
Abstract
IRX4 was the first identified cardiac transcription factor that is restricted to the ventricles at all stages of heart development. Irx4-deficient mice show ventricular dysfunction and develop cardiomyopathy. To study the potential impact of sequence variations in IRX4 on congenital heart disease (CHD) in humans, we examined the coding region of IRX4 in a cohort of 698 Chinese people with congenital heart disease and 250 healthy individuals as the controls. We found two potential disease-causing mutations, p. Asn85Tyr and p. Glu92Gly. A mammalian two-hybrid assay showed that both of the mutations significantly affected the interaction between IRX4 and RXRA. It demonstrated that IRX4 had a potential causative impact on the development of congenital heart disease, particularly ventricular septal defect.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart development is a complex, integrative process that is involved in a well-harmonized series of molecular and morphogenetic events (Srivastava and Olson 2000). Disturbance at any point of this process results in different types of congenital heart defects. Numerous genes that are responsible for both hereditary and sporadic congenital heart diseases have been identified. Most of these genes encode transcription factors that regulate major morphogenetic events in heart development (Bruneau 2002). A variety of heterozygous mutations in several transcription factors such as TBX5, NKX2.5 and GATA4 have been demonstrated to cause diverse kinds of congenital heart abnormalities (Garg et al. 2003; Li et al. 1997; Schott et al. 1998).
Homeodomain-containing transcription factors of the Iroquois (IRO) family are found in nearly all multicellular organisms and play an essential role in the regulation of many aspects of development (Cavodeassi et al. 2001). Irx4 is the earliest marker of the ventricular precursors and is predominantly expressed in the ventricles during all stages of cardiac development in mice, chickens and Xenopus (Bao et al. 1999; Bruneau et al. 2000; Garriock et al. 2001). In the heart of the Nkx2.5 −/− and dHand −/− mice embryos, the expression of Irx4 is markedly decreased, while the expression of eHand is altered in Irx4-deficient mice (Bruneau et al. 2001). IRX4 is a critical mediator of ventricular differentiation during cardiac development. In mice, Irx4 deficiency leads to abnormal ventricular gene expression and causes cardiomyopathy (Bruneau et al. 2001).
We hypothesize that IRX4 contributes to the development of congenital heart disease (CHD). Our study aimed to identify potential pathogenic mutations for IRX4 in 698 Chinese children with CHD and to provide insight into the etiology of CHD.
Materials and methods
Study population
A total of 698 non-syndromic CHD patients (Table 1) and 250 healthy control subjects with no reported cardiac phenotypes were recruited for this study from Lanzhou University, Beijing Children’s Hospital and Children’s Hospital of Fudan University. Informed consent was obtained from patients’ parents or guardians. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Ethics Committee of the National Research Institute for Family Planning.
Clinical assessment of the patients included anthropometric measurement, physical examination for dysmorphism and malformation, and radiological evaluation. The patients also underwent a chest X-ray examination, electrocardiogram and ultrasonic echocardiogram.
Mutational analysis and bioinformatics analysis
Genomic DNA was extracted from peripheral blood leukocytes using standard methods. The human IRX4 gene is located on 5p15.3 and is encoded by five exons. Five exons and splice sites of IRX4 were amplified by polymerase chain reaction (PCR) using three pairs of IRX4 gene-specific primers (Supplementary Table 1). PCR products were sequenced using the appropriate PCR primers and the BigDye Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA) and run on an automated sequencer, ABI 3730XL (Applied Biosystems), to perform mutational analysis.
The novelty of all variants found in sequencing was first determined from the National Centre for Biotechnology Information (NCBI) human SNP database (dbSNP) and the 1000 Genome Project database (http://browser.1000genomes.org/). The amino acid sequences of other species were obtained from NCBI GenBank, and conservation analysis was performed by CLC Main Workbench Software (Aarhus, Denmark).
Site-directed mutagenesis and plasmid construction
Human IRX4 cDNA was obtained from OriGene TrueClone and mutated by PCR-mediated mutagenesis with appropriate primers to generate human IRX4 bearing p. Asn85Tyr or p. Glu92Gly. DNA sequencing confirmed the produced mutation.
The TK promoter pGL3-basic vector with 4× Gal4 DNA-binding sites, the pCMX-Gal4 vector containing Gal4DBD, and the pCMX-VP16 vector with the potent transactivating domain of HSV VP16 were provided by Dr. Ronald M. Evans (The Salk Institute for Biological Studies, USA).
The open reading frames of wild-type and mutant IRX4 were amplified by PCR from cDNA and ligated into the pEGFP-N1 vector (BD Biosciences, Palo Alto, CA, USA) to create N-terminal GFP-IRX4 fusion proteins. To create VP16-IRX4 fusion proteins, the open reading frames of wild-type and mutant IRX4 were amplified by PCR and cloned into a BamHI and NheI digested pCMX-VP16 vector. The open reading frames of RXRA were amplified by PCR from human RXRA cDNA and inserted into an EcoRI and NheI digested pCMX-Gal4 vector to create the Gal4-RXRA fusion protein. All the clones were confirmed by sequencing. The nucleotide sequences of the PCR primers used above are shown in Supplementary Table 2.
Cell culture and transient transfection
Hela and 293T cells were maintained in Iscove’s modified Dulbecco’s medium supplemented with 10% fetal bovine serum, 100 mg/ml penicillin and 100 mg/ml streptomycin in a humidified atmosphere containing 5% CO2 at 37°C. Transfection was carried out using a standard calcium phosphate method or Lipofectamine 2000 (Invitrogen Corporation, Carlsbad, CA, USA).
Subcellular localization
Hela cells were seeded in six-well tissue culture plates 24 h prior to transfection at approximately 60% confluency. GFP-IRX4 expression constructs containing wild-type and mutant IRX4 was transfected using Lipofectamine 2000, according to the manufacturer’s instructions. The empty vector pEGFP-N1 was transfected as a control. At 48 h after transfection, the cells were fixed and permeabilized in 4% paraformaldehyde for 10 min and 0.1% Triton X-100 for 20 min at 4°C, and the DNA was stained with 0.5 μg/ml DAPI for 3 min at room temperature. The cells were analyzed by fluorescence microscopy.
Mammalian two-hybrid assay
The 293T cells were cotransfected with mammalian two-hybrid assay plasmids consisting of pCMX-VP16-IRX4 (wild-type or mutant), pCMX-Gal4-RXRA, TK promoter reporter plasmid and the Renilla luciferase control plasmid pREP7-RLu. Thirty hours post-transfection, cells were washed and lyzed in passive lysis buffer (Promega, Madison, WI, USA) and the transfection efficiency was normalized to paired Renilla luciferase activity by using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The values represent the means of three independent experiments performed in triplicate, and the bars denote the SD. The Student’s t test was used to determine statistical significance of unpaired samples.
Results
Genetic and bioinformatics analysis
Sequence analysis of IRX4 in 698 non-syndromic CHD patients identified two novel non-synonymous variants (Table 2). A 7-year-old male patient showed a substitution of c. A253T p. Asn85Tyr, with the phenotype of ventricular septal defect (VSD), and a substitution of c. A275G p. Glu92Gly was found in a 5-month-old male with the phenotype of VSD.
Both potential pathogenic mutations have not been previously reported in the NCBI dbSNP and are not contained in the 1000 Genome Project database (http://browser.1000genomes.org/).
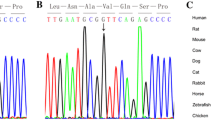
Both variants were located before the homeodomain, and resulted in the two substitutions, which were located at highly conserved regions among many species (humans, mice, rats, cattle and chickens; Fig. 1a, b).

a Sequence alignment of IRX4 proteins among several species. b Position of mutations in the IRX4 protein identified in CHD patients
Functional analysis
Subcellular localization was carried out to determine whether the mutations altered the cellular localization of IRX4. As shown in Fig. 2, wild-type IRX4 was detected only in the nucleus of Hela cells, while GFP (control) was located in both the nucleus and cytoplasm. However, neither of the IRX4 mutations altered their nuclear localization pattern.

Subcellular localization of IRX4. Localization of wild-type and mutant IRX4 GFP-fusion protein in transfected Hela cells was viewed by fluorescent microscope. The empty vector pEGFP-N1 was transfected as a control
A mammalian two-hybrid assay was used to evaluate whether the mutation affected the interaction between IRX4 and RXRA (Fig. 3). Cotransfection of both Gal4-RXRA and wild-type VP16-IRX4 with the TK promoter reporter plasmid resulted in a nearly threefold increase in luciferase activity compared with Gal4-RXRA alone (t test, p < 0.01). Cotransfection of Gal4-RXRA and the mutant VP16-IRX4 showed diminished luciferase activity compared with wild-type control. N85Y was only approximately 53% of wild type (t test, p < 0.01), while E92G was approximately 79% of wild type (t test, p < 0.05). These findings indicated that the mutated proteins showed weakened protein-protein interactions.

Effect of N85Y and E92G mutations on IRX4-RXRA interactions. We cotransfected 293T cells with pCMX-Gal4-RXRA, TK promoter reporter plasmid and the Renilla luciferase internal control plasmid, as well as empty vector pCMX-VP16, VP16-IRX4 wild-type and the N85Y mutant or E92G mutant. Firefly and Renilla luciferase activities were measured 30 h after transfection. Relative luciferase activity was determined by normalizing the firefly luciferase activity with Renilla luciferase activity. Data are shown as mean values from three independent experiments, with bars indicating SD. The significance of differences was calculated using the independent-samples t test. *p < 0.05, **p < 0.01 versus empty vector pCMX-Vp16; # p < 0.05, ## p < 0.01 versus wild type
Discussion
The present study screened the coding region and splice sites of the IRX4 gene in 698 Chinese CHD patients. Two potential pathogenic non-synonymous variants (p. Asn85Tyr and p. Glu92Gly) were identified. These two regional highly conserved substitutions (conserved among humans, cattle, mice, rats and chickens) were not identified in 500 healthy chromosomes or the variant databases. Therefore, we speculate that these two mutations were possibly causative. To the best of our knowledge, these are the first IRX4 mutations identified in CHD patients.
Both mutations lie in the N-terminal (encoding amino acids 1-135) of IRX4. Wang et al. demonstrated that this region is critical for the interaction between Irx4 and Rxra to regulate ventricular gene expression (Wang et al. 2001). Therefore, this suggests that the mutations may affect protein-protein interactions.
Mammalian two-hybrid analysis allows the semi-quantitative assessment of protein-protein interactions occurring within living cells (Mogensen et al. 2004). By cotransfecting wild-type or mutant IRX4 with RXRA into 293T cells, we found that the wild-type and mutant IRX4 could bind RXRA to activate reporter gene expression in vivo. Both mutated proteins showed significantly reduced reporter gene activation ability compared with wild type. Also, both the mutations did not affect the subcellular localization of IRX4, as their nuclear localization pattern was normal. Therefore, this demonstrated that the mutants reduce protein-protein interactions.
Notably, both of these mutations were found in VSD patients, which suggested that protein alterations of IRX4 possibly have an impact on the development of interventricular septum. As expected, the results of the functional study strongly support the speculation that the mutations are causative and IRX4 mutations might affect the formation of the ventricular septum.
The DNA-binding sites of IRX4 have not yet been defined. IRX4 may act via protein-protein interactions, similar to other classes of homeodomain proteins. A physical interaction between Irx4 and Rxra has been confirmed (Wang et al. 2001). Rxra plays a vital role in vitamin A signaling pathway, which is critical for various steps of cardiac morphogenesis (Sucov et al. 1994; Wilson et al. 1953). Rxra mutated mice display profound cardiac defects, especially hypoplastic development of the ventricular chambers with ventricular septal defect (Dyson et al. 1995; Sucov et al. 1994). It is possible that IRX4 is involved in retinoid-mediated cardiac gene regulation via RXRA, and that the mutations could disturb vitamin A signaling pathway to affect ventricular septation.
The expression pattern of Irx4 is very similar to Hey2, which is expressed in the ventricular myocardium and is involved in regulating gene expression in the ventricular chambers (Leimeister et al. 1999; Xin et al. 2007). Mice lacking Hey2 develop cardiomyopathy or ventricular septal defect depending on different genetic backgrounds (Gessler et al. 2002; Sakata et al. 2002). It is speculated that Irx4 works in synergy with Hey2 to suppress atrial gene expression in ventricular myocardium (Xin et al. 2007). The mutations of IRX4 may also affect its interaction with HEY2 and disturb the expression of genes important for the formation of ventricular septum.
Although the VSD phenotype was not seen in Irx4-deficient mice as observed in mutation carrying patients, we speculate that this could be due to species differences in the function of IRX4, or some other unidentified factors might interact with IRX4 and modify its phenotype. Alternatively, it is also possible that VSDs were present earlier in life, but spontaneously closed at a later time in Irx4 mutated mice.
In conclusion, our study suggests that Irx4 may be involved in the etiology of ventricular septal defect in the Chinese population. To the best of our knowledge, it is the first study to attempt identifying potential pathogenic mutations in the IRX4 gene in patients with VSD.
References
Bao ZZ, Bruneau BG, Seidman JG, Seidman CE, Cepko CL (1999) Regulation of chamber-specific gene expression in the developing heart by Irx4. Science 283(5405):1161–1164
Bruneau BG (2002) Transcriptional regulation of vertebrate cardiac morphogenesis. Circ Res 90(5):509–519
Bruneau BG, Bao ZZ, Tanaka M, Schott JJ, Izumo S, Cepko CL, Seidman JG, Seidman CE (2000) Cardiac expression of the ventricle-specific homeobox gene Irx4 is modulated by Nkx2–5 and dHand. Dev Biol 217(2):266–277
Bruneau BG, Bao ZZ, Fatkin D, Xavier-Neto J, Georgakopoulos D, Maguire CT, Berul CI, Kass DA, Kuroski-de Bold ML, de Bold AJ et al. (2001) Cardiomyopathy in Irx4-deficient mice is preceded by abnormal ventricular gene expression. Mol Cell Biol 21(5):1730–1736
Cavodeassi F, Modolell J, Gomez-Skarmeta JL (2001) The Iroquois family of genes: from body building to neural patterning. Development 128(15):2847–2855
Dyson E, Sucov HM, Kubalak SW, Schmid-Schonbein GW, DeLano FA, Evans RM, Ross J Jr, Chien KR (1995) Atrial-like phenotype is associated with embryonic ventricular failure in retinoid X receptor alpha−/− mice. Proc Natl Acad Sci USA 92(16):7386–7390
Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K et al. (2003) GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424(6947):443–447
Garriock RJ, Vokes SA, Small EM, Larson R, Krieg PA (2001) Developmental expression of the Xenopus Iroquois-family homeobox genes, Irx4 and Irx5. Dev Genes Evol 211(5):257–260
Gessler M, Knobeloch KP, Helisch A, Amann K, Schumacher N, Rohde E, Fischer A, Leimeister C (2002) Mouse gridlock: no aortic coarctation or deficiency, but fatal cardiac defects in Hey2−/− mice. Curr Biol 12(18):1601–1604
Leimeister C, Externbrink A, Klamt B, Gessler M (1999) Hey genes: a novel subfamily of hairy- and Enhancer of split related genes specifically expressed during mouse embryogenesis. Mech Dev 85(1–2):173–177
Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T et al. (1997) Holt–Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet 15(1):21–29
Mogensen J, Murphy RT, Shaw T, Bahl A, Redwood C, Watkins H, Burke M, Elliott PM, McKenna WJ (2004) Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol 44(10):2033–2040
Sakata Y, Kamei CN, Nakagami H, Bronson R, Liao JK, Chin MT (2002) Ventricular septal defect and cardiomyopathy in mice lacking the transcription factor CHF1/Hey2. Proc Natl Acad Sci USA 99(25):16197–16202
Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG (1998) Congenital heart disease caused by mutations in the transcription factor NKX2–5. Science 281(5373):108–111
Srivastava D, Olson EN (2000) A genetic blueprint for cardiac development. Nature 407(6801):221–226
Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR, Evans RM (1994) RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev 8(9):1007–1018
Wang GF, Nikovits W Jr, Bao ZZ, Stockdale FE (2001) Irx4 forms an inhibitory complex with the vitamin D and retinoic X receptors to regulate cardiac chamber-specific slow MyHC3 expression. J Biol Chem 276(31):28835–28841
Wilson JG, Roth CB, Warkany J (1953) An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat 92(2):189–217
Xin M, Small EM, van Rooij E, Qi X, Richardson JA, Srivastava D, Nakagawa O, Olson EN (2007) Essential roles of the bHLH transcription factor Hrt2 in repression of atrial gene expression and maintenance of postnatal cardiac function. Proc Natl Acad Sci USA 104(19):7975–7980
Acknowledgments
This work was supported by the National Basic Research Program of China (2010CB529505), the National Science and Technology Pillar Program of China (No.2008BAH24B05), the National Basic Research Program of China (2010CB529504) and the National Infrastructure Program of Chinese Genetic Resources (2006DKA21300).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Z. Cheng and J. Wang contributed equally to the work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Cheng, Z., Wang, J., Su, D. et al. Two novel mutations of the IRX4 gene in patients with congenital heart disease. Hum Genet 130, 657–662 (2011). https://doi.org/10.1007/s00439-011-0996-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-0996-7