
Abstract
Ventricular septal defects (VSDs) are recognized as one of the commonest congenital heart diseases (CHD), accounting for up to 40 % of all cardiac malformations, and occur as isolated CHDs as well as together with other cardiac and extracardiac congenital malformations in individual patients and families. The genetic etiology of VSD is complex and extraordinarily heterogenous. Chromosomal abnormalities such as aneuploidy and structural variations as well as rare point mutations in various genes have been reported to be associated with this cardiac defect. This includes both well-defined syndromes with known genetic cause (e.g., DiGeorge syndrome and Holt–Oram syndrome) and so far undefined syndromic forms characterized by unspecific symptoms. Mutations in genes encoding cardiac transcription factors (e.g., NKX2-5 and GATA4) and signaling molecules (e.g., CFC1) have been most frequently found in VSD cases. Moreover, new high-resolution methods such as comparative genomic hybridization enabled the discovery of a high number of different copy number variations, leading to gain or loss of chromosomal regions often containing multiple genes, in patients with VSD. In this chapter, we will describe the broad genetic heterogeneity observed in VSD patients considering recent advances in this field.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Ventricular septal defect
- VSD
- Atrial septal defect
- ASD
- Ventricular septum
- Down syndrome
- Trisomy 21
- Patau syndrome
- Trisomy 13
- Edwards syndrome
- Trisomy 18
- Pallister–Killian syndrome
- 22q11 deletion syndrome
- DiGeorge syndrome
- Velocardiofacial syndrome
- Wolf–Hirschhorn syndrome
- Cri-du-chat syndrome
- Sotos syndrome
- Williams–Beuren syndrome
- Kleefstra syndrome
- Jacobsen syndrome
- Potocki–Lupski syndrome
- Copy number variation
- CNV
- TBX1
- CRKL
- PRKAB2
- FMO5
- CHD1
- BCL9
- ACP6
- GJA5
- GATA4
- SOX7
- NTRK3
- TBX5
- Holt–Oram syndrome
- TBX3
- Ulnar–mammary syndrome
- Townes–Brock syndrome
- SALL1
- Okihiro syndrome
- SALL4
- Char syndrome
- TFAP2B
- Oculofaciocardiodental syndrome
- BCOR
- Ellis–van Creveld syndrome
- EVC1
- EVC2
- Blepharophimosis syndrome
- FOXL2
- GATA6
- Diaphragmatic hernia
- Mowat–Wilson syndrome
- ZFHX1B
- Pelger–Huet anomaly
- LBR
- Alagille syndrome
- NOTCH1
- NOTCH2
- JAG1
- Adams–Oliver syndrome
- Ras/MAPK
- Noonan syndrome
- PTPN11
- Noonan-like syndrome
- SHOC2
- Costello syndrome
- HRAS
- Robinow syndrome
- WNT5A
- DVL1
- ROR2
- Muenke syndrome
- FGFR3
- Cornelia de Lange syndrome
- NIPBL
- Opitz syndrome
- MID1
- CHARGE syndrome
- CHD7
- Sotos syndrome
- NSD1
- Kabuki syndrome
- MLL2
- Frank–ter Haar syndrome
- SH3PXD2B
- Simpson–Golabi–Behmel syndrome
- GPC3
- Marfan syndrome
- Hypophosphatemia
- FBN1
- Peters plus syndrome
- B3GALTL
- Larsen-like syndrome
- NKX2-5
- NKX2-6
- TBX20
- ZIC3
- IRX4
- CITED2
- PITX2
- TGFβ
- CRYPTIC
- CFC1
- GDF3
- Nodal
- NF1
- MYH7
- Noncompaction
- LVNC
- TNNI3
- MKRN2
- HAS2
- GWAS
- TBX15
- MAML3
- TDGF1
1 Introduction
Ventricular septal defects are recognized as one of the commonest congenital heart defects, accounting for up to 40 % of all cardiac malformations [1]. They can be classified according to their location, either within the muscular septum (muscular defects) or at its margins (perimembranous and supracristal defects) (see Chap. 22) [1]. Further, ventricular septal defect (VSD) is not only a common isolated congenital heart disease (CHD) but is often associated with other congenital cardiac defects, either in an individual or within a family. This heart defect also exists as an intrinsic component of several complex malformations, including tetralogy of Fallot (see Chap. 31) and univentricular heart (see Chap. 49) [1].
Epidemiologic studies strongly suggest that genetic factors play an important role in CHD etiology, although environmental exposures are also relevant (see Chap. 16) [2]. In a nationwide population-based study, Oyen et al. reported that CHDs in general show highly variable familial clustering in first degree relatives and indicated a threefold recurrence risk for isolated VSD [3]. In the following chapter, we will focus on the role of genetic factors associated with ventricular septal defects and describe their various genetic causes including chromosomal aberrations, structural variants, and single disease genes and mutations.
2 Isolated VSD
A number of studies have shown that isolated VSDs (without further cardiac and/or extracardiac congenital defects) can be associated with copy number variations as well as single gene mutations in patients and related families. Of note, the affected families often comprise individuals showing isolated VSDs as well as individuals with other CHDs (the latter will be presented in Sect. 23.3).
2.1 Copy Number Variation in Isolated VSD
A copy number variation (CNV) is a structural genomic variant that results in confined copy number changes in a specific chromosomal region that often contains multiple contiguous genes [4]. Commonly, CNVs are defined as any submicroscopic chromosomal changes affecting more than 1000 bases [5]. Frequently, microdeletions and microduplications are identified by high-resolution comparative genomic hybridization (array CGH) as changes of DNA quantity (see Chap. 18) [4].
Studies analyzing CNVs commonly used cohorts of patients with a broad range of different CHDs [6–10]. To date, in five studies analyzing a range of CHDs including isolated VSDs, a total of eight affected loci have been identified with the chromosomal region 22q11.2 and 8p23.1 being most frequently affected. At the 22q11.2 locus, the TBX1 gene (transcription factor T-box 1) known to be implicated in DiGeorge syndrome (see Sect. 23.4.2) was duplicated in one familial case [6], while the protein kinase CRKL gene (V-Crk avian sarcoma virus CT10 oncogene homolog-like) was affected in two independent patients [9, 10]. The locus 8p23.1 contains the candidate genes GATA4 (transcription factor GATA binding protein 4), playing an important role in cardiac development (see Sect. 23.2.2.1), and SOX7 (SRY (sex determining region Y)-box 7) [8, 9]. Rare copy number gains at the locus 11q25 were found in two patients but not associated with known risk genes [6, 7]. A summary of all CNVs found in isolated VSD cases is given in Table 23.1.
2.2 Single Gene Defects in Isolated VSD
In patients with isolated VSD, a number of different mutations have been found in genes encoding for transcription factors, signaling molecules, and proteins of other functions (see Table 23.2).
2.2.1 Transcription Factors
The development of the heart is orchestrated by transcription factor (TF) networks including members of the NK2 homeobox, T-box, and GATA binding families (see Chap. 12) [26]. GATA binding protein 4 (GATA4) is a transcriptional activator found to be affected in sporadic and familial cases of isolated VSD. Three different missense mutations in the GATA4 gene (p.Pro407Val, p.Ser175Cys, and p.Ala411Val) have been reported in sporadic cases [12–14]. In addition, two families carrying GATA4 missense mutations (p.Arg43Trp and p.Gly296Arg) were discovered in a follow-up analysis of index patients that participated in a screen of unrelated individuals (see also Sect. 23.3.2.1) [15, 16]. In the follow-up of affected family members, two and four additional cases of isolated VSD were found in the studies by Wang et al. [15] and Yang et al. [16], respectively.
So far, mutations affecting three members of the T-box family, namely, TBX1, TBX5, and TBX20, have been associated with isolated VSDs. Pan et al. screened 230 CHD cases and observed a heterozygous nonsense mutation (p.Gln277X) in the DNA-binding domain of TBX1 in one patient with double outlet right ventricle who had one affected relative with isolated VSD carrying the same mutation (see also Sect. 23.3.2.1) [19]. A missense mutation (p.Ile152Met) in TBX20 causing impaired DNA binding was found in a family affected by multiple septal defects including isolated VSD, atrial septal defect (ASD), and a large patent foramen ovale in different relatives [21]. In the case of TBX5, a non-coding variant in one of its enhancers was suggested to impact on the development of VSD [20]. The variant was found homozygous in a case of isolated VSD with unaffected heterozygous parents and supported by functional evidence based on transgenic expression studies in mouse and zebrafish [20].
A study focusing on the analysis of PITX2 (paired-like homeodomain 2) in a cohort of 170 unrelated neonates with CHD found two missense mutations (p.Arg91Gln and p.Thr129Ser) in two affected families [18]. Four mutation carriers presented with isolated VSD whereas two other relatives showed transposition of the great arteries (TGA) with VSD (see Sect. 23.3.2.1) [18].
Mutations have also been identified in the TF genes NKX2-6, CITED2, and IRX4 [11, 17, 27]. Screening a CHD cohort including 66 isolated VSD cases, Wang et al. identified a missense mutation (p.Lys152Gln) in the homeodomain of NKX2-6 (NK2 homeobox 6) [27]. Subsequent analysis identified the mutation in two further family members with isolated VSDs [27]. In a cohort of nearly 400 sporadic CHD cases, a nine amino acid deletion (p.Ser170_Gly178del) in CITED2 (Cbp/P300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain, 2), resulting in impaired activity, was detected by us in one patient with isolated perimembranous VSD [11]. For the ventricle-specific TF IRX4 (Iroquois homeobox 4), two missense mutations (p.Asn85Tyr and p.Glu92Gly) were reported in two unrelated patients with isolated VSD by direct sequencing of the gene in a cohort of about 700 CHD patients [17]. The two mutations affected the interaction with retinoid X receptor alpha, a nuclear receptor of the vitamin A signaling pathway important in cardiac morphogenesis.
2.2.2 Signaling Molecules
Various cellular processes in both the embryonic and adult organism are regulated via transforming growth factor beta (TGFβ) signaling pathways. Important developmental steps, such as the establishment of left–right asymmetry, are driven by the NODAL signaling pathway, which is named after the TGFβ superfamily member of the same name (see Chap. 7). Two genes encoding cofactors of the NODAL signaling pathway, TDGF1 (teratocarcinoma-derived growth factor 1 also known as CRIPTO) and CFC1 (Cripto, FRL-1, Cryptic family 1 also known as CRYPTIC), have been analyzed in a cohort of 500 CHD cases [22, 24]. Three missense mutations, p.Arg41Gly in TDGF1 [24] as well as p.Leu219Phe and p.Gly169Val in CFC1 [22], were identified in three patients with isolated VSD (see also Sect. 23.3.2.2). Another member of the NODAL signaling pathway, GDF3 (growth differentiation factor 3), was analyzed by Xiao et al. [23]. Direct sequencing of GDF3 in a cohort of 200 CHD patients led to the identification of a missense mutation (p.Ser212Leu) in a patient with isolated muscular VSD [23].
2.2.3 Other Genes
The gene HAS2 encodes hyaluronan synthase 2, an enzyme that synthesizes hyaluronic acid (a major component of the extracellular matrix) during embryogenesis [25]. Among 100 non-syndromic VSD cases, Zhu et al. detected a HAS2 missense mutation (p.Glu499Val) in one patient. The synthesis of hyaluronic acid was significantly impaired in the mutant enzyme as shown by in vitro assays [25].
3 Non-syndromic VSD
VSDs do not only occur as isolated malformations but most frequently are part of a more complex malformation. In the absence of extracardiac malformations, these VSDs are classified as “non-syndromic” (in contrast to syndromic VSD; see Sect. 23.4). Of note, VSD is an intrinsic component of complex malformations such as tetralogy of Fallot and double outlet right ventricle as well as univentricular heart (see Chaps. 31 and 49, respectively).
3.1 Copy Number Variation in Non-syndromic VSD
Two microduplications and two microdeletions were described in four cases of VSD with additional cardiac malformations (see Table 23.3) [6, 9]. Tomita-Mitchell et al. analyzed a cohort of several hundred CHD cases and identified among others one patient with VSD and pulmonary atresia who carried a duplication at the locus 1q21.1 comprising candidate genes such as CHD1 (chromodomain helicase DNA binding protein 1) and GJA5 (gap junction protein alpha 5, 40 kDa; also known as connexin 40) [9]. Screening a cohort of 105 CHD patients by array CGH, Erdogan et al. found a deletion at the 22q11.2 locus (including TBX1) in a case of VSD and aortic coarctation [6]. Further, they detected a duplication at chromosome 4q32.3 in one patient with VSD and PDA and a large 4 megabase deletion at chromosome 17p11.2 in a patient with VSD and ASD. No evident candidate genes have been identified in either of these regions [6].
3.2 Single Gene Defects in Non-syndromic VSD
Mutations have been reported in transcription factors, signaling molecules, and sarcomeric proteins in patients and families with non-syndromic VSD (see Table 23.4).
3.2.1 Transcription Factors
The evolutionary highly conserved homeobox factor NKX2-5 controls the expression of various cardiac genes during heart development [26]. Frameshift and missense mutations in the DNA-binding domain of NKX2-5 have been described in six families mainly affected by ASD and atrioventricular conduction block, but also showing other CHDs [29–32]. In total, 11 affected mutation carriers from those families showed additional VSDs [29–32]. Moreover, screening of 135 sporadic CHD cases revealed an NKX2-5 missense mutation (p.Pro283Gln) in a patient characterized by VSD, ASD, and PDA [12].
Direct interaction partners of NK2 homeobox 5 include the cardiac TFs T-box 5 and GATA binding protein 4. In a large pedigree with familial ASD, Garg et al. described a GATA4 missense mutation (p.Gly296Ser) located between the nuclear localization sequence and one of two GATA4 zinc fingers, which altered the interaction between GATA4 and TBX5 [28]. Three affected mutation carriers in that family presented with an additional VSD [28]. Furthermore, sequencing of GATA4 in two VSD cohorts revealed two missense mutations (p.Arg43Trp and p.Gly296Arg) in two families [15, 16]. Besides isolated VSDs, affected family members presented VSDs in combination with ASD in three cases and with PDA in one case (see also Sect. 23.2.2.1) [15, 16].
Additional cardiac TF genes mutated in patients with non-syndromic VSD comprise TBX1, ZIC3 (Zic family member 3), and PITX2. A nonsense mutation in TBX1 (p.Gln277X) has been found in a family including relatives with isolated VSD as well as one individual with VSD and PDA (see Sect. 23.2.2.1) [19]. ZIC3, a zinc finger TF known for its association with laterality defects (see Chap. 38), was mutated in one sporadic heterotaxy case showing VSD in combination with ASD, pulmonary stenosis, and TGA [33]. Wei et al. identified two subjects with VSD and TGA from two affected families who carried PITX2 missense mutations (see also Sect. 23.2.2.1) [18].
3.2.2 Signaling Molecules
As described before, various signaling pathways are active during cardiac development such as the NODAL signaling pathway (see Sect. 23.2.2.2). In a cohort of 362 severe CHD cases, the NF1 gene encoding neurofibromin 1, a negative regulator of the RAS signaling pathway, was found to be mutated in one case with VSD accompanied by pulmonary atresia and multiple aorticopulmonary collaterals [34].
3.2.3 Sarcomere Genes
Contraction of the heart involves the shortening of sarcomeres by the ATP-dependent interaction between thin (actin) and thick (myosin) filaments (see Chap. 17). Mutations in genes encoding sarcomeric proteins have been well established as disease-causing for different forms of cardiomyopathy (see Chap. 59). For MYH7 encoding cardiac specific β-myosin heavy chain, two mutations (p.Met362Arg and p.Glu1220del, respectively) in two families characterized by Ebstein’s anomaly (EA), left ventricular noncompaction cardiomyopathy (LVNC), and VSD were reported [35, 36]. Two mutation carriers from each family showed the phenotype with the combination of EA, LVNC, and VSD [35, 36]. Troponin I (encoded by TNNI3) is a cardiac specific thin filament component important for calcium sensing during contraction of the heart muscle. Yang et al. detected a de novo missense mutation in TNNI3 (p.Arg204His) in a patient first diagnosed with perimembranous VSD who then gradually developed restrictive cardiomyopathy [37].
4 Syndromic VSD
In the following section, we describe VSDs observed in patients showing additional congenital malformations in other organs. Those so-called syndromic forms include well-defined syndromes with known genetic cause (e.g., DiGeorge syndrome) as well as so far undefined syndromic forms with unspecific symptoms such as mental retardation and dysmorphic features of unknown genetic etiology.
4.1 Aneuploidy Syndromes
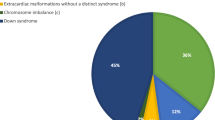
Chromosomal aneuploidy is the presence of an abnormal number of chromosomes in the cell leading to various syndromic genetic disorders. Aneuploidy syndromes can occur with almost any cardiac malformation [38]. CHD occurs in about 45–50 % of patients with Down syndrome (trisomy 21), the most common aneuploidy syndrome [39, 40]. Källen et al. showed in a large epidemiologic study with more than 5000 patients with trisomy 21 that VSD was present in 28 % of individuals with cardiac malformation [41]. In the National Down Syndrome Project cohort, Freeman et al. found a VSD rate of 19 % regarding all registered infants (65 % were membranous and 35 % were muscular VSDs) [40]. A retrospective cohort study including about 4300 Down syndrome patients undergoing CHD surgery was performed by Fudge et al. to examine postoperative outcomes [42]. VSD closure of any type was the second most common procedure (19 %) performed for patients with Down syndrome [42].
Further, CHD occurs in about 35 % of cases of Patau syndrome (trisomy 13) and about 45 % of cases with Edwards syndrome (trisomy 18) as Pont et al. described in a large epidemiologic study of hospitalizations of live-born infants with chromosomal abnormalities [43]. VSD was the most common heart defect in trisomy 13 (18 %) and in trisomy 18 (31 %) [43]. Pallister–Killian syndrome, a sporadic multisystem developmental disorder, is typically caused by the presence of a supernumerary isochromosome composed of the short arms of chromosome 12 resulting in tetrasomy 12p [44]. Tilton et al. evaluated 30 patients with this syndrome and CHD and found a VSD in 10 % of those [44].
4.2 Copy Number Variation in Syndromic VSD
A number of studies have shown the importance of CNVs, mainly microdeletions and microduplications, in syndromic CHD [38, 45]. Those studies reported subjects with well-known syndromes as well as so far undefined syndromic forms.
In Table 23.5, clinically delineated microdeletion and microduplication syndromes are listed in which a specific association to VSD was described. Of note, the most frequent genomic disorder associated with CHD is DiGeorge syndrome (DGS; 22q11 deletion or velocardiofacial syndrome) (see, e.g., Chap. 38). Cardiovascular anomalies are present in about 80 % of neonates with DGS [54]. Ryan et al. evaluated a cohort of 545 DGS patients and could show that VSDs were observed in 14 % of these patients (among other heart defects) [55]. Momma described in his review of several studies of DGS cohorts (ranging from 100 up to 222 patients) quite similar VSD rates [54]. All other syndromes show lower VSD rates. For example, in Williams–Beuren syndrome (caused by a deletion of about 1.5 megabases in chromosome 7q11.23), VSDs, mainly muscular ones (75 %), are present in 4–9 % of all patients [49]. A similar VSD rate (8 %) was described by Jefferies et al. in patients with Potocki–Lupski syndrome whereby most individuals harbor a common 3.7 megabase duplication within chromosome 17q11.2 [53].
Besides well-known syndromes, there are a number of studies describing syndromic patients with VSD who present different unspecific symptoms (see Table 23.6). Using array-based CGH, Syrmou et al. screened a cohort of 55 syndromic CHD patients and detected CNVs in 37 of them [57]. They found five patients with VSD showing either one CNV or a combination of up to three different deletions and duplications (ranging from 0.023 to 6.6 megabases) [57]. Through a genome-wide survey of two independent cohorts of CHD subjects with extracardiac abnormalities (700 subjects in total), Lalani et al. identified 16 CNV regions, present in two or more cases and absent in about 3000 controls [60]. Interestingly, one of the most frequent CNVs they found was a de novo copy number loss of 16q24.3 (affecting ANKRD11 encoding ankyrin repeat domain 11) in five subjects of whom four presented with VSD together with other CHDs (two with perimembranous and one each with muscular and conoventricular VSD) [60]. Screening a cohort of 60 syndromic CHD cases by array CGH, Thienpont et al. identified among others one patient with muscular VSD and various extracardiac manifestations showing a 3.8 megabase duplication at locus 19p13.12-13.11 [61]. Using the same detection method, a similar cohort of 90 patients was analyzed by Breckpot et al. [56]. They found two deletions at locus 1p36.33 (ranging from 3.5 to 5.9 megabases) in two patients with VSD and minor extracardiac malformations, one subject with additional hypertrophic cardiomyopathy and one with microcephaly [56]. Goldmuntz et al. analyzed 58 syndromic CHD cases and reported six different CNVs in six patients characterized by VSD and further congenital abnormalities and dysmorphic features such as cleft palate [59]. They detected copy number gains at loci 5q21.1-21.2 and 18p11.32, whereas losses were detected for the loci 9p23, 10p12.1-11.21, 15q26.1, and 22q11.21 (ranging from 0.4 to 7.1 megabases in length) [59]. Analyzing several hundred CHD trios (non-syndromic and syndromic cases) by SNP arrays and whole exome sequencing, a paternally inherited deletion of 35 kilobases at chromosome 4q34.1 was detected in a patient with VSD, TGA, and extracardiac manifestations [7]. Fakhro et al. genotyped a cohort of more than 250 heterotaxy cases by SNP arrays: they identified a heterozygous 1.5 megabase deletion at locus 3p24.1-23 (affecting TGFBR2 encoding TGFβ receptor II) in a patient characterized by VSD, ASD, partial anomalous pulmonary venous return, and situs inversus (see Chap. 38) [58].
4.3 Single Gene Defects in Syndromic VSD
In addition to structural genomic variations, syndromic CHD can be caused by point mutations influencing the dosage of genes functioning in developmental pathways that are broadly used in organogenesis and therefore affecting many organs. As patients covered in Sect. 23.4.2, the cases presented in the following section are also characterized by a variety of extracardiac manifestations. All described single gene syndromes are summarized in Table 23.7.
4.3.1 Transcription Factors
Mutations in the transcription factor TBX5 (T-box 5) cause Holt–Oram syndrome (HOS), which is characterized by upper-extremity malformations and cardiac defects including VSD [106] (see Chap. 20). Septal defects (VSD and ASD) are the most common cardiac malformations observed in HOS as Al-Qattan et al. showed in their meta-analysis of 16 different studies [106]. In total, 13 different TBX5 mutations (mostly missense ones) were associated with VSDs as part of complex cardiac malformations [106]. Borozdin et al. described VSDs in five out of 23 HOS patients with TBX5 mutations of which four were accompanied by ASD [75]. Another T-box gene, TBX3, is causative to ulnar–mammary syndrome characterized by limb malformations and sporadically associated with VSDs [73, 74].
Cardiac malformations, in particular VSDs, are described in about 14 % of Townes–Brocks syndrome patients, which are further characterized by hand and ear abnormalities and are caused by mutations in SALL1 encoding spalt-like transcription factor, a zinc finger TF [70]. In contrast, mutations of SALL4 (spalt-like transcription factor 4) cause Okihiro syndrome that is characterized by forearm and renal malformations (also known as Duane-radial ray syndrome) and rarely associated with VSD [71, 72].
VSDs are frequently seen in syndromes such as oculofaciocardiodental (OFCD), Ellis–van Creveld, Mowat–Wilson, and congenital diaphragmatic hernia with CHD. OFCD syndrome is an inherited X-linked dominant disorder caused by mutations in BCOR (BCL6 corepressor), a key transcriptional regulator during embryogenesis [62]. Ng et al. were the first to describe three OFCD patients who showed a VSD (one perimembranous, one subpulmonary, and one not further characterized VSD) [62]. In a later study, Hilton et al. presented six OFCD patients with VSD from 21 families showing that VSD was the second most common CHD in these patients [63].
Mutations in EVC1 and EVC2 (two leucine zipper transcription factors) lead to Ellis–van Creveld syndrome, which is mainly associated with atrioventricular septal defect and more sporadically with VSDs [64, 65]. Mowat–Wilson syndrome is caused by mutations in ZEB2 (ZFHX1B) encoding a zinc finger E-box binding factor and is frequently accompanied by cardiac malformations including VSD and ASD [77]. Mutations in GATA6 have been found in familial cases of congenital diaphragmatic hernia accompanied by VSD [68, 69].
Char and blepharophimosis syndrome infrequently present with VSD. Char syndrome is caused by mutations in the gene TFAP2B (transcription factor AP-2 beta) and associates with patent ductus arteriosus (PDA) [76]. However, in one case Char syndrome was accompanied by VSD [76]. Blepharophimosis syndrome is an autosomal dominant disorder that is characterized by a malformation of the eyelids and is caused by loss-of-function mutations in the forkhead TF FOXL2 (forkhead box L2) [66, 67]. VSDs are rare (about 1 %) in patients with this syndrome [66, 67].
4.3.2 Signaling Molecules
Inter- and intracellular communication via signaling molecules coordinates heart development, as has been shown, for example, for the Notch signaling pathway (see Chap. 11). Alagille syndrome, an autosomal dominant multisystem disorder, is associated with CHD in about 25 % of patients [80]. This syndrome is caused by mutations in either JAG1 or NOTCH2, with Jagged 1 being a ligand of the membranous Notch 2 receptor. Emerick et al. showed in a cohort of 73 Alagille syndrome patients a frequent association with cardiac malformation of which 32 % showed VSDs [80]. A further NOTCH signaling driven syndrome is Adams–Oliver syndrome characterized by limb defects and less frequent cardiac malformations including VSDs [82]. A study based on 11 familiar cases identified truncating mutations in NOTCH1 of which three cases showed VSDs [82].
Mutations of genes of the Ras/MAPK (Ras/mitogen-activated protein kinase) signaling pathway represent a frequent cause of Noonan syndrome, which is after Down syndrome the most common syndromic disorder involving cardiac malformations [83]. Noonan syndrome is genetically heterogenous and characterized by short stature, facial dysmorphism, and cardiac defects of different nature such as pulmonary stenosis, hypertrophic cardiomyopathy, ASD, and at a lower rate VSDs. PTPN11 (protein tyrosine phosphatase non-receptor type 11) causing Noonan syndrome 1 was described being mutated in about 50 % of cases [83]. Snayer et al. reported an incidence of 7 % of VSDs in Noonan syndrome 1 patients [84]. Single case reports showed VSDs in the less frequent Noonan syndrome 3 caused by mutations in KRAS (Kirsten rat sarcoma viral oncogene homolog) [87] and Noonan syndrome 4 caused by mutations in SOS1 (son of sevenless homolog 1) [81]. In 25 patients with a phenotype termed Noonan-like syndrome with anagen hair, the missense mutation p.Ser2Gly in SHOC2 (Soc-2 suppressor of clear homolog), a leucine-rich repeat-containing protein, was detected by Cordeddu et al. [86]. While the majority of patients have cardiac malformations, VSDs were found only in two cases [86]. Mutations in another member of the Ras/MAPK pathway, HRAS (Harvey rat sarcoma viral oncogene homolog), cause Costello syndrome, a disorder phenotypically closely related to Noonan syndrome. For Costello syndrome, one case report with VSD has been published [79].
Robinow syndrome is a rare skeletal dysplasia, inherited either in an autosomal dominant or recessive manner. Autosomal dominant Robinow syndrome, either caused by pathogenic variants in WNT5A (wingless-type MMTV integration site family member 5A) or DVL1 (dishevelled segment polarity protein 1), is frequently accompanied by VSDs [88, 89]. In contrast, VSDs are rarely present in autosomal recessive Robinow syndrome, caused by biallelic pathogenic variants in ROR2 (receptor tyrosine kinase-like orphan receptor 2) [85]. A further rare case with VSD has been documented for Muenke syndrome, which is characterized by fusion of cranial bones and is caused by mutations of FGFR3 encoding fibroblast growth factor receptor 3 [78].
4.3.3 Chromatin Regulators
In syndromes related to chromatin regulators, the VSD rates greatly vary ranging from about 40 % in Cornelia de Lange syndrome to about 4 % in Sotos syndrome. Cornelia de Lange syndrome 1 is caused by mutations in NIPBL encoding Nipped-B homolog with putative sister chromatid cohesion function [93, 94]. From a cohort of 24 typical Sotos cases, Cecconi et al. observed one individual with VSD [95]. Sotos syndrome is caused by haploinsufficiency of the NSD1 gene encoding a histone methyltransferase [95]. Microdeletions involving this gene are the major cause of the syndrome in Japanese patients (see Sect. 23.3) [48], whereas intragenic mutations are more frequent in non-Japanese patients [95].
About two-thirds of patients with CHARGE syndrome show cardiac malformations including 12 % with VSDs [90]. Genetically CHARGE syndrome is caused by mutations in CHD7 encoding a chromodomain helicase DNA binding protein. A far less frequent association of VSDs is given in Pelger–Huet anomaly, where one case was described in a cohort of 20 families harboring mutations in LBR encoding for the lamin B receptor, which mediates the interaction of chromatin and lamin B [96].
Finally, mutations in MLL2 encoding a DNA methyltransferase have been found in cases of Kabuki syndrome [91]. Half of these patients present with VSDs often accompanied by an ASD. In a meta-analysis, Yuan examined the cardiac phenotype of 76 published Kabuki cases and showed that about 20 % presented a VSD either isolated or as part of a complex cardiac malformation [92].
4.3.4 Structural and Cell Molecules
The integrity of the cell is maintained by structural proteins while cell adhesion factors are crucial for cell–cell contacts in a respective tissue. There are three different syndromes associated with VSD caused by mutations in structural and cell adhesion proteins. Mutations in SH3PXD2B (SH3 and PX domains 2B, involved in cell adhesion and migration of numerous cell types) cause Frank–ter Haar syndrome, an autosomal recessive skeletal dysplasia characterized among others by cardiovascular malformations [99]. VSD is commonly seen in patients with this syndrome (in 50 % of mutation carriers) as Iqbal et al. showed in their study [99]. Lin et al. reviewed 26 patients with genetically confirmed Simpson–Golabi–Behmel syndrome, a disorder characterized by high birth weight and length, which is caused by mutations in GPC3 encoding glypican 3, a cell surface proteoglycan [98]. A VSD in association with other CHDs was present in five of them (19 %) [98]. One case with Marfan syndrome (with the typical cardiovascular feature of aortic aneurysm) combined with X-linked hypophosphatemia showing VSD and ASD was described by Sheng et al. [97]. They found a de novo missense mutation in FBN1 encoding fibrillin 1, a large extracellular matrix glycoprotein, using whole exome sequencing [97].
4.3.5 Enzymes for Posttranslational Modification
Enzymes involved in posttranslational modifications of proteins such as glycosylation play a role in two syndromes associated with VSDs. Peters plus syndrome (named after the Peters anomaly, an eye-chamber defect) is caused by mutations in B3GALTL encoding a glycosyltransferase involved in addition of glycans to proteins and is associated with VSDs in about one-third of cases [100–102]. The rare Larsen-like syndrome is caused by mutations in B3GAT3 coding for glucuronyltransferase-I and is characterized by joint dislocations, short stature, craniofacial dysmorphism, and heart defects including VSDs [103].
4.3.6 Other Genes
Defects in ciliary structure cause Kartagener syndrome that is characterized by primary ciliary dyskinesia leading to situs inversus, the mirror image arrangement of all internal organs. It is caused by mutations in a number of genes encoding dynein arm components (see more details in Chap. 38). Kennedy et al. examined 21 Kartagener patients and found VSDs among other CHDs in three individuals (14 %) [104]. Zaidi et al. searched for de novo mutations in 362 severe CHD cases [34]. In one heterotaxy patient with abdominal situs inversus and CHD including VSD, they identified a missense mutation (p.Ala251Val) in MKRN2 (encoding makorin ring finger protein 2, a putative E3 ubiquitin ligase) [34]. The gene implicated in the X-linked form of Opitz syndrome, MID1 (midline 1), encodes a RING finger protein and is involved in the formation of multiprotein structures acting as anchor points to microtubules [105]. In a meta-analysis of several studies, Fontanella et al. showed that about 22 % of cases have heart defects with a high incidence of VSDs and ASDs [105].
5 Associations with Common Variations
Genome-wide associations studies (GWAS) look for associations between DNA sequence variants and phenotypes of interest [107]. They do so by studying many hundreds of individuals (affecteds and non-affecteds) and determining their genotype at the positions of hundreds of thousands of single nucleotide polymorphisms (SNPs) followed by statistical analysis and subsequent confirmation experiments in replication cohorts [107].
A small case–control association study genotyped 58 SNPs in the TBX5 region in 192 VSD cases and matched controls of Han Chinese origin and identified a significant association of SNP rs11067075 within intron eight of the TBX5 gene with VSD [108]. Hu et al. performed a large GWAS in 945 cases with septation defects (ASD, VSD, and ASD/VSD) and 1246 controls of Han Chinese ancestry followed by two-stage validation with further 2160 cases and 3866 controls [109]. They identified a highly significant association of two SNPs at chromosome 1p12 (SNP rs2474937 near TBX15) and 4q31.1 (SNP rs1531070 near MAML3 encoding mastermind like three involved in Notch signaling) [109]. A GWAS with 1995 CHD cases (including VSD) and 5159 controls of European Caucasian origin was carried out by Cordell et al. and failed to identify any SNPs with genome-wide significance [110]. A further replication study focusing on a subgroup analysis of about 200 VSD cases also did not find any significant association [110].
Conclusion
VSDs, one of the most common cardiac malformations in the general population, are commonly associated with a high number of well-known genetic syndromes caused by chromosomal aberrations or by smaller deletions and duplications as well as by single point mutations. Moreover, so far undefined syndromic disorders are often accompanied by VSDs.
As shown by a number of studies, mutations in genes encoding cardiac transcription factors (e.g., NKX2-5, GATA4, and TBX5) and signaling molecules (e.g., CFC1) have been most frequently found in VSD cases and families. This holds true for isolated VSD as well as for syndromic and non-syndromic forms. Of note, there is a great overlap of genes associated with other CHDs such as ASD (see Chap. 20), atrioventricular septal defect (see Chap. 26), tetralogy of Fallot, and double outlet right ventricle (see Chap. 32), as well as situs defects (see Chap. 38).
Over the last decade, tremendous effort has been made in the area of genetic technologies such as calling of CNVs and next-generation sequencing of whole exomes and genomes [111]. As a consequence, we better understand the genetic basis of VSDs and other CHDs. However, future studies are still required to unravel the effects of altered gene dosage or loss of function on formation of VSD. In addition, the interpretation of complex patterns of inheritance and phenotypic heterogeneity remains a difficult obstacle in individual VSD cases and families [45]. Nevertheless, these new technologies hold the potential to improve patient care. Especially in VSD cases with a growing number of surviving adults after successful cardiac surgery or intervention, the urgency for a better characterization of the CHD-related morbidities and mortality occurring late after surgery is evident [112]. Recently, Menting et al. reported on the outcomes in about hundred late survivors up to 40 years after VSD closure and demonstrated the ongoing clinical problems in this patient cohort such as arrhythmias and heart failure [113]. In fact, genetics may open new ways for identifying novel risk-stratifying factors in those patients and offer them earlier and better-tailored treatment.
References
Penny DJ, Vick GW 3rd (2011) Ventricular septal defect. Lancet 377:1103–1112
Gelb BD, Chung WK (2014) Complex genetics and the etiology of human congenital heart disease. Cold Spring Harb Perspect Med 4:a013953
Oyen N, Poulsen G, Boyd HA et al (2009) Recurrence of congenital heart defects in families. Circulation 120:295–301
Pollex RL, Hegele RA (2007) Copy number variation in the human genome and its implications for cardiovascular disease. Circulation 115:3130–3138
Redon R, Ishikawa S, Fitch KR et al (2006) Global variation in copy number in the human genome. Nature 444:444–454
Erdogan F, Larsen LA, Zhang L et al (2008) High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J Med Genet 45:704–709
Glessner JT, Bick AG, Ito K et al (2014) Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ Res 115:884–896
Soemedi R, Wilson IJ, Bentham J et al (2012) Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet 91:489–501
Tomita-Mitchell A, Mahnke DK, Struble CA et al (2012) Human gene copy number spectra analysis in congenital heart malformations. Physiol Genomics 44:518–541
Zhao W, Niu G, Shen B et al (2013) High‐resolution analysis of copy number variants in adults with simple‐to‐moderate congenital heart disease. Am J Med Genet A 161:3087–3094
Sperling S, Grimm CH, Dunkel I et al (2005) Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum Mutat 26:575–582
Peng T, Wang L, Zhou SF et al (2010) Mutations of the GATA4 and NKX2.5 genes in Chinese pediatric patients with non-familial congenital heart disease. Genetica 138:1231–1240
Salazar M, Consoli F, Villegas V et al (2011) Search of somatic GATA4 and NKX2.5 gene mutations in sporadic septal heart defects. Eur J Med Genet 54:306–309
Tomita-Mitchell A, Maslen CL, Morris CD et al (2007) GATA4 sequence variants in patients with congenital heart disease. J Med Genet 44:779–783
Wang J, Fang M, Liu XY et al (2011) A novel GATA4 mutation responsible for congenital ventricular septal defects. Int J Mol Med 28:557–564
Yang YQ, Li L, Wang J et al (2012) A novel GATA4 loss-of-function mutation associated with congenital ventricular septal defect. Pediatr Cardiol 33:539–546
Cheng Z, Wang J, Su D et al (2011) Two novel mutations of the IRX4 gene in patients with congenital heart disease. Hum Genet 130:657–662
Wei D, Gong XH, Qiu G et al (2014) Novel PITX2 loss-of-function mutations associated with complex congenital heart disease. Int J Mol Med 33:1201–1208
Pan Y, Wang ZG, Liu XY et al (2015) A novel TBX1 loss-of-function mutation associated with congenital heart disease. Pediatr Cardiol 36:1400–1410
Smemo S, Campos LC, Moskowitz IP et al (2012) Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum Mol Genet 21:3255–3263
Kirk EP, Sunde M, Costa MW et al (2007) Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet 81:280–291
Wang B, Wang J, Liu S et al (2011) CFC1 mutations in Chinese children with congenital heart disease. Int J Cardiol 146:86–88
Xiao J, Kang G, Wang J et al (2014) A novel variation of GDF3 in Chinese Han children with a broad phenotypic spectrum of non-syndromic CHDs. Cardiol Young 5:1–5
Wang B, Yan J, Peng Z et al (2011) Teratocarcinoma-derived growth factor 1 (TDGF1) sequence variants in patients with congenital heart defect. Int J Cardiol 146:225–227
Zhu X, Deng X, Huang G et al (2014) A novel mutation of Hyaluronan synthase 2 gene in Chinese children with ventricular septal defect. PLoS One 9:e87437
Olson EN (2006) Gene regulatory networks in the evolution and development of the heart. Science 313:1922–1927
Wang J, Mao JH, Ding KK et al (2015) A novel NKX2.6 mutation associated with congenital ventricular septal defect. Pediatr Cardiol 36:646–656
Garg V, Kathiriya IS, Barnes R et al (2003) GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424:443–447
Benson DW, Silberbach GM, Kavanaugh-McHugh A et al (1999) Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest 104:1567–1573
Gutierrez‐Roelens I, Sluysmans T, Gewillig M et al (2002) Progressive AV‐block and anomalous venous return among cardiac anomalies associated with two novel missense mutations in the CSX/NKX2‐5 gene. Hum Mutat 20:75–76
Pabst S, Wollnik B, Rohmann E et al (2008) A novel stop mutation truncating critical regions of the cardiac transcription factor NKX2-5 in a large family with autosomal-dominant inherited congenital heart disease. Clin Res Cardiol 97:39–42
Sarkozy A, Conti E, Neri C et al (2005) Spectrum of atrial septal defects associated with mutations of NKX2. 5 and GATA4 transcription factors. J Med Genet 42:e16
Ware SM, Peng J, Zhu L et al (2004) Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet 74:93–105
Zaidi S, Choi M, Wakimoto H et al (2013) De novo mutations in histone-modifying genes in congenital heart disease. Nature 498:220–223
Hirono K, Hata Y, Ibuki K et al (2014) Familial Ebstein’s anomaly, left ventricular noncompaction, and ventricular septal defect associated with an MYH7 mutation. J Thorac Cardiovasc Surg 148:e223–e226
Bettinelli AL, Mulder TJ, Funke BH et al (2013) Familial ebstein anomaly, left ventricular hypertrabeculation, and ventricular septal defect associated with a MYH7 mutation. Am J Med Genet A 161A:3187–3190
Yang SW, Hitz MP, Andelfinger G (2010) Ventricular septal defect and restrictive cardiomyopathy in a paediatric TNNI3 mutation carrier. Cardiol Young 20:574–576
Andersen TA, Troelsen Kde LL, Larsen LA (2014) Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci 71:1327–1352
Vis JC, Duffels MG, Winter MM et al (2009) Down syndrome: a cardiovascular perspective. J Intellect Disabil Res 53:419–425
Freeman SB, Bean LH, Allen EG et al (2008) Ethnicity, sex, and the incidence of congenital heart defects: a report from the National Down Syndrome Project. Genet Med 10:173–180
Källen B, Mastroiacovo P, Robert E (1996) Major congenital malformations in Down syndrome. Am J Med Genet 65:160–166
Fudge JC Jr, Li S, Jaggers J et al (2010) Congenital heart surgery outcomes in Down syndrome: analysis of a national clinical database. Pediatrics 126:315–322
Pont SJ, Robbins JM, Bird TM et al (2006) Congenital malformations among liveborn infants with trisomies 18 and 13. Am J Med Genet A 140:1749–1756
Tilton RK, Wilkens A, Krantz ID et al (2014) Cardiac manifestations of Pallister–Killian syndrome. Am J Med Genet A 164A:1130–1135
Lalani SR, Belmont JW (2014) Genetic basis of congenital cardiovascular malformations. Eur J Med Genet 57:402–413
Battaglia A, Filippi T, Carey JC (2008) Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet C Semin Med Genet 148C:246–251
Hills C, Moller JH, Finkelstein M et al (2006) Cri du chat syndrome and congenital heart disease: a review of previously reported cases and presentation of an additional 21 cases from the Pediatric Cardiac Care Consortium. Pediatrics 117:924–927
Kaneko H, Tsukahara M, Tachibana H et al (1987) Congenital heart defects in Sotos sequence. Am J Med Genet 26:569–576
Collins RT 2nd (2013) Cardiovascular disease in Williams syndrome. Circulation 127:2125–2134
Stewart DR, Kleefstra T (2007) The chromosome 9q subtelomere deletion syndrome. Am J Med Genet C Semin Med Genet 145C:383–392
Mattina T, Perrotta CS, Grossfeld P (2009) Jacobsen syndrome. Orphanet J Rare Dis 4:9
Girirajan S, Vlangos CN, Szomju BB et al (2006) Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet Med 8:417–427
Jefferies JL, Pignatelli RH, Martinez HR et al (2012) Cardiovascular findings in duplication 17p11.2 syndrome. Genet Med 14:90–94
Momma K (2010) Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol 105:1617–1624
Ryan AK, Goodship JA, Wilson DI et al (1997) Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet 34:798–804
Breckpot J, Thienpont B, Peeters H et al (2010) Array comparative genomic hybridization as a diagnostic tool for syndromic heart defects. J Pediatr 156:810–817
Syrmou A, Tzetis M, Fryssira H et al (2013) Array comparative genomic hybridization as a clinical diagnostic tool in syndromic and nonsyndromic congenital heart disease. Pediatr Res 73:772–776
Fakhro KA, Choi M, Ware SM et al (2011) Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc Natl Acad Sci U S A 108:2915–2920
Goldmuntz E, Paluru P, Glessner J et al (2011) Microdeletions and microduplications in patients with congenital heart disease and multiple congenital anomalies. Congenit Heart Dis 6:592–602
Lalani SR, Shaw C, Wang X et al (2013) Rare DNA copy number variants in cardiovascular malformations with extracardiac abnormalities. Eur J Hum Genet 21:173–181
Thienpont B, Mertens L, de Ravel T et al (2007) Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J 28:2778–2784
Ng D, Thakker N, Corcoran CM et al (2004) Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet 36:411–416
Hilton E, Johnston J, Whalen S et al (2009) BCOR analysis in patients with OFCD and Lenz microphthalmia syndromes, mental retardation with ocular anomalies, and cardiac laterality defects. Eur J Hum Genet 17:1325–1335
Digilio MC, Marino B, Ammirati A et al (1999) Cardiac malformations in patients with oral-facial-skeletal syndromes: clinical similarities with heterotaxia. Am J Med Genet 84:350–356
Ruiz-Perez VL, Tompson SW, Blair HJ et al (2003) Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am J Hum Genet 72:728–732
Beysen D, Raes J, Leroy BP et al (2005) Deletions involving long-range conserved nongenic sequences upstream and downstream of FOXL2 as a novel disease-causing mechanism in blepharophimosis syndrome. Am J Hum Genet 77:205–218
Beysen D, De Jaegere S, Amor D et al (2008) Identification of 34 novel and 56 known FOXL2 mutations in patients with Blepharophimosis syndrome. Hum Mutat 29:E205–E219
Yu L, Bennett JT, Wynn J et al (2014) Whole exome sequencing identifies de novo mutations in GATA6 associated with congenital diaphragmatic hernia. J Med Genet 51:197–202
Suzuki S, Nakao A, Sarhat AR et al (2014) A case of pancreatic agenesis and congenital heart defects with a novel GATA6 nonsense mutation: evidence of haploinsufficiency due to nonsense-mediated mRNA decay. Am J Med Genet A 164A:476–479
Surka WS, Kohlhase J, Neunert CE et al (2001) Unique family with Townes-Brocks syndrome, SALL1 mutation, and cardiac defects. Am J Med Genet 102:250–257
Kohlhase J, Heinrich M, Schubert L (2002) Okihiro syndrome is caused by SALL4 mutations. Hum Mol Genet 11:2979–2987
Kohlhase J, Schubert L, Liebers M et al (2003) Mutations at the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. J Med Genet 40:473–478
Linden H, Williams R, King J et al (2009) Ulnar mammary syndrome and TBX3: expanding the phenotype. Am J Med Genet A 149A:2809–2812
Meneghini V, Odent S, Platonova N et al (2006) Novel TBX3 mutation data in families with ulnar-mammary syndrome indicate a genotype-phenotype relationship: mutations that do not disrupt the T-domain are associated with less severe limb defects. Eur J Med Genet 49:151–158
Borozdin W, Bravo Ferrer Acosta AM, Bramshad MJ et al (2006) Expanding the spectrum of TBX5 mutations in Holt-Oram syndrome: detection of two intragenic deletions by quantitative real time PCR, and report of eight novel point mutations. Hum Mutat 27:975–976
Satoda M, Pierpont ME, Diaz GA et al (1999) Char syndrome, an inherited disorder with patent ductus arteriosus, maps to chromosome 6p12-p21. Circulation 99:3036–3042
Mowat DR, Wilson MJ, Goossens M (2003) Mowat-Wilson syndrome. J Med Genet 40:305–310
Escobar LF, Hiett AK, Marnocha A (2009) Significant phenotypic variability of Muenke syndrome in identical twins. Am J Med Genet A 149A:1273–1276
Zampino G, Pantaleoni F, Carta C et al (2007) Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum Mutat 28:265–272
Emerick KM, Rand EB, Goldmuntz E et al (1999) Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology 29:822–829
Roberts AE, Araki T, Swanson KD et al (2007) Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet 39:70–74
Southgate L, Sukalo M, Karountzos ASV et al (2015) Haploinsufficiency of the NOTCH1 receptor as a cause of Adams–Oliver syndrome with variable cardiac anomalies. Circ Cardiovasc Genet 8:572–581
Roberts AE, Allanson JE, Tartaglia M et al (2013) Noonan syndrome. Lancet 381:333–342
Sznajer Y, Keren B, Baumann C et al (2007) The spectrum of cardiac anomalies in Noonan syndrome as a result of mutations in the PTPN11 gene. Pediatrics 119:e1325–e1331
Schwabe GC, Tinschert S, Buschow C et al (2000) Distinct mutations in the receptor tyrosine kinase gene ROR2 cause brachydactyly type B. Am J Hum Genet 67:822–831
Cordeddu V, Di Schiavi E, Pennacchio LA et al (2009) Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet 41:1022–1026
Schubbert S, Zenker M, Rowe SL et al (2006) Germline KRAS mutations cause Noonan syndrome. Nat Genet 38:331–336
Al-Ata J, Paquet M, Teebi AS (1998) Congenital heart disease in Robinow syndrome. Am J Med Genet 77:332–333
Webber SA, Wargowski DS, Chitayat D et al (1990) Congenital heart disease and Robinow syndrome: coincidence or an additional component of the syndrome? Am J Med Genet 37:519–521
Jongmans MC, Admiraal RJ, van der Donk KP et al (2006) CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet 43:306–314
Ng SB, Bigham AW, Buckingham KJ et al (2010) Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42:790–793
Yuan SM (2013) Congenital heart defects in Kabuki syndrome. Cardiol J 20:121–124
Jackson L, Kline AD, Barr MA et al (1993) De Lange syndrome: a clinical review of 310 individuals. Am J Med Genet 47:940–946
Tsukahara M, Okamoto N, Ohashi H et al (1998) Brachmann-de Lange syndrome and congenital heart disease. Am J Med Genet 75:441–442
Cecconi M, Forzano F, Milani D et al (2005) Mutation analysis of the NSD1 gene in a group of 59 patients with congenital overgrowth. Am J Med Genet A 134:247–253
Hoffmann K, Dreger CK, Olins AL et al (2002) Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huët anomaly). Nat Genet 31:410–414
Sheng X, Chen X, Lei B et al (2015) Whole exome sequencing confirms the clinical diagnosis of Marfan syndrome combined with X-linked hypophosphatemia. J Transl Med 13:179
Lin AE, Neri G, Hughes-Benzie R et al (1999) Cardiac anomalies in the Simpson-Golabi-Behmel syndrome. Am J Med Genet 83:378–381
Iqbal Z, Cejudo-Martin P, de Brouwer A et al (2010) Disruption of the podosome adaptor protein TKS4 (SH3PXD2B) causes the skeletal dysplasia, eye, and cardiac abnormalities of Frank-Ter Haar syndrome. Am J Hum Genet 86:254–261
Lesnik Oberstein SA, Kriek M, White SJ et al (2006) Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am J Hum Genet 79:562–566
Hennekam RC, Van Schooneveld MJ, Ardinger HH (1993) The Peters’-Plus syndrome: description of 16 patients and review of the literature. Clin Dysmorphol 2:283–300
Maillette de Buy Wenniger-Prick LJ, Hennekam RC (2002) The Peters’ plus syndrome: a review. Ann Genet 45:97–103
Baasanjav S, Al-Gazali L, Hashiguchi T et al (2011) Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am J Hum Genet 89:15–27
Kennedy MP, Omran H, Leigh MW et al (2007) Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation 115:2814–2821
Fontanella B, Russolillo G, Meroni G (2008) MID1 mutations in patients with X-linked Opitz G/BBB syndrome. Hum Mutat 29:584–594
Al-Qattan MM, Abou Al-Shaar H (2015) Molecular basis of the clinical features of Holt-Oram syndrome resulting from missense and extended protein mutations of the TBX5 gene as well as TBX5 intragenic duplications. Gene 560:129–136
Donelly P (2008) Progress and challenges in genome-wide association studies in humans. Nature 456:728–731
Liu CX, Shen AD, Li XF et al (2009) Association of TBX5 gene polymorphism with ventricular septal defect in the Chinese Han population. Chin Med J (Engl) 122:30–34
Hu Z, Shi Y, Mo X et al (2013) A genome-wide association study identifies two risk loci for congenital heart malformations in Han Chinese populations. Nat Genet 45:818–821
Cordell HJ, Bentham J, Topf A et al (2013) Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat Genet 45:822–824
Andelfinger G (2014) Next-generation sequencing in congenital heart disease: do new brooms sweep clean? J Am Coll Cardiol 64:2507–2509
Hsu DT (2015) Closure is not correction: late outcomes of ventricular septal defect surgery. J Am Coll Cardiol 65:1952–1953
Menting ME, Cuypers JAAE, Opić P et al (2015) The unnatural history of the ventricular septal defect: outcome up to 40 years after surgical closure. J Am Coll Cardiol 65:1941–1951
Acknowledgments
This work was supported by the European Community’s Seventh Framework Programme contract (“CardioNeT”) grant 289600 to S.R.S and the German Research Foundation (Heisenberg professorship and grant 574157 to S.R.S.). This work was also supported by the Berlin Institute of Health (BIH-CRG2-ConDi to S.R.S.). K.B. was supported by Sonnenfeld-Stiftung.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer-Verlag Wien
About this chapter
Cite this chapter
Bellmann, K., Perrot, A., Rickert-Sperling, S. (2016). Human Genetics of Ventricular Septal Defect. In: Rickert-Sperling, S., Kelly, R., Driscoll, D. (eds) Congenital Heart Diseases: The Broken Heart. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1883-2_23
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1883-2_23
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1882-5
Online ISBN: 978-3-7091-1883-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)