
Abstract
In the last decade, osteocyte-produced sclerostin has emerged as a key regulator of bone remodeling. Sclerostin inhibits bone formation and may also stimulate bone resorption. Impaired sclerostin synthesis leads to generalized hyperostosis, particularly of the skull bones, in patients with sclerosteosis and van Buchem disease due to unrestrained bone formation. The synthesis of sclerostin is controlled by systemic and local factors, and aberrant sclerostin synthesis has been reported in several disorders of bone and mineral metabolism. The restricted expression of sclerostin in bone, the excellent quality of bone of patients with sclerosteosis and van Buchem disease and the lack of abnormalities in organs other than the skeleton in these patients have made sclerostin a promising target for new bone-building therapies for osteoporosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteocytes, the most abundant cells in bone, are terminally differentiated osteoblasts buried in the bone matrix which are key regulators of bone remodeling and have also important functions in the regulation of mineral metabolism [1]. Osteocytes act on both osteoclasts and osteoblasts. They synthesize receptor activator of nuclear factor kappa-B ligand (RANKL) [2, 3], which is essential for osteoclast proliferation, differentiation and survival, and are the main source of sclerostin which inhibits bone formation by the osteoblasts [4]. Whereas animal models were pivotal for the discovery of RANKL and its role in bone resorption [5], it is human studies of two rare bone sclerosing dysplasias, sclerosteosis [6, 7] and van Buchem disease (VBD) [8, 9] that have led to the discovery of sclerostin. Both these disorders are caused by deficient synthesis of sclerostin, resulting in unrestrained bone formation and progressive generalized hyperostosis.
We review here current knowledge of the mechanism of action and the regulation of synthesis of sclerostin, and of its role in the pathophysiology of sclerosteosis and VBD.
Sclerostin Synthesis
Osteocytes synthesize sclerostin in the late stages of their differentiation, after maturation and after the start of mineralization of the surrounding bone matrix [10] (Fig. 1). Newly synthesized sclerostin is then transported to the bone surface through the dendritic network of osteocytes, where it inhibits the activity of osteoblasts and stimulates their apoptosis [4, 11]. Recent evidence suggests that sclerostin has also an autocrine function [12].Furthermore, sclerostin upregulates RANKL synthesis thereby stimulating osteoclastogenesis [13]. Although osteocytes are the predominant source of sclerostin, other cell types embedded in mineralized matrices, such as chondrocytes [11] and cementocytes [14], have also been found to produce sclerostin.

Role of sclerostin in bone remodeling. Sclerostin is synthesized by matured osteocytes at the initiation of mineralization of new bone. Sclerostin act on the osteoblasts on the bone surface, by inhibiting their bone forming activity and life span. Sclerostin can also indirectly stimulate bone resorption by up regulating RANKL synthesis by osteocytes in an autocrine manner
Sclerostin is the product of the SOST gene, a relatively small gene comprising two exons, situated on chromosome 17q12-q21. The SOST gene is highly conserved among vertebrates, with the amino acid sequence of murine sclerostin being 88% homologous with the human protein [6]. Moreover, SOST knock-out mice develop a high-bone-mass phenotype similar to that of sclerosteosis patients [15], whereas mice overexpressing SOST become osteopenic [11]. In addition to bone, cartilage and cementum, transcripts of SOST have also been found in kidney, liver, and heart [6, 7], but no sclerostin expression could be detected in any of these tissues in human [16]. In keeping with this finding, patients with sclerostin deficiency have normal renal and liver function and no specific cardiac abnormalities [17–19].
Sclerostin Antagonizes the Canonical Wnt Signaling Pathway
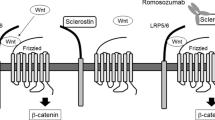
Sclerostin decreases bone formation by antagonizing the canonical Wnt signaling pathway in osteoblasts thereby inhibiting the proliferation, differentiation, and survival of these cells [20–22]. Secreted Wnt ligands bind to a co-receptor complex of the low-density lipoprotein receptor-related protein 5 or 6 (LRP5/6) receptor and a trans-membrane frizzled receptor. Upon binding, intracellular β-catenin is prevented from degradation and accumulates in the cytoplasm. β-catenin is translocated to the nucleus and triggers the transcription of target genes through the interaction with TCF/LEF-1 transcription factors [23] (Fig. 2a). Sclerostin binds to the first propeller domain of the LRP5/6 receptor [21], thereby disabling the formation of the co-receptor complex between LRP5/6 and the frizzled receptor, and inhibiting the Wnt pathway high up in the signaling cascade (Fig. 2b). Whereas the exact mechanism by which sclerostin interacts with the LRP5/6 receptor remains to be established, it is thought that it requires an as yet to be identified co-factor to inhibit the Wnt pathway similar to another Wnt antagonist, DKK1, which needs Kremen to inhibit the pathway [24]. LRP4 was recently proposed to be a mediator of sclerostin’s inhibitory function on bone formation, and mutations in LRP4 were identified in patients with a phenotype closely resembling that of sclerosteosis [25].

Mechanism of inhibition of canonical Wnt signaling by sclerostin. Sclerostin is an antagonist of the canonical Wnt signaling pathway. a The Wnt signaling pathway is initiated by the binding of Wnt ligands to a co-receptor complex of LRP5/6 and the frizzled receptor. Wnt signaling is essential for osteoblast proliferation, differentiation, and survival, and is thus the main stimulatory pathway of bone formation. b Sclerostin antagonizes the Wnt signaling pathway by inhibiting the formation of the LRP5/6-frizzled co-receptor complex, through binding to the LRP5/6 receptor, possibly facilitated by a (unknown) co-factor
Sclerosing Bone Disorders Associated with Sclerostin Deficiency
The significance of the role of sclerostin as a negative regulator of bone formation is highlighted by the characteristic phenotypes of patients with sclerosteosis and VBD, two bone sclerosing dysplasias belonging to the group of craniotubular hyperostosis [26] caused by genetically determined sclerostin deficiency, and characterized by very high bone mass.
Sclerosteosis (OMIM 269500) was described in 1958 by Trushwell as ‘osteopetrosis with syndactyly’ [27] and less than 100 cases have since been reported in the literature. Sclerosteosis is an autosomal recessive disorder caused by mutations in the SOST gene. Six different mutations have been reported so far resulting in either impaired synthesis of sclerostin [6, 7, 28], or synthesis of a non-functioning protein [29]. Although isolated cases of sclerosteosis have been reported in different parts of the world, the majority of patients are members of the Afrikaner community of South Africa, descendants of Dutch immigrants who settled in this country in the 17th century. Although the homozygous state is rare, the carrier rate of the SOST mutation in the Afrikaner population has been estimated to be as high as 1 in 140 individuals [30]. Van Buchem disease (OMIM 239100), first described by professor van Buchem and his colleagues in 1955 as ‘hyperostosis corticalis generalisata familiaris’ [31], is a sclerosing bone dysplasia also inherited as an autosomal recessive trait. In this disorder, the SOST gene is intact, but patients lack a regulatory element essential for the postnatal transcription of SOST in bone [32] due to a 52-kb deletion 35 kb downstream of the SOST gene [8, 9]. About 30 patients with VBD have been so far described, with the vast majority being inhabitants of a small village in north Holland. This village used to be an island off the coast until extensive land reclamation connected it to the mainland in the 1940s. Two siblings with VBD have been reported by German investigators, but the origin of the patients was not mentioned in the paper [33], so possible Dutch ancestry cannot be excluded.
The clinical features of sclerosteosis have been extensively described [17, 34, 35]. The most prominent of these features are due to overgrowth of the bones of the skull. Mandibular overgrowth and elongation of the forehead [17, 34] result in facial distortion, which becomes evident before the onset of puberty [17, 34] (Fig. 3). Excessive bone formation in skull bones eventually give rise to serious complications, the most common being cranial nerve entrapment syndromes due to obliteration of neural foramina (Fig. 4), with the facial nerve being the most frequently affected. This is often the first complication of the disease, occurring generally before the fifth year of life, although facial palsy may also be observed at birth [34, 35]. Unilateral or bilateral facial palsy eventually develops in almost all patients [17, 34, 35, 37]. Hearing loss is the second most frequently encountered complication of sclerosteosis. It usually starts in early childhood as pure conductive deafness, due to fixation of the ossicles in the inner ear, with a sensorineural component often developing later in life as a result of narrowing of the round and oval windows, or of impingement of the acoustic nerve in the internal acoustic canal [17, 34, 35, 37]. Although rare, symptoms associated with entrapment of other cranial nerves, such as loss of vision or sense of smell, have also been reported [34, 37]. However, the most severe, and life-threatening complication of sclerosteosis is increased intracranial pressure [17, 35]. This develops in the majority of patients as a result of a decreased intracranial volume due to considerable thickening of the calvaria and skull base. In the past, this has been a common cause of sudden death of patients with sclerosteosis [17] due to medullary compression. Although the course of sclerosteosis is evidently progressive during childhood and adolescence, the disease appears to stabilize after the third decade [34, 35, 38].

Chronological portraits of a patient with sclerosteosis from the age of 3 years onward. She was born with syndactyly at both hands and developed facial palsy, deafness, facial distortion, and maxillary overgrowth during childhood. By the age of 30, she had developed proptosis and elevated intracranial pressure due to overgrowth of the calvaria. Craniectomy was performed, but she died nevertheless because of elevated intracranial pressure at the age of 54 years (description of this case was previously published by Epstein et al. [36]). Reproduced with permission of Calcified Tissue International

Skull of a sclerosteosis patient (lower panel) compared with that of a normal subject (upper panel). Severely narrowed internal acoustic meatus in the patient (black arrow) compared with that of the normal subject (white arrow). The greatly thickened calvarium of the patient can also be noted (dotted line)
The phenotype of patients with VBD is highly similar to that of sclerosteosis except for two distinctive features of sclerosteosis, which have so far never been reported in patients with VBD, namely syndactyly and tall stature [39] (Table 1). There can be, however, a great variation in the severity of disease manifestations among patients with VBD [39]; van Lierop et al. in preparation]. While the phenotype of severe cases with VBD is highly similar to that of patients with sclerosteosis, other patients with VBD have mild abnormalities and only few lifelong complications. Facial distortion is a consistent feature of VBD [19] but less prominent than in sclerosteosis [39]. In the majority of patients, facial palsy also develops in the first years of life [18, 40], and had been also present at birth in some patients. The presence and severity of hearing loss varies greatly among VBD patients, being profound in some, but absent or mild in others, and can be purely conductive, purely sensorineural, or mixed [18, 41]. Increased intracranial pressure is a rare complication of VBD [42]. As mentioned above, patients with VBD do not have syndactyly, possibly because the deleted SOST enhancer element does not regulate embryonic SOST transcription [32]. It is noteworthy that apart from the characteristic skeletal changes, the general health of patients with sclerosteosis and VBD is otherwise very good [17, 19]. Remarkably the excessive bone formed in sclerosteosis and VBD is of very good quality, with patients sustaining no fractures even after severe trauma [17, 41].
Pathophysiologically, van Buchem had already suggested some 50 years ago that the disease may be caused by excessive bone formation, rather than decreased bone resorption, as is the case in osteopetrosis [19]. Excessive bone formation has indeed been histologically demonstrated in bone biopsies obtained from patients with sclerosteosis [35, 37] and biochemically by measurements of markers of bone turnover in patients with VBD [19, 43] and sclerosteosis [35–37]. Consistent with the identified genetic defect, no sclerostin was detected immunohistochemically in osteocytes of patients with either sclerosteosis [4] or VBD [14]. Particularly interesting is the finding of a normal pattern of bone markers during growth in these patients, increasing during childhood and adolescence, but declining after cessation of the growth spurt to levels around the upper limit of the adult reference range [35, 42] (Fig. 5). No abnormalities have been reported in serum calcium, phosphate and parathyroid hormone (PTH) concentrations [18, 36, 37] in either disease. There were no abnormalities in the pituitary hormonal axis when tested in a small number of patients with sclerosteosis [36].

Sequential measurement of serum alkaline phosphate activity (AP) in U/l, and urinary hydroxyproline to creatinine ratio (OHP/Cr) in μmol/mmol in a patient with van Buchem disease over a 10-year period. Interrupted lines indicate the upper limit of the normal range
Skeletal radiographs show generalized hyperdensity and increased endosteal thickening of the tubular bones, similar in patients with sclerosteosis and VBD [19, 41, 44]. These changes are reflected in measurements of bone mineral density (BMD) which is greatly increased at the hip and the spine with z-scores sometimes exceeding +10. On CT-scan obliteration of neural foramina, which can be decreased to less than 1 mm, is a common finding and the jugular canal was narrowed in a few cases [37, 45]. In both sclerosing dysplasias radiographic changes usually become evident at the end of the first decade and progress up to the third decade of life when they appear to slow, at least in patients with sclerosteosis [34], Van Hoenacker and colleagues showed, however, progressive radiographic changes in the metacarpals of patients with VBD with aging [41].
Treatment of Sclerosteosis and VBD
There is to date no specific therapy available for the management of patients with sclerosteosis and VBD which remains so far largely symptomatic. Decompressive surgery may be needed to free entrapped nerves, and hearing aids can help to improve hearing. In a patient with a severe case of VBD, the insertion of a ventriculo-peritoneal drain led to a reduction in intracranial pressure and improvement of symptoms [42] (Fig. 6). In sclerosteosis, placement of a ventriculoperitoneal or lumboperitoneal shunt do not give satisfactory results, in contrast to the improvement following anterior and/or posterior craniotomy [46]. We recently reported that glucocorticoids which decrease bone formation by reducing the number and function of osteoblasts may be a useful adjunct in the management of patients with severe disease [42].

CT scan of the skull of a patients with VBD, demonstrating severe thickening of the calvaria and presence of ventricular liquor drains (arrows) for the management of increased intracranial pressure
Measurement of Circulating Sclerostin
Assays for the measurement of sclerostin in serum of humans have recently become available. Using an assay developed by MSD (Gaithersburg, Maryland, USA), we could not detect (<1 pg/ml) any sclerostin in the serum of 19 patients with sclerosteosis. In contrast, sclerostin was detectable in serum of 77 healthy men and women tested. Serum samples from 7 patients with sclerosteosis were also measured with another, frequently used commercially available assay (Biomedica, Vienna, Austria). In 4 of the 7 patients, no sclerostin was detected, while the protein was detectable in the 3 other with values overlapping with reported low normal values [47–49]. We have further validated the MSD assay by epitope mapping, and we tested the reactivity of sclerostin fragments of different lengths in both the MSD and Biomedica assay. These fragments consisted of the 3 loops without the N- and C-terminus, the 1st and 2nd loop without the N- and C-terminus, and the 3rd loop alone (Fig. 7a). High concentrations of the fragments (10 ng/ml) were not detected by the MSD assay, while all were detected by the Biomedica assay (Fig. 7b). These findings illustrate the difficulties in the interpretation of data from different assays which do not detect the same sclerostin forms, as also demonstrated by McNulty et al. [52]. These authors showed that sclerostin values measured with the Biomedica assay correlated poorly with those measured with another commercial assay (TECO, Sissach, Switzerland). Until more is known about the secreted, circulating as well as bioactive forms of sclerostin, caution is called for in the clinical interpretation of results of circulating sclerostin.

Measurement of fragments of sclerostin molecule using 2 different commercially available sclerostin assays. a Simplified representation of the sclerostin molecule. Sclerostin is a glycoprotein consisting of 190 residues, which form a 3 loop-like structure, with a cystine knot (dotted line) at the base [50, 51]. The 3rd loop is considered to be the binding site to LRP5, while the 2nd loop was found to be the binding site of an inhibiting monoclonal antibody against sclerostin [51], implying an important function for this loop. The long C- and N-termini are also of importance for sclerostin action, for a sclerostin variant lacking only the C- and N-terminus had reduced activity [50]. b Detection of sclerostin fragments of different length by the MSD and Biomedica assay
Factors Affecting the Synthesis of Sclerostin
In animal studies, mechanical loading of bone has been shown to stimulate the expression of SOST and sclerostin in osteocytes while unloading had the opposite effect [53, 54]. Consistent with these findings, patients immobilized after a stroke, and thus deprived from mechanical load, were shown to have increased serum sclerostin levels, which were negatively correlated with the bone formation marker serum bone-specific alkaline phosphatase [55]. However, a more recent study of immobilized patients due to spinal cord injury reported opposite results, and it was suggested that mechanical unloading may have different, time-dependent effects on sclerostin production [56].
Hormonal factors have also been shown to modulate the synthesis of sclerostin both in animals and humans. In animals, continuous [57] or intermittent [58] administration of PTH downregulates SOST expression and sclerostin synthesis, and serum sclerostin levels are decreased in patients with primary hyperparathyroidism, increasing after successful parathyreoidectomy [59]. In addition, treatment of postmenopausal women with teriparatide for 14 days was associated with significant decreases in serum and bone marrow levels of sclerostin [60]. These results suggest that the anabolic effect of PTH on bone is exerted, at least in part, by a decrease in the production of sclerostin. Recent studies have also indicated that estrogens may also influence sclerostin synthesis. Postmenopausal women have been reported to have higher circulating sclerostin levels than premenopausal women; these were negatively associated with bone formation markers [48], and decreased by estrogen replacement therapy [47]. Estrogen deficiency may, therefore, adversely affect the skeleton not only by increasing bone resorption but also by directly reducing bone formation by increasing sclerostin synthesis, thus further deteriorating the imbalance between bone resorption and bone formation which is the pathophysiological basis of postmenopausal osteoporosis. In contrast, testosterone replacement therapy was reported to increase serum sclerostin levels in hypogonadal men [47].
Alterations in sclerostin synthesis may also be involved in the pathogenesis of bone lesions in inflammatory bone and joint disorders. The pro-inflammatory cytokines tumor necrosis factor (TNF) and TNF-related weak inducer of apoptosis (TWEAK) were reported to upregulate SOST expression in vitro and ex vivo [61]. Patients with ankylosing spondylitis have also been reported to have lower serum sclerostin levels compared with healthy subjects and to patients with rheumatoid arthritis and sclerostin levels were found to be associated with the formation of new syndesmophytes [62]. Sclerostin expression was also greatly reduced in joints of patients with ankylosing spondylitis compared with those of patients with rheumatoid arthritis or osteoarthritis. These results suggest a specific alteration of the function of osteocytes in patients with ankylosing spondylitis. Therapeutic agents known to have deleterious effects on bone by increasing bone loss and fracture risk, such as glucocorticoids and thiazolidinediones, were also reported to upregulate SOST expression in osteocytes in vitro [63, 64].
Sclerostin as a New Therapeutic Target for the Management of Osteoporosis
The restricted expression of sclerostin to the skeleton and its extracellular activity made this protein an attractive candidate for the development of a new bone forming therapy for the management of osteoporosis. This approach was further supported by the gene-dose effect suggested by findings in heterozygous carriers of sclerosteosis who demonstrate decreased serum sclerostin levels associated with increased levels of P1NP [35] and high normal or increased BMD [65] without any clinical symptoms, signs or complications of sclerosteosis. Over the past few years, neutralizing antibodies against sclerostin (Scl-ab) were developed and tested in several animal models. When given to overiectomized rats, an antibody significantly increased the rate of bone formation at all skeletal envelopes, increased bone mass, and improved bone strength [66]. The Scl-ab, importantly, also clearly increased bone formation in the periosteum, a site hardly affected by current treatments of osteoporosis, while it appeared to decrease bone resorption. When given to nonhuman primates, Scl-ab dose-dependently increased bone formation which was associated with increases in bone mass and strength [67]. In a Phase 1 clinical trial, a single subcutaneous or intravenous injection of Scl-ab administered to healthy men and women led to a rapid and dose-dependent increase in serum P1NP, and to an increase in BMD at the spine and hip within 3 months of administration [68]. Consistent with findings from animal studies, the administration of the Scl-ab also decreased serum CTX, a marker of bone resorption. Phase 2 clinical studies of the efficacy and tolerability of SCL-ab are currently underway.
Treatment with Scl-ab has also been shown to prevent inflammation-induced bone loss in mice with chronic colitis and glucocorticoid-induced bone loss in mice on dexamethasone treatment [69, 70]. The effect of Scl-ab on fracture healing was also recently studied in animal models and was shown to increase bone mass and strength at the site of fracture, to improve callus formation and maturation, and to reduce the incidence of non-union [71]. A Phase 2 clinical study of the effect of Scl-ab on fracture healing is in last stages of completion.
Conclusion
The story of sclerostin is a true example of how genetic studies of rare diseases can lead to ground-breaking new insights into molecular pathways, and how the understanding of the pathophysiology of a disorder may lead to the development of new therapies for another. Sclerostin has been shown to play a key role in bone metabolism, and sclerostin inhibition by neutralizing antibodies might prove to be a very successful therapy in restoring bone mass in low bone mass disorders such as osteoporosis. Unraveling the role of sclerostin in the pathophysiology of the disabling and sometimes life-threatening disorders sclerosteosis and VBD has, unfortunately, not yet led to the development of a satisfactory therapy for their management. Effective therapies to control the unrestrained bone formation associated with these disorders are clearly needed.
References
Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–38.
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–41.
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–4.
van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199(6):805–14.
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397(6717):315–23.
Balemans W, Ebeling M, Patel N, Van HE, Olson P, Dioszegi M, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10(5):537–43.
Brunkow ME, Gardner JC, Van NJ, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68(3):577–89.
Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, et al. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12–q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110(2):144–52.
Balemans W, Patel N, Ebeling M, Van HE, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39(2):91–7.
Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19(13):1842–4.
Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22(23):6267–76.
Chang M, Kramer I, Kneissel M. Sclerostin deficiency does not induce bone gain in mice lacking osteocyte beta-catenin. JBMR. 2011;26:S13.
Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One. 2011;6(10):e25900.
van Bezooijen RL, Bronckers AL, Gortzak RA, Hogendoorn PC, Wee-Pals L, Balemans W, et al. Sclerostin in mineralized matrices and van Buchem disease. J Dent Res. 2009;88(6):569–74.
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–9.
Moester MJ, Papapoulos SE, Lowik CW, van Bezooijen RL. Sclerostin: current knowledge and future perspectives. Calcif Tissue Int. 2010;87(2):99–107.
Hamersma H, Gardner J, Beighton P. The natural history of sclerosteosis. Clin Genet. 2003;63(3):192–7.
Van Buchem FS. Hyperostosis corticalis generalisata. Eight new cases. Acta Med Scand. 1971;189(4):257–67.
Van Buchem FS, Hadders HN, Hansen JF, Woldring MG. Hyperostosis corticalis generalisata. Report of seven cases. Am J Med. 1962;33:387–97.
van Bezooijen RL, Svensson JP, Eefting D, Visser A, van der Horst G, Karperien M, et al. Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res. 2007;22(1):19–28.
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280(20):19883–7.
Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280(29):26770–5.
Kubota T, Michigami T, Ozono K. Wnt signaling in bone metabolism. J Bone Miner Metab. 2009;27(3):265–71.
Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, et al. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature. 2002;417(6889):664–7.
Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286(22):19489–500.
de Vernejoul MC. Sclerosing bone disorders. Best Pract Res Clin Rheumatol. 2008;22(1):71–83.
Truswell AS. Osteopetrosis with syndactyly; a morphological variant of Albers-Schonberg’s disease. J Bone Joint Surg Br. 1958;40-B(2):209–18.
Balemans W, Cleiren E, Siebers U, Horst J, Van HW. A generalized skeletal hyperostosis in two siblings caused by a novel mutation in the SOST gene. Bone. 2005;36(6):943–7.
Piters E, Culha C, Moester M, Van BR, Adriaensen D, Mueller T, et al. First missense mutation in the SOST gene causing sclerosteosis by loss of sclerostin function. Hum Mutat. 2010;31(7):E1526–43.
Beighton P, Davidson J, Durr L, Hamersma H. Sclerosteosis—an autosomal recessive disorder. Clin Genet. 1977;11(1):1–7.
Van Buchem FS, Hadders HN, Ubbens R. An uncommon familial systemic disease of the skeleton: hyperostosis corticalis generalisata familiaris. Acta Radiol. 1955;44(2):109–20.
Loots GG, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, et al. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15(7):928–35.
Wengenroth M, Vasvari G, Federspil PA, Mair J, Schneider P, Stippich C. Case 150: Van Buchem disease (hyperostosis corticalis generalisata). Radiology. 2009;253(1):272–6.
Beighton P, Durr L, Hamersma H. The clinical features of sclerosteosis. A review of the manifestations in twenty-five affected individuals. Ann Intern Med. 1976;84(4):393–7.
van Lierop AH, Hamdy NA, Hamersma H, van Bezooijen RL, Power J, Loveridge N, et al. Patients with sclerosteosis and disease carriers: human models of the effect of sclerostin on bone turnover. J Bone Miner Res. 2011;26(12):2804–11.
Epstein S, Hamersma H, Beighton P. Endocrine function in sclerosteosis. S Afr Med J. 1979;55(27):1105–10.
Stein SA, Witkop C, Hill S, Fallon MD, Viernstein L, Gucer G, et al. Sclerosteosis: neurogenetic and pathophysiologic analysis of an American kinship. Neurology. 1983;33(3):267–77.
Barnard AH, Hamersma H, Kretzmar JH, Beighton P. Sclerosteosis in old age. S Afr Med J. 1980;58(10):401–3.
Beighton P, Barnard A, Hamersma H, van der Wouden A. The syndromic status of sclerosteosis and van Buchem disease. Clin Genet. 1984;25(2):175–81.
Van HW, Balemans W, Van HE, Dikkers FG, Obee H, Stokroos RJ, et al. Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12–q21. Am J Hum Genet. 1998;62(2):391–9.
Vanhoenacker FM, Balemans W, Tan GJ, Dikkers FG, De Schepper AM, Mathysen DG, et al. Van Buchem disease: lifetime evolution of radioclinical features. Skeletal Radiol. 2003;32(12):708–18.
van Lierop AH, Hamdy NA, Papapoulos SE. Glucocorticoids are not always deleterious for bone. J Bone Miner Res. 2010;25(12):2796–800.
Wergedal JE, Veskovic K, Hellan M, Nyght C, Balemans W, Libanati C, et al. Patients with Van Buchem disease, an osteosclerotic genetic disease, have elevated bone formation markers, higher bone density, and greater derived polar moment of inertia than normal. J Clin Endocrinol Metab. 2003;88(12):5778–83.
Beighton P, Cremin BJ, Hamersma H. The radiology of sclerosteosis. Br J Radiol. 1976;49(587):934–9.
Hill SC, Stein SA, Dwyer A, Altman J, Dorwart R, Doppman J. Cranial CT findings in sclerosteosis. AJNR Am J Neuroradiol. 1986;7(3):505–11.
du Plessis JJ. Sclerosteosis: neurosurgical experience with 14 cases. J Neurosurg. 1993;78(3):388–92.
Modder UI, Clowes JA, Hoey K, Peterson JM, McCready L, Oursler MJ, et al. Regulation of circulating sclerostin levels by sex steroids in women and in men. J Bone Miner Res. 2011;26(1):27–34.
Modder UI, Hoey KA, Amin S, McCready LK, Achenbach SJ, Riggs BL, et al. Relation of age, gender, and bone mass to circulating sclerostin levels in women and men. J Bone Miner Res. 2011;26(2):373–9.
Kirmani S, Amin S, McCready LK, Atkinson EJ, Melton LJ, III, Muller R, et al. Sclerostin levels during growth in children. Osteoporos Int. (2011) (epub).
Weidauer SE, Schmieder P, Beerbaum M, Schmitz W, Oschkinat H, Mueller TD. NMR structure of the Wnt modulator protein Sclerostin. Biochem Biophys Res Commun. 2009;380(1):160–5.
Veverka V, Henry AJ, Slocombe PM, Ventom A, Mulloy B, Muskett FW, et al. Characterization of the structural features and interactions of sclerostin: molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem. 2009;284(16):10890–900.
McNulty M, Singh RJ, Li X, Bergstralh EJ, Kumar R. Determination of serum and plasma sclerostin concentrations by enzyme-linked immunoassays. J Clin Endocrinol Metab. 2011;96(7):E1159–62.
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283(9):5866–75.
Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24(10):1651–61.
Gaudio A, Pennisi P, Bratengeier C, Torrisi V, Lindner B, Mangiafico RA, et al. Increased sclerostin serum levels associated with bone formation and resorption markers in patients with immobilization-induced bone loss. J Clin Endocrinol Metab. 2010;95(5):2248–53.
Morse LR, Sudhakar S, Danilack V, Tun C, Lazzari A, Gagnon DR, et al. Association between sclerostin and bone density in chronic SCI. J Bone Miner Res. (2011) (epub).
Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, et al. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146(11):4577–83.
Silvestrini G, Ballanti P, Leopizzi M, Sebastiani M, Berni S, Di VM, et al. Effects of intermittent parathyroid hormone (PTH) administration on SOST mRNA and protein in rat bone. J Mol Histol. 2007;38(4):261–9.
van Lierop AH, Witteveen JE, Hamdy NA, Papapoulos SE. Patients with primary hyperparathyroidism have lower circulating sclerostin levels than euparathyroid controls. Eur J Endocrinol. 2010;163(5):833–7.
Drake MT, Srinivasan B, Modder UI, Peterson JM, McCready LK, Riggs BL, et al. Effects of parathyroid hormone treatment on circulating sclerostin levels in postmenopausal women. J Clin Endocrinol Metab. 2010;95(11):5056–62.
Vincent C, Findlay DM, Welldon KJ, Wijenayaka AR, Zheng TS, Haynes DR, et al. Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFalpha induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J Bone Miner Res. 2009;24(8):1434–49.
Appel H, Ruiz-Heiland G, Listing J, Zwerina J, Herrmann M, Mueller R, et al. Altered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondylitis. Arthr Rheum. 2009;60(11):3257–62.
Mabilleau G, Mieczkowska A, Edmonds ME. Thiazolidinediones induce osteocyte apoptosis and increase sclerostin expression. Diabet Med. 2010;27(8):925–32.
Yao W, Cheng Z, Busse C, Pham A, Nakamura MC, Lane NE. Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: a longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthr Rheum. 2008;58(6):1674–86.
Gardner JC, van Bezooijen RL, Mervis B, Hamdy NA, Lowik CW, Hamersma H, et al. Bone mineral density in sclerosteosis; affected individuals and gene carriers. J Clin Endocrinol Metab. 2005;90(12):6392–5.
Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24(4):578–88.
Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25(5):948–59.
Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26(1):19–26.
Marenzana M, Greenslade K, Eddleston A, Okoye R, Marshall D, Moore A, et al. Sclerostin antibody treatment enhances bone strength but does not prevent growth retardation in young mice treated with dexamethasone. Arthr Rheum. 2011;63(8):2385–95.
Eddleston A, Marenzana M, Moore AR, Stephens P, Muzylak M, Marshall D, et al. A short treatment with an antibody to sclerostin can inhibit bone loss in an ongoing model of colitis. J Bone Miner Res. 2009;24(10):1662–71.
Ominsky MS, Li C, Li X, Tan HL, Lee E, Barrero M, et al. Inhibition of sclerostin by monoclonal antibody enhances bone healing and improves bone density and strength of nonfractured bones. J Bone Miner Res. 2011;26(5):1012–21.
Acknowledgments
Studies on sclerostin, sclerosteosis and van Buchem disease by the authors were carried out within the FP7 program TALOS, financied by the European Union (HEALTH-F2-2008-20199, TALOS).
Conflict of interest
All authors have no conflict of interest. All authors met the ICMJE criteria for authorship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
van Lierop, A.H., Hamdy, N.A.T., van Bezooijen, R.L. et al. The Role of Sclerostin in the Pathophysiology of Sclerosing Bone Dysplasias. Clinic Rev Bone Miner Metab 10, 108–116 (2012). https://doi.org/10.1007/s12018-011-9123-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12018-011-9123-5