
Abstract
Malaria is a major global parasitic disease responsible for tremendous health burden and mortality in tropical and subtropical regions of the world. Plasmodium falciparum is the causative agent of severe malaria, which accounts for most of the global malaria-related deaths, mainly in sub-Saharan Africa. Despite the enormous global efforts to curb the spread of the disease and significant decline in malaria-related deaths in the last decade, development of parasite resistance to currently used drugs is widespread, which necessitates the development of novel antimalarial targeting crucial parasite molecules. Parasite proteases are a group of molecules crucial for the development and propagation of the parasite inside the host cell. The major parasite-specific processes dependent on protease activity for their completion are hemoglobin degradation, merozoite egress from the host cell, and invasion of the host cells. A number of proteases of various classes are found in P. falciparum, many of which have the potential to be used as antimalarial drug targets. I this chapter, I have described the role of the proteases, which have the potential to be targeted for antimalarial drug development and the progresses made in the direction of drug development against these targets.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Malaria
- Plasmodium falciparum
- Asexual blood stages
- Proteases
- Hemoglobin degradation
- Merozoite egress
- Merozoite invasion
- Other cellular proteases
1 Introduction
Malaria is an ancient disease caused by protozoan parasite of the genus Plasmodium. Till date malaria remains a major health burden for impoverished regions of the world with limited sanitation and healthcare facilities. In humans, malaria is caused by five Plasmodium species, namely, P. falciparum, P. vivax, P. ovale, P. malariae, and P. cynomolgi. Of the five species, P. falciparum is the causative agent of severe malaria, responsible for most of the malaria-related deaths. Due to the combined global efforts to curb the menace of malaria, disease cases have come down by 37% globally and by 42% in Africa between 2000 and 2015. The more encouraging outcome of the efforts has been the drastic reduction in malaria-related deaths that have come down by 60% globally and 66% in the African regions in the same time span [1].
Despite these successes in malaria control, complete malaria eradication remains an ambitious goal for the global scientific community and healthcare providers. A completely protective malaria vaccine is still a dream for the scientists and the available arsenals of antimalarial drugs are limited. Till date major antimalarials are natural compounds or their derivatives and the knowledge about their mechanisms of action are limited. In addition, the ability of the parasite to develop resistance against antimalarials is common. The parasite quickly develops resistance against the extensively used antimalarials, especially when they are used as a monotherapy, although successes have been achieved to counter drug resistance using combination therapy [2,3,4,5,6,7,8,9,10,11]. Due to above-mentioned reasons, development of novel antimalarial drugs remains a high priority.
For development of new drugs, identification of crucial parasite-specific molecules is required, which could be used for specific targeting. One group of such molecules is parasite proteases. They constitute the major virulence factors in various parasitic diseases and are crucial for the pathogens for their survival and ability to cause the diseases [12]. Malarial proteases are crucial molecules for parasite development and virulence. Some of these proteases possess parasite-specific functions and structural features that could be specifically targeted by drugs. Potential of these molecules as antimalarial drug targets has been substantiated by the use of protease inhibitors as drugs against a number of diseases. Protease inhibitors are already in use as drugs against human immunodeficiency virus (HIV) [13], hepatitis C virus (HCV) [14], and in the treatment of hypertension [15] and coagulopathies [16].
These groups of molecules are widespread in organisms and carry out a variety of biological processes by regulating the fate, localization, and activity of the target proteins [17]. Through evolution they have adapted to a variety of physiological conditions found in complex organisms and employ different mechanisms for substrate hydrolysis. Depending on the mechanisms of action, proteases have been classified into different classes (serine, cysteine, metallo, aspartic, threonine, and glutamic proteases). Serine, threonine, and cysteine proteases rely upon the nucleophilic character of the side chains of serine, threonine, and cysteine, respectively, at the active site. These side chains directly attack the peptide bond to form a transient, covalent enzyme–substrate intermediate, which breaks to give rise to the product and the enzyme. Aspartic and metalloproteases employ water molecule as nucleophile and do not form a covalent intermediates [18]. A number of proteases of various classes are found in Plasmodium. This chapter describes the available information on P. falciparum proteases crucial for parasite development in the human host and the studies related to drug development efforts to target these molecules.
2 Proteases as Potential Antimalarial Drug Targets
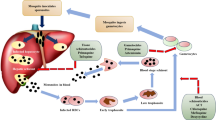
Malaria parasite completes its life cycle inside the vertebrate and invertebrate host cells. The host cell provides a rich source of nutrients for the parasite and a safe niche where the parasite is protected from the host immune responses. Asexual erythrocytic life cycle is responsible for the clinical manifestations of malaria, which starts when the merozoites released from liver infect the erythrocytes. The intracellular parasite feeds on erythrocyte hemoglobin and develops into more metabolically active trophozoite, which then undergoes nuclear division to transform into multinucleated schizont. After maturation, schizonts rupture to release the merozoites, which start a new wave of infection by invading new erythrocytes.
Completion of parasite life cycle in vertebrate and invertebrate hosts is dependent on a number of proteolytic activities. Inhibitor studies have shown that cysteine, aspartic, metallo, and serine proteases play critical roles in parasite-specific processes during the erythrocytic cycle such as hemoglobin digestion, merozoite release from infected erythrocytes, and invasion of fresh erythrocytes by the released merozoites. In addition a number of proteases are involved in metabolic processes necessary for parasite survival (Fig. 1). These processes have been the focus of drug development efforts [19]. In addition to erythrocytic stages, liver stages of the parasite are also attractive drug targets due to their low numbers and distinct metabolism [20, 21]. Liver stage parasites also employ proteolytic activities for their maturation and subsequent dispersal in the blood stream [22, 23]. Aspartate, cysteine, and metalloproteases are involved in hemoglobin degradation, a pathway necessary for parasite growth in the blood stages, whereas serine and cysteine proteases are crucial for the parasite egress and invasion at blood and liver stages. Table 1 summarizes the role of parasite proteases in P. falciparum development and propagation. Data mining revealed the presence of 92 putative proteases in P. falciparum genome [24]. Further analysis of P. falciparum genome database revealed the presence of 124 predicted proteases [25]. The roles of proteases in various parasite-specific processes and their potential as drug targets are described below.

Role of proteases in asexual erythrocytic life cycle of malaria parasite
3 Proteases Involved in Hemoglobin Digestion
P. falciparum has a limited capacity to synthesize amino acids [26]. In the blood stages, it relies on degradation of abundant host cell molecule hemoglobin for its utilization [27, 28]. During the trophozoite stage, parasite proteases degrade most of the hemoglobin [29] and supply the parasite with amino acids for protein synthesis and metabolism [30]. Plasmodium ingests hemoglobin by an invagination spanning from parasite plasma membrane to parasitophorous vascuolar membrane known as cytostome. The cytostome takes host cell cytoplasm and pinches off to form double-membrane transport vesicles loaded with hemoglobin, which fuse with food vacuoles to empty their contents there. Food vacuoles are acidic compartments with a pH between 5.0 and 5.4 [31, 32] and provide the site for hemoglobin degradation and heme detoxification [33].
Hydrolysis of hemoglobin is a semi-ordered process mediated by the action of a series of proteases. Aspartic [34,35,36] and metalloproteases [37] are involved in hemoglobin degradation in the food vacuole. Four proteases predominantly carry out hemoglobin degradation (aspartic proteases plasmepsin 1 and 2, cysteine protease falcipain 2, and metalloprotease falcilysin) [38]. Plasmepsin 1 and 2 possess the ability to cleave hemoglobin under non-denaturing conditions between Phe33 and Leu34 in the hinge region of the alpha globin chain, which probably allows the globin subunits to unwind that facilitates further proteolysis [33, 39]. After the initial cleavage by plasmepsins 1 and 2, falcipain 2 cleaves the globin fragments [38]. It has been demonstrated that plasmepsin 1 can degrade native hemoglobin efficiently under nonreducing conditions while plasmepsin 2 has a preference for denatured hemoglobin. Although falcipains cleave denatured globin but not native hemoglobin [39], under mild reducing conditions falcipain 2 cleaves both native and denatured globin [40]. Falcilysin further cleaves the globin fragments into smaller peptides [41]. The digested peptides are then transported to parasite cytosol, where cytosolic aminopeptidases hydrolyse these peptides to free amino acids [42].
Human hemoglobin lacks a single amino acid isoleucine. Liu et al. [43] demonstrated that P. falciparum could grow in a medium containing only a single amino acid isoleucine. This finding indicated that host hemoglobin is sufficient to provide the rest of the amino acid requirements of the parasite. Knockout parasite lines of the hemoglobin-degrading enzymes (falcipain-2, plasmepsins, alone or in combination) grown in this medium showed reduced growth. A potent inhibitor of plasmepsin pepstatin A did not significantly affect the growth of wild-type parasites but was able to kill the parasites lacking falcipain-2 at low concentrations. It was even more effective in killing the parasites triple knockout parasites lacking falcipain-2, plasmepsin 1, and 4. This study provided evidence that both plasmepsins and falcipain-2 are hemoglobinases with overlapping functions. While falcipain-2 was essential for parasite survival plasmepsins provided subsidiary roles and were dispensable for the parasite [43], different classes of hemoglobin-degrading proteases and their role in the parasite are summarized below.
3.1 Aspartic Proteases (Plasmepsins)
P. falciparum genome data predicted the presence of 10 plasmepsin genes. Expression, localization, and enzymatic data suggested that four of the plasmepsins (plasmepsin 1, plasmepsin 2, plasmepsin 4, and a histo-aspartic protease (HAP)) were found in the food vacuole and were capable to degrade hemoglobin [41, 44]. Gene knockout of P. falciparum food vacuole plasmepsins revealed that growth rates were reduced by 30–35% upon plasmepsin 1 and plasmepsin 4 disruption, whereas disruption of plasmepsin 2 did not cause any growth defect. Disruption of HAP caused a slight drop in growth rates that was not statistically significant [45]. In another similar study, targeted gene disruption by double crossover homologous recombination was carried out for plasmepsin 1, plasmepsin 2, HAP, and plasmepsin 4/plasmepsin 1. All the four knockout parasites were morphologically normal. In amino acid-limited medium, they exhibited slower growth rate as compared to the parental strain. Sensitivity of the knockout parasites to aspartic and cysteine protease inhibitors changed minimally, compared to wild-type 3D7 parasite, suggesting the functional redundancy of these proteases [46].
Among the two major hemoglobin-degrading plasmepsins, 1 and 2, plasmepsin 2 was found to be 5–10-fold more efficient on peptide substrates and also had 80-fold higher inhibition constant compared to plasmepsin 1 [47]. Because of their higher hemoglobin-degrading activities, a number of studies on drug screening have been focused on plasmepsins 1 and 2 [48,49,50,51,52,53,54]. Due to their redundant function, it is suggested that in order to achieve higher antiparasite activity, as many as of these enzymes have to been targeted by the inhibitors [55] and inhibitors targeting multiple plasmepsins could be used as antimalarials [56].
3.2 Cysteine Proteases (Falcipains)
P. falciparum genome encodes four clan CA cysteine proteases known as falcipains (falcipain 1, falcipain 2, falcipain 2′, and falcipain 3). These proteases have been biochemical and genetically characterized [57,58,59]. Genetic disruption of falcipain 2 caused block in hemoglobin degradation resulting in accumulation of undegraded hemoglobin in the food vacuole, similar to that caused by cysteine protease inhibitors [58]. Falcipain 3 was refractory to genetic deletion, although a tagged copy of the gene could readily be produced. Falcipain 1 and falcipain 2′ were dispensable for the parasite as their disruption did not cause any defect in parasite phenotype [59]. Based on these studies, falcipains 2 and 3 appear to be crucial hemoglobinases with redundant functions [57,58,59].
Activities of falcipain 2 and 3 have been assessed on peptide substrates in vitro. Falcipain 2 accounted for 90% of the cysteine protease activity on peptide substrates in trophozoite lysates [40], while falcipain 3 showed relatively lesser activity on peptide substrates [57]. Differential activities of these enzymes correlated with their timings of expression. Falcipain 2 was predominantly expressed in early trophozoites when parasite growth is maximum and hence maximum hemoglobin degradation is required while falcipain 3 showed maximum expressions in late trophozoites when the nutrient requirement is minimal and parasite prepares for division [58]. Falcipain 2′ did not exhibit any biochemical differences with falcipain 2. Gene knockout of falcipain 2′ did not cause any phenotypic defect in the parasite and the biological role of this enzyme remains unknown [60,61,62].
Due to the crucial role of falcipains 2 and 3, numerous studies have been focused on designing of their inhibitors [38, 63]. These inhibitors caused accumulation of undegraded hemoglobin in the food vacuole resulting in swollen morphology. They have also inhibited parasite development and have been effective in cure of experimental malaria [28, 40, 64,65,66,67].
3.3 Metalloproteases
Following degradation by aspartic and cysteine proteases, peptide fragments generated from hemoglobin are further hydrolyzed by metalloprotease falcilysin [37]. Specificity analysis using hydrolysis of a series of random peptide substrates revealed that the enzyme was highly active at acidic pH, consistent with its role in hemoglobin degradation. Surprisingly, the enzyme also showed robust activity at neutral pH and with slightly different substrate specificities [68]. Further studies revealed that in addition to food vacuole falcilysin was also translocated to the apicoplast. Based on in vitro studies, it was suggested that this enzyme might have a role in transit peptide hydrolysis [69].
3.4 Aminopeptidases
Nine aminopeptidases are annotated in P. falciparum genome. These enzymes catalyze the hydrolysis of amino acids from the N-terminus. Five aminopeptidases are involved in hydrolysis of peptides generated by hemoglobin digestion [70]. Two of these enzymes M1 alanyl aminopeptidase (PfM1AAP) and M17 leucine aminopeptidase (PfM17LAP) are essential for P. falciparum growth and development as inhibitors of these peptidases were found to be lethal to P. falciparum culture in vitro [71].
4 Proteases Involved in Parasite Egress from Erythrocytes
Erythrocyte rupture by malaria parasites is a temporally regulated process. Different models of erythrocyte rupture have been proposed based on live video microscopic and electron microscopic studies [72,73,74,75,76]. Although the exact mechanism of egress is obscure, the most widely experimentally supported model of erythrocyte rupture suggests that egress is a two-step process, in which the degradation of parasitophorous vacuole membrane takes place prior to RBC membrane rupture [74]. This model was supported by electron microscopy (EM)-based evidences demonstrating that highly mature schizonts occasionally lack a discrete parasitophorous vacuole membrane (PVM) [77, 78]. Similar results were obtained by immune electron microscopy of mature schizonts with antibody against parasitophorous vacuole (PV) resident protein S-antigen [74].
Mechanistically egress consists of destabilization of erythrocyte cytoskeleton, PVM, and the erythrocyte membrane. Studies based on serine, cysteine, and aspartic protease inhibitors suggest that egress is dependent on proteolytic activities [79,80,81,82,83]. Analysis on unruptured schizonts in the protease inhibitor-treated parasites revealed the defects in processing of merozoite surface protein (MSP1) [82] and PV protein p126 that is now known as SERA5 [83].
In vitro studies have suggested that hemoglobin-degrading proteases plasmepsin 2 and falcipain 2 could also have role in egress. Plasmepsin 2 digested erythrocyte cytoskeleton proteins spectrin, protein 4.1, and actin at neutral pH in vitro. Detection of this protease in the cytoplasm of schizont-infected erythrocytes was also suggestive of its role in destabilization of host cell membrane skeleton [84]. Falcipain 2 digested cytoskeleton proteins ankyrin and protein 4.1 at neutral pH in vitro [85, 86]. A peptide based on ankyrin cleavage site was found to be inhibitory to falcipain 2 activity and merozoite egress when added to schizont-stage parasites. Localization of falcipain 2 in PV and subcellular structures extending into the erythrocyte cytosol also provide strength to its possible involvement in erythrocyte cytoskeleton destabilization [87]. Surprisingly, plasmepsin 2 and falcipain 2 were dispensable for the parasite as shown by gene disruption studies and these knockout lines showed no defect in egress [43, 45, 46, 57, 58]. Although the role of these proteases in egress has not been conclusively ascertained, they seem to be redundant in function [88].
The role of a protease in egress was demonstrated in an exquisite study by Aly and Matuschewski [89] using reverse genetics approach. They showed that disruption of a P. berghei gene termed as “egress cysteine protease 1” (ecp1) inhibited the release of sporozoites from mosquito oocyst [89]. ecp1 belongs a family of cysteine proteases known as serine repeat antigens (SERAs). There are nine members of SERA proteases found in P. falciparum genome. Eight of them are arranged on chromosome 2 in a head-to-tail tandem orientation, whereas the ninth member SERA9 is located on chromosome 9 [90]. P. berghei ecp1 was found to be ortholog of P.falciparum SERA8.
The central regions of SERA proteins show homology to papain family cysteine proteases. However, in some SERAs active site cysteine is substituted with a serine residue [91,92,93]. In P. falciparum, SERAs 1–5 and SERA9 possess active site serine and SERAs 6–8 possess active site cysteine [93]. Proteolytic activity of P. falciparum SERA5, the most highly expressed member, has been demonstrated in vitro on peptide substrates [94]. To study the essentiality of P. falciparum SERAs, gene knockout studies were conducted. All but SERA5 and SERA6 could not be disrupted suggesting the essential function of these proteins for the parasite [90, 95].
SERA5, also known as SERA, is the most highly expressed member and a number of studies have suggested its involvement in merozoite egress from erythrocytes. It is synthesized as a 126 kDa precursor (p126) [96] and secreted into the lumen of the parasitophorous vacuole. Around the time of schizont rupture, it undergoes proteolytic processing into multiple fragments. These processing seem to be essential for its role in egress. The full-length p126 is processed into a 47-kDa N-terminal and a 73-kDa fragment (P73) C-terminal fragments. P73 is processed into 56-kDa and 18-kDa fragments and the 56-kDa fragment is further processed to form a 50-kDa central domain and a 6-kDa fragment [97]. The complex of N-terminal 47-kDa and C-terminal 18-kDa fragments is found associated with the merozoite surface and the central 50-kDa fragment is detected in the culture medium after schizont rupture [98, 99]. The cleavage that produces p47 and p73 from p126 and cleavage of p73 to p56 and p18 is carried out by PfSUB1. p56 is cleaved by an unknown protease to produce p50 and a 6-kDa fragment [88].
Evidence for involvement of SERA5 and other SERA family members came from inhibitor studies. A specific inhibitor of PfSUB1 was found to block the rupture of schizonts. Further biochemical analysis revealed that PfSUB1 mediated rupture of schizonts through the processing of SERA5 and other SERA family members including SERA4 and SERA6 [100]. In a reciprocal approach to identify the mediators of merozoite egress, using a chemical screen of 1200 covalent serine and cysteine protease inhibitors demonstrated that PfSUB1 and a cysteine protease DPAP3 as the regulators of this process. This study demonstrated that chemical blocking of PfSUB1 and DPAP3 resulted in blockade of SERA5 processing, correlating with the blockade in schizont rupture. Inhibition of DPAP3 resulted in reduced level of mature PfSUB1 suggesting that both the proteases regulate the efficient release of merozoites from the infected red blood cells by SERA5 processing [101]. Attempts to target the enzyme domain of SERA5 using peptides resulted in the block of merozoite egress [102]. In another study, incubation of P. falciparum culture with the SERA5 prodomain and a peptide derived from it resulted in block in schizont rupture [103]. These studies also suggested the role of SERA5 in merozoite egress and the possibility of targeting the protein for parasite growth inhibition. In addition to SERA5, SERA6 is also supposed to have role in merozoite egress. It is cleaved in the PV by PfSUB1 just before egress. Mutation of active site cysteine or the blockade in processing by PfSUB1 was lethal for the parasite. Processing of P. berghei ortholog of SERA6 (PbSERA3) by PfSUB1 converted it into an active cysteine protease. Taken together, these findings suggest the role of SERA6 in merozoite egress [104].
Based on these studies, PfSUB1 appears to be involved in maturation of multiple SERA proteases and hence is an important regulator of merozoite egress. Its role in egress at blood stages and expression in liver stages raised the possibility of similar role in liver stages. A conditional mutagenesis-based invalidation of P. berghei SUB1 in liver stages revealed that SUB1-deficient parasites failed to egress from hepatocytes [23]. Based on its role in both blood and liver stages, PfSUB1 qualifies an attractive multistage target against malaria. Molecular modeling, substrate specificity, and characterization of PfSUB1 and its homologs from Plasmodium vivax, Plasmodium knowlesi, and P. berghei revealed many unusual features in SUB1 substrate-binding cleft, although SUB1 from all the species cleaved the conserved parasite substrates. Two peptidyl alpha-ketoamides based on an authentic PfSUB1 substrate inhibited SUB1 from all the species [105]. In another study, high throughput screening of a proprietary library of compounds against PfSUB1 identified hydrazine 2 as an inhibitor of PfSUB1 [106]. Due to its potential as an attractive multistage druggable target, identification of potent inhibitors against PfSUB1 could pave the way for the development antimalarials acting at schizont rupture.
5 Proteases Involved in Erythrocyte Invasion by Merozoites
Malaria parasites efficiently invade their host cells to survive inside the host and protect themselves from the host immune responses. Apicomplexan parasites utilize their specialized invasion apparatuses to identify, penetrate, and establish themselves within the host cells. Host cell entry is a crucial checkpoint where parasite development can be blocked, and hence an important target for antimalarial drug development [107]. Evidences for the involvement of parasite proteases in invasion came from inhibitor studies. Protease inhibitors phenylmethylsulfonyl fluoride (PMSF) and chymostatin blocked merozoite invasion in P. falciparum and a number of other Plasmodium species [108, 109]. The chymostatin-sensitive effect was reversed by pretreatment of RBCs with chymotrypsin, suggesting that parasite-induced proteolytic modification of RBCs was involved in chymostatin-sensitive step [108, 110, 111]. Inhibitor studies on simian malaria parasite P. knowlesi revealed that treatment of isolated, invasive merozoites with N-tosyl phenylalanyl chloromethyl ketone, or N-tosyl lysyl chloromethyl ketone prevented primary attachment of parasites to host cells, whereas chymostatin blocked invasion at a later stage suggesting the involvement of multiple proteolytic activities in the pathway [81]. Consistent with this, a P. falciparum serine protease activity that mediated an essential processing and shedding of a major merozoite surface protein (merozoite surface protein 1; MSP1) at invasion was identified that was highly sensitive to inhibition by PMSF but not by chymostatin [112].
Invasion is a rapid process, taking about 60 s for the egressed merozoites to invade new erythrocytes. These merozoites distinguish between erythrocytes and other cells by initial low-affinity reversible contacts between merozoite and erythrocyte surface molecules [113]. The initial attachment occurs anywhere between the merozoite surface and erythrocyte; hence, the apical end of the merozoite orientates toward the erythrocyte surface in the subsequent steps. This orientation results in the formation of a tight junction between the merozoite apical end and the erythrocyte surface. This junction moves from anterior to the posterior end of the merozoite with the help of actin-myosin motor as it invades the erythrocytes [114]. The interaction between the merozoite and the erythrocyte molecules during the movement of the tight junction involves activity of parasite proteases [116,117,118]. Studies in Toxoplasma gondii and P. falciparum have revealed that micronemal secretion is induced upon initial contact. These secretory proteins are exposed at the parasite surface and mediate parasite orientation and formation of tight junction. Soon after microneme secretion, another apical organelles known as rhoptries discharge their contents, which are also supposed to have role in tight junction formation, although their precise role is not known [119, 120].
The initial interaction between the merozoite and the host cell is mediated by a protein complex on the merozoite surface known as merozoite surface protein (MSP1) complex MSP1 complex is composed of MSP1 and its associated proteins MSP6 and MSP7 [121,122,123]. This complex is processed in the parasitophorous vacuole by the serine protease PfSUB1. This processing termed as “primary processing” makes the merozoite competent for the initial interaction [124]. After initial interaction, micronemal secretions mediate the high affinity interaction resulting in the formation of tight junction. This tight junction moves from anterior to posterior pole of the parasite in actomyosin motor-dependent manner. During this movement, shedding of MSP1 complexes and another micronemal protein apical membrane antigen (PfAMA1) takes place that is essential for invasion. Cleavage of both these proteins takes place at the juxtamembrane site. MSP1 is cleaved at the site distal to the epidermal growth factor-like domain at its C-terminal, shedding the MSP1 complex except the C-terminal region known as MSP119 that enters into the host cell following invasion [125]. Similarly, cleavage of AMA1 29 residues away from the transmembrane domain releases the bulk of the ectodomain. In this way the juxtamembrane “stub” along with the transmembrane and cytoplasmic domains enters into the host cell after invasion [126]. In vitro studies on the shedding of these parasite proteins strongly suggest that micronemal subtilisin-like serine protease PfSUB2 is the most likely candidate for these processing events, and hence termed as “merozoite surface sheddase” (MESH) [116].
By reverse genetics approaches both PfSUB1 and PfSUB2 appear to be essential for the parasite [100, 105]. Based on the numerous studies on their role in merozoite egress and invasion, these two proteases appear to be promising drug targets against malaria. Recently, efforts have been made to identify PfSUB1 and PfSUB2 inhibitors. Characterization of PfSUB1 and its orthologs in P. knowlesi and P. berghei revealed that cleavage sites in the parasite substrates of these proteases in conserved despite the differences in the enzyme substrate-binding sites. Consistent with these features, two peptidyl alpha-ketoamide inhibitors of PfSUB1 also inhibited its orthologs [127]. A low toxic natural pentacyclic triterpene maslinic acid (MA) was inhibitory to P. falciparum transition from ring to schizont stage. MA also inhibited the processing of MSP1 complex and hence supposed to target PfSUB1 [128]. In an in vitro study, PfSUB2 prodomain selectively inhibited the shedding of MSP1 and AMA1, it could be used an attractive design for PfSUB2 inhibitors. Structural study of PfSUB2 prodomain using nuclear magnetic resonance (NMR) identified a likely catalytic domain-binding interface region in it, which could act as a design for peptidomimetic inhibitor against the enzyme [129].
In addition to subtilisin-like proteases, intramembrane rhomboid proteases also have role during parasite invasion by cleaving the adhesins inside the parasite membrane. Rhomboid proteases carry out intramembrane proteolysis, and hence have their catalytic triad embedded within the membrane bilayer that is surrounded by a hydrophilic cavity [130]. P. falciparum rhomboid proteases are largely uncharacterized till date with two members PfROM1 and PfPROM4 partially characterized. PfROM1 andPfROM4 carried out the intramembrane cleavage of the adhesins AMA1 [126, 131] and erythrocyte-binding antigen 175 (EBA-175), respectively [132]. In addition, they were able to cleave a variety of adhesins involved in host–parasite interaction within the transmembrane domains [131]. Although this group of proteases seems to be crucial for the invasion process, they need to be extensively characterized and evaluated for druggability.
Overall, proteolytic enzymes involved in invasion are considered to be attractive targets of antimalarial drug development. Invasion is a very rapid process, taking place within a minute of merozoite release; the access of parasite molecules involved in invasion is debatable. Dowse et al. [132] suggested that proteases involved in invasion have the potential to serve as drug targets as their biosynthesis and maturation start much earlier in the life cycle, making them available for targeting by drugs [132].
6 Other Cellular Proteases
In addition to the above-described proteases, a number of parasitic proteases are involved in regular metabolic processes and cell cycle regulation in the parasite, with unique parasite-specific features. The parasite possesses a single mitochondrion and a relict plant plastid-like organelle called apicoplast [133,134,135,136,137]. The mitochondrion possesses some unique biochemical features. It is defective in tricarboxylic acid cycle and does not appear to oxidize glucose to produce ATP in the blood stages of the parasite [138], although it seems to be involved in pyrimidine biosynthesis [139]. An intriguing P. falciparum protease is ClpQ threonine protease (heat shock loci V or HslV) is localized in the mitochondrion [140, 141]. Its ATPase partner is ClpY (heat shock loci U or HslU) [142]. These two proteins interact to form ClpQY machinery, a prokaryotic predecessor of the eukaryotic proteosomal machinery [142]. ClpQ appears to be critical for parasite survival as the disruption of the interaction between ClpQ and ClpY using peptide inhibitors resulted in the death of asexual blood stages in a phenotype resembling apoptosis [143].
The apicoplast is crucial for the biosynthesis of parasite heme, isopentenyl diphosphate, fatty acids, and isoprenoid precursors [144, 145]. The apicoplast possesses its own genome but 95% of its proteins are nuclear-encoded and transported to the organelle. Targeting of proteins to apicoplast through the secretory pathway is mediated by bipartite signal and transit peptide sequences. Two parasite proteases are involved in the targeting process; the stromal processing protease (SPP) cleaves the transit peptide to release the mature protein [146] and the metalloprotease falcilysin carries out the degradation of the transit peptide [69]. Another serine protease ClpP dependent on ATPase for functioning has been localized in the apicoplast and its inhibitor was found to significantly inhibit parasite growth in vitro [147].
Parasite signal peptide peptidases mediate protein trafficking to their destinations within the parasite and host cytosol [24, 147–149]. The P. falciparum signal peptide peptidase (PfSPP) is essential for parasite growth inside the erythrocytes and could be targeted by small molecule inhibitors [150]. Gamma secretase and signal peptide peptidase inhibitor LY411,575 was evaluated for the targeting Plasmodium berghei liver stages in human hepatoma cell lines and in mouse primary hepatocytes. LY411,575 was found to be inhibitory for normal liver stage development at nanomolar concentration (IC50 value of 80 nM). This inhibitor also decreased the liver stage parasite load in vivo and also conferred 55% resistance to cerebral malaria in mice [151].
The malaria parasite contains a minimal endoplasmic reticulum-associated degradation (ERAD) network relative to higher eukaryotes. P. falciparum is highly sensitive to the inhibition of protease component of this system (PfSSP). The compounds inhibiting PfSSP also showed low nanomolar activity against liver stage malaria parasites [152]. Many nuclear-encoded proteins are targeted to subcellular organelles like apicoplast and mitochondria using the signal and transit peptides. Upon reaching the target, the transit peptides are cleaved to release the proteins. Metalloprotease falcilysin is known to cleave the transit peptide in malaria parasite [68, 69]. Parasite exports a number of proteins to modify the host erythrocytes. These proteins contain a pentameric (RxLxE/Q/D) motif required for export into the erythrocytes known as PEXEL (Plasmodium EXport ELement) motif, which is responsible for trafficking of these proteins into the host cytosol [153, 154]. An ER-resident aspartic protease plasmepsin V is responsible for cleavage of PEXEL and facilitating trafficking of the proteins [155].
7 Conclusion
Despite the availability of drugs and significant successes in reducing malaria-related illnesses and deaths globally, malaria still poses a serious threat to human health. Due to the ability of the parasite to quickly acquire resistance against the drugs, development of novel drugs against parasite-specific molecules remains a priority. The parasite genome encodes proteases of many different classes, many of which carry out processes crucial for parasite survival and propagation. These molecules have been extensively studied and many of these have been shown promises as potential drug targets because they posses unique parasite-specific features that could be specifically targeted. The success of protease inhibitors as drugs has already been shown against other infectious agents like HIV and HCV, invoking interest in the use of malarial protease inhibitors as drugs. Overall, malarial proteases represent an intriguing group of molecules that could be utilized for specific targeting by the novel drugs.
References
World Malaria Report (2015) World Health Organization
Trape JF, Legros F, Ndiaye P, Konate L et al (1989) Chloroquine-resistant Plasmodium falciparum malaria in Senegal. Trans R Soc Trop Med Hyg 83:761
Zucker JR, Ruebush TK, Obonyo C et al (2003) The mortality consequences of the continued use of chloroquine in Africa: experience in Siaya, western Kenya. Am J Trop Med Hyg 68:386–390
Sibley CH, Hyde JE, Sims PF et al (2001) Pyrimethamine-sulfadoxine resistance in Plasmodium falciparum: what next? Trends Parasitol 17:582–588
Wongsrichanalai C, Pickard AL, Wernsdorfer WH et al (2002) Epidemiology of drug-resistant malaria. Lancet Infect Dis 2:209–218
TerKuile F, White NJ, Holloway P et al (1993) Plasmodium falciparum: in vitro studies of the pharmacodynamic properties of drugs used for the treatment of severe malaria. Exp Parasitol 76:85–95
White NJ (2008) Qinghaosu (artemisinin): the price of success. Science 320:330–334
Ittarat W, Pickard AL, Rattanasinganchan P et al (2003) Recrudescence in artesunate treated patients with falciparum malaria is dependent on parasite burden not on parasite factors. Am J Trop Med Hyg 68:147–152
Travassos MA, Laufer MK (2009) Resistance to antimalarial drugs: molecular, pharmacologic, and clinical considerations. Pediatr Res 65:64R–70R
Dondorp AM, Nosten F, Yi P et al (2009) Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467
Anderson TJ, Nair S, Nkhoma S et al (2010) High heritability of malaria parasite clearance rate indicates a genetic basis for artemisinin resistance in Western Cambodia. J Infect Dis 201:1326–1330
McKerrow JH, Caffrey C, Kelly B et al (2006) Proteases in parasitic diseases. Annu Rev Pathol 1:497–536
Flexner C, Bate G, Kirkpatrick P (2005) Tipranavir. Nat Rev Drug Discov 4:955–956
Melnikova I (2008) Hepatitis C therapies. Nat Rev Drug Discov 7:799–800
Smith CG, Vane JR (2003) The discovery of captopril. FASEB J 17:788–789
Melnikova I (2009) The anticoagulants market. Nat Rev Drug Discov 8:353–354
Turk B (2006) Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov 5:785–799
Neurath H (1989) in The diversity of proteolytic enzymes.In: Beynon RJ, Bond JS (eds) Proteolytic enzymes: a practical approach. IRL Press, Oxford, pp 1–13
Rosenthal PJ (2002) Hydrolysis of erythrocyte proteins by proteases of malaria parasites. Curr Opin Hematol 9:140–145
Gardner MJ, Hall N, Fung E et al (2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419:498–511
Mazier D, Rénia L, Snounou G (2013) A pre-emptive strike against malaria’s stealthy hepatic forms. Nat Rev Drug Discovery 8:854–864
Suarez C, Volkmann K, Gomes AR et al (2013) The malarial serine protease SUB1 plays an essential role in parasite liver stage development. PLoS Pathog 9:e1003811
Tawk L, Lacroix C, Gueirard P et al (2013) A key role for Plasmodium subtilisin-like SUB1 protease in egress of malaria parasites from host hepatocytes. J Biol Chem 288:33336–33346
Wu Y, Wang X, Liu X et al (2003) Data-mining approaches reveal hidden families of proteases in the genome of malaria parasite. Genome Res 13:601–616
Lilburn TG, Hong C, Zhan Zhou Z et al (2011) Protease-associated cellular networks in malaria parasite Plasmodium falciparum. BMC Genom 12(Suppl 5):S9
Sherman IW (1977) Amino acid metabolism and protein synthesis in malarial parasites. Bull World Health Organ 55:265–276
McKerrow JH, Sun E, Rosenthal PJ et al (1993) The proteases and pathogenicity of parasitic protozoa. Annu Rev Microbiol 47:821–853
Rosenthal PJ, Meshnick SR (1996) Hemoglobin catabolism and iron utilization by malaria parasites. Mol Biochem Parasitol 83:131–139
Wood PA, Eaton JW (1993) Hemoglobin catabolism and host-parasite heme balance in chloroquine-sensitive and chloroquine-resistant Plasmodium berghei infections. Am J Trop Med Hyg 48:465–472
Scheibel LW, Sherman IW (1988). In: Wernsdorfer WH, McGregor I (eds) Malaria. Churchill Livingstone, Edinburgh, vol 1, pp 219–252
Yayon A, Cabantchik ZI, Ginsburg H (1984) Identification of the acidic compartment of Plasmodium falciparum-infected human erythrocytes as the target of the antimalarial drug chloroquine. EMBO J 3:2695–2700
Krogstad DJ, Schlesinger PH, Gluzman IY (1985) Antimalarials increase vesicle pH in Plasmodium falciparum. J Cell Biol 101:2302–2309
Goldberg DE, Slater AF, Cerami A et al (1990) Hemoglobin degradation in the malaria parasite Plasmodium falciparum: an ordered process in a unique organelle. Proc Natl Acad Sci USA 87:2931–2935
Francis SE, Gluzman IY, Oksman A et al (1994) Molecular characterization and inhibition of a Plasmodium falciparum aspartic hemoglobinase. EMBO J 13:306–317
Hill J, Tyas L, Phylip LH et al (1994) High level expression and characterisation of Plasmepsin II, an aspartic proteinase from Plasmodium falciparum. FEBS Lett 352:155–158
Rosenthal PJ, McKerrow JH, Aikawa M et al (1988) A malarial cysteine proteinase is necessary for hemoglobin degradation by Plasmodium falciparum. J Clin Invest 82:1560–1566
Banerjee R, Liu J, Beatty W et al (2002) Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc Natl Acad Sci USA 99:990–995
Goldberg DE (2005) Hemoglobin degradation. Curr Top Microbiol Immunol 295:275–291
Gluzman IY, Francis SE, Oksman A et al (1994) Order and specificity of the Plasmodium falciparum hemoglobin degradation pathway. J Clin Invest 93:1602–1608
Shenai BR, Sijwali PS, Singh A (2000) Characterization of native and recombinant falcipain-2, a principal trophozoite cysteine protease and essential hemoglobinase of Plasmodium falciparum. J Biol Chem 275:29000–29010
Eggleson KK, Duffin KL, Goldberg DE (1999) Identification and characterization of falcilysin, a metallopeptidase involved in hemoglobin catabolism within the malaria parasite Plasmodium falciparum. J Biol Chem 274:32411–32417
Gavigan CS, Dalton JP, Bell A (2001) The role of aminopeptidases in haemoglobin degradation in Plasmodium falciparum-infected erythrocytes. Mol Biochem Parasitol 117:37–48
Liu J, Istvan ES, Gluzman IY et al (2006) Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc Natl Acad Sci USA 103:8840–8845
Liu K, Shi H, Xiao H et al (2009) Functional profiling, identification, and inhibition of Plasmepsins in intraerythrocytic malaria parasites. Angew Chem Int Ed 48:8293–8297
Omara-Opyene AL, Moura PA, Sulsona CR (2004) Genetic disruption of the Plasmodium falciparum digestive vacuole plasmepsins demonstrates their functional redundancy. J Biol Chem 279:54088–54096
Liu J, Gluzman IY, Drew ME (2005) The role of Plasmodium falciparum food vacuole plasmepsins. J Biol Chem 280:1432–1437
Moon RP, Bur D, Loetscher H et al (1998) Studies on plasmepsins I and II from the malarial parasite Plasmodium falciparum and their exploitation as drug targets. Adv Exp Med Biol 436:397–406
Ersmark K, Feierberg I, Bjelic S et al (2004) Potent inhibitors of the Plasmodium falciparum enzymes plasmepsin I and II devoid of cathepsin D inhibitory activity. J Med Chem 47:110–122
Johansson PO, Lindberg J, Blackman MJ et al (2005) Design and synthesis of potent inhibitors of plasmepsin I and II: X-ray crystal of inhibitor in complex with plasmepsin II. J Med Chem 48:4400–4409
Muthas D, Nöteberg D, Sabnis YA et al (2005) Synthesis, biological evaluation, and modeling studies of inhibitors aimed at the malarial proteases plasmepsins I and II. Bioorg Med Chem 13:5371–5390
Mohapatra SC, Tiwari HK, Single M (2010) Antimalarial evaluation of copper(II) nanohybrid solids: inhibition of plasmepsin II, a hemoglobin-degrading malarial aspartic protease from Plasmodium falciparum. J Biol Inorg Chem 15:373–385
Gupta D, Yedidi RS, Varghese S et al (2010) Mechanism-based inhibitors of the aspartyl protease plasmepsin II as potential antimalarial agents. J Med Chem 53:4234–4247
Dali B, Keita M, Megnassan E et al (2012) Insight into selectivity of peptidomimetic inhibitors with modified statine core for plasmepsin II of Plasmodium falciparum over human cathepsin D. Chem Biol Drug Des 79:411–430
Rasina D, Otikovs M, Leitans J et al (2016) Fragment-based discovery of 2-aminoquinazolin-4(3H)-ones as novel class nopeptidomimetic inhibitors of the plasmepsins I, II, and IV. J Med Chem 59:374–387
Ersmark K, Samuelsson B, Hallberg A (2006) Plasmepsins as potential targets for new antimalarial therapy. Med Res Rev 26:626–666
Nezami A, Kimura T, Hidaka K et al (2003) High-affinity inhibition of a family of Plasmodium falciparum proteases by a designed adaptive inhibitor. Biochemistry 42:8459–8464
Sijwali PS, Shenai BR, Gut J et al (2001) Expression and characterization of the Plasmodium falciparum haemoglobinase falcipain-3. Biochem J 360:481–489
Sijwali PS, Rosenthal PJ (2004) Gene disruption confirms a critical role for the cysteine protease falcipain-2in hemoglobin hydrolysis by Plasmodium falciparum. Proc Natl Acad Sci USA 101:4384–4389
Sijwali PS, Koo J, Singh N et al (2006) Gene disruptions demonstrate independent roles for the four falcipain cysteine proteases of Plasmodium falciparum. Mol Biochem Parasitol 150:96–106
Singh N, Sijwali PS, Pandey KC et al (2006) Plasmodium falciparum: biochemical characterization of the cysteineprotease falcipain-2. Exp Parasitol 112:187–192
Goh LL, Sim TS (2005) Characterization of amino acid variation at strategic positions in parasite and human proteases for selective inhibition of falcipains in Plasmodium falciparum. Biochem Biophys Res Commun 335:762–770
Jeong JJ, Kumar A, Hanada T et al (2006) Cloning and characterization of Plasmodium falciparum cysteine protease, falcipain-2B. Blood Cells Mol Dis 36:429–435
Rosenthal PJ (2004) Cysteine proteases of malaria parasites. Int J Parasitol l34:1489–1499
Rosenthal PJ, Wollish WS, Palmer JT (1991) Antimalarial effects of peptide inhibitors of a Plasmodium falciparum cysteine proteinase. J Clin Invest 88:1467–1472
Singh A, Rosenthal PJ (2001) Comparison of efficacies of cysteine protease inhibitors against five strains of Plasmodium falciparum. Antimicrob Agents Chemother 45:949–951
Batra S, Sabnis YA, Rosenthal PJ et al (2003) Structure-based approach to falcipain-2 inhibitors: synthesis and biological evaluation of 1,6,7-trisubstituted dihydroisoquinolines and isoquinolines. Bioorg Med Chem 11:2293–2299
Rosenthal PJ, Lee GK, Smith RE (1993) Inhibition of a Plasmodium vinckei cysteine proteinase cures murine malaria. J Clin Invest 91:1052–1056
Murata CE, Goldberg DE (2003) Plasmodium falciparum falcilysin: a metalloprotease with dual specificity. J Biol Chem 278:38022–38028
Ponpuak M, Klemba M, Park M et al (2007) A role for falcilysin in transit peptide degradation in the Plasmodium falciparum apicoplast. Mol Microbiol 63:314–334
Curley GP, O’Donovan SM, McNally J et al (1994) Aminopeptidases from Plasmodium falciparum, Plasmodium chabaudi chabaudi and Plasmodium berghei. J Eukaryot Microbiol 41:119–123
Mistry SN, Drinkwater N, Ruggeri C et al (2014) Two-pronged attack: dual inhibition of Plasmodium falciparum M1 and M17 metalloaminopeptidases by a novel series of hydroxamic acid-based inhibitors. J Med Chem 57:9168–9183
Winograd E, Clavijo CA, Bustamante LY et al (1999) Release of merozoites from Plasmodium falciparum-infected erythrocytes could be mediated by a non-explosive event. Parasitol Res 85:621–624
Glushakova S, Yin D, Li T et al (2005) Membrane transformation during malaria parasite release from human red blood cells. Curr Biol 15:1645–1650
Wickham ME, Culvenor JG, Cowman AF (2003) Selective inhibition of a two-step egress of malaria parasites from the host erythrocyte. J Biol Chem 278:37658–37663
Salmon BL, Oksman A, Goldberg DE (2001) Malaria parasite exit from the host erythrocyte: a two-step process requiring extraerythrocytic proteolysis. Proc Natl Acad Sci USA 98:271–276
Soni S, Dhawan S, Rosen KM et al (2005) Characterization of events preceding the release of malaria parasite from the host red blood cell. Blood Cells Mol Dis 35:201–211
Langreth SG, Jensen JB, Reese RT et al (1978) Fine structure of human malaria in vitro. J Protozool 25:443–452
Aikawa M (1971) Parasitological review. Plasmodium: the fine structure of malarial parasites. Exp Parasitol 30:284–320
Banyal HS, Misra GC, Gupta CM et al (1981) Involvement of malarial proteases in the interaction between the parasite and host erythrocyte in Plasmodium knowlesi infections. J Parasitol 67:623–626
Dutta GP, Banyal HS (1981) In vitro susceptibility of erythrocytes of Presbytis entellus (Indian Langur) to Plasmodium knowlesi & blocking of merozoite invasion process by certain protease inhibitors. Ind J Exp Biol 19:9–11
Hadley T, Aikawa M, Miller LH (1983) Plasmodium knowlesi: studies on invasion of rhesus erythrocytes by merozoites in the presence of protease inhibitors. Exp Parasitol 55:306–311
Lyon JA, Haynes JD (1986) Plasmodium falciparum antigens synthesized by schizonts and stabilized at the merozoite surface when schizonts mature in the presence of protease inhibitors. J Immunol 136:2245–2251
Delplace P, Bhatia A, Cagnard M et al (1988) Protein p126: a parasitophorous vacuole antigen associated with the release of Plasmodium falciparum merozoites. Biol Cell 64:215–221
Le Bonniec S, Deregnaucourt C, Redeker V et al (1999) Plasmepsin II, an acidic hemoglobinase from the Plasmodium falciparum food vacuole, is active at neutral pH on the host erythrocyte membrane skeleton. J Biol Chem 274:14218–14223
Dua M, Raphael P, Sijwali PS et al (2001) Recombinant falcipain-2 cleaves erythrocyte membrane ankyrin and protein 4.1. Mol Biochem Parasitol 116:95–99
Hanspal M, Dua M, Takakuwa Y et al (2002) Plasmodium falciparum cysteine protease falcipain-2 cleaves erythrocyte membrane skeletal proteins at late stages of parasite development. Blood 100:1048–1054
Dhawan S, Dua M, Chishti AH et al (2003) Ankyrin peptide blocks falcipain-2-mediated malaria parasite release from red blood cells. J Biol Chem 278:30180–30186
Blackman MJ (2008) Malarial proteases and host cell egress: an ‘emerging’ cascade. Cell Microbiol 10:1925–1934
Aly AS, Matuschewski K (2005) A malarial cysteine protease is necessary for Plasmodium sporozoite egress from oocysts. J Exp Med 202:225–230
Miller SK, Good RT, Drew DR et al (2002) A subset of Plasmodium falciparum SERA genes are expressed and appear to play an important role in the erythrocytic cycle. J Biol Chem 277:47524–47532
Bzik DJ, Li WB, Horii T et al (1988) Amino acid sequence of the serine-repeat antigen (SERA) of Plasmodium falciparum determined from cloned cDNA. Mol Biochem Parasitol 30:279–288
Gor DO, Li AC, Rosenthal PJ (1998) Protective immune responses against protease-like antigens of the murine malaria parasite Plasmodium vinckei. Vaccine 16:1193–1202
Kiefer MC, Crawford KA, Boley LJ et al (1996) Identification and cloning of a locus of serine repeat antigen (sera)-related genes from Plasmodium vivax. Mol Biochem Parasitol 78:55–65
Hodder AN, Drew DR, Epa VC et al (2003) Enzymic, phylogenetic, and structural characterization of the unusual papain-like protease domain of Plasmodium falciparum SERA5. J Biol Chem 278:48169–48177
McCoubrie JE, Miller SK, Sargeant T et al (2007) Evidence for a common role for the serine-type Plasmodium falciparum serine repeat antigen proteases: implications for vaccine and drug design. Infect Immun 75:5565–5574
Delplace P, Dubremetz JF, Fortier B et al (1985) A 50 kilodalton exoantigen specific to the merozoite release-reinvasion stage of Plasmodium falciparum. Mol Biochem Parasitol 17:239–251
Debrabant A, Maes P, Delplace P, Dubremetz JF, Tartar A, Camus D (1992) Intramolecular mapping of Plasmodium falciparum P126 proteolytic fragments by N-terminal amino acid sequencing. Mol Biochem Parasitol 53:89–95
Pang XL, Mitamura T, Horii T (1999) Antibodies reactive with the N-terminal domain of Plasmodium falciparum serine repeat antigen inhibit cell proliferation by agglutinating merozoites and schizonts. Infect Immun 67:1821–1827
Li J, Matsuoka H, Mitamura T et al (2002) Characterization of proteases involved in the processing of Plasmodium falciparum serine repeat antigen (SERA). Mol Biochem Parasitol 120:177–186
Yeoh S, O’Donnell RA, Koussis A et al (2007) Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell 131:1072–1083
Arastu-Kapur S, Ponder EL, Fonovic UP et al (2008) Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat Chem Biol 4:203–213
Fairlie WD, Spurck TP, McCoubrie JE et al (2008) Inhibition of malaria parasite development by a cyclic peptide that targets the vital parasite protein SERA5. Infect Immun 76:4332–4344
Alam A, Chauhan VS (2012) Inhibitory potential of prodomain of Plasmodium falciparum protease serine repeat antigen 5 for asexual blood stages of parasite. PLoS ONE, e30452
Ruecker A, Shea M, Hackett F et al (2012) Proteolytic activation of the essential parasitophorous vacuole cysteine protease SERA6 accompanies malaria parasite egress from its host erythrocyte. J Biol Chem 287:37949–37963
Withers-Martinez C, Suarez C, Fulle S et al (2012) Plasmodium subtilisin-like protease 1 (SUB1): insights into the active-site structure, specificity and function of a pan-malaria drug target. Int J Parasitol 42:597–612
Gemma S, Giovani S, Brindisi M et al (2012) Quinolylhydrazones as novel inhibitors of Plasmodium falciparum serine protease PfSUB1. Bioorg Med Chem Lett 22:5317–5321
Cowman AF, Crabb BS (2006) Invasion of red blood cells by malaria parasites. Cell 124:755–766
Dejkriengkraikhul P, Wilairat P (1983) Requirement of malarial protease in the invasion of human red cells by merozoites of Plasmodium falciparum. Z Parasitenkd 69:313–317
Breton CB, Blisnick T, Jouin H et al (1992) Plasmodium chabaudi p68 serine protease activity required for merozoite entry into mouse erythrocytes. Proc Natl Acad Sci USA 89:9647–9651
Dluzewski AR, Rangachari K, Wilson RJ et al (1986) Protease inhibitors and inhibition of erythrocyte invasion. Exp Parasitol 62:416–422
McPherson RA, Donald DR, Sawyer WH et al (1993) Proteolytic digestion of band 3 at an external site alters the erythrocyte membrane organisation and may facilitate malarial invasion. Mol Biochem Parasitol 62:233–242
Blackman MJ, Holder AA (1992) Secondary processing of the Plasmodium falciparum merozoite surface protein-1 (MSP1) by a calcium-dependent membrane-bound serine protease: shedding of MSP133 as a noncovalently associated complex with other fragments of the MSP1. Mol Biochem Parasitol 50:307–315
Bannister LH, Dluzewski AR (1990) The ultrastructure of red cell invasion in malaria infections: a review. Blood Cells 16:257–292
Keeley A, Soldati D (2004) The glideosome: a molecular machinepowering motility and host-cell invasion by Apicomplexa. TrendsCell Biol 14:528–532
Harris PK, Yeoh S, Dluzewski AR et al (2005) Molecular identification of a malaria merozoite surface sheddase. PLoS Pathog 1(e29):0241–0251
Brossier F, Jewett TJ, Sibley LD et al (2005) A spatiallylocalized rhomboid protease cleaves cell surface adhesins essentialfor invasion by Toxoplasma. Proc Natl Acad Sci USA 102:4146–4151
Dowse TJ, Pascall JC, Brown KD et al (2005) Apicomplexan rhomboids have a potential role in microneme protein cleavage during host cell invasion. Int J Parasitol 35:747–756
Alexander DL, Mital J, Ward GE et al (2005) Identification of the moving junction complex of Toxoplasma gondii: A collaboration between distinct secretory organelles. PLoS Pathog 1:e17
Singh S, Alam MM, Pal-Bhowick I et al (2010) Distinct external signals trigger sequential release of apical organelles during erythrocyte invasion by malaria parasites. PLoS Pathog 6:e1000746
Blackman MJ, Heidrich H-G, Donachie S et al (1990) A single fragment of a malaria merozoite surface protein remains on the parasite during red cell invasion and is the target of invasion-inhibiting antibodies. J Exp Med 172:379–382
Goel VK, Li X, Chen H et al (2003) Band 3 is a host receptor bindingmerozoite surface protein 1 during the Plasmodium falciparum invasion of erythrocytes. Proc Natl Acad Sc USA 100:5164–5169
Li X, Chen H, Oo TH et al (2004) A co-ligand complex anchors Plasmodium falciparum merozoites to the erythrocyte invasion receptor band 3. J Biol Chem 279:5765–5771
Child MA, Epp C, Bujard H et al (2010) Regulated maturation of malaria merozoite surface protein-1 is essential for parasite growth. Mol Microbiol 78:187–202
Barale J-C, Blisnick T, Fujioka H et al (1999) Plasmodium falciparum subtilisin-like protease 2, a merozoite candidate for the merozoite surface protein 1-42 maturase. Proc Natl Acad Sc USA 96:6445–6450
Howell SA, Hackett F, Jongco AM et al (2005) Distinct mechanisms govern proteolytic shedding of a key invasion protein in apicomplexan pathogens. Mol Microbiol 57:1342–1356
Uzureau P, Barale JC, Janse CJ et al (2004) Gene targeting demonstrates that the Plasmodium berghei subtilisin PbSUB2 is essential for red cell invasion and reveals spontaneous genetic recombination events. Cell Microbiol 6:65–78
Moneriz C, Mestres J, Bautista JM et al (2011) Multi-targeted activity of maslinic acid as an antimalarial natural compound. FEBS J 278:2951–2961
He Y, Chen Y, Oganesyan N et al (2012) Solution NMR structure of a sheddase inhibitor prodomain from the malarial parasite Plasmodium falciparum. Proteins 80:2810–2817
Urban S (2006) Rhomboid proteins: conserved membrane proteases with divergent biological functions. Genes Dev 20:3054–3068
Baker RP, Wijetilaka R, Urban S (2006) Two Plasmodium rhomboid proteases preferentially cleave different adhesins implicated in all invasive stages of malaria. PLoS Pathog 2:e113
O’Donnell RA, Hackett S, Howell SA et al (2006) Intramembrane proteolysis mediates shedding of a key adhesin during erythrocyte invasion by the malaria parasite. J Cell Biol 174:1023–1033
Dowse TJ, Koussis K, Blackman MJ, Soldati-Favre D (2008) Roles of proteases during invasion and egress by Plasmodium and Toxoplasma. In: Burleigh BA, Soldati-Favre D (eds) Molecular mechanism of parasite invasion. Landes Biosciences and Springer Science+Business Media
Waller RF, McFadden GI (2005) The apicoplast: a review of the derived plastid of apicomplexan parasites. Curr Issues Mol Biol 7:57–79
Goodman CD, Su V, McFAdden GI (2007) The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol Biochem Parasitol 152:181–191
Schlitzer M (2007) Malaria chemotherapeutics part I: History of antimalarial drug development, currently used therapeutics, and drugs in clinical development. ChemMedChem 2:944–986
Schlitzer M (2006) Selective enzyme inhibitor instead of an “iron-triggered cluster bomb”. Pharm Unserer Zeit 35:8–9
Dahl EL, Rosenthal PJ (2008) Apicoplast translation, transcription and genome replication: targets for antimalarial antibiotics. Trends Parasitol 24:279–284
Vaidya AB, Mather MW (2009) Mitochondrial evolution and functions in malaria parasites. Annu Rev Microbiol 63:249–267
Painter HJ, Morrisey JM, Mather MW, Vaidya AB (2007) Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446:88–91
Tschan S, Kreidenweiss A, Stierhof YD et al (2010) Mitochondrial localization of the threonine peptidase PfHslV, a ClpQ ortholog in Plasmodium falciparum. Int J Parasitol 40:1517–1523
Ramasamy G, Gupta D, Mohmmed A et al (2007) Characterization and localization of Plasmodium falciparum homolog of prokaryotic ClpQ/HslV protease. Mol Biochem Parasitol 152:139–148
Sousa MC, Trame CB, Tsuruta H et al (2000) Crystal and solution structures of an HslUV protease-chaperone complex. Cell 103:633–643
Rathore S, Jain S, Sinha D et al (2011) Disruption of a mitochondrial protease machinery in Plasmodium falciparum is an intrinsic signal for parasite cell death. Cell Death Dis 2:e231
Ralph SA, van Dooren GG, Waller RF et al (2004) Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat Rev Microbiol 2:203–216
Yeh E, DeRisi JL (2011) Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol 9:e1001138
van Dooren GG, Su V, D’Ombrain MC et al (2002) Processing of an apicoplast leader sequence in Plasmodium falciparum and the identification of a putative leader cleavage enzyme. J Biol Chem 277:23612–23619
Rathore S, Sinha D, Asad M et al (2010) A cyanobacterial serine protease of Plasmodium falciparum is targeted to the apicoplast and plays an important role in its growth and development. Mol Microbiol 77:873–890
Sharma S, Pradhan A, Chauhan VS et al (2005) Isolation and characterization of type I signal peptidase of different malaria parasites. J Biomed Biotechnol 2005:301–309
Tuteja R, Pradhan A, Sharma S (2008) Plasmodium falciparum signal peptidase is regulated by phosphorylation and required for intra-erythrocytic growth. Mol Biochem Parasitol 157:137–147
Li X, Chen H, Bahamontes-Rosa N et al (2009) Plasmodium falciparum signal peptide peptidase is a promising drug target against blood stage malaria. Biochem Biophys Res Commun 380:454–459
Parvanova I, Epiphanio S, Fauq A et al (2009) A small molecule inhibitor of signal peptide peptidase inhibits Plasmodium development in the liver and decreases malaria severity. PLoS ONE 4:e5078
Harbut MB, Velmourougane G, Dalal S et al (2011) Bestatin-based chemical biology strategy reveals distinct roles for malaria M1- and M17-family aminopeptidases. Proc Natl Acad Sci USA 108:E526–E534
Chang HH, Falick AM, Carlton PM et al (2008) N-terminal processing of proteins exportedby malaria parasites. Mol Biochem Parasitol 160:107–115
Boddey JA, Moritz RL, Simpson RJ et al (2009) Role of the Plasmodium export element intraffi cking parasite proteins to the infected erythrocyte. Traffi c 10:285–299
Boddey JA, Hodder AN, Gunther S et al (2010) An aspartyl protease directs malaria effectorproteins to the host cell. Nature 463:627–631
Acknowledgements
The author would like to acknowledge the scientists who contributed in the field of malarial protease research. The author would also like to thank the Japan Society for the Promotion of Science for financial support and Prof. Shigeto Yoshida for providing encouragement to write the article.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Alam, A. (2017). Plasmodium Proteases as Therapeutic Targets Against Malaria. In: Chakraborti, S., Chakraborti, T., Dhalla, N. (eds) Proteases in Human Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-10-3162-5_4
Download citation
DOI: https://doi.org/10.1007/978-981-10-3162-5_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3161-8
Online ISBN: 978-981-10-3162-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)