terminal, for a ratio of 50%. These results indicate that DNA replication is severely inhibited, even under circumstances in which most of the incorporated clofarabine is in internal positions in the DNA.
Clofarabine has also been shown to inhibit nucleotide excision DNA repair in chronic lymphocytic leukemia (CLL) lymphocytes isolated from patients that were induced by treatment with 4-hydroperoxycyclophosphamide [51]. Clofarabine was as potent as fludarabine in the inhibition of DNA repair in this model; however, clofarabine triphosphate demonstrated maximal inhibition at one-tenth the concentration of fludarabine triphosphate. Although not directly studied in this paper, the inhibition of nucleotide excision repair could be due to the inhibition of the DNA polymerases involved in the repair or to the inhibition of ribonucleotide reductase activity. In addition, inhibition of DNA repair could explain the activity of clofarabine against resting non-proliferating tumor cells [52].
Collectively, the results presented in the previous paragraphs indicate that DNA synthesis is inhibited in cells treated with clofarabine through two distinct but complementary actions: inhibition of ribonucleotide reductase and DNA polymerase activity. The potent inhibition of ribonucleotide reductase activity by clofarabine nucleotides enhances its inhibition of the replicative DNA polymerases (self-potentiation) by decreasing the intracellular concentration of the natural substrate, dATP, which competes with clofarabine triphosphate for use as a substrate by these enzymes (i.e., in the presence of ribonucleotide reductase inhibition, less clofarabine triphosphate is needed to inhibit DNA polymerase activity). Indeed, clofarabine combines the strong DNA-terminating activity of fludarabine (a weak inhibitor of ribonucleotide reductase) and the strong inhibition of ribonucleotide reductase by cladribine (a weak inhibitor of DNA polymerases) into one molecule that is a strong inhibitor of both DNA elongation and ribonucleotide reductase. The relative importance of clofarabine’s actions to the antitumor activity of clofarabine is not known, but it is likely that both targets significantly contribute to its cell-killing action.
5.2 Repair of DNA Containing Clofarabine
The removal of clofarabine from the 3′-end of DNA chains by 3′-exonucleases that are associated with the DNA replication complex is an important part of the mechanism of action of this compound that has not yet been evaluated. If clofarabine is quickly removed from the 3′-terminus of the DNA chain, DNA synthesis can continue normally, although the polymerase would still have a chance to incorporate another molecule of clofarabine in the repair site, resulting in a futile incorporation and removal cycle. The incorporation of two or more clofarabine residues sequentially in the DNA may be harder to repair than single incorporations and could represent a greater block to DNA synthesis. Once the DNA polymerase extends beyond the incorporated analog, DNA synthesis can continue normally. It is not known whether clofarabine that is successfully incorporated into the DNA chains (beyond the replication sites) results in DNA that cannot function normally. However, Hentosh and colleagues [53, 54] demonstrated that the random incorporation of cladribine into DNA can disrupt transcriptional regulation by altering DNA binding of TATA-binding protein and can have deleterious effects on cell function. It is likely that the random incorporation of clofarabine would have similar effects. However, if there are negative consequences to the incorporation of clofarabine into DNA chains, these actions are secondary in importance to the cell-killing mechanism of action of clofarabine.
5.3 Induction of Apoptosis
The inhibition of replication in cells normally leads to the turning on of replication checkpoint pathways. Stalled replication forks can threaten DNA replication fidelity, and cells respond to replication blocks by triggering checkpoint pathways that monitor replication fork progression [55]. This monitoring of DNA synthesis operates normally during low-intensity replication stress and is required for tumor cells to resume cell cycle progression. However, during chronic or high-intensity replication stress, stalled forks do not restart after removal of the stressor, resulting in irreversible S-phase arrest, possible mitotic catastrophe, and cell death.
The inhibition of DNA synthesis and the DNA damage caused by clofarabine resulted in the induction of apoptosis in CEM and HCT116 cells [56–58]. Replication stress induced by the treatment of CEM cells with 10 μM clofarabine leads to Chk1 phosphorylation, which is accompanied by Chk1 downregulation, concomitant apoptosis, and cell death [5]. Incubation with clofarabine resulted in a dose- and time-dependent downregulation of Bcl-XL, Mcl-1, and Cdc25A proteins, but it did not affect Cdc25C [57]. Treatment with clofarabine also resulted in the dephosphorylation of Akt and its downstream effectors (Bad and FKHRL1). In addition, there was a marked increase in the population of cells in G1/S and early S phase. Clofarabine led to more apoptotic cell death than did fludarabine, suggesting that DNA damage caused by clofarabine is more easily recognized by the surveillance systems that initiate apoptotic cascades [51].
5.4 Activity Against Non-proliferating Cancer Cells
Clofarabine is also active against quiescent human lymphocytes and monocytes [52]. Treatment of these cells with clofarabine induced DNA strand breaks, which were markedly reduced by supplementation of the culture medium with dCyd. Clofarabine also interfered with mitochondrial integrity in primary CLL cells [59], which led to the release of the proapoptotic mitochondrial proteins cytochrome C and apoptosis protein-activating factor (Apaf-1). The activity of clofarabine was similar to that of cladribine, and both agents induced mitochondrial damage more effectively than did fludarabine. The investigators proposed three mechanisms that may be responsible for the mitochondrial damage in non-proliferating CLL cells: (1) incorporation into nuclear DNA, resulting in DNA damage and inducing an apoptotic cascade; (2) binding to Apaf-1, resulting in the induction of apoptosis [60]; and (3) direct interference with mitochondrial function. They felt that mitochondrial damage caused by clofarabine or cladribine resulted from all three effects, whereas mitochondrial damage caused by fludarabine resulted from only nuclear DNA damage and Apaf-1 binding. They suggested that the direct activity against mitochondrial function could be due to the binding of clofarabine and cladribine nucleotides to proteins that are known to regulate mitochondrial functions. Clofarabine triphosphate was able to activate Apaf-1-dependent caspase activity in CLL cell extracts with kinetic parameters that were identical to those of dATP [60].
The caspase cascade is initiated by cytochrome c binding to Apaf-1; this induces it to undergo a conformational change in the presence of dATP, leading to the formation of Apaf-1 multimers and the recruitment of procaspase-9, forming a complex known as the apoptosome. The subsequent autocatalysis of procaspase-9 is followed by the proteolytic activation of procaspases 3, 6, and 7. Therefore, clofarabine triphosphate can contribute to the formation and activation of the apoptosome, which results in the activation of the caspase cascade. These studies notwithstanding, the mechanisms responsible for the disruption of mitochondrial integrity have not been fully elucidated, and it is possible that mitochondrial damage is the result of an apoptotic cascade resulting from the inhibition of ribonucleotide reductase or DNA polymerase activity.
Clofarabine has also been shown to inhibit DNA repair in quiescent cells [51]. Although clofarabine triphosphate is a weak inhibitor of DNA polymerase β [17], a polymerase that is involved in base excision repair, it is a potent inhibitor of DNA polymerase ε [45] and likely DNA polymerase δ, two enzymes that are also involved in DNA repair [61]. As noted, clofarabine is a potent inhibitor of ribonucleotide reductase, and the reduction in intracellular deoxynucleotides could inhibit DNA repair due to the lack of substrates. Because DNA is constantly being repaired in quiescent cells due to its spontaneous degradation, inhibition of DNA repair could induce the apoptotic response that has been observed in these cells.
5.5 Miscellaneous Activities
Clofarabine and cladribine were both found to bind to the A(2A) receptor, with Kis of 17 and 15 μM, respectively [62]. Clofarabine was the only adenosine analog to bind to the A(3) receptor, with a Ki of 10 μM. None of the adenosine analogs bound to the A(2B) receptor. Clofarabine was an agonist of the A(1) receptor. These results suggest that interaction with the adenosine receptors contributes to the toxicity observed with clofarabine and other deoxyadenosine analogs.
Treatment of breast cancer cell lines with clofarabine has been shown to reactivate silenced tumor suppressor genes such as APC, PTEN, and RARβ2 through hypomethylation and transcriptional upregulation [63, 64]. This action needs to be evaluated in liquid tumor cell lines as well as in primary tumor cells during therapy.
6 Cancer Selectivity of Clofarabine
Similar to other nucleoside analogs, clofarabine is not selective to cancer or to a specific cancer histological type. Nonetheless, as described above, most of the cell line studies are directed toward acute myelogenous or lymphoblastic diseases. The activity of the drug in the clinical setting also suggests that it has preferred activity toward ALL and AML. Within AML, cell line data suggest that in cells harboring FLT3 internal tandem duplication, prolonged treatment would be more effective, as there is a failure of the cell cycle checkpoint in FLT3-ITD mutant cells [65]. In addition to leukemias, other hematological malignancies, such as myeloma [41, 66, 67] and lymphoma [68–70], appear to show sensitivity to this analog. Among solid tumors, breast [63, 71], colon [9, 58], and brain tumors [72] have been evaluated.
In the clinical setting, described in detail in the next chapter (Chap. 17), most of clofarabine’s activity has been observed in acute leukemias. For these diagnoses, the drug has been approved for pediatric leukemias and is also effective in the treatment of adult leukemias. Solid tumors showed limited activity, perhaps due to myelosuppression as a dose-limiting toxicity. Unlike other drugs, clofarabine was approved first in pediatric leukemias. Clofarabine’s development, leading to its approval in acute leukemias, was reviewed in detail by Bonate et al. [73].
7 Clinical Pharmacology
To obtain investigational new drug approval and to use clofarabine clinically, MD Anderson conducted animal toxicology and pharmacology studies. On the basis of tolerated doses in mice and dogs, an IC10 of 15 mg/m2/day was selected for humans. Because of clofarabine’s similarity to fludarabine, the clinical trial design was based on this dAdo analog (i.e., every cycle consisted of a daily dose for 5 days, and the cycle was repeated every 28 days). The initial phase I investigation of clofarabine was performed in patients with solid tumors and hematological malignancies. As with fludarabine, the dose of clofarabine that was selected as the starting dose was high [74]. Myelosuppression was the dose-limiting toxicity; thus, the maximum tolerated dose in the solid tumor setting was much lower than the starting dose: 2 mg/m2/day for 5 days. For acute leukemias, where myelosuppression is not a concern, the dose was identified as 40 mg/m2/day [75].
7.1 Plasma Pharmacology
During phase I investigations, a de-escalation of dose in solid tumor patients and an increase in acute leukemia patients resulted in the evaluation of several doses (2, 4, 7.5, 11.25, 15, 22.5, 30, 40, and 55 mg/m2/day). The number of patients at each dose was low; however, there appeared to be a dose-dependent accumulation of plasma clofarabine (Fig. 16.4). The peak concentration occurred at the end of the 60-min infusion. For adult acute leukemias at the defined MTD, the plasma peak level was 1.5 μM (range, 0.4–3.2 μM) [75]. For pediatric acute leukemias, the doses were tested between 11.25 and 70 mg/m2/day for 5 days. The MTD was 52 mg/m2/day, which was slightly higher than what was identified for adult acute leukemia patients. At this MTD, the plasma concentration was a median of 1.1 μM [76]. On the basis of clofarabine’s clinical activity, identified specifically in adult acute leukemias during phase I clinical studies, a phase II study was conducted in 62 individuals with acute leukemias [23]. Plasma pharmacological studies in 25 patients elucidated that at the recommended phase II dose in acute leukemias (40 mg/m2/day × 5 days), the peak plasma concentration of clofarabine was 1 μM (range, 0.26–1.94 μM). Elimination kinetics in five patient samples suggested biphasic kinetics. With this pharmacokinetic profile, there was 0.04 μM clofarabine remaining in plasma at 24 h after infusion. This is when the second dose is administered.

Dose-dependent accumulation of plasma clofarabine. Blood samples were obtained on the first day from each patient at the end of clofarabine infusion. Plasma was separated, and clofarabine concentrations were quantitated as described by Gandhi et al. [20]. The numbers of samples were 2 at 7.5, 3 at 11.5, 3 at 15, 3 at 22.5, 2 at 30, 7 at 40, and 3 at 55 mg/m2/day. The dashed lines represent 95% confidence intervals (Obtained with permission from Gandhi et al. [20])
Clofarabine is eliminated renally. In the isolated perfused rat kidney model, clofarabine clearance was nonlinear. However, the concentration of drug was much higher than the physiological level achieved in humans [77]. The findings in a rat in vivo model system suggested that clofarabine is a substrate for the cimetidine-sensitive organic cation transporter (probably OCT2) system in kidneys [77].
Population pharmacokinetic studies, initially in pediatric patients [78] and then in both children and adults, demonstrated [79] that while there were no differences in plasma pharmacology by sex, tumor type, or race, age was a variant. For adults, the volume of distribution and β half-life of clofarabine elimination were much higher, resulting in a higher exposure of the drug. In addition to clofarabine, 6-ketoclofarabine, a metabolite of clofarabine, was also detected in the plasma; however, this was a minor metabolite of no known significance. Since clofarabine is also used in the clinic as an oral drug [68, 80–84], it is important to mention that the bioavailability of oral clofarabine is around 60%, suggesting that this is a viable formulation of this drug [79]. Clofarabine is resistant to purine nucleoside phosphorylase cleavage, which provides a rationale for an oral formulation of clofarabine that is not cleaved by microbial PNP in the gut. Clinically, oral clofarabine was well tolerated and had an MTD of 3 mg/m2/day for 21 days in relapsed and refractory non-Hodgkin lymphoma [68].
7.2 Cellular Pharmacology
As mentioned in the plasma pharmacology section, there were nine different doses used during the phase I clinical trial of clofarabine. The clofarabine triphosphate level analyzed in 40 patients suggested a dose-dependent increase in cellular triphosphate levels. However, there was heterogeneity at each dose level. For adult acute leukemias, at the MTD, the median triphosphate value was 19 μM (range, 3–52 μM) [75], which was similar to that observed in pediatric patients (range, 6 and 19 μM). Importantly, this level of cytotoxic triphosphate was sufficient in the complete and sustained inhibition of DNA synthesis [76].
The efficacy of clofarabine in acute leukemia resulted in a phase II investigation in relapsed refractory acute leukemias. At the MTD (40 mg/m2/day × 5 days), the peak clofarabine triphosphate level was 15 μM (range, 1–44 μM, n = 29). The accumulation was similar in different diagnoses, such as AML, ALL, and CML. The elimination profile of clofarabine triphosphate indicated that the triphosphate was retained, with a half-life of 24 h. Interestingly, the 5-day profile of triphosphate accumulation suggested that the levels of triphosphate had an incremental increase in responders, while they remained fairly similar in nonresponders (Fig. 16.5) [23]. A detailed comparison of clofarabine triphosphate pharmacological traits demonstrated that in responders, the peak value at the end of the first infusion was higher (18 μM; range, 5–44 μM; n = 16) than that in nonresponders (10 μM; range, 1–23 μM; n = 11). Furthermore, because of longer retention of triphosphate in leukemia blasts of responders, the second day end-of-infusion value was 30 μM (1–67 μM, n = 15) for responders and 9 μM (1–23 μM, n = 10) for nonresponders.

Incremental accumulation of clofarabine triphosphate in two representative patients. Pretreatment and end-of-infusion samples were obtained from two patients after several infusions of clofarabine to quantitate the intracellular levels of triphosphate. Rectangular bars on the abscissa indicate clofarabine infusions. a Nonresponder; b responder (Obtained with permission from Kantarjian et al. [23])
Clofarabine triphosphate competes with the endogenous dATP pool for incorporation into DNA and inhibition of DNA synthesis. The concentration of dATP was a median of 1.8 μM (range, 0.4–22 μM; n = 9). These data suggest that the ratio of clofarabine triphosphate to dATP favors incorporation of the analog into DNA. While DNA incorporation was not studied, DNA synthesis inhibition showed a dose-dependent [20] decrease in the DNA synthetic capacity of circulating leukemia cells in patients receiving this therapy. The decline was almost complete and was maintained at higher doses (Fig. 16.6).

Inhibition of DNA synthesis. Blood samples were obtained from two patients (patients 2 and 23) before and at the end of clofarabine infusion on the indicated days. Leukemia cells were isolated and incubated with thymidine, as described by Jeha et al. [76]. Data are expressed as a percentage of the untreated (control) value. Pre indicates, prior to infusion, and eoi, at the end of infusion (Obtained with permission from Jeha et al. [76])
8 Combination Rationales and Trials
Clinically, the maximum use of clofarabine was in pediatric and adult acute leukemias. The standard of care for AML is high-dose cytarabine (ara-C) or ara-C-containing regimens. Biochemically, it was demonstrated that clofarabine triphosphate modulated the accumulation of ara-C triphosphate (the cytotoxic metabolite of ara-C), resulting in two- to threefold higher levels of ara-C triphosphate in AML cell lines and AML primary blasts [33]. This is similar to the fludarabine- and cladribine-mediated modulation of ara-C triphosphate accumulation in AML [85–87]. The maximum augmentation of ara-C triphosphate accumulation was observed with 1 μM clofarabine, a concentration that is achievable at the MTD (40 mg/m2/day).
These observations were the basis of several clinical trials in which clofarabine was combined with ara-C. With these combinations, the plasma pharmacology of clofarabine was not impacted; however, clofarabine infusion prior to ara-C administration resulted in a 1.4-fold median increase in the ara-C triphosphate accumulation in five of eight patients [88]. Because of the phase I nature of this study, the doses of clofarabine varied. Systematic cellular pharmacokinetic studies of ara-C triphosphate are needed to fully evaluate the impact of MTD clofarabine on ara-C triphosphate accumulation in AML blasts. Clinically, a randomized study of ara-C, alone and in combination with clofarabine, demonstrated that the overall response rate was 47% in the combination arm compared to 23% in the single-agent arm. Importantly, the addition of clofarabine doubled the rate of complete remissions from 18% to 35%; however, toxicity was also increased with the combination [89]. A retrospective comparison of the clofarabine and ara-C combination with fludarabine and the ara-C couplet combined with granulocyte colony-stimulating factor suggested that there was an advantage to replacing fludarabine with clofarabine [90].
As described above, clofarabine’s actions on DNA replication are also mimicked during DNA repair. Both inhibition of ribonucleotide reductase and incorporation of analog triphosphate in DNA inhibit synthesis of the DNA repair patch [51]. For the initiation of a DNA damage response, alkylating agents such as cyclophosphamide and busulfan have been combined with clofarabine in the preclinical setting [91, 92] and during clinical trials in both adult AML [93] and ALL [94]. During these investigations, it was demonstrated that clofarabine administration, before or after cyclophosphamide, resulted in greater cytotoxicity as well as an increased damage response, measured as phosphorylation of H2AX (γH2AX).
In addition to these above-mentioned combination approaches, sorafenib has been combined with clofarabine and ara-C [95]. Pharmacodynamic endpoints suggested that this combination inhibited the phosphorylation of Akt, S6 ribosomal protein, and 4EBP-1 in leukemia blasts. Although this was a small study, it showed a high response rate. For example, all five patients with FLT3-ITD and four of six patients with wild-type FLT3 showed a response. Such combinations need to be evaluated in a large cohort of patients. Inhibition of the mTOR pathway by temsirolimus was also shown to be effective in combination with clofarabine in AML cell lines [96]. The opposite actions of clofarabine and decitabine on the R2 subunit of ribonucleotide reductase were evaluated and modeled in AML. This modeling study suggested that a synergistic interaction occurs between these two nucleoside analogs [97]. Low-dose decitabine and clofarabine have been combined in the treatment of MDS and AML [98]. In addition, clofarabine has been combined with other therapeutics, such as gemtuzumab ozogamicin [99].
9 Summary
Clofarabine’s actions, its similarities to and differences from cladribine and fludarabine, and its efficacy in acute leukemias led to its FDA approval. When combined with other chemotherapeutic drugs and targeted agents, it has resulted in clinical improvements in pediatric and adult acute leukemias. New combination strategies are being designed and tested for acute leukemias.
References
Jordheim LP, Durantel D, Zoulim F, Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov. 2013;12(6):447–64.
Parker WB. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev. 2009;109(7):2880–93.
Zimmerman TP, Gersten NB, Ross AF, Miech RP. Adenine as substrate for purine nucleoside phosphorylase. Can J Biochem. 1971;49(9):1050–4.
Jensen KF, Nygaard P. Purine nucleoside phosphorylase from Escherichia coli and Salmonella typhimurium. Purification and some properties. Eur J Biochem/FEBS. 1975;51(1):253–65.
Secrist 3rd JA, Thottassery J, Parker WB. Clofarabine: from design to approval. In: Herdewijn P, editor. Modified nucleosides: in biochemistry, biotechnology and medicine. Weinheim: WILEY-VCH Verlag GmbH & Co. KGaA; 2008. p. 631–46.
Maguire MH, Sim MK. Studies on adenosine deaminase. 2. Specificity and mechanism of action of bovine placental adenosine deaminase. Eur J Biochem/FEBS. 1971;23(1):22–9.
Watanabe KA, Reichman U, Hirota K, Lopez C, Fox JJ. Nucleosides. 110. Synthesis and antiherpes virus activity of some 2′-fluoro-2′-deoxyarabinofuranosylpyrimidine nucleosides. J Med Chem. 1979;22(1):21–4.
Montgomery JA, Shortnacy-Fowler AT, Clayton SD, Riordan JM, Secrist 3rd JA. Synthesis and biologic activity of 2′-fluoro-2-halo derivatives of 9-beta-D-arabinofuranosyladenine. J Med Chem. 1992;35(2):397–401.
Takahashi T, Kanazawa J, Akinaga S, Tamaoki T, Okabe M. Antitumor activity of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl) adenine, a novel deoxyadenosine analog, against human colon tumor xenografts by oral administration. Cancer Chemother Pharmacol. 1999;43(3):233–40.
Waud WR, Schmid SM, Montgomery JA, Secrist 3rd JA. Preclinical antitumor activity of 2-chloro-9-(2-deoxy-2-fluoro-beta-D- arabinofuranosyl)adenine (Cl-F-ara-A). Nucleosides Nucleotides Nucleic Acids. 2000;19(1–2):447–60.
Cass CE. Nucleoside transport. In: Georgopapadakou NH, editor. Drug transport in antimicrobial and cancer chemotherapy. New York: Marcel Dekker; 1995. p. 403–51.
King KM, Damaraju VL, Vickers MF, Yao SY, Lang T, Tackaberry TE, et al. A comparison of the transportability, and its role in cytotoxicity, of clofarabine, cladribine, and fludarabine by recombinant human nucleoside transporters produced in three model expression systems. Mol Pharmacol. 2006;69(1):346–53.
Elwi AN, Damaraju VL, Kuzma ML, Mowles DA, Baldwin SA, Young JD, et al. Transepithelial fluxes of adenosine and 2′-deoxyadenosine across human renal proximal tubule cells: roles of nucleoside transporters hENT1, hENT2, and hCNT3. Am J Physiol Renal Physiol. 2009;296(6):F1439–51.
de Wolf C, Jansen R, Yamaguchi H, de Haas M, van de Wetering K, Wijnholds J, et al. Contribution of the drug transporter ABCG2 (breast cancer resistance protein) to resistance against anticancer nucleosides. Mol Cancer Ther. 2008;7(9):3092–102.
Nagai S, Takenaka K, Nachagari D, Rose C, Domoney K, Sun D, et al. Deoxycytidine kinase modulates the impact of the ABC transporter ABCG2 on clofarabine cytotoxicity. Cancer Res. 2011;71(5):1781–91.
Fukuda Y, Schuetz JD. ABC transporters and their role in nucleoside and nucleotide drug resistance. Biochem Pharmacol. 2012;83(8):1073–83.
Parker WB, Shaddix SC, Chang CH, White EL, Rose LM, Brockman RW, et al. Effects of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl)adenine on K562 cellular metabolism and the inhibition of human ribonucleotide reductase and DNA polymerases by its 5′-triphosphate. Cancer Res. 1991;51(9):2386–94.
Xie C, Plunkett W. Metabolism and actions of 2-chloro-9-(2-deoxy-2-fluoro-beta-D- arabinofuranosyl)-adenine in human lymphoblastoid cells. Cancer Res. 1995;55(13):2847–52.
Avery TL, Rehg JE, Lumm WC, Harwood FC, Santana VM, Blakley RL. Biochemical pharmacology of 2-chlorodeoxyadenosine in malignant human hematopoietic cell lines and therapeutic effects of 2-bromodeoxyadenosine in drug combinations in mice. Cancer Res. 1989;49(18):4972–8.
Gandhi V, Kantarjian H, Faderl S, Bonate P, Du M, Ayres M, et al. Pharmacokinetics and pharmacodynamics of plasma clofarabine and cellular clofarabine triphosphate in patients with acute leukemias. Clin Cancer Res. 2003;9(17):6335–42.
Parker WB, Shaddix SC, Rose LM, Shewach DS, Hertel LW, Secrist 3rd JA, et al. Comparison of the mechanism of cytotoxicity of 2-chloro-9-(2-deoxy-2- fluoro-beta-D-arabinofuranosyl)adenine, 2-chloro-9-(2-deoxy-2-fluoro- beta-D-ribofuranosyl)adenine, and 2-chloro-9-(2-deoxy-2,2-difluoro- beta-D-ribofuranosyl)adenine in CEM cells. Mol Pharmacol. 1999;55(3):515–20.
Lotfi K, Mansson E, Spasokoukotskaja T, Pettersson B, Liliemark J, Peterson C, et al. Biochemical pharmacology and resistance to 2-chloro-2′-arabino-fluoro-2′-deoxyadenosine, a novel analogue of cladribine in human leukemic cells. Clin Cancer Res. 1999;5(9):2438–44.
Kantarjian H, Gandhi V, Cortes J, Verstovsek S, Du M, Garcia-Manero G, et al. Phase 2 clinical and pharmacologic study of clofarabine in patients with refractory or relapsed acute leukemia. Blood. 2003;102(7):2379–86.
Jordheim LP, Marton Z, Rhimi M, Cros-Perrial E, Lionne C, Peyrottes S, et al. Identification and characterization of inhibitors of cytoplasmic 5′-nucleotidase cN-II issued from virtual screening. Biochem Pharmacol. 2013;85(4):497–506.
Zhang Y, Secrist 3rd JA, Ealick SE. The structure of human deoxycytidine kinase in complex with clofarabine reveals key interactions for prodrug activation. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 2):133–9.
Leegwater PA, De Abreu RA, Albertioni F. Analysis of DNA methylation of the 5′ region of the deoxycytidine kinase gene in CCRF-CEM-sensitive and cladribine (CdA)- and 2-chloro-2′-arabino-fluoro-2′-deoxyadenosine (CAFdA)-resistant cells. Cancer Lett. 1998;130(1–2):169–73.
Mansson E, Flordal E, Liliemark J, Spasokoukotskaja T, Elford H, Lagercrantz S, et al. Down-regulation of deoxycytidine kinase in human leukemic cell lines resistant to cladribine and clofarabine and increased ribonucleotide reductase activity contributes to fludarabine resistance. Biochem Pharmacol. 2003;65(2):237–47.
Waud WR, Gilbert KS, Parker WB, Secrist JA. Isolation and characterization of a murine P388 leukemia line resistant to clofarabine. Nucleosides Nucleotides Nucleic Acids. 2011;30(11):826–38.
Yamauchi T, Uzui K, Nishi R, Shigemi H, Ueda T. Cytarabine-resistant leukemia cells are moderately sensitive to clofarabine in vitro. Anticancer Res. 2014;34(4):1657–62.
Shigemi H, Yamauchi T, Tanaka Y, Ueda T. Novel leukemic cell lines resistant to clofarabine by mechanisms of decreased active metabolite and increased antiapoptosis. Cancer Sci. 2013;104(6):732–9.
Lamba JK, Crews K, Pounds S, Schuetz EG, Gresham J, Gandhi V, et al. Pharmacogenetics of deoxycytidine kinase: identification and characterization of novel genetic variants. J Pharmacol Exp Ther. 2007;323(3):935–45.
Spasokoukotskaja T, Sasvari-Szekely M, Hullan L, Albertioni F, Eriksson S, Staub M. Activation of deoxycytidine kinase by various nucleoside analogues. Adv Exp Med Biol. 1998;431:641–5.
Cooper T, Ayres M, Nowak B, Gandhi V. Biochemical modulation of cytarabine triphosphate by clofarabine. Cancer Chemother Pharmacol. 2005;55(4):361–8.
Parker WB, Shaddix SC, Gilbert KS, Shepherd RV, Waud WR. Enhancement of the in vivo antitumor activity of clofarabine by 1-beta-D-[4-thio-arabinofuranosyl]-cytosine. Cancer Chemother Pharmacol. 2009;64(2):253–61.
Csapo Z, Sasvari-Szekely M, Spasokoukotskaja T, Talianidis I, Eriksson S, Staub M. Activation of deoxycytidine kinase by inhibition of DNA synthesis in human lymphocytes. Biochem Pharmacol. 2001;61(2):191–7.
Spasokoukotskaja T, Sasvari-Szekely M, Keszler G, Albertioni F, Eriksson S, Staub M. Treatment of normal and malignant cells with nucleoside analogues and etoposide enhances deoxycytidine kinase activity. Eur J Cancer. 1999;35(13):1862–7.
Amsailale R, Van Den Neste E, Arts A, Starczewska E, Bontemps F, Smal C. Phosphorylation of deoxycytidine kinase on Ser-74: impact on kinetic properties and nucleoside analog activation in cancer cells. Biochem Pharmacol. 2012;84(1):43–51.
Keszler G, Spasokoukotskaja T, Sasvari-Szekely M, Eriksson S, Staub M. Deoxycytidine kinase is reversibly phosphorylated in normal human lymphocytes. Nucleosides Nucleotides Nucleic Acids. 2006;25(9–11):1147–51.
Guo Y, Xu X, Qi W, Xie C, Wang G, Zhang A, et al. Synergistic antitumor interactions between gemcitabine and clofarabine in human pancreatic cancer cell lines. Mol Med Rep. 2012;5(3):734–8.
Valdez BC, Li Y, Murray D, Ji J, Liu Y, Popat U, et al. Comparison of the cytotoxicity of cladribine and clofarabine when combined with fludarabine and busulfan in AML cells: enhancement of cytotoxicity with epigenetic modulators. Exp Hematol. 2015;43(6):448–61.e2.
Valdez BC, Wang G, Murray D, Nieto Y, Li Y, Shah J, et al. Mechanistic studies on the synergistic cytotoxicity of the nucleoside analogs gemcitabine and clofarabine in multiple myeloma: relevance of p53 and its clinical implications. Exp Hematol. 2013;41(8):719–30.
Sjoberg AH, Wang L, Eriksson S. Substrate specificity of human recombinant mitochondrial deoxyguanosine kinase with cytostatic and antiviral purine and pyrimidine analogs. Mol Pharmacol. 1998;53(2):270–3.
Arner ES, Eriksson S. Mammalian deoxyribonucleoside kinases. Pharmacol Ther. 1995;67(2):155–86.
Lindemalm S, Liliemark J, Gruber A, Eriksson S, Karlsson MO, Wang Y, et al. Comparison of cytotoxicity of 2-chloro- 2′-arabino-fluoro-2′-deoxyadenosine (clofarabine) with cladribine in mononuclear cells from patients with acute myeloid and chronic lymphocytic leukemia. Haematologica. 2003;88(3):324–32.
Xie KC, Plunkett W. Deoxynucleotide pool depletion and sustained inhibition of ribonucleotide reductase and DNA synthesis after treatment of human lymphoblastoid cells with 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl) adenine. Cancer Res. 1996;56(13):3030–7.
Chen LS, Plunkett W, Gandhi V. Polyadenylation inhibition by the triphosphates of deoxyadenosine analogues. Leuk Res. 2008;32:1573–81.
Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706.
Aye Y, Stubbe J. Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc NatAcad Sci. 2011;108(24):9815–20.
Aye Y, Brignole EJ, Long MJ, Chittuluru J, Drennan CL, Asturias FJ, et al. Clofarabine targets the large subunit (alpha) of human ribonucleotide reductase in live cells by assembly into persistent hexamers. Chem Biol. 2012;19(7):799–805.
Fu Y, Lin HY, Wisitpitthaya S, Blessing WA, Aye Y. A fluorimetric readout reporting the kinetics of nucleotide-induced human ribonucleotide reductase oligomerization. Chem Biochem: Eur J Chem Biol. 2014;15(17):2598–604.
Yamauchi T, Nowak BJ, Keating MJ, Plunkett W. DNA repair initiated in chronic lymphocytic leukemia lymphocytes by 4-hydroperoxycyclophosphamide is inhibited by fludarabine and clofarabine. Clin Cancer Res. 2001;7(11):3580–9.
Carson DA, Wasson DB, Esparza LM, Carrera CJ, Kipps TJ, Cottam HB. Oral antilymphocyte activity and induction of apoptosis by 2-chloro-2′-arabino-fluoro-2′-deoxyadenosine. Proc Natl Acad Sci. 1992;89(7):2970–4.
Hartman WR, Hentosh P. The antileukemia drug 2-chloro-2′-deoxyadenosine: an intrinsic transcriptional antagonist. Mol Pharmacol. 2004;65(1):227–34.
Hartman WR, Walters DE, Hentosh P. Presence of the anti-leukemic nucleotide analog, 2-chloro-2′-deoxyadenosine-5′-monophosphate, in a promoter sequence alters DNA binding of TATA-binding protein (TBP). Arch Biochem Biophys. 2007;459(2):223–32.
Dimitrova DS, Gilbert DM. Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol. 2000;2(10):686–94.
Lee YJ, Hwang IS, Lee YJ, Lee CH, Kim SH, Nam HS, et al. Knockdown of Bcl-xL enhances growth-inhibiting and apoptosis-inducing effects of resveratrol and clofarabine in malignant mesothelioma H-2452 cells. J Korean Med Sci. 2014;29(11):1464–72.
Takahashi T, Shimizu M, Akinaga S. Mechanisms of the apoptotic activity of Cl-F-araA in a human T-ALL cell line. CCRF-CEM Cancer Chemother Pharmacol. 2002;50(3):193–201.
Wang X, Albertioni F. Effect of clofarabine on apoptosis and DNA synthesis in human epithelial colon cancer cells. Nucleosides Nucleotides Nucleic Acids. 2010;29(4–6):414–8.
Genini D, Adachi S, Chao Q, Rose DW, Carrera CJ, Cottam HB, et al. Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood. 2000;96(10):3537–43.
Genini D, Budihardjo I, Plunkett W, Wang X, Carrera CJ, Cottam HB, et al. Nucleotide requirements for the in vitro activation of the apoptosis protein-activating factor-1-mediated caspase pathway. J Biol Chem. 2000;275(1):29–34.
Miura S, Izuta S. DNA polymerases as targets of anticancer nucleosides. Curr Drug Targets. 2004;5(2):191–5.
Jensen K, Johnson LA, Jacobson PA, Kachler S, Kirstein MN, Lamba J, et al. Cytotoxic purine nucleoside analogues bind to A1, A2A, and A3 adenosine receptors. Naunyn Schmiedeberg’s Arch Pharmacol. 2012;385(5):519–25.
Lubecka-Pietruszewska K, Kaufman-Szymczyk A, Stefanska B, Cebula-Obrzut B, Smolewski P, Fabianowska-Majewska K. Clofarabine, a novel adenosine analogue, reactivates DNA methylation-silenced tumour suppressor genes and inhibits cell growth in breast cancer cells. Eur J Pharmacol. 2014;723:276–87.
Majda K, Kaufman-Szymczyk A, Lubecka-Pietruszewska K, Bednarek A, Fabianowska-Majewska K. Influence of clofarabine on transcriptional activity of PTEN, APC, RARB2, ZAP70 genes in K562 cells. Anticancer Res. 2010;30(11):4601–6.
Seedhouse C, Grundy M, Shang S, Ronan J, Pimblett H, Russell N, et al. Impaired S-phase arrest in acute myeloid leukemia cells with a FLT3 internal tandem duplication treated with clofarabine. Clin Cancer Res. 2009;15(23):7291–8.
Krett NL, Ayres M, Nabhan C, Ma C, Nowak B, Nawrocki S, et al. In vitro assessment of nucleoside analogs in multiple myeloma. Cancer Chemother Pharmacol. 2004;54(2):113–21.
Uy GL, Tomasson MH, Ruddell A, DiPersio JF, Vij R. The activity and toxicity of low dose clofarabine against relapsed or refractory myeloma. Haematologica. 2006;91(11):1581–2.
Abramson JS, Takvorian RW, Fisher DC, Feng Y, Jacobsen ED, Brown JR, et al. Oral clofarabine for relapsed/refractory non-Hodgkin lymphomas: results of a phase 1 study. Leuk Lymphoma. 2013;54(9):1915–20.
Blum KA, Hamadani M, Phillips GS, Lozanski G, Johnson AJ, Lucas DM, et al. Prolonged myelosuppression with clofarabine in the treatment of patients with relapsed or refractory, aggressive non-Hodgkin lymphoma. Leuk Lymphoma. 2009;50(3):349–56.
Nabhan C, Davis N, Bitran JD, Galvez A, Fried W, Tolzien K, et al. Efficacy and safety of clofarabine in relapsed and/or refractory non-Hodgkin lymphoma, including rituximab-refractory patients. Cancer. 2011;117(7):1490–7.
Rahmati-Yamchi M, Zarghami N, Nozad Charoudeh H, Ahmadi Y, Baradaran B, Khalaj-Kondori M, et al. Clofarabine has apoptotic effect on T47D breast cancer cell line via P53R2 gene expression. Adv Pharm Bull. 2015;5(4):471–6.
Patel YT, Jacus MO, Boulos N, Dapper JD, Davis AD, Vuppala PK, et al. Preclinical examination of clofarabine in pediatric ependymoma: intratumoral concentrations insufficient to warrant further study. Cancer Chemother Pharmacol. 2015;75(5):897–906.
Bonate PL, Arthaud L, Cantrell Jr WR, Stephenson K, Secrist 3rd JA, Weitman S. Discovery and development of clofarabine: a nucleoside analogue for treating cancer. Nat Rev Drug Discov. 2006;5(10):855–63.
Collins JM, Grieshaber CK, Chabner BA. Pharmacologically guided phase I clinical trials based upon preclinical drug development. J Natl Cancer Inst. 1990;82(16):1321–6.
Kantarjian HM, Gandhi V, Kozuch P, Faderl S, Giles F, Cortes J, et al. Phase I clinical and pharmacology study of clofarabine in patients with solid and hematologic cancers. J Clin Oncol. 2003;21(6):1167–73.
Jeha S, Gandhi V, Chan KW, McDonald L, Ramirez I, Madden R, et al. Clofarabine, a novel nucleoside analog, is active in pediatric patients with advanced leukemia. Blood. 2004;103(3):784–9.
Ajavon AD, Bonate PL, Taft DR. Renal excretion of clofarabine: assessment of dose-linearity and role of renal transport systems on drug excretion. Eur J Pharm Sci. 2010;40(3):209–16.
Bonate PL, Craig A, Gaynon P, Gandhi V, Jeha S, Kadota R, et al. Population pharmacokinetics of clofarabine, a second-generation nucleoside analog, in pediatric patients with acute leukemia. J Clin Pharmacol. 2004;44(11):1309–22.
Bonate PL, Cunningham CC, Gaynon P, Jeha S, Kadota R, Lam GN, et al. Population pharmacokinetics of clofarabine and its metabolite 6-ketoclofarabine in adult and pediatric patients with cancer. Cancer Chemother Pharmacol. 2011;67(4):875–90.
Rudrapatna VK, Morley K, Boucher KM, Pierson AS, Shull CT, Kushner JP, et al. Phase I trial of low-dose oral Clofarabine in myelodysplastic syndromes patients who have failed frontline therapy. Leuk Res. 2015;39(8):835–9.
Jacoby MA, Martin MG, Uy GL, Westervelt P, Dipersio JF, Cashen A, et al. Phase I study of oral clofarabine consolidation in adults aged 60 and older with acute myeloid leukemia. Am J Hematol. 2014;89(5):487–92.
Faderl S, Garcia-Manero G, Estrov Z, Ravandi F, Borthakur G, Cortes JE, et al. Oral clofarabine in the treatment of patients with higher-risk myelodysplastic syndrome. J Clin Oncol. 2010;28(16):2755–60.
Al Ustwani O, Greene JD, Wetzler M. The use of low-dose protracted oral clofarabine in a patient with myelodysplastic syndrome after failing 5-azacitidine. Leuk Res Rep. 2013;2(1):34–5.
Buckley SA, Mawad R, Gooley TA, Becker PS, Sandhu V, Hendrie P, et al. A phase I/II study of oral clofarabine plus low-dose cytarabine in previously treated acute myeloid leukaemia and high-risk myelodysplastic syndrome patients at least 60 years of age. Br J Haematol. 2015;170(3):349–55.
Gandhi V, Plunkett W. Modulation of arabinosylnucleoside metabolism by arabinosylnucleotides in human leukemia cells. Cancer Res. 1988;48(2):329–34.
Gandhi V, Estey E, Keating MJ, Chucrallah A, Plunkett W. Chlorodeoxyadenosine and arabinosylcytosine in patients with acute myelogenous leukemia: pharmacokinetic, pharmacodynamic, and molecular interactions. Blood. 1996;87(1):256–64.
Gandhi V, Estey E, Keating MJ, Plunkett W. Fludarabine potentiates metabolism of cytarabine in patients with acute myelogenous leukemia during therapy. J Clin Oncol. 1993;11(1):116–24.
Faderl S, Gandhi V, O’Brien S, Bonate P, Cortes J, Estey E, et al. Results of a phase 1-2 study of clofarabine in combination with cytarabine (ara-C) in relapsed and refractory acute leukemias. Blood. 2005;105(3):940–7.
Faderl S, Wetzler M, Rizzieri D, Schiller G, Jagasia M, Stuart R, et al. Clofarabine plus cytarabine compared with cytarabine alone in older patients with relapsed or refractory acute myelogenous leukemia: results from the CLASSIC I Trial. J Clin Oncol. 2012;30(20):2492–9.
Becker PS, Kantarjian HM, Appelbaum FR, Petersdorf SH, Storer B, Pierce S, et al. Clofarabine with high dose cytarabine and granulocyte colony-stimulating factor (G-CSF) priming for relapsed and refractory acute myeloid leukaemia. Br J Haematol. 2011;155(2):182–9.
Valdez BC, Li Y, Murray D, Champlin RE, Andersson BS. The synergistic cytotoxicity of clofarabine, fludarabine and busulfan in AML cells involves ATM pathway activation and chromatin remodeling. Biochem Pharmacol. 2011;81(2):222–32.
Valdez BC, Murray D, Nieto Y, Li Y, Wang G, Champlin RE, et al. Synergistic cytotoxicity of the DNA alkylating agent busulfan, nucleoside analogs and suberoylanilide hydroxamic acid in lymphoma cell lines. Leuk Lymphoma. 2012;53(5):973–81.
Karp JE, Ricklis RM, Balakrishnan K, Briel J, Greer J, Gore SD, et al. A phase 1 clinical-laboratory study of clofarabine followed by cyclophosphamide for adults with refractory acute leukemias. Blood. 2007;110(6):1762–9.
Faderl S, Balakrishnan K, Thomas DA, Ravandi F, Borthakur G, Burger J, et al. Phase I and extension study of clofarabine plus cyclophosphamide in patients with relapsed/refractory acute lymphoblastic leukemia. Clin Lymph Myelo Leuk. 2014;14(3):231–8.
Inaba H, Rubnitz JE, Coustan-Smith E, Li L, Furmanski BD, Mascara GP, et al. Phase I pharmacokinetic and pharmacodynamic study of the multikinase inhibitor sorafenib in combination with clofarabine and cytarabine in pediatric relapsed/refractory leukemia. J Clin Oncol. 2011;29(24):3293–300.
Chiarini F, Lonetti A, Teti G, Orsini E, Bressanin D, Cappellini A, et al. A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget. 2012;3(12):1615–28.
Thudium KE, Ghoshal S, Fetterly GJ, Haese JP, Karpf AR, Wetzler M. Synergism between clofarabine and decitabine through p53R2: a pharmacodynamic drug-drug interaction modeling. Leuk Res. 2012;36(11):1410–6.
Faderl S, Ravandi F, Huang X, Wang X, Jabbour E, Garcia-Manero G, et al. Clofarabine plus low-dose cytarabine followed by clofarabine plus low-dose cytarabine alternating with decitabine in acute myeloid leukemia frontline therapy for older patients. Cancer. 2012;118(18):4471–7.
Foster MC, Amin C, Voorhees PM, van Deventer HW, Richards KL, Ivanova A, et al. A phase I dose-escalation study of clofarabine in combination with fractionated gemtuzumab ozogamicin in patients with refractory or relapsed acute myeloid leukemia. Leuk Lymphoma. 2012;53(7):1331–7.
Acknowledgments
The authors thank Hima Vangapandu, PhD, for her help with the references. The authors also thank Ms. Ann Sutton for scientific editing.
Conflict of Interest
Clofarabine was discovered at Southern Research Institute, which receives licensing fees and royalty payments from its commercial development. Some of this money is distributed to Dr. Parker. Dr. Gandhi has no conflicts of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Parker, W.B., Gandhi, V. (2017). Clofarabine: Structure, Mechanism of Action, and Clinical Pharmacology. In: Ueda, T. (eds) Chemotherapy for Leukemia. Springer, Singapore. https://doi.org/10.1007/978-981-10-3332-2_16
Download citation
DOI: https://doi.org/10.1007/978-981-10-3332-2_16
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3330-8
Online ISBN: 978-981-10-3332-2
eBook Packages: MedicineMedicine (R0)