
Abstract
Malaria remains a global threat with millions of deaths annually. Emergence of parasite strains resistant to widely used antimalarials, including the artemisinin combination therapy (ACT), and the absence of an effective vaccine makes treatment of malaria difficult than ever before. The need of the hour is to re-evaluate the chemotherapeutic approach and to identify new drug targets and develop new pharmacophores against the parasite. An important approach for antimalarial drug discovery is to understand critical metabolic pathways in the parasite which may help us to identify critical targets in the parasites and design specific inhibitors for these targets. Here, we have discussed proteins and pathways in different parasite organelles, i.e. apicoplast, mitochondrial and food vacuole, which have been suggested as potential drug targets; these unique parasite proteins can be targeted to develop new and novel antimalarials. In addition, we have also discussed several antimalarial projects currently under different stages of drug development pipeline. These promising antimalarial compounds have the potential to overcome multidrug resistance. Ongoing global efforts to develop new antimalarials and to identify drug targets suggest a promising future on malaria elimination and eradication.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Malaria is one of the oldest infectious diseases responsible for socio-economic burden on mankind. Malaria situation in India has undergone different phases from high prevalence to controllable condition with the use of insecticide and control measures, to resurgence in the era of insecticide and chloroquine resistance, to the use of artemisinin combination therapies from 1953 to 2008 [1,2,3]. One of the biggest challenges for malaria treatment is the rise of resistance against available antimalarials in the last decade [4]. Discovered in the early 1930s, chloroquine became the drug of choice for treatment against malaria [5]; however, the drug faced trouble of resistance in the late 1950s when reports of its ineffectiveness started to come from Colombia to Southeast Asia [6,7,8]. Later, discovery of artemisinin has helped treatment of malaria very effectively, although recently in 2009, P. falciparum resistant to artemisinin-based combination therapy (ACT) was reported [9]. Therefore, there is a constant requirement for the development of new lead molecules in the drug development pipeline. In this chapter, we have discussed parasite’s metabolic pathways which could be novel key targets for drug development, recent advances in drug development and promising lead molecules which are in pipeline towards development of new antimalarials.
Antimalarials Targeting Haemoglobin Uptake, Degradation and the Food Vacuole
During the life cycle of parasite in red blood cell, the parasite internalizes a huge amount of haemoglobin for degradation, i.e. ring, trophozoite and schizont, internalizes a huge amount of haemoglobin for degradation. Haemoglobin uptake and its degradation has remained one of the most essential pathways for the survival of parasite and thus has been targeted with small molecule inhibitors as antimalarials. The haemoglobin uptake itself is being defined by four different processes including “Big Gulp”, which is internalization of huge chunk of RBC cytoplasm with haemoglobin by ring-stage parasite. Big Gulp involves a cuplike shape formation by ring-stage parasite which does not involve actin polymerization. Another pathway defined for the uptake is somewhat similar to the classical endocytosis having cytostome, small vesicle-like structures marked with Rab5a, the early endosomal regulatory protein [10, 11]. There are reports of thin long tube-like structures formed through actin polymerization, termed as cytostomal tubes, which is considered as third pathway for the uptake by the parasite. Fourth process is termed as phagotrophic as it resembles phagocytosis-like process, but it lacks actin polymerization which is key event in the case of phagocytosis [12]. All of the defined four pathways of haemoglobin uptake are good targets for antimalarial therapy as they have some unique aspects with Plasmodium.
However, there are little efforts on development of new antimalarials targeting the endocytosis pathways. Antimalarials, like chloroquine, mefloquine and artemisinin, have been shown to have various effects on the endocytosis and endocytic pathways of the different mammalian cells [13, 14]. Mefloquine and artemisinin have been shown to inhibit phagocytosis in immune cells [14, 15], whereas chloroquine is shown to play role as lysosomotropic agents disrupting trafficking in the endocytic pathways, along with secretory pathway in different mammalian cells [16,17,18]. Similarly, the 8-aminoquinoline primaquine is shown to disturb endocytic pathway in hep-G2 cells [19]. Quinolones and artemisinin have been also shown to disrupt haemoglobin uptake by inhibiting endocytosis mechanism in the parasite. Chloroquine can inhibit endocytosis-based uptake on 12 h treatment to the ring-stage parasite and leads to accumulation of small vesicles containing haemoglobin [20]. After the ingestion of host cytoplasm with haemoglobin by parasite, cytosomal-like vesicles, which are double membrane-bound structures, carry the ingested material to the food vacuole. Subsequently, haemoglobin gets degraded in the food vacuole by the action of several proteases under acidic conditions. Proteolytic digestion in the food vacuole leads to accumulation of toxic amounts of ferriprotoporphyrin IX prosthetic group of the protein (heme). Parasite crystallizes this heme into insoluble hemozoin crystals, which are nontoxic to the parasite. A number of proteases are suggested to play a role in haemoglobin degradation in food vacuole of Plasmodium; these include aspartic proteases (plasmepsins), cysteine proteases (falcipains), histo-aspartic proteases and some of diamino peptidyl aminopeptidases (DPAP1). As the haemoglobin degradation is one of the most important pathways for survival of the parasite, these sets of proteases have remained a target for development of various small molecule inhibitors and drugs. A number of small molecule compounds based on different scaffolds such as fluoromethyl ketones, leupeptin, vinyl sulfones and chalcones have been developed against these proteases [21,22,23,24,25,26].
Plasmepsins, aspartic proteases, are good targets for development of antimalarials as they have been associated with important metabolic functions during the parasite life cycle. Plasmepsin V has been shown to play a role in export of parasite proteins to the host cytoplasm and beyond and thus is an important molecule which can be targeted [27, 28]. Plasmepsins I, II, III and IV have been shown to be present in food vacuole and playing roles in the degradation of haemoglobin and thus poised as drug targets [29, 30]. However, initial efforts to develop antimalarial against these proteases have faced roadblocks due to redundant action of these proteases [29]. 2-Aminoquinazolin-4(3H)-ones are a novel class of malaria digestive vacuole plasmepsin inhibitors which are identified using NMR-based fragment screening against PM II. These compounds show antiplasmodial activity targeting plasmepsins I, II and IV although they show almost tenfold higher activity towards cathepsin D [29]. Recently, non-peptidomimetic inhibitors are being designed based on these scaffolds which specifically target plasmepsin IV [31]. Another approach targeting plasmepsins includes development of compounds on the basis of “double-drug” approach: primaquine, which has been linked to statine-based inhibitors of plasmepsins (PMs) [32]. Retroviral aspartic proteases RS370 and RS367 demonstrated inhibition against plasmepsin II at sub-micromolar concentrations. The 7-azabicyclo[2.2.1]heptane-based inhibitors have been reported to show high potency against PM I and IV. Recently, Dominique et al. showed multistage antimalarial activity of aspartic protease inhibitor hydroxyl-ethyl-amine-based scaffold compound, 49c [33].
Falcipains, which is a family of four papain-family proteases, are another set of cysteine proteases which are important for haemoglobin degradation [34,35,36,37,38]. Falcipain-2 and 3 work in combination with other proteases to hydrolyse haemoglobin in food vacuole. Falcipain-2 disruption has led to decrease in the rate of haemoglobin degradation, whereas falcipain-3 deletion was not successful pointing towards essential role of this protease for the parasite survival [38]. Due to their important role and no known homologue in humans, falcipains are valid drug targets, and constant efforts are going on to develop inhibitors against them. Small inhibitors ranging from peptidyl fluoromethyl ketone [39, 40], vinyl sulfone [22, 24] and aldehydic inhibitors are developed against falcipain and shown to have potent inhibitory activity against the parasite [25]. Drug development against falcipains has now been facilitated much with available structures of falcipains complexed with small molecule inhibitors, which leads to more potent compounds being developed [41]. Dihydroartemisinin derivative against Falcipain-2 has been developed and shown to have potent activity against parasite [42]. Similarly, some non-peptidomimetic inhibitors are also designed and used against falcipain-2 which showed a potential activity against the parasite [43]. E-64 which is a known cysteine protease inhibitor and has an important role in elucidating the role of falcipains in haemoglobin degradation has also been derivatized, and epoxy succinate E-64 compounds also have been developed [44].
Recently, the Plasmodium signal peptide peptidase (PfSPP), an aspartic protease, which is a member of family of intramembrane cleaving proteases, has been shown to be essential for parasite survival in both blood and liver stages [45]. In addition, some of the HIV protease inhibitors, e.g. saquinavir and ritonavir, are also being employed with chloroquine and mefloquine for the treatment [46].
Degradation of haemoglobin in the parasite food vacuole leads to generation of a large amount of toxic by product, i.e. heme, which can damage membranes due to its peroxidative properties. Parasite has devised a strategy of combating this toxic effect by converting heme to hemozoin crystals. This process involves oxidation of heme to hematin and then conversion to hemozoin. It has been shown recently that the process is carried out in coordination with the haemoglobin degradation through a protein complex, “degradosome” [47]. Chloroquine, one of the well-known antimalarials, is known to disrupt the process of hemozoin development. The heme-detoxification protein, HDP, and Histidine-rich protein (HRP-2) are the key proteins involved in the process, and these are suggested to be potential drug targets in the food vacuole linked pathways. In addition, interphase regions of protein-protein interaction in the haemoglobin degradation and hemozoin development complex can also be targeted to design new antimalarials.
Mitochondrial Metabolic Pathways as Targets for Anti-malarial Development
Mitochondria, vividly called as “powerhouse of the cell”, are present in all eukaryotic organisms from protists to mammals. The origin of these conserved organelles is traced back to a single endosymbiotic event involving alpha proteobacteria, and all the mitochondria of eukaryotes have evolved as divergence from these early mitochondria [48]. Major role of mitochondria in most eukaryotic cells from fungi to mammals is the generation of ATPs by oxidative phosphorylation by complete oxidation of pyruvate generated from glycolysis pathway. During the process of glycolysis in cytosol, glucose is converted in to pyruvate, and two ATP equivalents are generated. The mitochondria harbours metabolic pathways for energy metabolisms including: TCA cycle for utilizing pyruvate to release CO2 and high-energy electron carriers NADH or FADH2; and electron transport chain for harnessing energy of NADH/FADH2 to create a electrochemical potential across inner mitochondrial membrane and mitochondrial matrix called as mitochondrial membrane potential; membrane potential is then used to synthesize ATP by ATP synthase complex present in inner mitochondrial membrane. Along with energy metabolism, mitochondria also perform various other functions including, synthesis of mitochondrial proteins, replication of mitochondrial genome, provision of precursors and metabolites for various cellular processes, maintaining redox balance of the cell and making life or death decision by regulating PCD pathway and synthesis of Fe-S cluster complexes [49].
Similar to all the apicomplexan organisms, Plasmodium species also harbour a single tubular mitochondrion [50]. Although cristae are absent in plasmodial mitochondria, sometimes a few tubular membrane whorls are reported [51]. The mitochondrion of Plasmodium harbours multiple copies of ~6 Kb genome [52], encoding only three proteins, cytochrome b, cytochrome c oxidase subunit II and cytochrome c oxidase subunit III, all of which are components of electron transport chain residing in inner mitochondrial membrane. It also contains genes for rRNAs as fragments of 20–200 nucleotides distributed all over the mitochondrial genome [52, 53]. However, it doesn’t encode for any tRNAs which are required for translation in mitochondrion. All the other mitochondrial proteins are encoded by nuclear genome of the parasite. During asexual erythrocytic cycle, the plasmodial mitochondria divides in synchrony with nuclear division, thus transferring a single mitochondrion with each nucleus to daughter cells (merozoites). Similar to all other eukaryotes, in Plasmodium also, only the female gametocyte provides mitochondrion to the zygote during sexual stages of parasite in mosquito vector [54]. Biochemical studies have revealed that during asexual intraerythrocytic stages of Plasmodium, CO2 is not produced from oxidation of glucose [55]. Further studies revealed that indeed asexual stages of parasite derive its energy majorly from glycolysis which occurs in cytosol. This is in contrast to the mitochondria of other eukaryotes where mitochondrial oxidative phosphorylation is the main energy harnessing pathway. This reduces the role of Plasmodium mitochondrion to functions other than energy generation. Plasmodium mitochondria host whole repertoire of proteins and pathway components such as TCA cycle, ETC, ATP synthase complex, pyrimidine biosynthesis, Fe-S cluster biosynthesis, heme biosynthesis and protein turnover mechanisms. Some of these pathways/mechanisms and their potential as drug targets for development of new antimalarials are being discussed below.
Electron Transport Chain (ETC)
Plasmodium mitochondria have functional ETC machinery in the inner mitochondrial membrane. Components of its ETC are complex II, complex III, complex IV and a type II NADH:ubiquinone oxidoreductase as complex I instead of NADH dehydrogenase [55]. ETC is proved to be indispensable for the parasite survival. Hence, a number of compounds are targeted to the ETC of Plasmodium mitochondria to contain malaria [56]. As NADH:ubiquinone oxidoreductase is more bacteria like and different from host’s complex I, a number of compounds specifically target this complex [55]):
-
(a)
Atovaquone: A hydroxynaphthoquinone derivative targets cytochrome bc1 of complex III in ETC and was the first mitochondria targeting drug for malaria [57]. Atovaquone binds to Qo site of cytochrome b active site where it acts as Ubiquinone analogue and ablates the binding of ubiquinone thus impairing the ETC [58]. It proved to be a potent antimalarial drug. Later on, in clinical settings resistant parasites appeared which limit the use of atovaquone alone as an antimalarial drug. Lack of good DNA editing mechanism in mitochondria together with drug pressure escalates the chances of resistance conferring mutations in cytochrome b gene which is encoded by mitochondrial genome itself and is the target of atovaquone [59, 60]. Although mutant cytochrome b is found to be less active, these parasites are still able to thrive through asexual stages, as it relies mostly on glycolysis for energy metabolism. However, in sexual stages of Plasmodium, where the availability of nutrients is limited, mitochondrial ETC and oxidative phosphorylation are of immense importance, and thus such mutant parasites are unable to pass through these stages, thereby limiting their transfer to another human host [61]. This restricts the resistance and does not allow it to spread any further. This kind of containment is unique to mitochondrial genes because it is only the female gametocyte, which transmits mitochondrion to the zygote and thus ablating any chance of crossover with wild-type gene of another gamete, which is common in nuclear-encoded genes [59]. Thus atovaquone also helps uniquely in containment of malaria within a patient [62].
-
(b)
Malarone: To overcome the problem of resistance and bring down the dose of atovaquone, it is used in combination with a biguanide drug called proguanil under the trade name of “Malarone”. Proguanil is a prodrug which in the liver is converted to active DHFR inhibitor, cycloguanil. Proguanil in monotherapy is not effective at all; however, in combination with atovaquone it significantly reduces the effective dose of atovaquone. The exact mechanism of action of proguanil is not yet known, but some studies point towards inhibition of reverse ATP synthase activity where ATP synthase pump is operated in opposite direction utilizing cytosolic ATPs to generate electrochemical potential, thereby maintaining the MOMP. As, in absence of atovaquone, the ETC is quite effective in maintaining the MOMP thus rendering the proguanil ineffective against the Plasmodium [62].
-
(c)
ELQ-300 (in combination with atovaquone), pyridones, quinolones, acridones and acridinediones are other classes of compounds which target complex III of mitochondrial ETC in Plasmodium [63].
Components of TCA Cycle
Plasmodium mitochondria have most of the components of TCA cycle, but pyruvate dehydrogenase is exclusively localized in apicoplast, thereby limiting its ability to utilize pyruvate as precursor for acetyl-CoA [64]. Aconitase enzyme, along with its function in TCA cycle, also acts as cytosolic iron-response element-binding protein regulating mRNAs of iron homeostasis [65]. Isocitrate dehydrogenase is found to be upregulated under oxidative stress condition in parasite indicating its involvement in redox balance of the parasite [66]. However, some isolates of parasite from patients are found to have higher levels of TCA cycle components, which point towards nutrient limitation-induced expression of these proteins in parasite [67]. However, no potent compound has so far been found to be targeting TCA cycle that could be used as antimalarial.
Pyrimidine Biosynthesis Pathway
Plasmodium lacks salvage pathway for meeting its need for pyrimidine; hence, it solely depends on in vivo pyrimidine biosynthesis pathway making it indispensable for the parasite [68]. Mitochondria resident type II DHODH is needed to convert dihydroorotate to orotate which is a precursor for pyrimidine biosynthesis. Plasmodium DHODH transfers its electron to ubiquinone of mitochondrial ETC. Studies have pointed out that to act as sink for these electrons from DHODH is the sole essential purpose of Plasmodium mitochondrial ETC. This is further supported by reducing the effectiveness of ETC inhibitors such as atovaquone on parasites complemented with cytosolic type I DHODH from yeast, which uses fumarate instead of ubiquinone and thus does not require mitochondrial ETC [69]. DSM1 was the first drug found to be active against Plasmodium DHODH and proven antimalarial; however, resistance appeared quickly in DSM1 treated parasites as in case of atovaquone. A derivative of it, DSM 268 is a more potent compound which targets multiple stages and confers single-dose therapy [70, 71].
Protein Synthesis and Degradation Pathways in Mitochondria
The mitochondrial genome of Plasmodium encodes for three essential proteins of ETC, which needs to be translated inside mitochondrion itself [53]. Mitochondrial genome encodes for rRNAs which along with nuclear-encoded rRNAs form a mitoribosome for protein translation in mitochondrion. Recent reports have emphasized the role of ribosomal proteins in normal functioning of mitochondrion in Plasmodium. One of the well-characterized proteins of Plasmodium mitochondria is mitochondrial ribosomal protein L13 (PfMRPL13). PfMRPL13 is shown to be essential for parasite survival [72]. Its knockdown caused significant reduction in parasite survival beyond three cycles in asexual erythrocytic stages. Although under knockdown of PfMRPL13, the parasite shows increased sensitivity to proguanil similar to atovaquone treatment, but the parasite growth under PfMRPL13 knockdown could not be rescued by providing either decylubiquinone or by ectopically expressing type I yDHODH. The study suggested a domino effect of PfMRPL13 ablation in Plasmodium: (1) reduced translation efficiency in mitochondrial ribosomes; (2) shortage of cyt b, COXI and COXIII proteins; (3) failure to assemble functional mtETC complexes; (4) significant reduction of mitochondrial membrane potential; (5) inability to complete pyrimidine biosynthesis; and (6), ultimately, parasite death. This PfMRPL13 or mitochondrial ribosomes of the Plasmodium are potential drug targets for development of new, more potent antimalarials [72].
Mitochondria also maintain their proteostasis by utilizing a number of mitochondria-resident proteases. Most of the proteases in mitochondria are of prokaryotic-like including Lon protease FtsH and ClpQY protease systems. In Plasmodium, FtsH was shown to be present in mitochondria and suggested to be involved in mitochondrial division [73]. However, some recent studies suggested its role in apicoplast maintenance too [74]. The Clp (caseinolytic protease) proteases are prokaryotic counterparts of eukaryotic 26S proteasome. In Plasmodium ClpQY machinery is localized in mitochondrial matrix. PfClpQY is multimeric unit having six units of ClpQ and six units of ClpY. ClpY acts as chaperone to unfold the substrate protein by utilizing ATPs. This unfolded protein is then passed through the proteolytic barrel formed by ClpQ hexamer where ClpQ cleaves the protein by its protease activity. In Plasmodium falciparum, disruption of ClpQY activity is lethal for the parasite, while it is refractory to ClpQ knockout. This suggests the essential nature of ClpQY machinery in the parasite [75]. Further, a 12-amino acid synthetic peptide of PfClpY C-terminus specifically inhibits the interaction of PfClpQ with PfClpY and thus affects the overall activity of PfClpQY system. Same peptide when added to in vitro culture of Plasmodium falciparum asexual erythrocytic stages significantly abolishes the growth of parasite with signs of mitochondrial dysregulation such as loss of mitochondrial membrane potential and fragmentation of mitochondrion as a whole. PfClpQY system is thus essential for the parasite survival in asexual erythrocytic cycle. PfClpQY system poses as a good drug target for antimalarial development. One possible approach is designing peptide mimetic compounds which could ablate association of ClpQ with ClpY making it non-functional.
Fe-S Cluster Biosynthesis
The iron-sulphur cluster biosynthesis occurs in mitochondria. These are needed as cofactor of ABC cassette RNase L inhibitor for the maturation and incorporation of rRNAs into the ribosome. There are 18 putative proteins of Fe-S cluster synthesis pathway identified in Plasmodium, which could be potential drug targets owing to the essential nature of this pathway [50].
Metabolic Pathways in Apicoplast for Antimalarial Development
Apicoplast remains one of the most intriguing organelle of the parasite ever since it was reported [76]. The four-membrane-bound organelle is a site of various important pathways, which are essential for parasite survival. The prokaryotic nature of its genome and high homology of various apicoplast proteins to cyanobacteria has made it excellent target site for various antimalarials. Further, high sequence similarity between the plastid genomes of apicomplexan parasites P. falciparum and Toxoplasma gondii strongly suggests that the apicoplast biology is strongly conserved across all species of intracellular parasites and is inherited from a common ancestor [77]. Reverse genetics approaches helped to ascertain the essential role of this organelle in the parasite life cycle. It has been shown that blocking the apicoplast development does not inhibit parasite division and production of viable daughter merozoitese, which are able to invade fresh host cell; however, these parasites are no longer able to divide; since the effect is transferred to next cell cycle, the phenomenon has been termed as “delayed death” phenotype.
There are three key metabolic pathways which are functional in the apicoplast, viz. synthesis of fatty acids, de novo heme biosynthesis and isoprenoids biosynthesis. These are the three pathways which are majorly been targeted for drug development against the parasite.
Apicoplast-Associated Fatty Acid Biosynthesis
Fatty acids are required for the various essential features of the parasite life cycle including synthesis of lipids. Earlier it was believed that Plasmodium relies totally on the uptake of fatty acids from the host [78, 79]. Identification of three important nuclear-encoded apicoplast-targeted proteins, ACP, KAS III and β-hydroxyacyl-ACP dehydratase, provided the first-time evidence for the presence of FAS pathway in the apicoplast [77]. Fatty acid synthesis in Plasmodium apicoplast was found to be type II fatty acid synthesis which is altogether different from the type III fatty acid synthesis in eukaryotes or humans and thus has remained a good target for development of antimalarials for long time. Fatty acid synthesis starts by the carboxylation of acetyl-CoA to malonyl-CoA using bicarbonates as source of carboxyl group. Acetyl coenzyme carboxylase (ACC) carries out this reaction and initiates the fatty acid synthesis pathway. For a long time, Plasmodium ACC has remained as promising drug target as it is a discrete multidomain enzyme as compared to human ACC which is a part of multifunctional enzyme complex and thus became a suitable target for the action of aryloxyphenoxypropionate-based herbicides, e.g. fops and dims. Aryloxyphenoxypropionate-based herbicides are potent inhibitors of Toxoplasma ACC which harbours the same multidomain ACC as of Plasmodium [80]. As both fops and dims showed activity against blood-stage Plasmodium, the compounds were assumed to target the multifunctional apicoplast ACC [81]. However genetic manipulation studies highlighted the dispensability of FASII pathway in blood stage of Plasmodium [82,83,84,85], which suggested both herbicides have their effect on some other targets. Following the discovery that FASII was required for liver stage development in the rodent models, interest in the pathway then shifted towards its disruption for malaria prophylaxis. Several compounds thought to target FASII were tested against Plasmodium liver stages and showed promising activities in vitro [86,87,88]. FabD (malonyl-CoA:ACP transacylase) has also been confirmed to be localized to apicoplast and proposed to be a good target [89]. FabH, which carries out the ACP condensation with acetyl-CoA, was considered as target of thiolactomycin and its analogues [90, 91]. FabZ (β-hydroxyacyl-ACP dehydratase) which carries out formation of enoyl-ACP from β-hydroxyacyl-ACP are being targeted by synthesis NAS compounds [92]. Enoyl-ACP reductase, the enzyme carrying out reduction of enoyl-ACP to acyl-reductase, is also being targeted using compounds such as pyrazoles [93], rhodamines [94] and flavonoids [95].
Isoprenoids Biosynthesis as Target and Use of Antibiotics
As apicoplast have evolved from blue-green algae, it is seen as analogous to bacteria, and thus various important pathways of apicoplast are being targeted using antibiotics. Plasmodium differs from humans in synthesizing isoprenoids via 1-deoxy-D-xylulose 5-phosphate (DOXP). This non-mevalonate pathway resembles one form of bacteria and plants and thus has become an attractive target for designing inhibitors against the apicoplast development. Fosmidomycin has been shown to target DOXP reductoisomerase in isoprenoid biosynthesis pathway of apicoplast [96]. FR900098, a synthetic derivative of fosmidomycin, is found to be twice as active and is under clinical trials where it shows effective parasite clearance in human subjects in Gabon and Thailand [97, 98].
Inhibitors of Transcription and Translation Machinery in the Apicoplast
Families of antibiotics with antimalarial activity include tetracyclines, lincosamides, macrolides and ketolides. Similarly, ciprofloxacin, a DNA gyrase inhibitor, can specifically block plastid DNA synthesis of apicoplast by inhibiting the linearization of 35-Kb circular DNA [99]. However, in the clinics norfloxacin and ciprofloxacin have not shown any substantial effects during the trials [100, 101]. Some of the fluoroquinolones, e.g. grepafloxacin and norfloxacin, are effective against the parasite in vitro, and further development of the compounds based on these can be very effective [102]. Similarly, antibiotics inhibiting the transcription, e.g. rifampicin, a potent inhibitor of multisubunit RNA polymerase, show potential when used in combination therapy [103]. However, rifampicin was found not to be very effective in case of Plasmodium vivax infected patients [104]. Tetracycline-based antibiotic minocycline has been shown to reduce the transcript levels of rpoB and rpoC from plastid origin, while it imparts no effect on the nuclear-encoded rpoB/rpoC [105]. Clindamycin is also been used successfully in combination with other known antimalarials, e.g. quinine and atovaquone, to treat uncomplicated malaria [106, 107]. The site of action for clindamycin is shown to be domain V of 23S rRNA, in toxoplasma [108]. Another possible explanation for the antimalarial activity of clindamycin is that interruption of protein synthesis in the plastid blocks production of the Clp protein encoded on the plastid genome, which is required for the development of apicoplast. Protein translation inhibitors are also been shown as effective antimalarials when used in combinations. Treatment with prokaryotic translation inhibitor, doxycycline, targeted apicoplast [109, 110]. Doxycycline is now been used worldwide in prophylactic and combination chemotherapy against malaria [111]. Inhibitors targeting aminoacyl-tRNA synthetases, e.g. indolmycin and mupirocin, are recently being shown to inhibit parasite growth [112].
Heme Biosynthesis
The heme biosynthesis is one of the important pathways for the parasite development. Heme synthesis doesn’t seem to be essential in the blood stages, but it seems to be important for the liver stages [113, 114]. Inhibitors targeting heme pathway includes succinylacetone which inhibits aminolevulinic acid dehydratase (ALAD). High concentration usage of succinylacetone (1–2 mM) points towards off-target effect of this compound [114].
Iron-Sulphur (Fe-S) Biosynthesis
Iron-sulphur (Fe-S) complex is known to play a role in electron transport and acts as a cofactor to enzymes in a variety of pathways, including fatty acid and isoprenoid synthesis. Disruption of Fe-S synthetic pathways (iron-sulphur cluster) leads to death of the parasite, which can be rescued by supplying isoprenoid by-product [115]. Fe-S cluster synthesis is also very essential in the sexual stage development of Plasmodium. Inhibition of SufS by D-cycloserine is the only known inhibitor of Suf pathway against the parasite till now [116].
As majority of proteins in apicoplast are nuclear encoded, transport of these proteins towards apicoplast can be a suitable target for antimalarials. A number of autophagy-related proteins, including ATG3 and ATG8 complex, are also suggested to be involved in protein trafficking and apicoplast maintenance. Thus protein import machineries can also be utilized to develop apicoplast targeting drugs. Till date only known inhibitor targeting the apicoplast protein transport machinery is deoxyspergualin (DSG), which has been shown to inhibit the interaction between transit peptide and HSP-70 [117, 118].
Apicoplast Proteases
Parasite proteases have remained as the front runner for the development of inhibitors in various systems. Plasmodium proteases have also been targeted in various studies for the development of antimalarials. Some of the important proteases playing role in the development of the apicoplast are caseinolytic proteases (i.e. PfClpP, PfClpC and PfClpA), Ftsh1 (mitochondria), ERAD(ER) and OTU. The ATP-dependent protease systems of Clp family have been shown to have essential role in the development of the parasite apicoplast. Disruption of PfClpP using chemical knockdown approach with a lactone-based inhibitor highlights the essential role of the protease in the parasite life cycle [119]. Further, a novel pyrimidine series of compounds inhibiting P. falciparum ClpP protease activity have been shown to be effective compounds which can be further developed into lead antimalarials [120].
Another important protease shown to be essential for the development of apicoplast is PfOTU. PfOTU is a cysteine protease which belongs to the family of deubiquitinating enzyme (DUB) family. It is localized in vesicular-like structures, which are found to be close to apicoplast. Downregulation of PfOTU has been shown to inhibit apicoplast protein transport along with regulation of apicoplast-bound PfATG8 [121]. The important role of PfOTU in apicoplast-directed protein import highlights the suitability of this protein as drug target.
As malaria parasite is highly evolved eukaryote, a number of proteins have different functions as is of their homologues in other systems. Endoplasmic reticulum (ER)-associated protein degradation (ERAD) has been “rewired” to provide a conduit for protein transport to the apicoplast [122]. LY-411575, an inhibitor targeting signal peptide peptidase component of this ERAD system, has been shown to be potent molecule for the parasite killing [123]. Further screening of libraries based on LY-411575 scaffold, two more compounds, e.g. NITD731 and NITD697, have been identified which are potent against P. falciparum at 17 nm and 65 nm, respectively. ERAD pathway in Plasmodium has also been targeted using HIV protease inhibitors. These inhibitors are two general aspartyl protease inhibitors and three AAA-p97 ATPase inhibitors which inhibit the zygote to ookinete transition of the Plasmodium parasite [124].
FtsH1 is an important metalloprotease in thylakoid membrane which plays a crucial role in the maintenance of thylakoid membranes [125]. In Plasmodium PfFtsH1 has been shown to be localized in mitochondria [73], but a recent report targeting TgFtsh1 and PfFtsH1 using actinonin drug points out towards its role in the development of apicoplast [74]. In addition, IPP-mediated “Chemical Rescue” of actinonin-treated P. falciparum resulted in “apicoplast-minus parasites” [126, 127].
Lipid Metabolism in Plasmodium: Potential Targets for Drug Development
During its development in the host cell, the parasite requires enormous amount of lipids to develop organelle (e.g. nucleus, mitochondria, food vacuole, apicoplast), tubulovesicular network (TVN) and also for subsequent replication. Additionally, emergence of new membranous structures (Maurer’s cleft and transported vesicles) that involves trafficking of proteins and other factors to RBC surface require lipids [128]. Further, there is accumulation of lipid in form of lipid bodies in asexual stage and osmophilic bodies in sexual stage. There is huge dynamic change in the lipid content and composition in both parasite and in infected RBC. Phospholipids and fatty acid synthesis are the major pathways in lipid metabolism of the parasite. So the enzymes involved in these pathways are potential drug targets.
One of the key pathways of lipid homeostasis includes phospholipid metabolism in the cell. Earlier phospholipids were recognized as structural components only, but now phospholipids and their by-products are emerging as major signalling molecules [129] which control parasite development and differentiation in host cell. During the schizogony, lipid metabolism gets dramatically increased because parasite progeny requires huge amount of lipids for membrane synthesis and results in a sixfold increase in phospholipids in iRBCs. P. falciparum is dependent on phospholipids for multiplication, and there is some uniqueness in phospholipid metabolism which creates opportunities for identification of novel drug targets for the parasite. Parasite uses both exogenous source and also synthesizes phospholipids de novo to acquire the necessary lipids. About 300 different lipid species were identifies in asexual blood stages and gametocytes of Plasmodium falciparum which are essential for its growth, proliferation, transmission and sexual reproduction [130]. The major membrane lipid components are phosphatidylcholine (PC), phosphatidylethanolamine (PE) and phosphatidylserine (PS). In uninfected RBCs, PC, PE and PS constitute 30–40%, 25–35% and 10–20% of the total phospholipids, respectively [131], whereas in infected RBCs, these major phospholipids constitute 20–55%, 15–40% and 4–15% of the total phospholipids, respectively [132,133,134].
Plasmodium genome database and bioinformatic tools played an important role in identifying the components of PC, PE and PS biosynthetic machineries. Phosphatidylcholine (PC) is the major phospholipid making the parasite membranes and erythrocytes. Hydrolysis of PC by phospholipase leads to formation of signalling molecules; Diacylglycerols (DAG) and phosphatidic acid (PA) etc. One of the pathways for the PC synthesis is cytidine diphosphate- or CDP-choline branch of the Kennedy pathway. Metabolic studies have shown that 89% PC is synthesized from choline by using this pathway [135]. First, choline has to be transported to the parasite by a yet unidentified transporter, and then it is converted into phosphocholine after phosphorylation by choline kinase which is then used as a precursor for CDP-choline and finally phosphatidylcholine, by the 1,2-diacylglycerol cholinephosphotransferase [136]. Inhibition of choline kinase by hexadecyltrimethylammonium bromide (HDTAB) leads to decrease in PC synthesis and ultimately leads to parasite death [135]. Remaining 11% PC is synthesized via PMT pathway in which phosphoethanolamine (PE) is used as a substrate for the synthesis of PC. This phosphoethanolamine methyltransferase (PfPMT) does three sequential methylations of PE (via S-adenosyl methionine donors) to form PC [137]. PfPMT is also essential for the parasite; deletion of PfPMT affects gametogenesis in erythrocytes, their transmission to mosquitoes and oocysts development in mosquito gut. This essentiality of PfPMT for the parasite creates opportunities for identification of the novel drug targets against Plasmodium falciparum. Screening of more than 3000 molecules identified 28 lead compounds blocking the PfPMT activity at micromolar range, and 11 of them also inhibit asexual replication of the parasite. Out of these 11 compounds, NSC-158011 was found to be a competitive inhibitor of PfPMT [138]. Another study showed that 4-aminoquinoline, amodiaquine, was able to inhibit PfPMT activity [139].
Phosphatidylethanolamine (PE) and phosphatidylserine (PS) – PE is the second major membrane phospholipid essential for the cytokinesis and for membrane fusion and fission which increases the membrane curvature [140,141,142]. PE metabolism has been altered in various human diseases such as Parkinson’s disease, Alzheimer’s disease and non-alcoholic liver disease [143]. Mainly two pathways are responsible for PE biosynthesis. First is via Kennedy pathway in which parasite generates PE de novo from ethanolamine [144] and another is by decarboxylation of PS [131, 145, 146]. Recently, a detailed study shows the mode of action of two choline kinase inhibitors. Surprisingly these compounds inhibit the ethanolamine kinase activity of choline kinase leading to decrease in PE level in P. falciparum [147].
Recent Developments in Antimalarial Discovery
Artemisinin combination therapy has been the mainstay of malaria treatment in recent years; however, it has been threatened with the emergence and spread of resistance against the available drugs [148, 149]. Development and innovation of new, cheap, safe and effective drugs targeting various novel biochemical pathways and better mechanism of action are need of the hour. Over the past few years, vigorous effort in antimalarial drug discovery has generated several promising antimalarials, which are active across different stages of the parasite life cycle, offering hope for new treatments. Various organizations like Novartis, GSK and MMV are involved with malaria drug discovery. The Medicines for Malaria Venture (MMV) is one of the largest organizations which are involved in development of new antimalarial drugs. Today, several leading antimalarial candidates are compounds from Malaria Box and Pathogen Box of MMV [150, 151]. A number of antimalarial candidates are discussed below.
DSM265
Several antimalarial drugs like pyrimethamine and atovaquone target DNA synthesis by inhibiting the pyrimidine nucleotide biosynthesis. A vital step of this pathway is catalyzed by dihydroorotate dehydrogenase (DHODH). DSM 265 is a triazolopyrimidine-based inhibitor of DHODH [152]. It is the first DHODH inhibitor targeting Plasmodium DHODH to reach clinical development for treatment of malaria. DSM265 is highly selective towards Plasmodium DHODH and shows high efficacy against both blood and liver stage parasites [71, 153]. Pharmacokinetic studies of DSM 265 shows it to have a lengthy half-life in humans, thus making it a promising candidate for single-dose chemotherapy [154].
KAF 156
The KAF 156 or GNF156 is jointly developed by Novartis and STPHI with support from the Bill and Melinda Gates Foundation (MMV) and is currently undergoing phase IIb combination study with lumefantrine. It belongs to a novel class of antimalarial agent, imidazolopiperazines. It has been found to be effective against both asexual and liver-stage parasites and artemisinin-resistant parasites. Some studies have showed it to be effective against the mature gametocytes too [155,156,157]. The mode of action of the KAF156 is currently unknown because in vitro resistance to KAF156 is associated with mutations in three P. falciparum genes, acetyl-CoA transporters, UDP-galactose and CARL (cyclic amine resistance locus) which are not thought to be the target of KAF156 [155, 158, 159]. First in human pharmacokinetic data found it to be safe and well tolerated at even 1200 mg. Clinical trials showed KAF 156 to have a cure rate of around 67%. Clearance of parasitemia was reported from patients with P. falciparum as well as those with P. vivax. Even patients suffering with infections from artemisinin-resistant parasites were cured. However, many patients were reported to suffer from various adverse events like asymptomatic sinus bradycardia, thrombocytopenia, anaemia and hyperbilirubinemia [160].
Tafenoquine
The tafenoquine or WR238605 is a new antimalarial drug jointly developed by the GlaxoSmithKline Pharmaceuticals (GSK) and Walter Reed Army Institute of Research (WRAIR) in association with MMV [161]. Tafenoquine, an 8-aminoquinoline, was developed as a possible alternative for primaquine. In vivo studies have shown it to be ten times more efficient and less toxic than primaquine when used as a prophylactic. It is active against the liver and blood and also blocks the sporozoite development in the mosquitoes [162,163,164]. It has completed phase III clinical trials and has recently been approved by the US FDA for prevention of relapse in the case of P. vivax malaria under the commercial name of Krintafel. PK studies for tafenoquine show that it has rapid absorption and a prolonged half-life of about 16 days, thus ideal for a single-dose regimen. The dose of tafenoquine has not been established. In general, a “fire and forget” strategy suggests three doses of 200 mg are sufficient to provide protection for up to 11 weeks idea for short-term travellers [165]. A single dose of 300 mg has been used for treatment of P. vivax under global P. vivax radical cure programme [166]. However, like primaquine, tafenoquine is reported to cause haemolysis in people with G6PD deficiency thus suggesting that care should be taken while administering drug to such patients [167]. Adverse effects include mild GI upset, headaches, nausea and myalgia.
Artefenomel
OZ439 or artefenomel is a novel trioxolane being developed as a partner drug with ferroquine jointly by MMV and Sanofi [71]. It is one of the MMV’s front-runner compounds with single-dose cure potential and is currently in phase IIb of clinical trials. Artefenomel is a synthetic ozonide which is based upon the 1, 2, 4-trioxolane pharmacophore which are similar to artemisinin. The first trioxolane, OZ277/arterolane, was developed by MMV and Ranbaxy India and was marketed in combination with piperaquine under the brand name Synriam [168]. Lately, it has now been licensed for clinical use in several African countries. However, studies have shown loss of potency against artemisinin-resistant parasites.
Artefenomel is the second trioxolane after arterolane to advance the clinical trials. Studies by Phyo et al. [9] showed artefenomel has high parasite clearance rate against both P. falciparum and P. vivax and was active against artemisinin-resistant parasites too. It offers a number of advantages like ease of synthesis, better ADME properties and a prolonged half-life of around 46–62 h making it ideal for single-dose therapy in combination with other drugs [169,170,171,172]. Combination studies with ferroquine are in the patient exploratory [173].
MMV0048
The MMV0048 or MMV 390048 is another promising drug candidate being developed by MMV in collaboration with H3D Cape Town. In 2014 it became the first drug to enter phase I in Africa; by 2017 FPFV phase IIa was completed in Ethiopia. MMV0048 belongs to a novel class of inhibitors called 2-aminopyridine which was developed on the basis of a series of hits identified from a high-throughput screening of a commercially available BioFocus library [174].
Chibale et al., through various in vitro and in vivo studies, showed this compound to be highly efficacious against multiple stages of the parasite in host and mosquitoes (except liver hypnozoites) which not only provides protection against infection but also has the potential for transmission-blocking activity [175]. In order to understand the mechanism of action of MMV0048, Chibale et al. used various genomic and chemo-proteomic approaches; PI4k (a membrane-associated kinase, involved in cell signalling and trafficking, essential for parasite) was identified as the possible target of MMV0048. PKPD studies showed it to have a long half-life and good absorption which makes it a promising antimalarial which can further develop for single-dose combination therapy.
UCT943
The UCT943 is the second preclinical candidate developed jointly by H3D and Medicines for Malaria Venture (MMV). It was developed as a next-generation PI4K inhibitor, to address the issues of low aqueous solubility and anti-Plasmodium potency associated with MMV048 [176]. Modification of 2-aminopyridine to 2-aminopyrazine core along with incorporation of a piperazinylamide group not only improved the physiochemical properties of the compound but also significantly improved the efficacy across the parasite stages. Pharmacokinetic studies found it to be slow-acting compound with long half-life similar to mefloquine. Based on the studies by Chibale et al., UCT943 has potent activity against all stages of the malaria parasite and thus has the potential to form part of a single-exposure radical cure and prophylaxis treatment [177].
Spiroindolone/KAE609 (Cipargamin)
The spiroindolone is a novel class of antimalarials discovered in a whole-cell screen by Novartis and STPHI [178]. KAE609 is a synthetic analogue of spiroindolone, which has shown to possess better antimalarial activity against multiple stages of malaria parasite than artemisinin [179]. The phase IIa trail of KAE609 has been completed, with a target set for next milestone – completion of phase II in 2019/2020. The likely molecular target of KAE609 has been identified as P-type Na+ATpase, PfATP4p due to emergence of resistance mutations in the gene coding for the ATPase [180]. In vitro studies in Plasmodium falciparum show it to be active in nanomolar ranges including gametocytes stages [181]. Clinical trials data disclosed no safety concerns regarding KAE609 as well as had favourable pharmacokinetic properties needed for single-dose cure. Further studies showed it to have a fast parasite clearance time in patients with uncomplicated Plasmodium falciparum and Plasmodium vivax malaria [182]. It was also found to be highly effective in treatment of resistant malaria.
SJ733
The SJ733 is another clinical candidate identified through high-throughput screening that acts upon pfATP4 [180]. It is a dihydroisoquinoline (DHIQ) and is being developed as single-exposure radical cure and prophylaxis (SERCaP) drug by MMV in collaboration with St. Jude Children’s Research Hospital and Rutgers University. In vitro and in vivo studies have shown it to be potent against both asexual and sexual blood stages in nanomolar ranges, with a rapid clearance rate [183]. Further studies showed it to be safe and highly orally bioavailable. Currently it is in phase I trial of clinical development.
Fosmidomycin
Fosmidomycin is an antibacterial compound which was developed to treat urinary tract infections; however, it was later discontinued due to its lack of effectiveness against recurrent infections [184, 185]. It is a potent inhibitor of 1-deoxy-d-xylulose 5-phosphate reductoisomerase, an essential enzyme of the non-mevalonate pathway of isoprenoid pathway. The MEP pathway is shown to be essential for malarial parasites [186]. As this pathway is absent in humans, MEP pathway enzymes represent an attractive target for drug development [96, 187]. Clinical studies have showed that the drug is well tolerated and is effective in the treatment of clinical malaria. However, because of its highly charged nature, it has a poor pharmacokinetics. It also has a short plasma half-life of 3.5 h which is likely related to high rate of recrudescent malaria in children treated with the FSM combinations [188]. Few episodes of mild gastrointestinal side effects have also been reported. Currently the drug in phase IIb studies in combination with piperaquine [189].
AQ-13
Chloroquine was one of the most widely used antimalarials, but the emergence and spread of resistance against the drug has led to its abandonment. In recent years, various chemical approaches have been followed to develop new 4-aminoquinolines that are having similar chloroquine antimalarial potency as well as are active against CQ-resistant Plasmodium parasites. AQ-13 alternatively named Ro47–0543 is a CQ analogue with a shortened diaminoalkane side chain and is in development for treatment of resistant P. falciparum malaria. The molecule was discovered simultaneous by two research groups one at Roche, Switzerland, and other at Tulane University, USA [190, 191]. Because of the structural analogy, antiplasmodial activity of AQ-13 is probably similar to CQ, i.e. interferes with heme detoxification pathway [191]. Pk studies of AQ-13 showed it to have fast absorption and a half-life of about 13 days. Maximal concentration (Cmax) was achieved within 3–4 h. Phase 2 of clinical trial showed to it to be well tolerated and a safe drug [192, 193]. Thus, it possess all the excellent properties of being an ideal drug, namely, the prolonged coverage of protection provided by the long half-life leading to sustained cure and a post-treatment prophylactic effect. It is currently being considered for combination therapy.
Sevuparin
Mortality due to severe malaria remains high. Death of severe malaria is characterized by hyperparasitaemia and microcirculatory flow obstruction in vital organs because of cytoadherence of infected RBCs and rosette formation [194, 195]. Sevuparin is being developed as an alternate treatment in severe P. falciparum malaria. It acts as a decoy receptor of heparan sulphate, during malaria infection, affecting both merozoite invasion and sequestration of infected erythrocytes [196, 197]. It is an acidic, negatively charged, anti-adhesive polysaccharide derived from heparin with eliminated antithrombin (AT) binding domain [198, 199]. A phase I/II clinical study showed sevuparin to be safe and well tolerated in the malaria patients. It is currently being tested for sickle cell disease.
SC83288
The SC83288 is an amicarbalide derivative which is being developed for the treatment of severe malaria. Amicarbalide was used in veterinary medicine as an antiprotozoal drug but was discontinued due to slow parasite clearance rates, high relapse frequencies, poor oral bioavailability, undesirable mutagenic and toxic side effects and manufacturing safety issues [200, 201]. Optimization of pharmacological and antiparasitic properties of amicarbalide yielded compound SC83288, which is fast-acting and is able to clear P. falciparum parasites at low nanomolar concentrations in vitro (IC50 valueso10nM). Studies in humanized NOD/SCID mouse model system showed it cured P. falciparum infection within 48 h following a dose of 2.5 mg/kg once per day over a period of 3 days. Preclinical pharmacokinetic and toxicological studies support the clinical development of SC83288. The compound is primarily active against trophozoites blood stage but also target early stage (I–III) gametocytes [202]. Considering its fast antiplasmodial activity and unique chemotype that does not reveal cross resistance to currently used antimalarials, SC83288 could be used in combination therapy against severe malaria.
Methylene Blue (Proveblue)
The methylene blue is an old antimalarial introduced in the late nineteenth century which was later replaced by mepacrine and chloroquine because of the presence of heavy metals in its preparation. Recently, Provence Technologies have synthesized Proveblue which is a new methylene blue formulation that complies with the European Pharmacopoeia and contains limited organic impurities and heavy metals of recognized toxicity.
Methylene blue has an unusually high antimalarial potency (IC50 = 4 nM) and selectivity (with a cytotoxicity index of 450) [203]. Methylene blue is pleiotropic, interferes with heme metabolism and also inhibits the activity of Pf glutathione reductase leading into cytosolic depletion of glutathione which favours the activity of chloroquine [204]. Thus, methylene blue might be able to sensitize the parasite for chloroquine and even revert chloroquine resistance. Synergistic effects were observed when Proveblue was used with mefloquine, quinine and dihydroartemisinin [205]. Proveblue when combined with atorvastatin, showed high efficacy in preventing cerebral malaria [206]. However, further studies are needed to define its role in malaria treatment.
Summary
Malaria is an important disease in the aspect of mortality and socio-economic burden. Treatment strategies against malaria went through phases of success and failure for long time, and thus a constant effort for antimalarial development has remained a priority. Lack of effective vaccine for protection and the artemisinin and its derivatives as the only option has made the situation rather dangerous. Increase in drug resistance in recent times once again highlighted the utmost importance of newer small molecule inhibitor identification and development. Although lower success rates of lead molecules in the trials highlight the need for identification of more drug targets and rational drug design. Different organelles in parasite harbour some of the important pathways which are required for the survival of the parasite and thus pose as suitable drug targets. With advent of new technologies in genomics, proteomics, structure biology and medicinal chemistry, identification of such targets gained pace and the field of antimalarials development has progressed significantly in recent past. In recent past, a number of novel compounds targeting pathways related to haemoglobin degradation, lipid synthesis, fatty acid synthesis and pyrimidine synthesis have been developed as potential antimalarial. Development of these new candidates and designing of combination therapy will help to combat the drug resistance in malaria (Fig. 1 and Table 1).

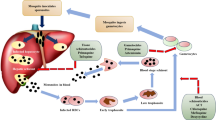
Schematic showing site of action of various antimalarials
References
Sharma, V. P. (1996). Re-emergence of malaria in India. The Indian Journal of Medical Research, 103, 26–45.
Sharma, V. P. (1999). Current scenario of malaria in India. Parassitologia, 41, 349–353.
Dash, A. P., Valecha, N., Anvikar, A. R., & Kumar, A. (2008). Malaria in India: Challenges and opportunities. Journal of Biosciences, 33, 583–592.
Cui, L., Mharakurwa, S., Ndiaye, D., Rathod, P. K., & Rosenthal, P. J. (2015). Antimalarial drug resistance: Literature review and activities and findings of the ICEMR network. The American Journal of Tropical Medicine and Hygiene, 93, 57–68.
Greenwood, D. (1992). The quinine connection. The Journal of Antimicrobial Chemotherapy, 30, 417–427.
Sweeney, A. W. (1996). The possibility of an “X” factor. The first documented drug resistance of human malaria. International Journal for Parasitology, 26, 1035–1061.
Baird, J. K. (2009). Resistance to therapies for infection by Plasmodium vivax. Clinical Microbiology Reviews, 22, 508–534.
Baird, J., & Chloroquine, K. (2004). Resistance in Plasmodium vivax. Antimicrobial Agents and Chemotherapy, 48, 4075–4083.
Phyo, A. P., et al. (2016). Antimalarial activity of artefenomel (OZ439), a novel synthetic antimalarial endoperoxide, in patients with Plasmodium falciparum and Plasmodium vivax malaria: An open-label phase 2 trial. The Lancet Infectious Diseases, 16, 61–69.
Milani, K. J., Schneider, T. G., & Taraschi, T. F. (2015). Defining the morphology and mechanism of the hemoglobin transport pathway in Plasmodium falciparum-infected erythrocytes. Eukaryotic Cell, 14, 415–426.
Bakar, N. A., Klonis, N., Hanssen, E., Chan, C., & Tilley, L. (2010). Digestive-vacuole genesis and endocytic processes in the early intraerythrocytic stages of Plasmodium falciparum. Journal of Cell Science, 123, 441–450.
Elliott, D. A., et al. (2008). Four distinct pathways of hemoglobin uptake in the malaria parasite Plasmodium falciparum. Proceedings of the National Academy of Sciences, 105, 2463–2468.
Labro, M. T., & Babin-Chevaye, C. (1988). Effects of amodiaquine, chloroquine, and mefloquine on human polymorphonuclear neutrophil function in vitro. Antimicrobial Agents and Chemotherapy, 32, 1124–1130.
Tietze, C., Schlesinger, P., & Stahl, P. (1980). Chloroquine and ammonium ion inhibit receptor-mediated endocytosis of mannose-glycoconjugates by macrophages: Apparent inhibition of receptor recycling. Biochemical and Biophysical Research Communications, 93, 1–8.
Wenisch, C., Parschalk, B., Zedwitz-Liebenstein, K., Wernsdorfer, W., & Graninger, W. (1997). The effect of artemisinin on granulocyte function assessed by flow cytometry. The Journal of Antimicrobial Chemotherapy, 39, 99–101.
Schwartz, A. L., Bolognesi, A., & Fridovich, S. E. (1984). Recycling of the asialoglycoprotein receptor and the effect of lysosomotropic amines in hepatoma cells. The Journal of Cell Biology, 98, 732–738.
Moore, H.-P., & Kelly, R.B. Secretory protein targeting in a pituitary cell differential transport of foreign secretory proteins to distinct secretory pathways line. https://doi.org/10.1083/jcb.101.5.1773.
Smith, R. M., & Jarett, L. (1982). Ultrastructural basis for chloroquine-induced increase in intracellular insulin in adipocytes: Alteration of lysosomal function. Proceedings of the National Academy of Sciences of the United States of America, 79, 7302–7306.
Vanweert, A., Geuze, H. J., Groothuis, B., & Stoorvogel, W. (2000). Primaquine interferes with membrane recycling from endosomes to the plasma membrane through a direct interaction with endosomes which does not involve neutralisation of endosomal pH nor osmotic swelling of endosomes. European Journal of Cell Biology, 79, 394–399.
Rosenthal, P. J., Wollish, W. S., Palmer, J. T., & Rasnick, D. (1991). Antimalarial effects of peptide inhibitors of a Plasmodium falciparum cysteine proteinase. The Journal of Clinical Investigation, 88, 1467–1472.
Rosenthal, P. J., McKerrow, J. H., Rasnick, D., & Leech, J. H. (1989). Plasmodium falciparum: Inhibitors of lysosomal cysteine proteinases inhibit a trophozoite proteinase and block parasite development. Molecular and Biochemical Parasitology, 35, 177–183.
Rosenthal, P. J., et al. (1996). Antimalarial effects of vinyl sulfone cysteine proteinase inhibitors. Antimicrobial Agents and Chemotherapy, 40, 1600–1603.
Rockett, K. A., et al. (1990). Inhibition of intraerythrocytic development of Plasmodium falciparum by proteinase inhibitors. FEBS Letters, 259, 257–259.
Shenai, B. R., et al. (2003). Structure-activity relationships for inhibition of cysteine protease activity and development of Plasmodium falciparum by peptidyl vinyl sulfones. Antimicrobial Agents and Chemotherapy, 47, 154–160.
Lee, B. J., et al. (2003). Antimalarial activities of novel synthetic cysteine protease inhibitors. Antimicrobial Agents and Chemotherapy, 47, 3810–3814.
Mane, U. R., et al. (2013). Falcipain inhibitors as potential therapeutics for resistant strains of malaria: A patent review. Expert Opinion on Therapeutic Patents, 23, 165–187.
Russo, I., et al. (2010). Plasmepsin V licenses Plasmodium proteins for export into the host erythrocyte. Nature, 463, 632–636.
Boonyalai, N., Collins, C. R., Hackett, F., Withers-Martinez, C., & Blackman, M. J. (2018). Essentiality of Plasmodium falciparum plasmepsin V. PLoS One, 13, e0207621.
Rasina, D., et al. (2016). Fragment-based discovery of 2-aminoquinazolin-4(3 H )-ones as novel class nonpeptidomimetic inhibitors of the plasmepsins I, II, and IV. Journal of Medicinal Chemistry, 59, 374–387.
Ersmark, K., Samuelsson, B., & Hallberg, A. (2006). Plasmepsins as potential targets for new antimalarial therapy. Medicinal Research Reviews, 26, 626–666.
Wyatt, D. M., & Berry, C. (2002, Feb 27). Activity and inhibition of plasmepsin IV, a new aspartic proteinase from the malaria parasite, Plasmodium falciparum FEBS Letters, 513(2–3), 159–162.
Dell’Agli, M., et al. (2006). High antiplasmodial activity of novel plasmepsins I and II inhibitors. Journal of Medicinal Chemistry, 49, 7440–7449.
Nasamu, A. S., et al. (2017). A multistage antimalarial targets the plasmepsins IX and X essential for invasion and egress. Science (80-.), 528, 522–528.
Shenai, B. R., Sijwali, P. S., Singh, A., & Rosenthal, P. J. (2000). Characterization of native and recombinant falcipain-2, a principal trophozoite cysteine protease and essential hemoglobinase of Plasmodium falciparum. The Journal of Biological Chemistry, 275, 29000–29010.
Sijwali, P. S., Shenai, R. B., Gut, J., Singh, A., & Rosenthal, P. J. (2001). Expression and characterization of the Plasmodium falciparum haemoglobinase falcipain-3. Biochemical Journal, 360, 481.
Shenai, B. R., & Rosenthal, P. J. (2002). Reducing requirements for hemoglobin hydrolysis by Plasmodium falciparum cysteine proteases. Molecular and Biochemical Parasitology, 122, 99–104.
Sijwali, P. S., Koo, J., Singh, N., & Rosenthal, P. J. (2006). Gene disruptions demonstrate independent roles for the four falcipain cysteine proteases of Plasmodium falciparum. Molecular and Biochemical Parasitology, 150, 96–106.
Rosenthal, P. J. (2011). Falcipains and other cysteine proteases of malaria parasites. Advances in Experimental Medicine and Biology, 712, 30–48.
Olson, J. E., Lee, G. K., Semenov, A., & Rosenthal, P. J. (1999). Antimalarial effects in mice of orally administered peptidyl cysteine protease inhibitors. Bioorganic & Medicinal Chemistry, 7, 633–638.
Scheidt, K. A., et al. (1998). Structure-based design, synthesis and evaluation of conformationally constrained cysteine protease inhibitors. Bioorganic & Medicinal Chemistry, 6, 2477–2494.
Hernández González, J. E., Hernández Alvarez, L., Pascutti, P. G., & Valiente, P. A. (2017). Predicting binding modes of reversible peptide-based inhibitors of falcipain-2 consistent with structure-activity relationships. Proteins: Structure, Function, and Bioinformatics, 85, 1666–1683.
Liu, Y., et al. (2011). Synthesis and antimalarial activity of novel dihydro-artemisinin derivatives. Molecules, 16, 4527–4538.
Coterón, J. M., et al. (2010). Falcipain inhibitors: Optimization studies of the 2-pyrimidinecarbonitrile lead series †. Journal of Medicinal Chemistry, 53, 6129–6152.
Kerr, I. D., et al. (2009). Structures of falcipain-2 and falcipain-3 bound to small molecule inhibitors: Implications for substrate specificity ‡. Journal of Medicinal Chemistry, 52, 852–857.
Li, X., et al. (2009). Plasmodium falciparum signal peptide peptidase is a promising drug target against blood stage malaria. Biochemical and Biophysical Research Communications, 380, 454–459.
Mishra, L. C., Bhattacharya, A., Sharma, M., & Bhasin, V. K. (2010). HIV protease inhibitors, indinavir or nelfinavir, augment antimalarial action of artemisinin in vitro. The American Journal of Tropical Medicine and Hygiene, 82, 148–150.
Jani, D., et al. (2008). HDP—a novel heme detoxification protein from the malaria parasite. PLoS Pathogens, 4, e1000053.
Richards, T. A., & van der Giezen, M. (2006). Evolution of the Isd11–IscS complex reveals a single α-proteobacterial endosymbiosis for all eukaryotes. Molecular Biology and Evolution, 23, 1341–1344.
Kaul, S., & Wadhwa, R. (2012). Mortalin biology: Life, stress and death. Springer: Dordrecht.
Mather, M. W., & Vaidya, A. B. (2008). Mitochondria in malaria and related parasites: Ancient, diverse and streamlined. Journal of Bioenergetics and Biomembranes, 40, 425–433.
Krungkrai, J., Prapunwattana, P., & Krungkrai, S. R. (2000). Ultrastructure and function of mitochondria in gametocytic stage of Plasmodium falciparum. Parasite, 7, 19–26.
Vaidya, A. B., Akella, R., & Suplick, K. (1989). Sequences similar to genes for two mitochondrial proteins and portions of ribosomal RNA in tandemly arrayed 6-kilobase-pair DNA of a malarial parasite. Molecular and Biochemical Parasitology, 35, 97–107.
Feagin, J. E., Werner, E., Gardner, M. J., Williamson, D. H., & Wilson, R. J. M. (1992). Homologies between the contiguous and fragmented rRNAs of the two Plasmodium falciparum extrachromosomal DNAs are limited to core sequences. Nucleic Acids Research, 20, 879–887.
van Dooren, G. G., et al. (2005). Development of the endoplasmic reticulum, mitochondrion and apicoplast during the asexual life cycle of Plasmodium falciparum. Molecular Microbiology, 57, 405–419.
Biagini, G. A., Viriyavejakul, P., O’Neill, P. M., Bray, P. G., & Ward, S. A. (2006). Functional characterization and target validation of alternative complex I of Plasmodium falciparum mitochondria. Antimicrobial Agents and Chemotherapy, 50, 1841–1851.
Stocks, P. A., et al. (2014). Novel inhibitors of the Plasmodium falciparum electron transport chain. Parasitology, 141, 50–65.
Peters, J. M., et al. (2002). Mutations in cytochrome b resulting in atovaquone resistance are associated with loss of fitness in Plasmodium falciparum. Antimicrobial Agents and Chemotherapy, 46, 2435–2441.
Srivastava, I. K., Morrisey, J. M., Darrouzet, E., Daldal, F., & Vaidya, A. B. (1999). Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Molecular Microbiology, 33, 704–711.
Lynch, M. (2010). Evolution of the mutation rate. Trends in Genetics, 26, 345–352.
Wilson, R. J., Fry, M., Gardner, M. J., Feagin, J. E., & Williamson, D. H. (1992). Subcellular fractionation of the two organelle DNAs of malaria parasites. Current Genetics, 21, 405–408.
Goodman, C. D., et al. (2016). Parasites resistant to the antimalarial atovaquone fail to transmit by mosquitoes. Science (80-.), 352, 349–353.
Goodman, C. D., Buchanan, H. D., & McFadden, G. I. (2017). Is the mitochondrion a good malaria drug target? Trends in Parasitology, 33, 185–193.
Miley, G. P., et al. (2015). ELQ-300 prodrugs for enhanced delivery and single-dose cure of malaria. Antimicrobial Agents and Chemotherapy, 59, 5555–5560.
Foth, B. J., et al. (2004). The malaria parasite Plasmodium falciparum has only one pyruvate dehydrogenase complex, which is located in the apicoplast. Molecular Microbiology, 55, 39–53.
Hodges, M., et al. (2005). An iron regulatory-like protein expressed in Plasmodium falciparum displays aconitase activity. Molecular and Biochemical Parasitology, 143, 29–38.
Wrenger, C., & Müller, S. (2003). Isocitrate dehydrogenase of Plasmodium falciparum. European Journal of Biochemistry, 270, 1775–1783.
Daily, J. P., et al. (2007). Distinct physiological states of Plasmodium falciparum in malaria-infected patients. Nature, 450, 1091–1095.
Gutteridge, W. E., Dave, D., & Richards, W. H. (1979). Conversion of dihydroorotate to orotate in parasitic protozoa. Biochimica et Biophysica Acta, 582, 390–401.
Painter, H. J., Morrisey, J. M., Mather, M. W., & Vaidya, A. B. (2007). Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature, 446, 88–91.
Llanos-Cuentas, A., et al. (2018). Antimalarial activity of single-dose DSM265, a novel plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: A proof-of-concept, open-label, phase 2a study. The Lancet Infectious Diseases, 18, 874–883.
Phillips, M. A., et al. (2015). A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Science Translational Medicine, 7, 296ra111–296ra111.
Ke, H., Dass, S., Morrisey, J. M., Mather, M. W., & Vaidya, A. B. (2018). The mitochondrial ribosomal protein L13 is critical for the structural and functional integrity of the mitochondrion in Plasmodium falciparum. The Journal of Biological Chemistry, 293, 8128–8137.
Tanveer, A., et al. (2013). An FtsH protease is recruited to the mitochondrion of Plasmodium falciparum. PLoS One, 8, e74408.
Amberg-Johnson, K., et al. (2017). Small molecule inhibition of apicomplexan FtsH1 disrupts plastid biogenesis in human pathogens. eLife, 6, e29865.
Jain, S. et al. (2013, Oct). The prokaryotic ClpQ protease plays a key role in growth and development of mitochondria in Plasmodium falciparum. Cell Microbiology, 15(10), 1660–1673.
McFadden, G. I., Reith, M. E., Munholland, J., & Lang-Unnasch, N. (1996). Plastid in human parasites. Nature, 381, 482–482.
Waller, R. F., et al. (1998). Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proceedings of the National Academy of Sciences of the United States of America, 95, 12352–12357.
Matesanz, F., Durán-Chica, I., & Alcina, A. (1999). The cloning and expression of Pfacs1, a Plasmodium falciparum fatty acyl coenzyme a synthetase-1 targeted to the host erythrocyte cytoplasm. Journal of Molecular Biology, 291, 59–70.
Maguire, P. A., & Sherman, I. W. (1990). Phospholipid composition, cholesterol content and cholesterol exchange in Plasmodium falciparum-infected red cells. Molecular and Biochemical Parasitology, 38, 105–112.
Zuther, E., Johnson, J. J., Haselkorn, R., McLeod, R., & Gornicki, P. (1999). Growth of Toxoplasma gondii is inhibited by aryloxyphenoxypropionate herbicides targeting acetyl-CoA carboxylase. Proceedings of the National Academy of Sciences of the United States of America, 96, 13387–13392.
Waller, R. F., et al. (2003). A type II pathway for fatty acid biosynthesis presents drug targets in Plasmodium falciparum. Antimicrobial Agents and Chemotherapy, 47, 297–301.
Yu, M., et al. (2008). The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell Host & Microbe, 4, 567–578.
Vaughan, A. M., et al. (2009). Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cellular Microbiology, 11, 506–520.
Pei, Y., et al. (2010). Plasmodium pyruvate dehydrogenase activity is only essential for the parasite’s progression from liver infection to blood infection. Molecular Microbiology, 75, 957–971.
López-Barragán, M. J., et al. (2011). Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics, 12, 587.
Scala, F., et al. (2010). Bromopyrrole alkaloids as lead compounds against protozoan parasites. Marine Drugs, 8, 2162–2174.
Tarun, A. S., et al. (2008). A combined transcriptome and proteome survey of malaria parasite liver stages. Proceedings of the National Academy of Sciences of the United States of America, 105, 305–310.
Singh, A. P., Surolia, N., & Surolia, A. (2009). Triclosan inhibit the growth of the late liver-stage of Plasmodium. IUBMB Life, 61, 923–928.
Prigge, S. T., He, X., Gerena, L., Waters, N. C., & Reynolds, K. A. (2003). The initiating steps of a type II fatty acid synthase in Plasmodium falciparum are catalyzed by pfACP, pfMCAT, and pfKASIII. Biochemistry, 42, 1160–1169.
Jones, S. M., et al. (2004). Analogues of thiolactomycin as potential anti-malarial and anti-trypanosomal agents. Bioorganic & Medicinal Chemistry, 12, 683–692.
Jones, S. M., et al. (2005). Analogues of thiolactomycin as potential antimalarial agents. Journal of Medicinal Chemistry, 48, 5932–5941.
Sharma, S. K., et al. (2003). Identification, characterization, and inhibition of Plasmodium falciparum beta-hydroxyacyl-acyl carrier protein dehydratase (FabZ). The Journal of Biological Chemistry, 278, 45661–45671.
Kumar, S., Guha, M., Choubey, V., Maity, P., & Bandyopadhyay, U. (2007). Antimalarial drugs inhibiting hemozoin (beta-hematin) formation: A mechanistic update. Life Sciences, 80, 813–828.
Kumar, G., Banerjee, T., Kapoor, N., Surolia, N., & Surolia, A. (2010). SAR and pharmacophore models for the rhodanine inhibitors of Plasmodium falciparum enoyl-acyl carrier protein reductase. IUBMB Life, 62, 204–213.
Tasdemir, D., et al. (2006). Inhibition of Plasmodium falciparum fatty acid biosynthesis: Evaluation of FabG, FabZ, and FabI as drug targets for flavonoids. Journal of Medicinal Chemistry, 49, 3345–3353.
Jomaa, H., et al. (1999). Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science, 285, 1573–1576.
Missinou, M. A., et al. (2002). Fosmidomycin for malaria. Lancet (London, England), 360, 1941–1942.
Kremsner, P. G., et al. (2003). Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrobial Agents and Chemotherapy, 47, 735–738.
Weissig, V., Vetro-Widenhouse, T. S., & Rowe, T. C. (1997). Topoisomerase II inhibitors induce cleavage of nuclear and 35-kb plastid DNAs in the malarial parasite Plasmodium falciparum. DNA and Cell Biology, 16, 1483–1492.
Watt, G., et al. (1991). Ciprofloxacin treatment of drug-resistant falciparum malaria. The Journal of Infectious Diseases, 164, 602–604.
McClean, K. L., Hitchman, D., & Shafran, S. D. (1992). Norfloxacin is inferior to chloroquine for falciparum malaria in northwestern Zambia: A comparative clinical trial. The Journal of Infectious Diseases, 165, 904–907.
Mahmoudi, N., et al. (2003). In vitro activities of 25 quinolones and fluoroquinolones against liver and blood stage Plasmodium spp. Antimicrobial Agents and Chemotherapy, 47, 2636–2639.
Goerg, H., Ochola, S. A., & Goerg, R. (1999). Treatment of malaria tropica with a fixed combination of rifampicin, co-trimoxazole and isoniazid: A clinical study. Chemotherapy, 45, 68–76.
Pukrittayakamee, S., et al. (1994). Antimalarial effects of rifampin in Plasmodium vivax malaria. Antimicrobial Agents and Chemotherapy, 38, 511–514.
Lin, Q., Katakura, K., & Suzuki, M. (2002). Inhibition of mitochondrial and plastid activity of Plasmodium falciparum by minocycline. FEBS Letters, 515, 71–74.
Adehossi, E., et al. (2003). Three-day quinine-clindamycin treatment of uncomplicated falciparum malaria imported from the tropics. Antimicrobial Agents and Chemotherapy, 47, 1173.
Lell, B., & Kremsner, P. G. (2002). Clindamycin as an antimalarial drug: Review of clinical trials. Antimicrobial Agents and Chemotherapy, 46, 2315–2320.
Fichera, M. E., & Roos, D. S. (1997). A plastid organelle as a drug target in apicomplexan parasites. Nature, 390, 407–409.
Dahl, E. L., et al. (2006). Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrobial Agents and Chemotherapy, 50, 3124–3131.
McConkey, G. A., Rogers, M. J., & McCutchan, T. F. (1997). Inhibition of Plasmodium falciparum protein synthesis. Targeting the plastid-like organelle with thiostrepton. The Journal of Biological Chemistry, 272, 2046–2049.
Gaillard, T., Madamet, M., & Pradines, B. (2015). Tetracyclines in malaria. Malaria Journal, 14, 445.
Pasaje, C. F. A., et al. (2016). Selective inhibition of apicoplast tryptophanyl-tRNA synthetase causes delayed death in Plasmodium falciparum. Scientific Reports, 6, 27531.
Nagaraj, V. A., et al. (2013). Malaria parasite-synthesized heme is essential in the mosquito and liver stages and complements host heme in the blood stages of infection. PLoS Pathogens, 9, e1003522.
Ke, H., et al. (2014). The heme biosynthesis pathway is essential for Plasmodium falciparum development in mosquito stage but not in blood stages. The Journal of Biological Chemistry, 289, 34827–34837.
Gisselberg, J. E., Dellibovi-Ragheb, T. A., Matthews, K. A., Bosch, G., & Prigge, S. T. (2013). The suf iron-sulfur cluster synthesis pathway is required for apicoplast maintenance in malaria parasites. PLoS Pathogens, 9, e1003655.
Charan, M., et al. (2017). [Fe-S] cluster assembly in the apicoplast and its indispensability in mosquito stages of the malaria parasite. The FEBS Journal, 284, 2629–2648.
Ramya, T. N. C., Mishra, S., Karmodiya, K., Surolia, N., & Surolia, A. (2007). Inhibitors of nonhousekeeping functions of the apicoplast defy delayed death in Plasmodium falciparum. Antimicrobial Agents and Chemotherapy, 51, 307–316.
Murty, U., Banerjee, A., & Arora, N. (2012). Analyzing a potential drug target N-myristoyltransferase of Plasmodium falciparum through in silico approaches. Journal of Global Infectious Diseases, 4, 43.
Rathore, S. et al. (2010). A cyanobacterial serine protease of Plasmodium falciparum is targeted to the apicoplast and plays an important role in its growth and development. Molecular Microbiology, 77, no–no.
Mundra, S., et al. (2017, Oct 15). A novel class of Plasmodial ClpP protease inhibitors as potential antimalarial agents. Bioorganic & Medicinal Chemistry, 25(20), 5662–5677.
Datta, G., Hossain, M. E., Asad, M., Rathore, S., & Mohmmed, A. (2017). Plasmodium falciparum OTU-like cysteine protease (PfOTU) is essential for apicoplast homeostasis and associates with noncanonical role of Atg8. Cellular Microbiology, 19, e12748.
Spork, S., et al. (2009). An unusual ERAD-like complex is targeted to the apicoplast of Plasmodium falciparum. Eukaryotic Cell, 8, 1134–1145.
Harbut, M. B., et al. (2012). Targeting the ERAD pathway via inhibition of signal peptide peptidase for antiparasitic therapeutic design. Proceedings of the National Academy of Sciences of the United States of America, 109, 21486–21491.
Goulielmaki, E., et al. (2018). Distinct effects of HIV protease inhibitors and ERAD inhibitors on zygote to ookinete transition of the malaria parasite. Molecular and Biochemical Parasitology, 220, 10–14.
Kato, Y., Kouso, T., & Sakamoto, W. (2012). Variegated tobacco leaves generated by chloroplast FtsH suppression: Implication of FtsH function in the maintenance of thylakoid membranes. Plant & Cell Physiology, 53, 391–404.
Yeh, E., & DeRisi, J. L. (2011). Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biology, 9, e1001138.
He, C. Y., Striepen, B., Pletcher, C. H., Murray, J. M., & Roos, D. S. (2001). Targeting and processing of nuclear-encoded apicoplast proteins in plastid segregation mutants of toxoplasma gondii. The Journal of Biological Chemistry, 276, 28436–28442.
Ward, G. E., Miller, L. H., & Dvorak, J. A. (1993). The origin of parasitophorous vacuole membrane lipids in malaria-infected erythrocytes. Journal of Cell Science, 106(Pt 1), 237–248.
Bobenchik, A. M., et al. (2013). Plasmodium falciparum phosphoethanolamine methyltransferase is essential for malaria transmission. Proceedings of the National Academy of Sciences of the United States of America, 110, 18262–18267.
Gulati, S., et al. (2015). Profiling the essential nature of lipid metabolism in asexual blood and gametocyte stages of Plasmodium falciparum. Cell Host & Microbe, 18, 371–381.
Wein, S., et al. (2018). Contribution of the precursors and interplay of the pathways in the phospholipid metabolism of the malaria parasite. Journal of Lipid Research, 59, 1461–1471.
Flammersfeld, A., Lang, C., Flieger, A., & Pradel, G. (2017). Phospholipases during membrane dynamics in malaria parasites. International Journal of Medical Microbiology, 308, 129–141.
Flammersfeld, A., Lang, C., Flieger, A., & Pradel, G. (2018). Phospholipases during membrane dynamics in malaria parasites. International Journal of Medical Microbiology, 308, 129–141.
Vial, H. J., & Ancelin, M. L. (1992). Malarial lipids. An overview. Sub-Cellular Biochemistry, 18, 259–306.
Choubey, V., et al. (2007). Inhibition of Plasmodium falciparum choline kinase by hexadecyltrimethylammonium bromide: A possible antimalarial mechanism. Antimicrobial Agents and Chemotherapy, 51, 696–706.
Fagone, P., & Jackowski, S. (2013). Phosphatidylcholine and the CDP–choline cycle. Biochimica et Biophysica Acta – Molecular and Cell Biology of Lipids, 1831, 523–532.
Pessi, G., Choi, J.-Y., Reynolds, J. M., Voelker, D. R., & Mamoun, C. B. (2005). In vivo evidence for the specificity of Plasmodium falciparum phosphoethanolamine methyltransferase and its coupling to the Kennedy pathway. The Journal of Biological Chemistry, 280, 12461–12466.
Bobenchik, A. M., Augagneur, Y., Hao, B., Hoch, J. C., & Ben Mamoun, C. (2011). Phosphoethanolamine methyltransferases in phosphocholine biosynthesis: Functions and potential for antiparasite therapy. FEMS Microbiology Reviews, 35, 609–619.
Garg, A., et al. (2015). Structure, function and inhibition of the phosphoethanolamine methyltransferases of the human malaria parasites Plasmodium vivax and Plasmodium knowlesi. Scientific Reports, 5, 9064.
Emoto, K., et al. (1996). Redistribution of phosphatidylethanolamine at the cleavage furrow of dividing cells during cytokinesis. Proceedings of the National Academy of Sciences, 93, 12867–12872.
McMahon, H. T., & Boucrot, E. (2015). Membrane curvature at a glance. Journal of Cell Science, 128, 1065–1070.
Vance, J. E., & Tasseva, G. (2013). Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochimica et Biophysica Acta – Molecular and Cell Biology of Lipids, 1831, 543–554.
Calzada, E., Onguka, O., & Claypool, S. M. (2016). Phosphatidylethanolamine metabolism in health and disease. International Review of Cell and Molecular Biology, 321, 29–88.
Elabbadi, N., Ancelin, M. L., & Vial, H. J. (1992). Use of radioactive ethanolamine incorporation into phospholipids to assess in vitro antimalarial activity by the semiautomated microdilution technique. Antimicrobial Agents and Chemotherapy, 36, 50–55.
Choi, J.-Y., et al. (2016). Characterization of Plasmodium phosphatidylserine decarboxylase expressed in yeast and application for inhibitor screening. Molecular Microbiology, 99, 999–1014.
Elabbadi, N., Ancelin, M. L., & Vial, H. J. (1997). Phospholipid metabolism of serine in Plasmodium-infected erythrocytes involves phosphatidylserine and direct serine decarboxylation. The Biochemical Journal, 324, 435–445.
Serrán-Aguilera, L., et al. (2016). Plasmodium falciparum choline kinase inhibition leads to a major decrease in phosphatidylethanolamine causing parasite death. Scientific Reports, 6, 33189.
For the treatment of malaria guidelines. (2015).
Dondorp, A. M., et al. (2009). Artemisinin resistance in Plasmodium falciparum malaria. The New England Journal of Medicine, 361, 455–467.
Duffy, S., et al. (2017). Screening the medicines for malaria venture pathogen box across multiple pathogens reclassifies starting points for open-source drug discovery. Antimicrobial Agents and Chemotherapy, 61, e00379–e00317.
Antonova-Koch, Y., et al. (2018). Open-source discovery of chemical leads for next-generation chemoprotective antimalarials. Science (80-. ), 362, eaat9446.
Phillips, M. A., & Rathod, P. K. (2010). Plasmodium dihydroorotate dehydrogenase: A promising target for novel anti-malarial chemotherapy. Infectious Disorders Drug Targets, 10, 226–239.
Coteron, J. M., et al. (2011). Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. Journal of Medicinal Chemistry, 54, 5540–5561.
Sulyok, M., et al. (2017). DSM265 for Plasmodium falciparum chemoprophylaxis: A randomised, double blinded, phase 1 trial with controlled human malaria infection. The Lancet Infectious Diseases, 17, 636–644.
Meister, S., et al. (2011). Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science (80-.), 334, 1372–1377.
Wu, T., et al. (2011). Imidazolopiperazines: Hit to lead optimization of new antimalarial agents. Journal of Medicinal Chemistry, 54, 5116–5130.
Kuhen, K. L., et al. (2014). KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrobial Agents and Chemotherapy, 58, 5060–5067.
LaMonte, G. et al. (2016). Mutations in the Plasmodium falciparum cyclic amine resistance locus (PfCARL) confer multidrug resistance. MBio, 7.
Lim, M. Y.-X., et al. (2016). UDP-galactose and Acetyl-CoA transporters as Plasmodium multidrug resistance genes. Nature Microbiology, 1, 16166.
Leong, F. J., et al. (2014, Nov). A first-in-human randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose study of novel imidazolopiperazine KAF156 to assess its safety, tolerability, and pharmacokinetics in healthy adult volunteers. Antimicrobial Agents and Chemotherapy, 58(11), 6437–6443.
Kitchener, S., Nasveld, P., & Edstein, M. D. (2007). Tafenoquine for the treatment of recurrent Plasmodium vivax malaria. The American Journal of Tropical Medicine and Hygiene, 76, 494–496.
Fisk, T. L., Millet, P., Collins, W. E., & Nguyen-Dinh, P. (1989). In vitro activity of antimalarial compounds on the exoerythrocytic stages of Plasmodium cynomolgi and P. knowlesi. The American Journal of Tropical Medicine and Hygiene, 40, 235–239.
Pradines, B., et al. (2006). In vitro activity of tafenoquine against the asexual blood stages of Plasmodium falciparum isolates from Gabon, Senegal, and Djibouti. Antimicrobial Agents and Chemotherapy, 50, 3225–3226.
Coleman, R. E. (1990). Sporontocidal activity of the antimalarial WR-238605 against Plasmodium berghei ANKA in Anopheles stephensi. The American Journal of Tropical Medicine and Hygiene, 42, 196–205.
Fda. Highlights of prescribing information.
Lell, B., et al. (2000). Malaria chemoprophylaxis with tafenoquine: A randomised study. Lancet, 355, 2041–2045.
Rueangweerayut, R., et al. (2017). Hemolytic potential of tafenoquine in female volunteers heterozygous for glucose-6-phosphate dehydrogenase (G6PD) deficiency (G6PD Mahidol variant) versus G6PD-normal volunteers. The American Journal of Tropical Medicine and Hygiene, 97, 702–711.
Patil, C., Katare, S., Baig, M., & Doifode, S. (2014). Fixed dose combination of arterolane and piperaquine: A newer prospect in antimalarial therapy. Annals of Medical and Health Sciences Research, 4, 466–471.
Dong, Y, † et al. (2005). Spiro and dispiro-1,2,4-trioxolanes as antimalarial peroxides: Charting a workable structure−activity relationship using simple prototypes. https://doi.org/10.1021/JM049040U
Wang, X., et al. (2013). Comparative antimalarial activities and ADME profiles of ozonides (1,2,4-trioxolanes) OZ277, OZ439, and their 1,2-dioxolane, 1,2,4-trioxane, and 1,2,4,5-tetraoxane isosteres. Journal of Medicinal Chemistry, 56, 2547–2555.
Moehrle, J. J., et al. (2013). First-in-man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials. British Journal of Clinical Pharmacology, 75, 535–548.
McCarthy, J. S., et al. (2016). Efficacy of OZ439 (artefenomel) against early Plasmodium falciparum blood-stage malaria infection in healthy volunteers. The Journal of Antimicrobial Chemotherapy, 71, 2620–2627.
Held, J., et al. (2015). Ferroquine and artesunate in African adults and children with Plasmodium falciparum malaria: A phase 2, multicentre, randomised, double-blind, dose-ranging, non-inferiority study. The Lancet Infectious Diseases, 15, 1409–1419.
Younis, Y., et al. (2012). 3,5-diaryl-2-aminopyridines as a novel class of orally active antimalarials demonstrating single dose cure in mice and clinical candidate potential. Journal of Medicinal Chemistry, 55, 3479–3487.
Paquet, T., et al. (2017). Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Science Translational Medicine, 9, eaad9735.
Brunschwig, C., et al. (2018). UCT943, a next-generation Plasmodium falciparum PI4K inhibitor preclinical candidate for the treatment of malaria. Antimicrobial Agents and Chemotherapy, 62, 1–16.
Le Manach, C., et al. (2016). Identification of a potential antimalarial drug candidate from a series of 2-aminopyrazines by optimization of aqueous solubility and potency across the parasite life cycle. Journal of Medicinal Chemistry, 59, 9890–9905.
Yeung, B. K. S., et al. (2010). Spirotetrahydro β-carbolines (spiroindolones): A new class of potent and orally efficacious compounds for the treatment of malaria. Journal of Medicinal Chemistry, 53, 5155–5164.
van Pelt-Koops, J. C., et al. (2012). The spiroindolone drug candidate NITD609 potently inhibits gametocytogenesis and blocks Plasmodium falciparum transmission to Anopheles mosquito vector. Antimicrobial Agents and Chemotherapy, 56, 3544–3548.
Spillman, N. J., & Kirk, K. (2015). The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. International Journal for Parasitology: Drugs and Drug Resistance, 5, 149–162.
Rottmann, M., et al. (2010). Spiroindolones, a potent compound class for the treatment of malaria. Science (80-.), 329, 1175–1180.
White, N. J., et al. (2014). Spiroindolone KAE609 for falciparum and vivax malaria. The New England Journal of Medicine, 371, 403–410.
Jiménez-Díaz, M. B., et al. (2014). (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proceedings of the National Academy of Sciences of the United States of America, 111, E5455–E5462.
Kuemmerle, H. P., Murakawa, T., & De Santis, F. (1987). Pharmacokinetic evaluation of fosmidomycin, a new phosphonic acid antibiotic. Chemioterapia, 6, 113–119.
Kuemmerle, H. P., et al. (1985). Fosmidomycin, a new phosphonic acid antibiotic. Part II: 1. Human pharmacokinetics. 2. Preliminary early phase IIa clinical studies. International Journal of Clinical Pharmacology, Therapy, and Toxicology, 23, 521–528.
Odom, A. R., & Van Voorhis, W. C. (2010). Functional genetic analysis of the Plasmodium falciparum deoxyxylulose 5-phosphate reductoisomerase gene. Molecular and Biochemical Parasitology, 170, 108–111.
Hale, I., O’Neill, P. M., Berry, N. G., Odom, A., & Sharma, R. (2012). The MEP pathway and the development of inhibitors as potential anti-infective agents. Medchemcomm, 3, 418.
Na-Bangchang, K., Ruengweerayut, R., Karbwang, J., Chauemung, A., & Hutchinson, D. (2007). Pharmacokinetics and pharmacodynamics of fosmidomycin monotherapy and combination therapy with clindamycin in the treatment of multidrug resistant falciparum malaria. Malaria Journal, 6, 70.
Wells, T. N. C. (2011). New medicines to combat malaria: An overview of the global pipeline of therapeutics. In Treatment and prevention of malaria (pp. 227–247). Basel: Springer. https://doi.org/10.1007/978-3-0346-0480-2_12.
Ridley, R. G., et al. (1996). 4-aminoquinoline analogs of chloroquine with shortened side chains retain activity against chloroquine-resistant Plasmodium falciparum. Antimicrobial Agents and Chemotherapy, 40, 1846–1854.
De, D., Krogstad, F. M., Cogswell, F. B., & Krogstad, D. J. (1996). Aminoquinolines that circumvent resistance in Plasmodium falciparum in vitro. The American Journal of Tropical Medicine and Hygiene, 55, 579–583.
Mzayek, F., et al. (2007). Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clinical Trials, 2, e6.
Koita, O. A., et al. (2017). AQ-13, an investigational antimalarial, versus artemether plus lumefantrine for the treatment of uncomplicated Plasmodium falciparum malaria: A randomised, phase 2, non-inferiority clinical trial. The Lancet Infectious Diseases, 17, 1266–1275.
Udomsangpetch, R., et al. (1989). Plasmodium falciparum-infected erythrocytes form spontaneous erythrocyte rosettes. The Journal of Experimental Medicine, 169, 1835–1840.
Deen, J. L., von Seidlein, L., & Dondorp, A. (2008). Therapy of uncomplicated malaria in children: A review of treatment principles, essential drugs and current recommendations. Tropical Medicine and International Health, 13, 1111–1130.
Saiwaew, S., et al. (2017). Effects of sevuparin on rosette formation and cytoadherence of Plasmodium falciparum infected erythrocytes. PLoS One, 12, 1–15.
Leitgeb, A. M., et al. (2017). Inhibition of merozoite invasion and transient de-sequestration by sevuparin in humans with Plasmodium falciparum malaria. PLoS One, 12, e0188754.
Nde, P., et al. (2011). Low anticoagulant heparin disrupts Plasmodium falciparum rosettes in fresh clinical isolates. The American Journal of Tropical Medicine and Hygiene, 84, 390–396.
International Society on Thrombosis and Haemostasis. Journal of Thrombosis and Haemostasis. (Blackwell Pub., c2003-).
Ashley, J. N., Berg, S. S., & Lucas, J. M. (1960). 3:3′-diamidinocarbanilide: A new drug active against Babesial infections. Nature, 185, 461.
De Vos, A. J., Barrowman, P. R., Coetzer, J. A., & Kellerman, T. S. (1978). Amicarbalide: A therapeutic agent for anaplasmosis. The Onderstepoort Journal of Veterinary Research, 45, 203–208.
Pegoraro, S., et al. (2017). SC83288 is a clinical development candidate for the treatment of severe malaria. Nature Communications, 8, 3–4.
Pascual, A., et al. (2011). In vitro activity of Proveblue (methylene blue) on Plasmodium falciparum strains resistant to standard antimalarial drugs. Antimicrobial Agents and Chemotherapy, 55, 2472–2474.
Farber, P. M., Becker, K., Muller, S., Schirmer, R. H., & Franklin, R. M. (1996). Molecular cloning and characterization of a putative glutathione reductase gene, the PfGR2 gene, from Plasmodium falciparum. European Journal of Biochemistry, 239, 655–661.
Dormoi, J., et al. (2012). Proveblue (methylene blue) as an antimalarial agent: In vitro synergy with dihydroartemisinin and atorvastatin. Antimicrobial Agents and Chemotherapy, 56, 3467–3469.
Pradines, B., et al. (2007). Atorvastatin is 10-fold more active in vitro than other statins against Plasmodium falciparum. Antimicrobial Agents and Chemotherapy, 51, 2654–2655.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Asad, M. et al. (2019). Road Towards Development of New Antimalarial: Organelle Associated Metabolic Pathways in Plasmodium as Drug Targets and Discovery of Lead Drug Candidates. In: Hameed, S., Fatima, Z. (eds) Pathogenicity and Drug Resistance of Human Pathogens. Springer, Singapore. https://doi.org/10.1007/978-981-32-9449-3_10
Download citation
DOI: https://doi.org/10.1007/978-981-32-9449-3_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-32-9448-6
Online ISBN: 978-981-32-9449-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)