
Abstract
Sclerostin is a negative regulator of bone formation, which is produced by osteocytes and acts on osteoblasts where it binds the LRP5/6 co-receptors and antagonizes canonical Wnt signaling in these cells. The availability of commercial assays to measure sclerostin in blood initiated numerous clinical studies to elucidate the role of this protein in physiological and disordered bone metabolism. In interpreting results of such studies it is important to consider that neither the active form of sclerostin nor its metabolism are currently known and values obtained with available assays are moderately correlated. However, measurement of circulating sclerostin can assist the differential diagnosis of patients with rare bone sclerosing disorders and high bone mass such as sclerosteosis, van Buchem disease, and the high bone mass phenotype.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
Key Facts of Osteocytes
-
They constitute more than 95% of bone cells in the adult skeleton.
-
They are one of the longest living cells in the body (half-life 25 years).
-
They reside in lacunae within the mineralized bone matrix and communicate with neighboring osteocytes and cells at the bone surface and vasculature through cytoplasmic processes (dendrites) that run through tiny channels (canaliculi).
-
They sense mechanical signals (mechanosensors), and they control the adaptive responses of the skeleton to mechanical loading.
-
They orchestrate bone remodeling by regulating the function of osteoclasts and osteoblasts and, hence, bone resorption and bone formation.
-
They integrate hormonal and mechanical signals in the regulation of bone mass.
-
They are the target cells for PTH action on bone, and they mediate the anabolic actions of the canonical Wnt signaling pathway.
-
They have an endocrine function and produce FGF23, a factor, which acts in the kidney and controls phosphate homeostasis.
-
Osteocyte-produced molecules are targets of therapeutics for bone disorders (e.g., RANKL, sclerostin, FGF23).
Definition of Words and Terms
- DKK1:
-
An inhibitor of the canonical Wnt signaling pathway that binds to LRP5/6, co-receptors preventing the activation of Wnt signaling
- High bone mass phenotype:
-
A bone sclerosing disorder due to mutations of the LRP5 gene, rendering the LRP5 receptor resistant to the antagonist actions of sclerostin and DKK1
- LRP5 receptor:
-
A transmembrane receptor that forms a complex with the Frizzled receptor to transduce signals by Wnt proteins through the canonical Wnt pathway
- Sclerosteosis:
-
A bone sclerosing disorder due to loss-of-function mutations of the SOST gene that encodes sclerostin
- Sclerostin:
-
A 22.5-KDa secreted glycoprotein that is synthesized in bone by osteocytes and functions as a negative regulator of bone formation by inhibiting the canonical, β catenin-dependent, Wnt signaling pathway
- Van Buchem disease:
-
A bone sclerosing disorder due to a large genomic deletion downstream of the SOST gene resulting in impaired synthesis of sclerostin
Introduction
During the past two decades, studies of rare bone disorders have greatly enhanced our understanding of the local regulation of bone metabolism. The finding of gain-of-function mutations in the low density lipoprotein receptor-related protein 5 (LRP5) gene in patients with the high bone mass (HBM) phenotype (Boyden et al. 2002; Little et al. 2002) and loss-of-function mutations of LRP5 in patients with the osteoporosis-pseudoglioma syndrome (Gong et al. 2001) revealed the crucial role of canonical Wnt signaling pathway in osteoblast development and function. Subsequently, a number of regulators of the Wnt signaling pathway in bone was identified of which the most extensively studied is sclerostin. Commercial assays for the measurement of circulating sclerostin were developed and are used in studies of patients with metabolic bone disorders. We review here the value of circulating sclerostin as biochemical marker of bone metabolism in patients with rare skeletal disorders associated with high bone mass.
Sclerostin Molecule and Actions
Sclerostin is a secreted glycoprotein with sequence similarity to the DAN (differential screening-selected gene aberrative in neuroblastoma) family of proteins. It has 190 amino acid residues, which form a three loop-like structure with a cystine knot at its central core (Fig. 1). While the first and third loops are well structured, the second loop appears disordered on nuclear molecular resonance imaging. The long N- and C-termini are also flexible but appear to have a functional role (Weidauer et al. 2009; Veverka et al. 2009). The third loop of the sclerostin molecule is the presumed binding site to its target receptor LRP5/6 (Veverka et al. 2009), although parts of the cystine knot might also be involved (Boschert et al. 2013). A neutralizing monoclonal antibody against sclerostin binds the second loop, suggesting that this loop is also important for sclerostin activity.

Left panel shows a ribbon representation of the backbone topology of the structured core of the protein (residues 52 to 147) in the same orientation. Right panel shows a schematic representation of the protein, highlighting the positions of disulphide bonds, the resulting 3 loop structure, and residues involved in regular sheets (Reproduced with permission from Veverka et al. 2009)
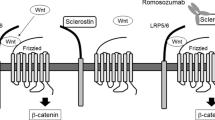
Sclerostin is produced in the skeleton exclusively by osteocytes and is transported by the osteocyte canaliculi to act on osteoblasts at the bone surface but also on neighboring osteocytes (van Bezooijen et al. 2004; Poole et al. 2005). At the bone surface, sclerostin inhibits bone formation by osteoblasts while it increases bone resorption by stimulating the production of RANKL by osteocytes. Sclerostin antagonizes the canonical Wnt signaling pathway in osteoblasts through binding to the first propeller of LRP5/6 (Li et al. 2005; Bourhis et al. 2011), thereby preventing the binding of Wnt ligands to the LRP5/6 and frizzled co-receptor complex and blocking Wnt signaling. The action of sclerostin on Wnt signaling pathway is facilitated by LRP4, a negative regulator of LRP5/6 signaling which acts as co-receptor for sclerostin (Choi et al. 2009; Leupin et al. 2011; Chang et al. 2014; Fig. 2).

Osteocyte-produced sclerostin inhibits the proliferation, differentiation, and survival of osteoblasts and reduces bone formation; it also stimulates the production of RANKL by neighboring osteocytes and bone resorption. In osteoblasts, sclerostin binds to LRP5/6 and inhibits the Wnt signaling pathway, an action facilitated by LRP4. Production of sclerostin is decreased by mechanical loading, PTH, estrogens, and antisclerostin antibodies. LRP low-density lipoprotein receptor-related protein, PTH parathyroid hormone, RANKL receptor activator of nuclear factor kappa-B ligand (Reproduced from Appelman-Dijkstra and Papapoulos 2015)
Sclerostin Assays
During the past few years, there has been mounting interest in the role of sclerostin in bone remodeling, and the availability of assays to measure this protein in blood facilitated clinical studies to understand its role in disorders of bone and mineral metabolism (reviewed by Clarke and Drake 2013). These studies have mainly been conducted with three commercially available sclerostin assays: the enzyme-linked immunosorbent sclerostin assays of Biomedica Medizinprodukt (Wien, Austria), TECO medical (Sissach, Switzerland), and the electro-chemiluminescence sclerostin assay of Meso-Scale diagnostics (Rockville, USA). In interpreting results obtained with these assays it should be noted that the active form of sclerostin is not known and there is no information about its metabolism and, hence, of circulating fragments that may be immunoreactive.
The Biomedica assay uses a polyclonal goat antihuman sclerostin capture antibody and a murine monoclonal antisclerostin detection antibody. It has a reported detection range between 3.2 pmol/ml (72 pg/ml) and 240 pmol/ml (5400 pg/ml) and a lower limit of quantification of 7.5 pmol/l (170 pg/ml). Validation of the assay showed intra- and interassay coefficients of variation of 5% and 6%, respectively, and linearity of values of serially diluted samples. Recovery of sclerostin in serum spiked with the protein was 92–108%. Serum and EDTA plasma samples showed stable results when kept at room temperature for up to 24 h and after four freeze/thaw cycles (Biomedica n.d.). In earlier developed assays, sclerostin levels were higher in EDTA and heparin plasma compared with serum samples (McNulty et al. 2011), but later assays showed no difference between sample matrixes (Biomedica n.d.). The Biomedica assay detects recombinant sclerostin fragments of different length (the three loop core without the N- and C-termini, the 1st and 3rd loop without the N- and C-termini, and the 2nd loop alone; Fig. 3) (van Lierop et al. 2012a). This suggests that the assay does not recognize the entire sclerostin molecule. Although the assay did not detect structurally related proteins such as noggin and wise (Biomedica n.d.), sclerostin was detectable in serum of 3 of 7 patients with sclerosteosis, suggesting possible cross-reactivity with other proteins (van Lierop et al. 2012a).

Detection of fragments of the sclerostin molecule using different sclerostin assays. (a) Simplified representation of the sclerostin molecule. (b) Detection of sclerostin fragments of different sizes with the Biomedica, TECO, and the MSD sclerostin assays. All fragments were undectable in the TECO and MSD assay, but were detected by the Biomedica assay (Modified from van Lierop et al. 2012a)
The TECO sclerostin assay uses a polyclonal goat antihuman sclerostin capture antibody and a monoclonal antihuman detection antibody. Antibodies recognize epitopes that reside at the amino terminus and the midregion of the protein (Durosier et al. 2013). The detection range is between 50 pg/ml and 3000 pg/ml, with a lower limit of quantification of 130 pg/ml (Garnero et al. 2013). The intra- and interassay coefficients of variation range between 3.8 and 8.0% and 3.3–9.0%, respectively (Durosier et al. 2013). Serial dilutions of samples showed good linearity, and recovery in serum spiked with sclerostin was 96–102% (Garnero et al. 2013). Sclerostin levels are stable at room temperature for more than 24 h and after three freeze/thaw cycles (TECOmedical n.d.). The sclerostin fragments described in the previous paragraph were not detected by the TECO assay. Sclerostin was measurable, however, in serum samples of all six patients with sclerosteosis tested but values were close to the limit of detection (own unpublished observations).
The electro-chemiluminescence sclerostin assay of MSD uses a polyclonal goat capture antibody against murine sclerostin and a polyclonal goat antihuman sclerostin detection antibody. Epitope mapping of the capture and detection antibodies revealed binding sites for the capture antibody at the N-terminus, and one at the distal end of the C-terminus of human sclerostin, while for the detection antibody three epitopes were identified, at the N-terminus, at the third loop, and at the C-terminus, respectively (van Lierop et al. 2011; Fig. 4). The MSD sclerostin assay has a very broad detection range from 1 pg/ml to 10.000 pg/ml and a very low limit of quantification (5 pg/ml). The intra- and interassay coefficients of variation are 4 and 7%, respectively. The recovery of added sclerostin in serum samples was 82–93%. The measured concentrations of serial dilutions of recombinant sclerostin were highly concordant with that of the sclerostin standard provided in the assay (ratio 1.01, range 0.8–1.3) (van Lierop et al. 2011). When samples of the sclerostin standard provided with the Biomedica assay were measured by the MSD assay, sclerostin values were on average six-fold lower than expected (own unpublished observations). Sclerostin levels remained stable after multiple freeze/thawing cycles, but levels declined by about 50% when samples were kept at room temperature for more than 24 h (Durosier et al. 2013). The MSD sclerostin assay did not detect any of the above described recombinant sclerostin fragments and did not detect any immunoreactive sclerostin in the serum of 19 patients with sclerosteosis (van Lierop et al. 2011). These findings suggest that this assay is specific for the entire sclerostin molecule and does not appear to cross react with other circulating proteins.

Epitope mapping of the MSD capture antibody (upper panel) and detection antibody (lower panel). Spikes in the baseline represent increased binding affinity of the antibodies for the sclerostin molecule. Above each panel is a schematic linear representation of the sclerostin molecule (Unpublished)
Differences Between Sclerostin Assays
Comparison of measured sclerostin levels in the same samples with the three assays showed considerable differences (McNulty et al. 2011; Durosier et al. 2013). The Biomedica assay detected about two-fold and 30-fold higher levels than the TECO and the MSD assays, respectively, and serum sclerostin levels were moderately correlated (Spearman’s correlation coefficient between 0.53 and 0.71) (Durosier et al. 2013). Observed differences between measured values by the three assays are presumably the result of different epitope recognition. The lower stability of the N- and C-terminus compared to the central core of the protein, and their easy cleavage, may explain the diminished stability of measured sclerostin with the MSD assay in samples kept at room temperature for more than 24 h. Furthermore, this may also explain the much lower levels of circulating sclerostin measured by the MSD assay compared with the Biomedica and TECO assays. In addition, binding of circulating sclerostin to proteins, that may obscure binding sites crucial for antibody detection, may also contribute to differences in measured values. Sclerostin was found to have a binding site for heparin (Veverka et al. 2009), and the addition of heparin to sclerostin-producing osteoblastic cell cultures increased the amount of sclerostin in the supernatant, without affecting its production. In line with these observations, measurement of sclerostin with the Biomedica and TECO assays revealed higher levels in heparin samples than serum samples (McNulty et al. 2011). Since the presence of heparin in the samples matrix results in higher sclerostin concentrations, it has been suggested that the heparin displaces sclerostin from other proteins, which obscured the binding sites to the detection or capture antibodies. Differences in reported sclerostin values with the three assays may, therefore, also result from differences in the sensitivity of specific epitopes of antibodies to protein-bound and free-circulating sclerostin. Notably, Biomedica in their last generation of the sclerostin ELISA has resolved the differences in measured sclerostin levels between sample matrixes.
Circulating Sclerostin in Sclerosing Bone Disorders
Sclerosteosis
Sclerosteosis is an autosomal recessive bone sclerosing disorder caused by loss-of-function mutations in SOST, the gene encoding sclerostin (Balemans et al. 2001; Brunkow et al. 2001). So far, eight different mutations have been identified resulting either in impaired synthesis of sclerostin or in the synthesis of a nonfunctioning protein (Balemans et al. 2001, 2005; Brunkow et al. 2001; Kim et al. 2008; Piters et al. 2010; Bhadada et al. 2013; Belkhribchia et al. 2014; He et al. 2016). The majority of patients are members of the Afrikaner population in South Africa, but isolated cases have been reported in other countries around the world. In the absence of sclerostin, bone formation is increased in these patients, while bone resorption is mostly unaffected or decreased (van Lierop et al. 2011). As a result, patients develop generalized osteosclerosis, with bone mineral density Z-scores sometimes exceeding +10 (Gardner et al. 2005). The excess bone formed in patients with sclerosteosis is of excellent quality (Hassler et al. 2014) evidenced also by the lack of any reports of fractures in these patients (Hamersma et al. 2003; van Lierop et al. 2011). Serious complications of sclerosteosis are due to overgrowth of the skull bones and include facial distortion with elongation of the forehead and mandibular overgrowth and nerve entrapment syndromes, such as facial palsy due to narrowing of cranial foramina. Another frequently encountered complication is hearing loss, resulting from narrowing of the external auditory canal, fixation of ossicles, or impingement of the acoustic nerves. Moreover, the thickening of calvaria and skull base can lead to increased intracranial pressure, prompting the need for craniotomy in a substantial number of patients, being also a cause of sudden death (Hamersma et al. 2003; van Lierop et al. 2011). Other characteristic findings of sclerosteosis include tall stature and syndactyly, which can be used to clinically differentiate these patients from those with van Buchem disease and HBM (Beighton et al. 1988). Although carriers of sclerosteosis mutations have increased bone formation and a higher bone mineral density compared with healthy controls, they do not develop any of the complications of sclerosteosis (Gardner et al. 2005; van Lierop et al. 2011).
In line with the underlying genetic defect, no sclerostin could be detected in serum of 19 South African patients with sclerosteosis with the MSD sclerostin assay (van Lierop et al. 2011). In contrast, sclerostin was detected in serum of all 26 measured disease carriers, but mean levels were about 50% lower than those of healthy controls (16 pg/ml vs. 40 pg/ml), indicating a gene-dose effect. There was, however, considerable overlap between values of carriers and healthy controls (Fig. 5). As already discussed, sclerostin was also measured in a few patients with sclerosteosis with the Biomedica and TECO assays.

Serum levels of sclerostin in patients and carriers with sclerosteosis and healthy controls. Sclerostin levels were measured with the MSD sclerostin assay. No sclerostin was detected in serum samples of 19 patients with sclerosteosis (Reproduced from van Lierop et al. 2011)
As expected by the inhibitory action of sclerostin on bone formation, patients with sclerosteosis and sclerostin deficiency have elevated levels of the bone formation marker procollagen type 1 N-terminal propeptide (P1NP), while P1NP levels in carriers are within the reference range but higher than those of healthy controls (Fig. 6). There were no differences in levels of the bone resorption marker carboxy-terminal collagen crosslinks (CTX) between patients, carriers, and controls (van Lierop et al. 2011). Interestingly, serum levels of Dickkopf 1 (DKK1), another Wnt signaling inhibitor which also acts on the LRP5 receptor, is increased in patients with sclerosteosis (van Lierop et al. 2014), possibly as a compensatory response to sclerostin deficiency.

Serum sclerostin and P1NP levels in patients and heterozygous carriers of sclerosteosis (SCL), van Buchem disease (VBD), and healthy subjects (Reproduced from Appelman-Dijkstra and Papapoulos 2014)
Van Buchem Disease
Van Buchem disease (VBD), or “hyperostosis corticalis generalisata familiaris,” is an autosomal recessive disorder due to a 52 kb deletion downstream of the SOST gene, which contains an essential regulatory element for postnatal SOST transcription (Balemans et al. 2002; Staehling-Hampton et al. 2002). About 30 cases of VBD have been described, the majority inhabitants of a small village in the Netherlands. Clinical manifestations of patients with VBD are very similar to those of patients with sclerosteosis including complications due to bone overgrowth of the skull, such as facial distortion, facial nerve palsy, and hearing loss. However, increased intracranial pressure is a rare finding (van Lierop et al. 2010), and patients have normal stature and no digit abnormities (van Lierop et al. 2013). Bone mass is greatly increased to values comparable to those of patients with sclerosteosis, with bone mineral density Z-scores sometimes exceeding +10 (van Lierop et al. 2013).
In patients with VBD, circulating sclerostin levels measured with the MSD assay were low, but detectable in nearly all patients (range <5 to 17 pg/ml). This is in agreement with the slightly milder phenotype of these patients compared with that of patients with sclerosteosis. Carriers of VBD had levels lower than controls (29 vs. 40 pg/ml) with considerable overlap of values (Fig. 6; van Lierop et al. 2013). There are no reported values of circulating sclerostin in carriers or patients with VBD with either the Biomedica or the TECO assays.
Patients with VBD had increased levels of P1NP in serum, while in carriers these were slightly higher than in healthy controls; in contrast, serum CTX levels were similar in patients and carriers (van Lierop et al. 2013). As in sclerosteosis, patients with VBD had increased levels of DKK1 in serum (van Lierop et al. 2014).
High Bone Mass Phenotype
In contrast to sclerosteosis and VBD, the HBM phenotype is inherited as an autosomal dominant trait (Boyden et al. 2002). Twelve different mutations have been identified, all clustered within the region encoding for the first propeller domain of LRP5 (Balemans et al. 2007; Boyden et al. 2002; Gregson et al. 2015; Kwee et al. 2005; Little et al. 2002; Rickels et al. 2005; van WesenBeeck et al. 2003; Whyte et al. 2004). These mutations render the LRP5 receptor resistant to the inhibitory action of sclerostin and DKK1 (Ai et al. 2005; Semenov et al. 2005; Balemans et al. 2007; Niziolek et al. 2015). About one hundred cases have been identified in different parts of the world.
HBM is characterized by generalized endosteal hyperostosis, although to a lesser extent than sclerosteosis or VBD, with bone mineral density Z- scores ranging between +5 and +10 (Boyden et al. 2002; Little et al. 2002; Rickels et al. 2005; Balemans et al. 2007). Enlargement of the jaw is a frequent finding but complications resulting from cranial nerve entrapment are rare (Kwee et al. 2005). Furthermore, patients have normal stature and no digit abnormalities but almost all have torus pallatinus, a bone protrusion of the palate, which has also been described in some patients with sclerosteosis but not in patients with VBD (Beighton et al. 1988).
Three studies reported sclerostin levels measured with the Biomedica assay in patients with HBM. In 19 patients with HBM due to a T253I mutation, Frost et al. found higher sclerostin levels than in 19 control subjects (22 vs. 13 pmol/l or 500 vs. 296 pg/ml) (Frost et al. 2011). Similarly, Gregson et al. reported higher serum sclerostin levels in six HBM patients compared with 196 controls (129 vs. 66 pmol/l; 2900 vs. 1500 pg/ml) (Gregson et al. 2014). In contrast, Simpson and colleagues found comparable serum sclerostin levels in 16 HBM patients with G171V or N198S LRP5 mutations and 24 controls (53 vs. 55 pmol; 1204 vs. 1250 pg/ml) (Simpson et al. 2014).
Boyden et al. reported increased serum osteocalcin levels in HBM patients (Boyden et al. 2002) while others found normal levels of bone formation markers (bone specific alkaline phosphatase, osteocalcin, P1NP) in the serum of such patients; no differences in serum levels of bone resorption markers between patients and healthy controls have been observed (Frost et al. 2011; Boyden et al. 2002; Gregson et al. 2014; Simpson et al. 2014). In addition, and in contrast to sclerosteosis and VBD, patients with HBM have normal levels of DKK1 (Simpson et al. 2014). In population studies, serum sclerostin levels are positively correlated with bone mass (Durosier et al. 2013), reflecting probably the total number of functioning osteocytes. Increased circulating sclerostin levels in HBM might, therefore, result either from increased synthesis as a compensatory response to sclerostin resistance and/or to increased osteocyte numbers due to the high bone mass.
Sclerosing Bone Dysplasia due to Mutations of LRP4
In two cases with generalized osteosclerosis, syndactyly, facial palsy, and hearing loss, no mutations in either SOST or LRP5 could be identified. Instead, in these patients homozygous and heterozygous mutations of LRP4 were found. These mutations diminish the inhibitory function of sclerostin on LRP5/6, underscoring the important role of LRP4 for sclerostin action (Leupin et al. 2011). In one patient, serum sclerostin levels were increased (Fijalkowski et al. 2016). In addition, in mice harboring LRP4 mutation, sclerostin levels were highly increased due to increased expression and decreased binding of sclerostin (Chang et al. 2014). Because of these findings and the mechanism of action of LRP4, it is expected that patients with loss-of-function mutations of LRP4 will have increased circulating sclerostin levels.
Practical Guidance
Circulating sclerostin can be measured in the initial assessment of patients with similar bone phenotypes associated with high bone mass to distinguish patients with defective sclerostin production from those with resistance to sclerostin action (Table 1). Undetectable sclerostin levels in serum are pathognomic for sclerosteosis, while very low levels are suggestive of VBD. Because the MSD sclerostin assay has a very low limit of detection with appropriate accuracy, even at the lower end of the reference range, it is presently the preferred assay to distinguish between absent or low sclerostin levels in serum. This initial screening can direct genetic analyses to specific genes avoiding examination of a panel of candidate genes, a cost-effective approach. In addition, because disease carriers of sclerosteosis have lower sclerostin levels than controls, serum sclerostin measurements can also be used to detect carriers in family members of patients. On the other hand, high serum levels of sclerostin exclude the diagnoses of sclerosteosis and VBD and point to different genetic defects involving the Wnt signaling pathway. Standardization of the assays and appropriate determination of reference range for sclerostin levels in the normal population are essential for proper interpretation of measured values.
Potential Application to Prognosis, Other Diseases or Conditions
There is no recognized value of serum sclerostin measurements to the prognosis of bone sclerosing dysplasias. Serum sclerostin has been measured in various disorders affecting the skeleton but consistent changes were not always detected precluding its use as biomarker in routine clinical practice (reviewed by Clarke and Drake 2013). Examples include osteoporosis (Ardawi et al. 2012b; Lapauw et al. 2013, Garnero 2014; Guañabens et al. 2014), hyper- and hypoparathyroidism (van Lierop et al. 2010; Costa et al. 2011; Ardawi et al. 2012a), hypercortisolism (van Lierop et al. 2012b; Belaya et al. 2013), Paget’s disease of bone (Yavropoulou et al. 2012), osteogenesis imperfecta (Kocijan et al. 2016), diabetes mellitus type 2 (van Lierop et al. 2012c; Garcia-Martin et al. 2012), chronic renal disease (Moysés and Schiavi 2015), multiple myeloma (Terpos et al. 2012), and prostate cancer (Yavropoulou et al. 2012; García-Fontana et al. 2014).
Summary Points
-
Sclerostin, a protein produced in bone by osteocytes, decreases bone formation by inhibiting the Wnt signaling pathway in osteoblasts.
-
Deficient production or impaired action of sclerostin are associated with distinct clinical phenotypes associated with high bone mass.
-
Sclerostin measurements in blood are not standardized and available assays provide different results.
-
Sclerostin is undetectable in serum of patients with sclerosteosis while patients with van Buchem disease have very low levels.
-
Phenotypically healthy carriers of these diseases have serum sclerostin levels lower than healthy controls.
-
Patients with the high bone mass phenotype reported serum sclerostin levels are either normal or increased.
-
Measurement of circulating sclerostin can be used in the differential diagnosis of bone sclerosing disorders.
Abbreviations
- CTX:
-
Carboxyterminal collagen crosslinks
- DAN:
-
Differential screening-selected gene abberative in neuroblastoma
- DKK1:
-
Dickkopf-related protein 1
- HBM:
-
High bone mass
- LRP:
-
Low-density lipoprotein receptor-related protein
- P1NP:
-
Procollagen type 1 N-terminal propeptide
- PTH:
-
Parathyroid hormone
- RANKL:
-
Receptor activator of nuclear factor kappa-B ligand
- SCL:
-
Sclerostin
- VBD:
-
Van Buchem disease
- Wnt:
-
Wingless-related integration site
References
Ai M, Holmen SL, van Hul W, Williams BO, et al. Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass-associated missense mutations in LRP5 affect canonical Wnt signaling. Mol Cell Biol. 2005;25:4946–55.
Appelman-Dijkstra NM, Papapoulos SE. Novel approaches to the treatment of osteoporosis. Best Pract Res Clin Endocrinol Metab. 2014;28:843–57.
Appelman-Dijkstra NM, Papapoulos SE. Modulating bone resorption and bone formation in opposite directions in the treatment of postmenopausal osteoporosis. Drugs. 2015;75:1049–58.
Ardawi MS, Al-Sibiany AM, Bakhsh TM, et al. Decreased serum sclerostin levels in patients with primary hyperparathyroidism: a cross-sectional and a longitudinal study. Osteoporos Int. 2012a;23:1789–97.
Ardawi MS, Rouzi AA, Al-Sibiani SA, et al. High serum sclerostin predicts the occurrence of osteoporotic fractures in postmenopausal women: the Center of Excellence for Osteoporosis Research Study. J Bone Miner Res. 2012b;27:2592–602.
Balemans W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10:537–43.
Balemans W, Patel N, Ebeling M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–7.
Balemans W, Cleiren E, Siebers U, et al. A generalized skeletal hyperostosis in two siblings caused by a novel mutation in the SOST gene. Bone. 2005;36:943–7.
Balemans W, Devogelaer JP, Cleiren E, et al. Novel LRP5 missense mutation in a patient with a high bone mass phenotype results in decreased DKK1-mediated inhibition of Wnt signaling. J Bone Miner Res. 2007;22:708–16.
Beighton P. Sclerosteosis. J Med Genet. 1988;25:200–3.
Belaya ZE, Rozhinskaya LY, Melnichenko GA, et al. Serum extracellular secreted antagonists of the canonical Wnt/β-catenin signalling pathway in patients with Cushing’s syndrome. Osteoporos Int. 2013;24:2191–9.
Belkhribchia MR, Collet C, Laplanche JL, et al. Novel SOST gene mutation in a sclerosteosis patient from Morocco, a case report. Eur J Med Genet. 2014;57:133–7.
Bhadada SK, Rastogi A, Steenackers E, et al. Novel SOST gene mutation in a sclerosteosis patient and her parents. Bone. 2013;52:707–10.
Biomedica. Sclerostin ELISA-assay performance and characteristics, viewed 1 December 2015, from http://www.bmgrp.com/fileadmin/user_upload_immunoassays/downloads/Validation_Data/BI-20492_SCLEROSTIN_Validation_Data.pdf
Boschert V, van Dinther M, Weidauer S, et al. Mutational analysis of sclerostin shows importance of the flexible loop and the cystine-knot for Wnt-signaling inhibition. PLoS One. 2013;29:e81710.
Bourhis E, Wang W, Tam C, et al. Wnt antagonists bind through a short peptide to the first β-propeller domain of LRP5/6. Structure. 2011;19:1433–42.
Boyden LM, Mao J, Belsky J, et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–21.
Brunkow ME, Gardner JC, Van Ness J, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–89.
Chang MK, Kramer I, Huber T, et al. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc Natl Acad Sci U S A. 2014;111:E5187–95.
Choi HY, Dieckmann M, Herz J, et al. Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PLoS One. 2009;4:e7930.
Clarke BL, Drake MT. Clinical utility of serum sclerostin measurements. BoneKEy Rep. 2013;2:361.
Costa AG, Cremers S, Rubin MR, et al. Circulating sclerostin in disorders of parathyroid gland function. J Clin Endocrinol Metab. 2011;96:3804–10.
Durosier C, van Lierop A, Ferrari S, et al. Association of circulating sclerostin with bone mineral mass, microstructure, and turnover biochemical markers in healthy elderly men and women. J Clin Endocrinol Metab. 2013;98:3873–83.
Fijalkowski I, Geets E, Steenackers E, et al. A novel domain-specific mutation in a sclerosteosis patient suggests a role of LRP4 as an anchor for sclerostin in human bone. J Bone Miner Res. 2016;31:874–81.
Frost M, Andersen T, Gossiel F, et al. Levels of serotonin, sclerostin, bone turnover markers as well as bone density and microarchitecture in patients with high-bone-mass phenotype due to a mutation in Lrp5. J Bone Miner Res. 2011;26:1721–8.
García-Fontana B, Morales-Santana S, Varsavsky M, et al. Sclerostin serum levels in prostate cancer patients and their relationship with sex steroids. Osteoporos Int. 2014;25:645–51.
García-Martín A, Rozas-Moreno P, Reyes-García R, et al. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97:234–41.
Gardner JC, van Bezooijen RL, Mervis B, et al. Bone mineral density in sclerosteosis; affected individuals and gene carriers. J Clin Endocrinol Metab. 2005;90:6392–5.
Garnero P. New developments in biological markers of bone metabolism in osteoporosis. Bone 2014;66:46–55.
Garnero P, Sornay-Rendu E, Munoz F, et al. Association of serum sclerostin with bone mineral density, bone turnover, steroid and parathyroid hormones, and fracture risk in postmenopausal women, the OFELY study. Osteoporos Int. 2013;24:489–94.
Gong Y, Slee RB, Fukai N, et al. Osteoporosis-Pseudoglioma Syndrome Collaborative Group, LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–23.
Gregson CL, Poole KE, McCloskey EV, et al. Elevated circulating sclerostin concentrations in individuals with high bone mass, with and without LRP5 mutations. J Clin Endocrinol Metab. 2014;99:2897–907.
Gregson CL, Wheeler L, Hardcastle SA, et al. Mutations in known monogenic high bone mass loci only explain a small proportion of high bone mass cases. J Bone Miner Res. 2015; [Epub ahead of print].
Guañabens N, Gifre L, Peris P. The role of Wnt signalling and sclerostin in the pathogenesis of glucocorticoid-induced osteoporosis. Curr Osteoporos Rep. 2014;12(1):90–7.
Hamersma H, Gardner J, Beighton P. The natural history of sclerosteosis. Clin Genet. 2003;63:192–7. Erratum in: Clin Genet 2003;64:176.
Hassler N, Roschger A, Gamsjaeger S, et al. Sclerostin deficiency is linked to altered bone composition. J Bone Miner Res. 2014;29:2144–51.
He WT, Chen C, Pan C, et al. Sclerosteosis caused by a novel nonsense mutation of SOST in a consanguineous family. Clin Genet. 2016;89:205–9.
Kicijan R, Dinu S, Muschitz C, 2016. Sclerostin as biomarker in osteogenesis imperfecta. In: Patel VB, Preedy VR editors. Biomarkers in disease: methods, discoveries and applications. Doordrecht: Springer.
Kim CA, Honjo R, Bertola D, et al. A known SOST gene mutation causes sclerosteosis in a familial and an isolated case from Brazilian origin. Genet Test. 2008;12:475–9.
Kwee ML, Balemans W, Cleiren E, et al. An autosomal dominant high bone mass phenotype in association with craniosynostosis in an extended family is caused by an LRP5 missense mutation. J Bone Miner Res. 2005;20:1254–60.
Lapauw B, Vandewalle S, Taes Y, et al. Serum sclerostin levels in men with idiopathic osteoporosis. Eur J Endocrinol. 2013;168:615–20.
Leupin O, Piters E, Halleux C, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286:19489–500.
Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–7.
Little RD, Carulli JP, Del Mastro RG, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70:11–9.
McNulty M, Singh RJ, Li X, et al. Determination of serum and plasma sclerostin concentrations by enzyme-linked immunoassays. J Clin Endocrinol Metab. 2011;96:E1159–62.
Moysés RM, Schiavi SC. Sclerostin, osteocytes, and chronic kidney disease – mineral bone disorder. Semin Dial. 2015;28(6):578–86.
Niziolek PJ, MacDonald BT, Kedlaya R, et al. High bone mass-causing mutant LRP5 receptors are resistant to endogenous inhibitors in vivo. J Bone Miner Res. 2015;30:1822–30.
Piters E, Culha MM, et al. First missense mutation in the SOST gene causing sclerosteosis by loss of sclerostin function. Hum Mutat. 2010;31:E1526–43.
Poole KE, van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–4.
Rickels MR, Zhang X, Mumm S, et al. Oropharyngeal skeletal disease accompanying high bone mass and novel LRP5 mutation. J Bone Miner Res. 2005;20:878–85.
Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–5.
Simpson CA, Foer D, Lee GS, et al. Serum levels of sclerostin, Dickkopf-1, and secreted frizzled-related protein-4 are not changed in individuals with high bone mass causing mutations in LRP5. Osteoporos Int. 2014;25:2383–8.
Staehling-Hampton K, Proll S, Paeper BW, et al. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110:144–52.
TECOmedical. Sclerostin TECO® high sensitive product information, viewed 1 December 2015, from http://www.tecomedical.com/en/laboratory-ivd-kits-reagents/bone-and-cartilage-parameters/bone-metabolism/Sclerostin-TECO-High-sensitive
Terpos E, Christoulas D, Katodritou E, et al. Elevated circulating sclerostin correlates with advanced disease features and abnormal bone remodeling in symptomatic myeloma: reduction post-bortezomib monotherapy. Int J Cancer. 2012;131:1466–71.
van Bezooijen RL, Roelen BA, Visser A, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–14.
van Lierop AH, Hamdy NA, Papapoulos SE. Glucocorticoids are not always deleterious for bone. J Bone Miner Res. 2010a;25:2796–800.
van Lierop AH, Witteveen J, Hamdy N, et al. Patients with primary hyperparathyroidism have lower circulating sclerostin levels than euparathyroid controls. Eur J Endocrinol. 2010b;163:833–7.
van Lierop AH, Hamdy NA, Hamersma H, et al. Patients with sclerosteosis and disease carriers, human models of the effect of sclerostin on bone turnover. J Bone Miner Res. 2011;26:2804–11.
van Lierop AH, Hamdy NA, van Bezooijen RL, et al. The role of sclerostin in the pathophysiology of sclerosing bone dysplasias. Clin Rev Bone Miner Metab. 2012a;10:108–16.
van Lierop AH, van der Eerden AW, Hamdy NA, et al. Circulating sclerostin levels are decreased in patients with endogenous hypercortisolism and increase after treatment. J Clin Endocrinol Metab. 2012b;97:E1953–7.
van Lierop AH, Hamdy NAT, van der Meer RW, et al. Distinct effects of pioglitazone and metformin on circulating sclerostin and biochemical markers of bone turnover in men with Type 2 Diabetes Mellitus. Eur J Endocrinol. 2012c;166:711–6.
van Lierop AH, Hamdy NA, van Egmond ME, et al. Van Buchem disease, clinical, biochemical, and densitometric features of patients and disease carriers. J Bone Miner Res. 2013;28:848–54.
van Lierop AH, Moester MJ, Hamdy NA, et al. Serum Dickkopf 1 levels in sclerostin deficiency. J Clin Endocrinol Metab. 2014;99:E252–6.
Van Wesenbeeck L, Cleiren E, Gram J, et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet. 2003;72:763–71.
Veverka V, Henry AJ, Slocombe PM, et al. Characterization of the structural features and interactions of sclerostin, molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem. 2009;284:10890–900.
Weidauer SE, Schmieder P, Beerbaum M, et al. NMR structure of the Wnt modulator protein Sclerostin. Biochem Biophys Res Commun. 2009;380:160–5.
Whyte MP, Reinus WH, Mumm S. High-bone-mass disease and LRP5. N Engl J Med. 2004;350:2096–9.
Yavropoulou MP, van Lierop AH, Hamdy NA, et al. Serum sclerostin levels in Paget’s disease and prostate cancer with bone metastases with a wide range of bone turnover. Bone. 2012;51:153–7.
Acknowledgement
Supported by a grant from the European Commission (HEALTH-F2-2008-20199, TALOS).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media Dordrecht
About this entry
Cite this entry
van Lierop, A.H., Papapoulos, S.E. (2017). Circulating Sclerostin in Bone Sclerosing Disorders. In: Patel, V., Preedy, V. (eds) Biomarkers in Bone Disease. Biomarkers in Disease: Methods, Discoveries and Applications. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7693-7_39
Download citation
DOI: https://doi.org/10.1007/978-94-007-7693-7_39
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7692-0
Online ISBN: 978-94-007-7693-7
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences