
Abstract
Germ cell tumors (GCT) of the ovary and other locations in the female genital tract are analyzed from a clinicopathological viewpoint and reconsidered in the light of the latest genetic knowledge of their origin and information from new pluripotency and developmental stage markers.
Ovarian malignant primitive GCT have a well-defined phenotype. The expression of characteristic pluripotency markers in dysgerminoma is useful in the diagnosis of its various histologic variants, including artifacts. Dysgerminoma has a pure germ cell origin, in contrast to yolk sac tumors (YST), which may develop at any age from both germ and somatic cells at various stages of pluripotentiality. The histologically heterogeneous YST are reconsidered as primitive endodermal tumors applying a diagnostic immunohistochemical panel able to distinguish between extraembryonal and somatic variants.
Type II GCT of postpubertal testicular type such as embryonal carcinoma, mixed GCT, and choriocarcinomas are rare in females; they often occur associated with disorders of sex development.
Ovarian mature teratomas are the paradigm of a parthenogenetic origin (type IV), and their morphology and complications are exhaustively reviewed. Prognostically relevant histologic grading of immature are reanalyzed, taking into account the presence of immature endodermal structures and expression of pluripotency markers SALL4 and SOX2.
An emerging category of highly malignant GCT originating from somatic Müllerian tumors in older patients (endometrioid carcinomas and clear cell tumor) is considered. They probably arise from induced pluripotential tumor stem cells (type VI GCT) and often represent a diagnostic challenge. The comparative immunophenotypic expression of various pluripotency and tissue-specific proteins is a useful tool for their identification.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Germ cells
- Germ cell tumors
- Ovary
- Uterus
- Pluripotency markers
- Parthenogenesis
- Immunohistochemistry
- iPSC
- Teratoma
- Dysgerminoma
- Yolk sac tumor
- Choriocarcinoma
- Embryonal carcinoma
- Embryoids
6.1 Introduction and General Considerations
In the female genital tract, germ cell tumors (GCT) are found mainly in the ovary, being merely anecdotal in other locations. They occur twice as often in the ovary than in the testis in both children and adults [1]. Most are benign teratomas [2]; malignant GCT are infrequent. Since epidemiologic data from tumor registries deal almost exclusively with malignant tumors, it is difficult to establish a general incidence for benign forms despite their being considered the most frequent cause of ovarian mass in adolescents and young adults [3]. GCT are rarely congenital, and their incidence begins to rise during late infancy and early adulthood [4, 5].
As analyzed in Chap. 3, the origin of female genital tract GCT is far from univocal. Most ovarian teratomas originate from parthenogenetically activated oocytes in various stages of meiosis [6, 7], corresponding to type IV pluripotential tumors in Oosterhuis and Looijenga’s classification, discussed in Chap. 3 and followed here. This is reflected in the ability to differentiate into practically any embryonal or adult tissue and an outstanding capacity of organization, reproducing complex organs such as cerebellum and conforming tissue relationships that may even have a certain axial symmetry, caricaturizing fetal appearances [8].
The parthenogenetic origin of ovarian type IV GCT is, however, different from most testicular and extragonadal GCT, which arise from totipotent-state, probably type II, primordial germ cells. However, the exception to this would be GCT in female fetuses, neonates, and infants, where ovarian GCT such as yolk sac tumors and immature teratoma probably originate from pluripotent primed embryonal stem cells derived from germ cells prior to maternal imprinting, corresponding to type I tumors in the Oosterhuis and Looijenga’s classification. Nevertheless, distinction between parthenogenetically originated (type IV) and infantile type I GCT is by no means clear-cut.
Finally, in elderly women, GCT are found in association with somatic tumors of Müllerian nature and possibly arise from malignant stem cells analogous to induced pluripotential stem cells (iPSC) corresponding to type VI GCT.
As a consequence of their different origin, type IV ovarian teratomas and type I tumors rarely exhibit genetic markers such as 12p isochromosome and chromosome 12 overrepresentation [9]. For this reason, although equating ovarian and testicular germ cell tumors is not biologically correct, their morphology and most of their diagnostic immunohistochemical features are similar, the exception being the expression of some proteins in male but not in female teratomas [10]. Interestingly, some tumor types, such as strumal carcinoid, are unique to the ovary.
In the ovary, GCT are only bilateral in 5 % [11] and lack a familial distribution [12] with some rare exceptions [13, 14]. When they do occur in multiple members of the same family or in families with testicular GCT, a gene conferring susceptibility to GCT may exist [15].
In further contrast with testicular tumors, usual parthenogenetic ovarian GCT do not have a precursor or in situ lesion. Indeed, no parthenotes, similar to those found in the oocytes from the LT/Sv mice strain which spontaneously develop teratoma [16], are found in the human ovary. However, gonadoblastoma, a characteristic tumor of disorders of sex development (DSD), behaves as the equivalent of the germ cell neoplasia in situ (GCNIS), the precursor lesion of most testicular tumors that is almost invariably associated with a background of gonadal Y-chromosome mosaicism; consequently, it should not be considered as true ovarian tissue. Indeed, tumors originated from gonadoblastoma are similar to type II testicular GCT in their histologic types and their relative proportions.
The WHO classification (Table 6.1) [17] of ovarian GCT has remained unchanged for more than a quarter of a century (see Chap. 1) and in many ways overlaps with that of older classifications of testicular tumors. It is a descriptive, histopathologic classification that does not take into account the different cellular developmental origins of GCT during different ages of life that are relevant to their behavior. Indeed, it does not include distinct bio- and clinicopathologic categories such as GCT associated with disorders of sex differentiation (DSD) or those originating from somatic neoplasms. It lists together both common tumors and extremely unusual lesions in the ovary (i.e., embryonal carcinoma, sebaceous tumors). Moreover, the term monodermal teratoma comprises a mixed bag of ectopic tissue lesions of debatable histogenesis that may have either germ- or stem cell origins. However, the introduction of new stem cell pluripotency antibodies and genetic markers provides a new understanding of the identification and taxonomy of malignant GCT types [18] that may help their reclassification according to other criteria than mere histologic ones (see Chap. 3).
6.2 Primitive Germ Cell Tumors of the Ovary
Primitive GCT are defined as malignant neoplasms that caricaturize early embryonal developmental stages. Primitive ovarian GCT represent only 2 to 5 % of all ovarian cancers [20, 21], but they constitute the most common malignant ovarian tumor in women under the age of 20 [5]. Recent epidemiologic data [22, 23] have demonstrated a trimodal age distribution of malignant GCT in women: a high incidence peak at the end of the second decade of life and two of lower amplitude (in the immediate postmenopause and at age 70). These peaks are possibly related to different pathogeneses of ovarian GCT in each age group: the first one is represented by parthenogenetic tumors, the second includes malignant degenerations of benign teratomas, and the third is comprised of GCT patterns originated from somatic tumors.
Malignant GCT represent a model of progressive neoplastic differentiation from pluripotent stem cells where every stage of development and tissue is caricaturized by a well-defined tumor type [24]. Thus, dysgerminomas reproduce the morphology of primordial germ cells, embryonal carcinoma mimics inner mass embryonic stem cells, polyembryoma duplicates the trilaminar embryo, yolk sac tumors the primitive endoderm and its differentiations, choriocarcinoma the early placenta, and both immature and mature teratoma, the embryonal somatic organs. However, malignancy is not excluded by full differentiation, as mature tissues may ultimately undergo malignant change, giving rise to a malignant neoplasm within a mature teratoma (see Chap. 12) [25].
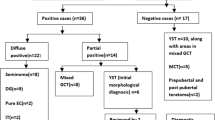
There are over 23 instances of malignant testicular GCT for every malignant ovarian one [22]. As a reflection of their different pathogeneses, the proportion of histologic types of germ cell tumors varies widely between testis and ovary. Compared with the testis, the ovary shows a substantially lower proportion of seminoma/dysgerminomas (2:50) and mixed GCT (33:<1) and a negligible presence of embryonal carcinoma compared to the testis, while yolk sac tumors (YST) and choriocarcinomas share similar proportions in each organ (1:1 and 0.3: <0.1, respectively) [26]. An unknown, but significant, proportion of malignant tumors in phenotypic females originates in underdiagnosed DSD cases with Y-chromosome mosaicism, which further narrows the relative number of malignant GCT originating from ovarian tissue.
In the ovary, the proportion of the different histologic types of GCT is variable. For some [27, 28], dysgerminoma is the more common malignant GCT. However, immature teratoma seems to be the more frequent GCT in various statistics [20, 29–31]. Since parthenogenetic benign teratomas are the most frequent ovarian GCT, the same would be true for immature teratomas, some of which may share a similar origin.
Malignant ovarian GCT have highly aggressive behavior. Before the introduction of modern combination chemotherapy in the 1970s and 1980s, the prognosis for patients with malignant ovarian germ cell tumors was extremely poor [32]. In non-dysgerminomas, the survival rate for patients with apparent stage I disease was only 5–20 %, and the survival for those with advanced-stage disease was insignificant [33]. Nowadays, GCT are fortunately a paradigm of tumor response, with a 5-year cause-specific survival of 97 % for dysgerminoma and 92 % for non-dysgerminoma [34]. Adverse prognostic factors are represented by a stage higher than the International Federation of Gynecology and Obstetrics (FIGO) IA, the presence of metastases, and an age at diagnosis greater than 40–45 years [34, 35]. Since malignant GCT often involve young females, conservative surgery with preservation of fertility is paramount [32]. A high proportion of women treated with this type of surgery and chemotherapy retain menstrual function and fertility [36]; this may be related to the extent of chemotherapy administered [37]. Monitoring anti-Müllerian hormone levels as a biomarker of gonadal function and ovarian oocyte reserve may be helpful [37].
Although malignant ovarian GCT incidence seems to be stable or decreasing in some statistics [20], others show an increasing trend [22] (see Chap. 2).
6.2.1 Dysgerminoma
This tumor of the ovary and dysgenetic gonads is the histologic equivalent of the more common testicular seminoma and extragonadal germinomas, with which it shares a high rate of 12p abnormalities [38] and KIT mutations and amplifications [39]. Due to its frequent association to DSD, it was named disgerminoma (sic) in Robert Meyer’s 1930 seminal paper on ovarian tumors and sexuality [40]. Its frequency in phenotypic females is less than that of teratomas and yolk sac tumors combined, comprising 5–10 % of malignant ovarian tumors occurring in the first two decades of life and being less frequent than immature teratoma [20, 29, 34]. Its well-known precursor lesion is gonadoblastoma, although in an unknown number of cases, it may arise de novo.
6.2.1.1 Clinical Features and Treatment
Practically, dysgerminoma involves only adolescents and young adults, being rare in children and in older patients. A coexisting subclinical DSD, such as androgen insensitivity syndrome, should be unmasked by sex-determining region Y (SRY) analysis [41].
Although 30–50 % of dysgerminoma patients are fertile [42, 43], few series studied karyotypic features [44] of malignant ovarian GCT patients, thus not differentiating the relative numbers of true 46, XX females from those with a Y-containing karyotype. Dysgerminomas originated from gonadoblastoma have a different set of chromosomal abnormalities than de novo dysgerminomas in true 46, XX females. However, it must be borne in mind that rarely gonadoblastomas may occur in fertile females [45, 46] and in true hermaphrodites [47], where a mosaicism cannot be excluded. Occasionally, DSD-associated dysgerminomas can be familial [48].
Since dysgerminoma involves young patients, abdominal mass may be associated with pregnancy and can cause fetal death and dystocia. Rare cases of pseudopregnancy have been reported [49]. Dysgerminoma biomarkers include human chorionic gonadotropin (hCG), which may be present in dysgerminomas with focal syncytiotrophoblastic differentiation [50]. AFP is usually secreted when dysgerminoma is part of a mixed GCT with primitive endodermal components of YST. Among paraneoplastic manifestations, there are many reports of hypercalcemia [51] that should be differentiated, especially in the older literature, from ovarian small cell carcinoma with hypercalcemia, which is a histological look-alike. Rarely, it may be associated with systemic lupus erythematosus [52] and cholestasis [53].
In over half the cases, tumors are found at FIGO stage I [54, 55]. Nevertheless, there is a high rate of undetected deposits in para-aortic lymph nodes that may recur [54], implying that clinical stage I tumors are often understaged [35]. Combination platin-based chemotherapy cure rates approach 98 % in early stages and 75 % in advanced stages [31]. No adjuvant chemotherapy is recommended for true FIGO IA stage. Consequently, surgery should be fertility sparing with complete staging procedures, including omentectomy, peritoneal washings, and peritoneal biopsies. Only when karyotypic features or abdominal findings of gonadal dysgenesis are present, bilateral adnexectomy is indicated in order to avoid possible development of a metachronic GCT in the contralateral gonad.
6.2.1.2 Pathology
6.2.1.2.1 Macroscopy
Although it is the GCT with the highest rate of bilaterality, this only occurs in 10 % of cases [28]. Tumors are usually solid and well encapsulated; on cut section they are homogeneous or lobulated (Fig. 6.1) but rarely cystic. They are usually white or tan in color, except in congestive, torsioned tumors. Heterogeneous and gritty areas should be sampled in order to demonstrate other histologic components. Punctate red, hemorrhagic foci are often associated with syncytiotrophoblastic differentiation. Marked necrosis should prompt an extensive sampling of the specimen.

Dysgerminoma in Swyer syndrome. Tumor is attached to a fallopian tube and is homogeneous and rubbery. A contralateral gonadal streak was present
6.2.1.2.2 Microscopy
An extensive microscopic description of its testicular counterpart, seminoma, is found in Chap. 7. Here we will review histopathologic features present in dysgerminomas in females and dysgenetic gonads that are relevant to differential diagnosis with other ovarian tumors.
At low power, it is necessary to search for coarse or morular microcalcifications that may remain from a preexisting gonadoblastoma, overgrown by the malignant germ cells of dysgerminoma. Dysgerminoma cells are arranged in a variety of patterns: diffuse or solid, lobular, trabecular, etc. They have uniform nuclei with prominent nucleoli and well-defined membranes. A variable degree of atypia is present but lacks any prognostic significance.
Cells of dysgerminoma have a labile structure due to a poorly developed cytoskeleton, and consequently dysgerminoma histology is often subject to rapid autolytic changes being very sensitive to frozen section artifacts. Retraction of cytoplasmic gels during fixation is often reflected in the formation of clear cells. Invariably, the intervening septa show lymphocytes and, less often, histiocytic, epithelioid non-caseating granulomas, the consequence of an enhanced T-lymphocytic response induced by neoplastic germ cells [56]. This inflammatory response is an invaluable diagnostic aid in poorly fixed cases where identification of the large, clear cells is difficult. Only in rare instances may extensive chronic inflammatory infiltrates complicate diagnosis by effacing the dysgerminoma architecture.
Distortion due to autolysis may produce large empty tubular or pseudofollicular colloid-filled spaces that may mimic other ovarian tumors in the young, such as small cell carcinoma, hypercalcemic type (SCCH), and even struma ovarii (Figs. 6.2a, b). Differential diagnoses also include ovarian lymphoma and clear cell carcinoma (CCC). Dysgerminoma shares many similarities with SCCH since it involves the same age group and may also present with serum hypercalcemia. Especially in poorly fixed cases that imitate pseudofollicular spaces, the diagnosis is made by the absence of lymphocytic infiltrate in SCCH and the characteristic immunophenotype of dysgerminoma. CCCs involve an older age group but can show solid areas with cytoplasmic clearing and even plasma cell stromal infiltrates [57]. Usually, a thorough sampling will reveal the tubulocystic areas of CCCs. Lymphoma in young patients is a more remote differential and when an adequate immunohistochemical panel is used, diagnosis should not present any problems, although in rare cases, ovarian lymphomas can be negative for B and T-lymphocyte (CD3) markers [58] and thus may mimic an undifferentiated tumor.

Artifactual changes in dysgerminoma complicating diagnosis: (a) pseudofollicular change mimics small cell carcinoma with hypercalcemia. (b) Large pools of proteinaceous fluid separated by septa mimic struma ovarii
Syncytiotrophoblastic focal differentiation occurs in about 3–5 % of cases. Syncytiotrophoblasts are found focally either alone or accompanied by isolated mononuclear cells (Fig. 6.3a). Often they are surrounded by erythrocytes from burst capillaries.

Unusual forms of dysgerminoma. (a) Syncytiotrophoblastic differentiation. (b) Dysgerminoma displaying somatic malignant change (fibrosarcoma)
In the same way as germinomas of other locations (see Chap. 12), rarely dysgerminoma may be complicated by somatic malignancies (see Chap. 12). A case of coexisting fibrosarcoma [59] has been reported in the ovary (Fig. 6.3b).
6.2.1.2.3 Immunohistochemistry
In the past, diagnostic immunohistochemistry for dysgerminomas has relied on the classic membrane staining of placental-like alkaline phosphatase (PLAP). In our experience, however, PLAP is relatively unreliable, especially in poorly fixed material; furthermore, it may stain positively in a high percentage of cases of both embryonal carcinoma and YST [18]. CD117 (c-kit) [60], however, is a conspicuous membrane marker, and c-kit mutations have been shown to occur in a third of dysgerminoma cases [39]. Clone D2–40 of podoplanin is a stable cytoplasmic and membrane marker [61], working well in poorly fixed and necrotic material [62]. OCT4 is regularly expressed in dysgerminoma and in gonadoblastoma, often a precursor lesion, but also in embryonal carcinoma [63]. However, considering the exceptionality of embryonal carcinoma in the ovary, OCT4 can be considered as a selective marker of ovarian dysgerminoma [18], especially in cases of poorly fixed tissue with tubular structures, helping to differentiate them from SCCH and even struma ovarii. OCT4 also identifies isolated dysgerminoma cells engulfed by fibrosis or effaced by inflammation [18] (Fig. 6.4). SALL4 represents a sensitive marker for all malignant ovarian GCT being also expressed in dysgerminoma. Cytokeratin expression does not exclude the diagnosis of dysgerminoma, since positives ranging from strongly diffuse to focal and dot-like can be seen in up to a third of cases [18]. Frequently used cocktails such as CAM 5.2 and AE1/AE3 are positive in 19.2 % and 7.7 %, respectively [64]. Cytokeratin 7 is occasionally focally positive; consequently, it is not advisable to differentiate dysgerminoma from embryonal carcinoma by cytokeratin expression alone, as initially proposed [65]. Expression of cytokeratins has unknown significance, but it may be related to an eventual differentiation of primitive germ cells into somatic-type cells. This assumption is partly supported by the focal positivity of blood group-related antigens in the cytoplasm of dysgerminomas [66], which may reflect a differentiation into somatic-type tissues or yolk sac tumor. Additionally, trophoblastic cell differentiation in dysgerminoma is always cytokeratin positive. Finally, rare cases may be positive for neuron-specific enolase (NSE) [67].

OCT4 expression identifies scattered dysgerminoma cells among a dense lymphocytic inflammatory background
6.2.2 Yolk Sac Tumors: Primitive Endodermal Tumors
Yolk sac tumors (YST) do not represent a discrete histopathologic entity but rather a morphologically heterogeneous group of neoplasms capable of differentiating into various types of endodermal tissues, ranging from the primitive stages of primary and secondary yolk sac to endodermal somatic embryonal and mature adult tissues. Different developmental stages often coexist in the same neoplasm, although usually primitive-type ones predominate. As in any other tumor reproducing embryonic structures, YST are potentially able to differentiate into fully mature tissues [68] such as mucinous and hepatoid neoplasms. Although it is typically a true germ cell-derived tumor in the majority of cases, it can also be a heterogeneous component of somatic Müllerian neoplasms.
Due to its changing multifaceted histology and histogenesis, few tumors have attracted so much historical interest, as summarized in pertinent reviews [69–71].
YST are found at all ages in both the ovary and testis. It is possible that tumors have a different pathogenesis according to the age group in which they present: in neonates, infants, and young adults, they correspond to type I GCT, while in sexually mature women and the elderly, they would belong to type VI. In DSD with a female phenotype, they represent type II GCT.
The current terminology includes both endodermal sinus and yolk sac tumor. The former, which is not recommended [27], is used mostly by non-pathologists without a clear understanding of its meaning: the term resulted from comparative pathological findings between similar morphological structures (endodermal sinuses) in the murine placenta and human tumors. The human placenta and yolk sac, however, are totally different from their murine counterpart. This, together with the relatively rare presence of endodermal sinuses in human tumors, makes this term obsolete [71]. However, neither is the recommended term yolk sac tumor devoid of shortcomings, as it may imply that the actual yolk sac is the origin of the tumor. Furthermore, it is often translated into many other languages, with the name Yolk believed to be an eponym [71]. The recently proposed term primitive endodermal tumors [71, 72] defines more accurately the various endodermal and mesenchymal differentiations found in these neoplasms [27]. This broader term would also include some somatic clear cell, AFP-secreting neoplasms from the stomach, liver, bladder, and other areas which have an embryonal endodermal glandular appearance. This approach is analogous to the use of the term primitive neuroectodermal tumor (PNET), which includes all possible and complex types of differentiation from primitive neuroectoderm: neuronal, glial, melanotic, etc. [71].
6.2.2.1 Clinical Features and Treatment
Most YST are found at 16–19 years of age. Although only 10 % of cases occur before age 10, they are the most frequent malignant gonadal GCT at this early age [73]. Their frequency is almost identical to that of dysgerminoma. Although in adult and adolescents (possibly belonging to type II) YST share with dysgerminoma a similar rate of 12p abnormalities [44], in children (type I GCT), 12p is not represented [73]. However, YST and dysgerminomas present substantial clinicopathologic differences:
-
(a)
While dysgerminoma shows a 10 % bilaterality, YST are invariably unilateral.
-
(b)
YST occur mostly in 46, XX females, being less frequently originated in gonadoblastoma than dysgerminomas [74, 75].
-
(c)
Dysgerminoma is rarely found in children and postmenopausal patients; [76] in contrast, YST may occur in older patients [77, 78].
-
(d)
In the ovary, YST associate with dysgerminoma and less frequently with other malignant GCT [28]. Indeed, some reports point out to a possible histogenetic continuum of transformation between dysgerminoma and YST [66]. However, their most frequent association with another GCT is with mature teratoma [79, 80]. All these data make difficult to ascribe adult ovarian YST to a particular pathogenetic GCT type.
-
(e)
Type VI GCT arising in somatic Müllerian neoplasms are nearly always YST patterns; only infrequently are they teratomatous structures, such as neurogenic tissues or muscle, but are never dysgerminomas. These points are summarized in Table 6.2.
Tumors present as a rapidly growing abdominal mass. Clinical features include a relatively frequent association with pregnancy that may behave as hormonally functioning, due to stromal luteinization of the tumor [81]. Association with ataxia-telangiectasia syndrome, a condition associated with an abnormal production of alpha-fetoprotein (AFP) has been reported [82]. AFP elevation is usual [83], showing raised levels between 51,100 ± 12,400 ng/ml. Nevertheless, in a differential clinical diagnosis, AFP is also a marker for tumors harboring immature endodermal tissues such as mediastinal, hepatic, gastric, and urinary bladder tumors, hepatoid carcinoma, immature teratoma, and Sertoli-Leydig cell tumors with the liver as heterologous component [84], which exceptionally reach the high serum levels usually associated with YST.
Older series preceding modern chemotherapy [75] reveal their high grade of malignancy: 70 % of tumors were found at stage I at the time of diagnosis but presented subclinical metastases in 84 % [80]. Untreated cases spread rapidly, involving preferentially the liver, lymph nodes, and peritoneum: brain metastases usually occur in advanced stages of the disease [85].
6.2.2.2 Pathology
6.2.2.2.1 Macroscopy
Tumors are unilateral and large, with an average size of 15 cm; their weight may reach 5 kg [25]. They are usually well encapsulated, although large herniations and rupture may be present. On cut section, the tumor tissue is gray or yellow with numerous irregular zones of hemorrhage and liquefaction. Cysts are usually situated at the periphery and contain mucinous or gelatinous material. Rarely, the tumors are unicystic [86] or uniformly multicystic; the latter appearance correlates with a histological pattern of polyvesicular type [87, 88] (Fig. 6.5a). Solid tumors with a fibrous or elastic consistency correspond to YST with an abundant mesenchymal component (Fig. 6.5b). One-seventh of cases are associated with an otherwise typical mature cystic teratoma in the ipsi- or contralateral ovary [89].

Unusual macroscopic findings in YST. (a) Multicystic mass corresponding to a polyvesicular vitelline tumor. (b) Fibrous, elastic mass corresponding histologically to a mesenchymal overgrowth
6.2.2.2.2 Microscopy [71]
Tumors usually present an admixture of endodermal extraembryonal and somatic patterns in variable proportions.
-
1.
Extraembryonal endodermal differentiations. The primitive extraembryonal areas represent their most characteristic diagnostic feature and their identification facilitates the diagnosis of YST in cases where other complex morphological patterns predominate. These patterns resemble visceral and parietal type of murine yolk sac carcinoma and represent the most primitive attempt of endodermal differentiation including primitive yolk sac cavity and mesenchyme. Broadly, the following histological patterns are described:
-
(a)
Reticular-microcystic (Fig. 6.6a). This is the most frequent and characteristic, having a basophilic loose mesenchymal background containing a labyrinth of anastomosing microcysts lined by a flattened epithelium, often presenting with hyaline globules which are also present within the lumina. Masses of amorphous eosinophilic basement membrane material are found in the stroma.
-
(b)
Endodermal sinus. They are tubulopapillary sinusoidal structures with vascular cores lined by cuboidal or columnar endodermal epithelium that protrude into a space that frequently communicates with the microcystic network (Fig. 6.6b). This structure, however, occurs in only 20 % of tumors and should be differentiated from papillary formations present in clear cell tumors. Experimentally, this visceral structure is only present in neoplasms derived from displaced rat visceral yolk sac [90]. Similar papillary structures are found in the endodermal component of teratomas developed from xenotransplanted human induced pluripotent stem cells [71].
-
(c)
Parietal. This highly unusual pattern closely resembles mouse parietal yolk sac carcinoma; AFP-negative tumor cells are embedded in an amorphous hyaline material [91] (Fig. 6.6c). This feature can occur as a post-chemotherapy conversion [92]. Certainly, this morphology is difficult to explain in humans, since the secondary human yolk sac only has a tenuous, transitory basement membrane [93], which does not resemble the thick Reichert’s membrane of the rat yolk sac.
-
(d)
Polyvesicular. This rare variant is composed of numerous cystic spaces (Figs. 6.6d, e) lined by a mesothelial-like epithelium that merges with columnar, clear vacuolated cells. It is more frequent in the ovary, where it may rarely represent the predominant pattern [87, 88], being readily confused with other multicystic tumors (clear cell, endometrioid adenofibromas, cystic mesotheliomas, etc.). We believe that these vesicles reproduce the morphology of the allantois, which has similar lining of a biphasic epithelium.
-
(e)
Cribriform-tubular . This exceptional YST growth pattern is the only one that caricaturizes the histology of a 6–7-week human yolk sac (also see Fig. 6.7a). Histologically, it has polygonal cells arranged in tubular formations (Figs. 6.6f, g) that, in a similar way to the secondary human yolk sac, may even display hematopoiesis (Fig. 6.6h). In mediastinal and lung YST, these hematopoietic cells may act as precursors of some hematological malignancies (see Chap. 12). Hematopoietic cells should be differentiated from apoptotic epithelial cells.
-
(f)
Solid. This pattern [94] has no correspondence with any embryonal structures and consists of a solid sheet of polygonal, often vacuolated, cells (Fig. 6.6i) with frequent hyaline globules. They have eosinophilic or clear cytoplasm, often negative for AFP [95], and may also present abortive tubular formations. Solid YST lacks the plump cytoplasm and characteristic nuclei of embryonal carcinomas and the consistent chronic lymphocytic infiltrates of dysgerminoma.
Fig. 6.6 


Heterogeneous histology of YST. (a) Characteristic microcystic pattern. (b) Endodermal sinuses. (c) Parietal variant. (d, e) Polyvesicular vitelline pattern showing a cystic pattern. Vesicles have a biphasic lining similar to allantois. (f, g) Unusual cribriform variant with tubular formations and hemopoiesis (h) reproducing human yolk sac histology. (i) Solid pattern with microcysts and hyaline globules. (j) Glandular papillary variant imitates intestinal tube formation and is lined by clear cells surrounding a space (k) filled with dense eosinophilic material. (l) Columnar cells in glandular pattern with characteristic apical and basal vacuolation, often surrounded by marked periglandular stromal rarefaction (m). In rare occasions glandular YST shows a tubular pattern lined by compact non-vacuolated cells (n). Hepatoid pattern (o) showing a trabecular arrangement. Hepatic cells have moderate atypia and exhibit numerous hyaline globules (p). Mesenchymal overgrowth of a YST (q), composed by a loose mesenchyme with only few isolated epithelial elements present. Glandular YST (top) coexisting with an insular type carcinoid (r). Mucinous tumor of the ovary (s) presenting with an aggressive clinical course. Immunohistochemistry revealed a full YST immunophenotype
-
(a)
-
2.
Somatic endodermal differentiations. They reproduce endodermal somatic derivatives such as respiratory, intestinal, and liver tissues. These morphological variations can mimic many other tumors among which glandular patterns are the most frequent.
-
(a)
Glandular patterns [96]. We prefer to use this broader term rather than others such as enteroblastic, enteroid, intestinal, endometrioid-like, etc. They represent a differentiation into somatic endodermal epithelia of the embryonal gut. Glandular patterns consist of a complex network of cysts and glandular spaces that often have papillary projections (Fig. 6.6j). Occasionally, the glandular spaces are filled with a dense eosinophilic material (Fig. 6.6k) and lined by columnar epithelial cells with apical or subnuclear vacuolation (Fig. 6.6l), similar to those of the embryonal gut, that may differentiate into isolated goblet and neuroendocrine cells. Often, a marked periglandular stromal rarefaction is present (Fig. 6.6m). Unusually, somatic glandular patterns may have gland-like spaces with empty lumina lined by compact, closely packed columnar cells lacking vacuoles but set in a loose stroma (Fig. 6.6n). Due to their similarities with many adenocarcinomas, glandular patterns are frequently misinterpreted [97]. Similar areas may also occur in AFP-positive gastric clear cell carcinomas and fetal-type lung adenocarcinomas. Gastric tumors may metastasize to the ovary and may mimic a primary ovarian YST, especially if the metastasis is unilateral. In these cases, the absence of concurrent primitive YST patterns in the ovarian mass and the presence of angioinvasion may support the diagnosis of metastasis.
-
(b)
Hepatic differentiation [98] is a relatively common focal differentiation. True hepatocytes are found in a solid or trabecular arrangement (Fig. 6.6o), often presenting with hyaline globules (Fig. 6.6p). The hepatic nature of these cells has been documented ultrastructurally [99] and may reveal bile secretion and even have associated hematopoiesis. This growth pattern is a frequent source of misinterpretation [97] as it can be confused with dysgerminomas, hepatoblastoma, hepatoid carcinoma, and clear cell carcinomas, but usually the presence of primitive YST foci provides the clue to their identity.
-
(a)
-
3.
Other secondary mesenchymal or endodermal epithelial tumor patterns in YST. They represent either overgrowths or full differentiations of endodermal lineages present in YST.
-
(a)
Mesenchymal overgrowth occurs [100] as a mesenchymal expansion of loose, myxoid tissue stimulated by endodermal epithelium, taking place in periglandular areas and thus mimicking a similar epithelial-mesenchymal transition seen in early embryogenesis. Primitive mesenchyme may differentiate into any derivatives such as smooth or striated muscle and cartilage. Rarely, mesenchymal tissues constitute the bulk of tumor with the presence of only a few engulfed epithelial foci (Fig. 6.6q). In these cases, the diagnosis of YST is only possible after extensive sampling.
-
(b)
Carcinoids. Since the neuroendocrine cells of the gut have an endodermal origin, YST may, albeit rarely, differentiate into an extensive epithelial secondary pattern resembling mucinous carcinoid (adenocarcinoid), displaying prominent goblet cells and neuroendocrine components [101]. They should be differentiated from metastases of gastric carcinomas. Foci of insular type of carcinoid originate from YST, particularly from glandular, intestinal-type patterns [102] (Fig. 6.6r), and may produce insulin [103].
-
(a)
-
4.
Endodermal differentiations occurring in other ovarian tumors. Immature teratomas may differentiate endodermal foci that have prognostic significance [97, 104].
-
(a)
Immature endodermal teratomas [68], (type I GCT) in children, reproduce endodermal and mesenchymal tissues in the absence of neuroectodermal differentiation and represent a neoplasm developmentally related to YST.
-
(b)
Mucinous tumors of the ovary. Some histologically well-differentiated mucinous tumors occurring in young patients rarely may have an aggressive clinical course and present a YST immunophenotype expressing SALL4, villin, AFP, and Glypican-3 (Fig. 6.6s) [105]. They could represent a mature mucinous differentiation of a YST. This hypothesis is partly supported by recent genetic findings proposing that some intestinal-type mucinous tumors may have a germ cell origin [106].
-
(a)



6.2.2.2.3 Immunohistochemistry [96, 107]
The heterogeneous immunophenotype of YST is a consequence of the various differentiations and developmental stages that coexist in these tumors. For this reason, the use of a broad immunohistochemical diagnostic panel covering various endodermal phenotypes is advised [96].
Comparative expression of various proteins in the human yolk sac (HYS) and YST has been attempted. As a referential point, the HYS immunophenotype has been recently studied by analysis of the expression of pluripotentiality and endodermal proteins [107]. The HYS (Fig. 6.7a) has a consistent expression of immunohistochemical markers associated with hepatic (HepPar-1 and GPC3) and intestinal (villin and CDX2) functions. In addition, it expresses SALL4 during all its functional period. In contrast, LIN28, another pluripotentiality protein, is only expressed in early yolk sacs up to the 5th week of development. Additionally, AFP is secreted by endodermal cells via a transient canalicular network that represents the substrate of a transport system functioning during the period of maximum activity of the HYS [107].


Immunohistochemistry of YST. (a) A human yolk sac of the eighth week showing characteristic tubules. Heterogeneous epithelial expression of AFP (b). Epithelial expression of GPC3 (c). HepPar-1 positivity in the columnar components of a polyvesicular vitelline tumor (d). CDX2 expression in microcysts of YST (e). Strong, diffuse epithelial expression of villin (f), SALL4 (g), and LIN28 (h). GATA3 is only positive in the primitive endodermal component of YST, while the differentiated gut structures are negative (i)
Alpha-fetoprotein (AFP) has been considered a gold standard for the diagnosis of YST, although it is known that this embryonal protein can be expressed, among others, in ovarian clear cell carcinoma [108] and in ovarian metastases of gastric carcinoma. In the normal yolk sac, AFP is present as early as the 5th week showing a granular cytoplasmic positivity, which is concentrated in the intra- and intercellular lumina and tubular surfaces that constitute a complex transfer system. In classic YST patterns, AFP expression is constant but heterogeneous in the epithelium, (Fig. 6.7b) where it also delineates occasional cellular lumina. In somatic glandular or solid [95] patterns, however, its distribution tends to be focal or absent [109]. Thus, it can be said that AFP negativity does not necessarily preclude a diagnosis of YST.
Glypican-3 (GPC3). In the normal yolk sac, GPC3 displays a cytoplasmic or membranous expression (Fig. 6.7c) that also highlights inter- and intracellular tubules. GPC3 is also extensively or patchily expressed in YST [110]. GPC3 is diffusely expressed in classical YST patterns but has a heterogeneous distribution, and it is even absent in somatic glandular variants. Similar in distribution to AFP, it is a more sensitive antibody, although not as specific as AFP [111], being also expressed in tumors of the female genital tract such as the primitive neural tubules of immature teratoma, hepatocellular carcinoma, squamous cell carcinomas, carcinosarcomas, and placental site trophoblastic tumors, among many others. Some clear cell carcinomas may also express GPC3, and consequently its demonstration may not be that useful in the differential diagnosis [112] of YST originating from somatic tumors.
Hepatocyte paraffin-I (HepPar-1) is expressed throughout the lifespan of the human yolk sac, reflecting its vicarious hepatic role. HepPar-1 positivity has been reported in both the hepatoid areas of YST and in hepatoid carcinomas [113]. Our study suggests that the frequent, but focal, HepPar-1 positivity in YST does not necessarily mean that it identifies a hepatic tissue, but that it can also reflect a HYS differentiation. Well-differentiated glandular YST of intestinal type show an intense and diffuse HepPar-1 expression (Fig. 6.7d) similar to that found in the small intestine [114].
CDX2 expression is consistently present in the human yolk sac [107]. In classical patterns, CDX2 expression is focal [115, 116] (Fig. 6.7e), but in somatic glandular patterns, it displays a stronger, diffusely positive, staining [96]. Therefore, it would seem that CDX2 positivity will highlight both areas of HYS and intestinal differentiation, the latter being more evident in somatic glandular patterns, especially those with vacuolated epithelia resembling the embryonal gut.
Villin is consistently expressed during early embryogenesis in both HYS and early endoderm [107] and is a highly sensitive endodermal epithelial component marker in all cases of YST, both in classical and somatic glandular patterns (Fig. 6.7f), being absent in pure embryonal carcinoma and seminoma/dysgerminoma. Its diffuse cytoplasmic expression parallels that of the HYS [107] and intestinal adenocarcinomas, where both diffuse and apical staining patterns occur [117]. We believe that villin is an excellent marker of both primitive and differentiated YST variants. To our knowledge, villin has not been previously used as a marker for the epithelial components of YST.
SALL4, as a marker of cells retaining pluripotency, is a highly sensitive marker for YST (Fig. 6.7g), but with a low specificity, since it is also consistently expressed by all malignant primitive germ cell tumors of both gonadal and extragonadal locations [118–120]. It is also present throughout the life cycle of the HYS, reflecting that this temporary organ retains a degree of pluripotency in its endodermal cell component. This could explain the eventual differentiation of ectopic mature endodermal tissues such as liver cell and enteric tissue in the placenta, which may originate from displaced yolk sac remnants [121, 122].
LIN28 is also a highly sensitive marker for malignant GCT [108, 123–125] (Fig. 6.7h). However, its expression is not specific for YST, although it has been proposed that it might be useful for immunohistochemical detection of YST metastases and for differential diagnoses with clear cell carcinoma of the ovary [108, 124, 125], where both AFP and GPC3 can also be positive. Other stemness markers, such as OCT4 and SOX2, are not expressed in YST [126].
GATA3 [127] exhibits nuclear expression only in primitive patterns of YST being, however, often negative in glandular and hepatic variants [128]. Conversely, NUT (nuclear protein in the testis) expression is absent in primitive patterns but is positive in differentiated variants, as it seems to play a role in intestinal or hepatoid differentiations [129].
As a summary, a diagnostic antibody panel, including both markers of pluripotentiality (SALL4 and LIN28) and endodermal identity (AFP, GPC3, and villin), is useful in recognizing the multiple differentiations present in YST. The overlapping immunophenotypes of primitive and differentiated YST areas lend support to the newly proposed term of primitive endodermal tumors [71]. This diagnostic panel will also prove useful in the differential diagnosis of unusual histological variants of YST in uncommon locations [130], as well as in the absence of classical diagnostic patterns and AFP expression. Furthermore, it helps both to identify YST arising from somatic neoplasms [131] and differentiate them from clear cell carcinomas, especially in elderly patients. Additionally, the negativity of CK7 and EMA is characteristic of primitive YST and discriminates them from endometrioid and clear cell carcinomas [132, 131]. In contrast, glandular somatic patterns often show an incomplete YST immunophenotype, being potentially negative for AFP or GPC3 and unexpectedly positive for CK7, EMA [133], HepPar-1, and CDX2, especially when they occur in association with somatic neoplasms (type VI GCT). Consequently, AFP or GPC3 negativity in the presence of other markers such as villin, SALL4, or LIN28 should not preclude the diagnosis of YST.
6.2.3 Postpubertal-Type Testicular GCT (Type II) in Phenotypic Females
Apart from the relatively common dysgerminoma, embryonal carcinoma, polyembryoma, mixed germ cell tumors, and choriocarcinoma are highly unusual in the ovary but frequent in the testis where they are associated with germ cell neoplasia in situ (GCNIS) [134–136] and, consequently, are dealt with at length in Chap. 7. It is possible that many reports of these tumors in women, especially in the older literature, may correspond to phenotypic females with a Y-chromosome-containing DSD [137], where the precursor lesion would be gonadoblastoma and therefore analogous in pathogenesis to type II testicular tumors. Hence, such diagnoses in phenotypic females should prompt karyotypic or molecular genetic studies [18]. Their real incidence in true 46,XX females is difficult to assess, since many cases reported in the past do not indicate presumptive clinical or genetic data relating to DSDs such as pubertal, menstrual, and gestational status; negative sex chromatin; or karyotype, which may help to classify them as true female neoplasms. As stated in Chap. 3, it is possible that type II GCT of the ovary may have their origin in mild forms of ovarian dysgenesis, possibly transient, which leave no obvious final phenotypic traces.
6.2.3.1 Embryonal Carcinoma (EC)
It represents the archetypal stem cell neoplasm, where every cell is pluripotential [138, 139]. Due to their many histopathologic similarities, it is possible that some EC reported in older series were misinterpretations of solid [140] or hepatoid forms of YST [141] or even dysgerminomas. The characteristic immunophenotype of EC, expressing OCT4, CD30, and SOX2, should facilitate diagnosis. However, EC may be associated with yolk sac or trophoblastic differentiation and thus may reveal focal expression for AFP or GPC3. Exceptionally, we have seen EC in true female children and adolescents in association with YST and dysgerminoma, but never in a pure form. In the older literature, there is only one large histopathologic series [142] with limited clinical details, as well as a few isolated case reports, often only partially illustrated or supported by modern immunohistochemistry. One recent study analyzed six ovarian mixed GCT with a component of EC with characteristic immunophenotype of pluripotential markers and presence of 12p chromosome alteration, but the Y-chromosome or testis-specific protein Y-encoded-1 (TSPY-1) status was not mentioned [140].
6.2.3.2 Polyembryoma
This recently reviewed, fascinating GCT pattern [143] caricaturizes the morphology of the trilaminar embryo, forming blastocyst-like embryoid bodies scattered throughout the tumor. They are often associated with EC, YST, or trophoblast. Embryoids are surrounded by a circumferential halo of rarefied loose stroma. Their embryonic disks delineate a small amniotic cavity over the ectodermal plate and, more prominently, a cystic primitive yolk sac cavity from the endoderm (Fig. 6.8). Foci of liver cells or primitive embryonic gut are often related to the primitive yolk sac area. A rarefied, basophilic mesoblast surrounds the embryoid, which may present isolated syncytiotrophoblasts. The identity and position of polyembryoma among primitive GCT is still debated. For some, it represents the “most immature of teratomas” [28, 143] due to its organoid appearance and coexistence of teratoid tissues, while for others, it is a form of mixed GCT [144]. We believe, however, that considering its situation in the embryonal developmental pathway, where stem cells become embryonal germ cells in the naïve pluripotent state (see Chap. 3) and its frequent association with EC, polyembryoma represents an attempt of organoid differentiation of a stem cell proliferation.

An embryoid body showing a primitive yolk sac cavity, positive for AFP
6.2.3.3 Mixed Germ Cell Tumors
They consist of the admixture of various patterns of malignant GCT, analogous to those commonly found in the postpubertal testis. However, the rare mixed ovarian GCT have the following particular features that makes them different from testicular and some extragonadal neoplasms:
-
1.
In the ovary, the most characteristic combinations of malignant GCT are YST and dysgerminoma [28], and immature teratoma and dysgerminoma (Fig. 6.9), as opposed to the usual combinations of testicular tumors, where EC is common, whereas it is exceptional in ovarian mixed GCT.
-
2.
In the ovary, malignant GCT patterns can be associated to benign, differentiated ones. In particular, YST may associate with mature cystic teratoma in the ipsi- or contralateral ovary [79], both of them lacking 12p alteration. This possibly indicates a pathogenetic relationship in type I or IV GCT between both neoplasms [89]. This mixed form is characteristic in the ovary.
-
3.
Another situation involving coexistence of tumor types is represented by the neoplastic transformation of mature components of cystic teratoma, a situation characteristic in the ovary but infrequent in other sites (see Chap. 12). This combination should not be considered a mixed GCT.
-
4.
In high-grade immature teratoma of the ovary, immature neural structures coexist with immature endodermal ones [97]. This should be interpreted as a manifestation of the pluripotentiality of the teratoma rather than an associated tumor.
-
5.
Teratomas in mixed ovarian GCT have a pathogenesis akin to postpubertal testicular types (type II tumors), different to typical parthenogenetic (type IV) benign teratomas of the ovary [145]. Thus, this type of neoplasm is likely to occur in DSD with a Y-chromosome component. IMP3 expression, characteristic of male teratomas and absent in ovarian ones [10], has not been studied in ovarian mixed GCT.

A rare example of mixed GCT (dysgerminoma and immature teratoma) in a 46, XX female
6.2.3.4 Choriocarcinoma
Choriocarcinoma of non-gestational germ cell origin is also rare and truly exceptional as a pure neoplasm, being usually DSD associated and a component of a mixed GCT [146]. Rarely, it may develop in a mature cystic teratoma (Fig. 6.10). In fertile patients, it should be differentiated both clinically and histopathologically from gestational choriocarcinoma originated from an ovarian pregnancy [147], as both share similar histology, symptoms, image findings, and high serum levels of β-hCG. However, in cases where the differentiation between both types is difficult, genetic analysis can demonstrate either a monospermic complete mole as a precursor of gestational choriocarcinoma [148] or the paternal DNA sequences of an androgenetic mole.

Unusual coexistence of a mature cystic teratoma with non-gestational choriocarcinoma
Macroscopically they are solid or cystic hemorrhagic masses, and histologically they show extensive hemorrhage and necrosis that often makes identification of the trophoblast difficult. Both mononuclear cyto- and extravillous (intermediate) trophoblast are intimately admixed in a plexiform arrangement. Numerous tissue blocks should be taken in order to demonstrate other GCT patterns such as mature somatic tissues.
Although non-gestational choriocarcinoma has different chemosensitivity from gestational type, methotrexate-based regimens can be used [149]. However, it has to be taken into account that these neoplasms are likely to be associated with other GCT types and, consequently, cisplatin regimens are preferable.
6.3 Ovarian Teratomas
The usual definition of teratoma implies the differentiation of tissues derived from all three germ layers. Teratomas imitate somatic embryogenesis and a histologic correlation with developmental stages has been proposed [150]. However, this correlation is certainly more accurate when the sequential expression of pluripotentiality markers is analyzed [18]. There are exceptions, as some teratomas only develop one particular tissue foreign to those normally differentiated in any given organ, representing derivations from a single germ cell layer (monophyletic or monodermal teratomas), such as the endoderm in struma ovarii or ectoderm in an ovarian neurogenic cyst. Teratomas show a gradient of maturation and organoid arrangement that includes coexisting embryonal tissues.
Pathogenetically (see Chap. 3), mature, cystic ovarian teratomas, are GCT of parthenogenetic origin (type IV) and constitute the most frequent GCT in 46, XX females. However, there seems to exist a continuum between the type IV mature, cystic ovarian teratomas and type I immature teratomas. While the tissues of most teratomas are differentiated (mature teratomas, cystic or solid), a small proportion show varying amounts of immature tissues, which are responsible for their aggressive behavior. Some may show the presence of secondary malignancies originated from the terminally differentiated tissues. While in children the proportion of mature to immature teratomas is relatively similar (2:1) [3], in adult women mature teratomas show an overwhelming predominance over immature ones.
6.3.1 Immature Teratomas (IT)
Immature teratomas (IT) reproduce progressive stages of tissue organization reflected in variable amounts of immature tissues, usually of neural origin. Xenotransplant of human embryonal stem cells or iPSC produces experimental immature teratomas identical to spontaneous ones [151]. Most patients are 46, XX with the exception of rare bilateral IT, some of which may occur associated with a Y-chromosome genotype [137]. 12p alterations are absent, but gains from 1p, 16p, 19, and 22q are reported [44]. As mentioned in Chap. 3, immature teratomas of infancy belong to type I GCT; however, in adult women, boundaries between type I and IV tumors are not clear-cut, since both immature and mature (dermoid-like)areas may coexist in the same tumor [158].
6.3.1.1 Clinical Features and Treatment
Most IT occur in adolescent and young females and are exceptional in the peri- or postmenopause [152]. Symptoms are nonspecific, with an average duration of 3 months before surgery, and consist of abdominal mass, pain, vaginal bleeding, and fever in a quarter of patients [153]. Paraneoplastic limbic autoimmune encephalitis [154] may occur. Although the tumors are usually solid, preoperative ultrasonograms may reveal a multicystic neoplasm, in which case a careful surgical incision should be made to avoid surgical rupture and spillage [155].
A third of tumors can exhibit AFP elevation and less than 10 % may show increased serum hCG levels [156]. A unilateral stage I mass is found in up to 69 % of cases [155, 157]. Bilaterality is rare [11]; however, 10 % may be associated with a mature cystic teratoma (MCT) in the contralateral ovary, and, exceptionally, they can be preceded by a previous resection of a MCT in the same ovary [158].
Abdominal extension is present at the time of surgery in a third of cases. Rarely they may present with metastases in soft tissues [159] as well as in the brain, lung, and liver [160].
Surgery with preservation of fertility is the procedure of choice since IT involves young patients. In stage 1A, grade 1 tumors, adjuvant therapy is not necessary. Resection of stage 1A tumors has been reported as curative in pediatric IT [156]. More advanced stages and grades should be treated with current combination chemotherapy. Gliomatosis peritonei is not treated except for recurrent cases or in the development of growing teratoma syndrome [161]; it remains stable for many years and a possible complication is abdominal hemorrhage [162].
6.3.1.2 Pathology
6.3.1.2.1 Macroscopy
IT are usually bulky, solid tumors, rarely bilateral, that can be associated with MCT in a fourth of cases [158]. Three quarters show a smooth capsule, and only a third may present with rupture or herniations. On cut section, most tumors are solid, white, encephaloid (Fig. 6.11) and may present small cysts. Small foci of bone or cartilage may be present. A multilocular appearance occurs in a third of cases. Only rarely are tumors parvilocular or pedunculated within a large, solitary cyst, and up to a fourth of cases may show embedded or peripheral dermoid-like cysts containing sebum and hairs [158]. Hemorrhage and necrosis are often found and are especially evident in cases of torsion. Sampling must include a tissue block for every centimeter in diameter; it is mandatory that the mass should be adequately sampled as this is crucial for grading the tumor.

Characteristic gross appearance of immature ovarian teratoma with abundant encephaloid nodular formations
6.3.1.2.2 Microscopy
A mixture of mature and immature tissues with predominance of neural ones characterizes this tumor. Grading, assessed in the relative amounts and degree of immaturity of the neural tissues, is prognostically related as demonstrated in prechemotherapy series of IT [155, 157]. Grade 0, or mature solid teratomas, do not show the usual dermoid features and are composed of mature tissues of predominant neural origin. Grade 1 tumors contain rare foci of immature neural tissue (<1 low-power field (LPF) in any one slide), while grade 2 and grade 3 tumors contain 2–3 LPFs or 4 or more LPFs of immature neural tissue, respectively [28]. This system can be simplified into a two-tier classification with low grades referring to grade 1 teratomas and high grades encompassing grades 2 and 3 [163].
Structures relevant to histologic grading. The characteristic immature neural structures evaluated in grading are neural blastematous nodules containing rosettes and neuroepithelial tubules (Fig. 6.12a) with a crowded cell lining with abundant mitoses that may be occasionally pigmented. Their sharp-edged apical borders are well delineated by an inner limiting membrane (Fig. 6.12b) which differentiates them from other immature tubules of nonneural origin. It is important to take into account some other immature tubular structures that can mimic neuroepithelial tubules. Endodermal tubules lined by tall columnar vacuolated epithelium (Fig. 6.12c), similar to glandular YST, are, in our experience, intimately admixed with immature neural tubules in grade 3 tumors, especially in pediatric IT (type I GCT), where they are found in a third of cases [156]. While some authors may regard these tubules in high-grade IT as microscopic foci of YST, we believe that the presence of these does not necessarily signify a mixed GCT but yet another immature embryologic structure differentiated in a high-grade tumor and thus may influence prognosis [97]. Microscopic areas of primitive, microcystic YST can be found, especially in high-grade IT in children (Fig. 6.12d). Endodermal tubules are often identified by marked periglandular stromal basophilic rarefaction (Fig. 6.12e). Hence, the finding of endodermal tubules should also be taken into account when grading a teratoma. Solid, undifferentiated areas, rarely forming embryoid bodies, may be present in high-grade tumors and resemble EC, even sharing a similar immunophenotype (see below).

Structures relevant to histologic grading in IT. Neural tubules in a high-grade tumor (a) with a basophilic cellular lining with numerous mitoses. The luminal edge is usually sharp (b). Coexistence of endodermal tubules (top) with a neural rosette (bottom) (c). Neural tubules admixed with a small focus of reticular YST in a grade III teratoma (d). Endodermal tubules with periglandular stromal rarefaction coexist with a small focus of neuroectoderm (arrow) (e). Neural tubules mimicry: metanephric tubules resemble neural structure but also display abortive glomeruli (f)
Elements which are not relevant to grading include metanephric tubules. Nevertheless, these may mimic neuroepithelial tubules as they are lined by crowded cells with scanty cytoplasm. However, they lack the inner limiting membrane characteristic of neurotubules and are associated with abortive glomerular structures (Fig. 6.12f). Other areas showing developmental immature features which are not relevant to grading are mesenchymal derivatives, such as immature skeletal muscle, which should be differentiated from true overgrowth of sarcoma [28]. Immature mesenchymal areas often resemble myxoid liposarcoma with a characteristic vascular arrangement. Other immature areas such as tooth buds, lack any importance other than providing a beautiful microphotograph. Marked vascular hyperplasia with both endothelial and adventitial proliferations, similar to that occurring in central nervous system tumors, is frequently found in any grade. However, it may be quite prominent in high-grade IT, where it may be associated with a malignant overgrowth of primitive neuroectodermal tumors (PNET), including malignant retinal anlage-type tumors [164].
Only when IT elements are a component of a type II mixed GCT, they may it show 12p chromosome abnormalities [145]. Some teratomas are almost exclusively composed of both mature and immature endodermal structures in various stages of differentiation. For these, the name immature endodermal teratoma has been used, and their relationship with YST is open to speculation [68].
6.3.1.3 Complications and Unusual Features
Malignant overgrowth of some immature tissue components in IT is a rare event, being less frequent than the development of secondary somatic malignancies in testicular GCT. Since neural tissues predominate in IT, overgrowth by astrocytoma [165] and highly malignant PNET (Fig. 6.13a, b) may occur. Cases reported as neuroblastoma, neurocytoma, etc., arising in IT, would belong to this category. GCT-associated PNET often have central genotype [166], lacking the ESWR-1 rearrangement, and consequently diagnosed as medulloblastoma-like [167, 168]. Ependymoma is only rarely associated with teratoma (Fig. 6.13c).

PNET overgrowths in immature teratoma. Nested pattern with vascular hyperplasia (a). adamantiform pattern (b) and ependymal differentiation (c)
Rhabdomyosarcoma has also been reported as an overgrowth from IT presenting with peritoneal and retroperitoneal involvement [169].
Mature implants and metastases from IT . Immature tissues from ovarian teratoma can achieve full differentiation in abdominal and less frequently, extra-abdominal deposits, either spontaneously or subsequent to treatment. This phenomenon takes place in both growing teratoma syndrome and gliomatosis peritonei and may occur simultaneously [170].
Growing teratoma syndrome . The presence of expansile masses of mature teratoid tissues in the abdomen, pelvis, retroperitoneum, liver, lung, or pleura after chemotherapy for ovarian IT was initially reported by DiSaia [171] and later found in testicular tumors [172] (see Chap. 7). Only rarely may it also be present in untreated patients. Resected masses are usually nodular and comprised of cystic structures that expand by continued secretion from their serous or mucinous epithelia, causing compression of various organs and structures. Long-term follow-up [173] often shows no changes in the benign tissues, although eventually they may rarely develop endodermal type VI GCT, such as glandular YST and carcinoid [174, 173] or intestinal-type adenocarcinomas [170].
Gliomatosis peritonei (GP) [175] is a relatively frequent phenomenon associated to ovarian IT, being rare in teratomas of other organs. GP presents as widespread small solid nodules of mature glia in the peritoneum (Fig. 6.14a, b) and abdominal lymph nodes. Rarely, it may occur outside the abdomen [176]. Its behavior is benign, although it may recur [177]. Implants grow rapidly but remain unchanged for life, undergoing only involutive phenomena of an ischemic nature. Astrocytes represent their main component, but other neural lineage elements, such as neurons and ependyma, and mesenchymal tissues such as cartilage can be found [178]. Secondary changes in glial nodules may include (a) degenerative astrocytic changes; (b) granulomatous and follicular chronic inflammatory response; (c) association with hormonally related changes, such as decidual peritoneal metaplasia; (d) endothelial and adventitial vascular hyperplasia [162] (Fig. 6.14c); (e) complete regression [179, 180]; and (f) development of glioma [181]. Association with endometriosis has been reported, where endometrium and glia are intimately admixed (Fig. 6.14d). This intriguing phenomenon may either imply a common origin for each component or a possible glial induction of endometrial differentiation [182].

Gliomatosis peritonei. Glial nodules in the ovarian surface (a) and peritoneum (b) coexisting with a marked granulomatous response to embedded hairs. Marked hyperplastic vascular change in gliomatosis peritonei (c) may be responsible for hemoperitoneum. Rarely glial nodules coexist with endometriosis (d) (center)
Two pathogenetic mechanisms are considered in GP: (a) direct seeding of immature neural cells from a primary tumor with subsequent differentiation and (b) metaplasia from peritoneal stem cells.
An implantation mechanism is supported by the following clinicopathologic data:
-
(i)
The GP nodules present with a wide cellular heterogeneity of neural and nonneural components, showing coexistence of mature astroglia with neural blastemal areas and other non-ectodermal tissues such as cartilage. This would favor an implantative origin from pluripotential teratoma cells that undergo multiple differentiations in the peritoneal environment.
-
(ii)
The frequent presence of shed keratin and embedded hairs within the nodules (Fig. 6.14b), possibly originating from the primary ovarian neoplasm, is a strong clue for an implantative origin.
-
(iii)
The presence of lymphovascular involvement and even extraperitoneal deposits [176].
-
(iv)
There are rare cases of GP associated with ventriculoperitoneal shunts which would constitute a natural experiment of the implantative capacity of glial cells present in the cerebrospinal fluid into the peritoneum [183].
However, a metaplastic origin is sustained by a heterozygosity pattern of GP nodules, identical to the normal tissue but different from the coexistent ovarian teratoma [184, 185]. SOX2 is expressed by gliomatosis [175] and is one of the key factors for the maintenance of pluripotency in stem cells. Its expression is required for inducing stem cells to differentiate toward the neural lineage, and, consequently, SOX2 may play a key role in the pathogenesis of gliomatosis peritonei [181]. Thus, GP would constitute a peritoneal change in response to growth factors secreted from teratoma or macrophages.
While an implantative origin from ovarian teratoma remains the more probable mechanism in most cases, metaplastic glial differentiation from peritoneal stem cells could explain cases of GP with a monomorphic astrocytic cell population, as well as those associated with endometriosis [186].
6.3.1.4 Immunohistochemistry
The main role of immunohistochemistry in these otherwise diagnostically straightforward tumors lies in identifying features relevant to grading that may be not conclusively recognized with hematoxylin eosin stains. This could be achieved by distinguishing various tissue components as well as their degree of immaturity [96]. Identification of neural areas can be complemented by characteristic neural makers such as glial fibrillary acidic protein, nestin, and others. In immaturity assessment, SALL4 (Fig. 6.15a) is expressed in immature components that may be present in grade 3 tumors, not being expressed in lower grades. On the other hand, SOX2 is only expressed in immature neural areas (Fig. 6.15b), while any of the endodermal markers used in diagnosis of YST (AFP, GPC3, villin, HepPar-1, CDX2, etc.) would be expressed in immature endodermal areas. It is worth remembering that GPC3 is also expressed in neuroepithelium and could highlight both endo- and neuroectodermal components. A combination of SALL4, SOX2, and markers such as villin, AFP, and GPC3 will manifest both immature endodermal and neural areas and facilitate semiquantitative grading. Finally, SOX2 marking is particularly useful in the demonstration of the rare PNET overgrowths of teratoma, including glioma arising from gliomatosis peritonei [181].

Immunohistochemical support for diagnosis of immature areas of IT. SALL4 stains diffusely all immature elements (a), while SOX2 (b) only highlights immature neural areas
In some grade 3 tumors, we have identified [105] focal solid areas with an EC immunophenotype, coexpressing OCT4, SOX2, and CD30 (Fig. 6.16a, b), which would represent the stem cell population of some IT. In these unusual cases, EC-like areas are not associated with the usual admixture of tumor types present in testicular-type mixed GCT but may develop into organoid structures of embryoid bodies (Fig. 6.17) [28]. This finding may explain reports of OCT4 positivity in IT [187].

Rare foci of embryonal carcinoma-like areas (a) in high-grade IT. The cellular, fenestrated areas that coexist with neural elements (right) coexpress OCT4 (b) as well as CD30 and SOX2

Isolated embryoid found in a high-grade IT
6.3.2 Mature Teratomas
The most frequent GCT in females represent the final stages of multidifferentiation of a parthenogenetically induced germ cell [7, 188] after meiosis I [189]. These fascinating ovarian tumors are capable of reproducing practically any adult tissue and organ, even replicating a rudimentary external human form (fetiform teratoma).
Ovarian teratomas that can also originate from supernumerary ovaries [190] present with fully differentiated tissues with a cystic appearance (dermoids) and are only rarely solid or macroscopically similar to IT. Mature cystic teratomas (MCT) constitute the most common ovarian tumor in the young and, consequently, are prevalent in countries with a young average population. They can be diagnosed at any age but only a small proportion is found in postmenopausal women. Familial incidence, including presentation in identical twins, is rare [191–193].
6.3.2.1 Clinical Features and Treatment
They present with the usual symptoms of abdominal mass, mostly when they are associated with pregnancy. Preoperative diagnosis is usually performed due to their characteristic ultrasound and MRI appearances [193].
Torsion seems to be the most frequent complication of MCT. In rare cases it may lead to amputation and eventual parasitic implantation of tumor in the abdominal cavity. During pregnancy, MCT may be a mechanical obstacle to uterine expansion and delivery and cause ectopic pregnancies. Rupture, either spontaneous or iatrogenic, is relatively uncommon but may produce granulomatous serositis and hemoperitoneum. Rupture may be followed by fistulization into adjacent organs. Exceptional cases of peritoneal melanosis may be associated with chronic hemorrhagic spillage of pigmentary substances including melanin from the MCT. As any other tissue, it may harbor various infectious bacteria or parasites.
MCT may associate with generalized autoimmune disorders triggered by various components of the cyst; hemolytic anemia, dermatomyositis, polyarthritis, pruritus, urticarial vasculitis, etc. are occasionally reported. All symptoms disappear after removal of the teratoma. However, the most frequently reported autoimmune complication of MCT is limbic encephalitis with antibodies against N-methyl-D-aspartate receptor (NMDAR). This rare, often fatal, form of encephalitis has psychiatric symptoms, particularly psychosis, and was first reported in 2005 [194–196]. It has now become a clinically recognizable disease, diagnosed by the presence of anti-NMDAR antibodies in the cerebrospinal fluid, only treatable with removal of the ovarian teratoma and subsequent immunosuppressive treatment [197]. More than half of the ovarian teratomas reported in association with anti-NMDAR encephalitis are MCT, while a quarter of them correspond to IT. Although also associated with teratomas of other organs, the lower frequency of teratomas in extraovarian locations and the smaller proportion of neural tissues present in them would partly explain their rarity. Often, MCT in this syndrome are small and thus difficult to diagnose by ultrasound [198], making higher resolution MRI studies necessary. A similar autoimmune phenomenon, but without systemic consequences, occurs in teratoid thyroid tissue, where Hashimoto’s thyroiditis has been reported.
Endocrine symptoms may be associated with the presence of functioning endocrine glands developed in teratomas. Examples include hypophysis able to develop pituitary adenomas, producing Cushing’s syndrome or hyperprolactinemia. Hypersecretion of androgens is not unusual and is related, not to the teratoid tissues themselves but to the induced ovarian stromal cells at the periphery of the expanding mass (peripheral luteinization).
Other rare complications include metastases of breast tumors to teratoma.
Treatment must take into account the future fertility of the patient, with preservation of ovarian tissue and limitation of the risk of adhesion formation [199]. Laparoscopic treatment has less postoperative complications and morbidity than oophorectomy, but, on the other hand, it may produce spillage into the abdominal cavity and subsequent adhesions. Furthermore, incomplete resections of multifocal tumors may lead to recurrence, which can occur in up to 4 % of cases, especially in rare multiple tumors within the ovary. A comprehensive summary of reported complications (except malignant transformation) occurring in MCT is shown in Table 6.3.
6.3.3 Pathology
6.3.3.1 Macroscopy [153]
Bilaterality occurs in 15 % of cases and not infrequently contralateral ovaries harbor small, occult MCT. Rarely, tumors are multifocal in the same ovary [264]. Average size is 8 cm, although it may vary remarkably, the record size being awarded to a 42 kg tumor [265]. Tumors are round or ovoid with a prominent vascular network and have a smooth capsule with occasional adhesions. Their consistency immediately after excision is fluid becoming doughy at room temperature. Tumors are usually cystic and contain yellow sebum mixed with hairs in 98 % of cases; only on rare occasions are they predominantly solid. Sometimes the sebum becomes aggregated in sebum and hair spheroids (“golf balls” or “jelly beans”) (Fig. 6.18a, b) that have a peculiar MRI appearance [266]. Infrequently, contents are exclusively fluid: serous, mucinous, or resembling cerebrospinal fluid. The wall is usually elastic, although irregular calcified plaques can be present in older patients.

Unusual macroscopic findings in mature cystic teratomas. Hair and sebum aggregate to form “golf balls” (a) or “jelly beans” (b). (c) This rare hairy teratoma replicates the illustration of the first description of a teratoma: Johannes Sculteto: Trichiasis admiranda siue morbus pilaris mirabilis noribergae (Nürnberg, 1658)
A dermoid protuberance, often named in the older literature after Wilms or Rokitansky, is found in 87 % of cases. Although singular in most instances, multiple protuberances can occur. Hairs may constitute the main bulk of cystic contents (Fig. 6.18c), and although their color is often unrelated to that of the patient, they may become gray with age. Teeth are found embedded in the protuberance, usually in groups of two or three, but up to more than 300 teeth have been counted in a single specimen [267]. Only seldom are they set in a well-formed mandible surrounded by gingiva [268]. The teeth are of permanent type, usually canines, incisors, or molars, and are often dysplastic [269].
Unusual macroscopic findings include eversion of the inner lining of the cyst [270]. Highly organized structures resembling fetuses (fetiform teratoma) [271] may be found within cysts, and some may even have metameric, axial, and symmetrical organization [272] with nails, vertebral bodies, lung, limbs, and even external genitalia [273], thus deserving the name of homunculus, latin for little man (or perhaps better muliercula: little woman, taking into account the presence of female external genitalia in some tumors) [273, 274].
6.3.3.2 Microscopy [153]
Typically, the inner surface of the cyst is lined by epidermis and skin adnexa which desquamate into the lumen. Most tissues are found within the protuberance and mostly reproduce those corresponding to the rostral part of embryos. They are haphazardly mixed, although an attempt to maintain a tissue relationship similar to that found in adult organs (polarity of growth) is frequent: bronchi lined with respiratory epithelium with bronchial-type glands and cartilage, intestinal, or gastric mucosa with corresponding layers and even harboring Cajal’s cells as pacemarkers in the muscle. Teeth buds imitate normal odontogenesis and thyroid tissue is often in proximity to thymus. Practically any type of tissue except perhaps gonads or adrenal has been reported. Relative percentages of tissues and various curiosities are shown in Table 6.4, and the presence of some unusual tissues is depicted in Fig. 6.19a–k.


Unusual tissues in mature cystic teratoma. (a) Retinal structures may simulate the immature neural tubules of IT. (b) Conjunctival epithelium, (c) breast lobules, (d) fetal fat, (e) PSA-positive prostate, (f) vascular structures similar to corpora cavernosa, (g) thymus, (h) Schneiderian epithelium, (i) gemistocytic transformation of astrocytes, (j) Rosenthal fibers, and (k) well-formed cerebellum
Functioning luteinized stroma, occasionally associated with virilization [256], may occur in the ovarian tissue at the periphery of the cysts [257].
Ageing of the various tissue components is mainly reflected in degenerative phenomena of the nervous tissue, such as microcalcification, microcystic degeneration, gliosis, and gemistocytic change (Fig. 6.19i); Rosenthal fibers (Fig. 6.19j) may also occur [297] and are possibly related to an impaired vascular supply. Chronic lymphocytic inflammation of neural structures has been related to autoimmune anti-NMDAR encephalitis [28].
Reactive phenomena, such as a foreign body reaction to keratin and hair as well as an oleogranulomatous response to sebum, are often frequent in older patients although they may occur at any age. When prominent, they may efface teratoid tissues and only stable structures such as hair or bones remain. In the tumor wall, mesovarium and mesosalpinx, cystoid oleogranulomas occur and represent an indirect, but pathognomonic, sign of the presence of a neighboring teratoma [153]. Coagulative necrosis, unrelated to torsion, is rare and in one instance [198] has occurred in relationship with autoimmune anti-NMDAR encephalitis. Myospherulosis, as a response to oleous contents, develops occasionally in surrounding tissues [298]. A summary of reported secondary lesions in MCTs is shown in Table 6.3.
6.3.3.3 Secondary Neoplasms Arising in Mature Teratomas
The presence of secondary tumors developing from the differentiated tissues of MCT is a phenomenon that occurs in the ovary in a much higher proportion than in teratomas of any other location (see Chap. 12). Mature teratoid tissues can potentially develop into any tumor type found in the adult. Malignant change occurs in patients in the fifth and sixth decades in 1.5–3 % of MCT [220, 299], often being related to senescence of the teratoid tissues, although neoplasms typical of childhood, such as primitive neuroectodermal tumors, may also occur. Local factors are possibly involved in tumorigenesis, as there are neither associated multicentric tumors in the rest of the female genital tract nor simultaneous malignant change in cases of contralateral mature cystic teratomas, the only exception being bilateral mucinous tumor associated with bilateral teratomas [300].
Squamous cell carcinomas constitute the overwhelming majority of secondary tumors in older patients. Due to their relative rarity, they are seldom diagnosed preoperatively on imaging workups. It has been proposed that elevated serum markers of SCC (squamous cell carcinoma antigen) and CA19.9 may be helpful in diagnosis and even in staging of the tumor [301]. At the time of diagnosis, the tumor often extends beyond the ovary adherent to pelvic organs in over half of cases and ascites is present [302]. Omental, lymph node, and distant metastases are frequent. Due to their advanced stage at the time of diagnosis and their resistance to chemo- or radiotherapy, survival is poor [220, 299].
6.3.3.3.1 Macroscopy
The usually unilateral cysts show irregular, thickened, or ulcerated plaques and/or fungating intracavitary growths. Since they involve postmenopausal women, any teratoma in this age group warrants extensive sampling, especially when necrosis, hemorrhage, or thickened or pigmented areas (Fig. 6.20a, b) are present.


Secondary tumors in teratoma. Squamous cell carcinoma presenting as a thickened wall in a cystic teratoma (a). Pigmented lesion in a teratoma that corresponded to a malignant melanoma (330) (b). Surface squamous cell carcinoma (c) in teratoma with early invasion (d). A squamous papillary growth simulating Schneiderian papilloma (e). Stromal and vascular invasion by squamous cell carcinoma (f). Intestinal mucinous overgrowth in teratoma with pseudomyxoma ovarii. Pilomatrixoma in an ovarian teratoma (g) with coexisting neural tissues (right). Sebaceous carcinoma (h) in teratoma, positive for adipophilin (i). A calcified growth corresponding to an odontoma (j)
6.3.3.3.2 Microscopy
Squamous cell carcinoma is the most common histologic type of secondary malignant tumor found in MCT. Most originate from the epidermis, as demonstrated by the presence of in situ lesions (Fig. 6.20c) [153], but are not related to human papilloma virus. An origin from bronchogenic structures is also contemplated. A squamous papillomatous pattern (Fig. 6.20d), reminiscent of a schneiderian papilloma of the upper respiratory tract, should be differentiated from a proliferative Brenner tumor.
Differential diagnosis from rare primary squamous cell ovarian carcinomas [303], either of pure histological type or associated with endometrioid lesions or Brenner tumor, should be made if teratoid structures are not found after extensive sampling. Squamous cell carcinoma often invades the capsule and lymphatic vessels and has a poor prognosis (Fig. 6.20e).
Benign, malignant, and borderline intestinal-type mucinous tumors [300, 304] originating from mature teratoma exhibit well-differentiated intestinal epithelium and are often associated with pseudomyxoma ovarii (Fig. 6.20f). A recent study [106] has demonstrated the homozygosity of these tumors, suggesting that ovarian intestinal-type mucinous tumors may have a teratoid origin. Those showing appendiceal-type histology [305] may associate with a form of pseudomyxoma peritonei less aggressive than that originated in appendiceal tumors. Immunohistochemically, these tumors show a variable expression of CK20 and 7. In mucinous cystadenomas, a CK7+/CK20− phenotype occurs as commonly as a CK7−/CK20+ phenotype [300], being diffusely CK20 positive in over a third of cases and, more often, only focal or partially positive [304]. Borderline or malignant mucinous tumors tend to have a lack of expression of cytokeratin 7 [300]. Mucin (Muc) family antibodies, although often nonspecific, may be helpful in establishing an origin from teratoid tissues [306].
Melanoma (Fig. 6.20b). Although most ovarian melanomas are metastatic in nature, origin from teratoid tissues may occur, representing one of the rarest origins of metastatic melanoma. The only series dealing with ovarian melanoma originating in teratoma [307] revealed a wide age range from 18–72 years. All were unilateral and histologically showed large epithelioid cells with eosinophilic cytoplasm, small cells, spindle-shaped cells, or a combination of these. Melanin pigment was not always present, but their melanocytic nature was shown by electron microscopic demonstration of melanosomes and positivity for one or more melanocytic markers.
Potentially, practically any tumor pattern found in adults and children can occur in the tissues of a mature teratoma. An updated list of reported sarcomas, cutaneous, neural, and miscellaneous tumors, and their rare combinations is presented in Table 6.5.
6.3.3.4 Monodermal (Monophyletic) Teratomas
This imprecise term refers to tumors revealing a one-sided differentiation of a tissue not native to the ovary, derived from one of the germ cell layers, usually ecto- or endoderm, since the ovary is an organ of mesodermal derivation. Thus, tumors originating from tissues of mesodermal origin such as chondroma [394] are not included. Neither are those of celomic origin such as epidermoid cysts [395] and some rare ectopic tumor patterns such as ovarian extrarenal Wilms’ tumor [396], extra-axial ependymomas [361], hepatoid carcinomas [397], and PNET that may result from tumor stem cell univocal differentiation. The same would apply to rare heterologous differentiations such as hepatic or intestinal areas in granulosa and Sertoli-Leydig cell tumors [84, 398].
6.3.3.4.1 Struma Ovarii (SO)
SO is a term applied to teratomas which contain thyroid tissue as the unique or predominant component, usually 50 % or more. SO occurs in an older age group than simple MCT and presents as an abdominal mass that is unusually associated with hyperthyroidism or pseudo Meigs’ syndrome [399, 400]. The great majority of cases are benign. Malignant change is uncommon but may represent a challenge in the understanding of its clinical behavior and treatment. Ultrasonographic findings are nonspecific, usually revealing a heterogeneous, predominantly solid mass [401].
6.3.3.4.2 Macroscopy
Most cases are unilateral and associated with teratoma and can reach up to 20 cm in size, although a small SO is not an unusual incidental finding (Fig. 6.21a). Larger tumors often have a pure thyroidal histology and may be malignant. Tumors are well encapsulated and multicystic and their contents are usually brown or green colloid. Rarely, they may present as a unicystic tumor that is only diagnosed in the microscopic study (Fig. 6.21b, c).

Struma ovarii. (a) Cystic struma with colloid contents. Thyroidal lining is reduced to a single cell line (b) positive for TTF-1 (c). Pseudopapillary (d), Hürthle cell (e), and mucinous (f) changes
6.3.3.4.3 Microscopy
Thyroid tissue is readily identified with colloid-filled cystic follicles grouped in nodules. The full range of microscopic changes seen in the eutopic thyroid including microfollicular, solid, tubular, trabecular, Hürthle cell, pseudopapillary and mucinous [305] changes (Fig. 6.21d–f), hyperplasia, atrophy, lymphocytic infiltration, etc. can be present in the teratomatous thyroid. Cases exhibiting densely packed cellular follicles or pseudopapillary change have been termed proliferative struma [399], and although benign, they represent the main differential diagnosis with histologically malignant struma. Luteinized stromal cells are not uncommon in the periphery.
The enigmatic association with Brenner tumor (Fig. 6.22a, b) [402, 403] and mucinous cystadenoma suggests that, in these cases, the urothelial and mucinous components so intimately admixed with thyroid tissue would represent a combination of teratoid endodermal tissue differentiations rather than a coincidental association of a common epithelial neoplasm with teratoma.

Mixed Brenner tumor and struma. Brenner nests are intimately admixed with thyroid follicles
The pathology of malignant SO parallels that of thyroid carcinomas [399, 404, 405]. Papillary carcinomas of both classic, tall cell, and follicular variants (Fig. 6.23a–e) are the most frequent, and they should be differentiated from proliferative struma using the same criteria applied in the eutopic thyroid to differentiate between papillary carcinomas of follicular variant and hyperplastic nodules (ground glass nuclei, vascular invasion (Fig. 6.23f), etc.) [399, 405]. True follicular carcinomas are unusual and poorly differentiated carcinomas (Fig. 6.23d, e) are even more infrequent. It must be borne in mind that true medullary carcinomas do not occur in the ovary, their closest histological relative possibly being represented by strumal carcinoid.

Malignant struma ovarii. Papillary carcinoma in SO (a). Rare cases may exhibit a tall cell pattern (b). Follicular variant of papillary carcinoma with characteristic nuclei (c). Poorly differentiated thyroid carcinoma (d) with a patchy expression of thyroglobulin (e). Vascular invasion (f) in malignant struma
Malignant SO are usually large masses measuring over 10 cm in size, often with adhesions and ascites. Their behavior is indolent, with a good survival rate at both 10 (89 %) and 25 years (84 %) [406]; extraovarian spread or recurrences rarely develop. The hormonal milieu of pregnancy apparently may provoke more aggressive behavior [405, 407]. Slow growing peritoneal implants, previously called benign strumosis, are only examples of well-differentiated metastases. Histologically, both a papillary pattern and a poor degree of differentiation are associated with a more aggressive course [406].
The expression of specific markers such as thyroglobulin (Fig. 6.23e) and thyroid transcription factor-1 (TTF-1) is useful in identifying the thyroidal nature of the neoplastic tissue in cases when it is difficult to recognize, such as the thin cuboidal lining of the cavity of cystic SO (Fig. 6.21b, c) and in solid, poorly differentiated thyroid carcinomas with minimal follicular formation. These markers also help in establishing a thyroidal phenotype in the differential diagnosis with other ovarian tumors with tubular or trabecular arrangement, such as Sertoli-Leydig cell and carcinoid tumors.
Classic papillary carcinoma is a relatively straightforward diagnosis, but the more complex diagnosis of the follicular variant of papillary carcinomas may be facilitated by the expression of HMBE-1 and CK19 positivity [408]. BRAF point mutations of the type commonly observed in papillary carcinomas of the eutopic thyroid have also been reported in malignant struma [407, 409].
Histologic diagnosis of malignancy occurs in most cases without any evidence of extraovarian spread. In these, a conservative approach with salpingo-oophorectomy and analysis of thyroglobulin serum levels to exclude occult disease is advised [399, 404, 405]. Postoophorectomy treatment should follow similar protocols to those used in eutopic thyroid carcinoma, potentially including thyroidectomy or radioiodine ablation, and hormonal suppressive therapy. Long-term follow-up for both metastases and abdominal recurrences can be accomplished by serial serum thyroglobulin measurements and radioiodine scans [410].
6.3.3.5 Carcinoid Tumors
Ovarian carcinoids are infrequent, well-differentiated neuroendocrine tumors with restricted malignant potential and a good long-term prognosis. They are often unilateral and small and coexist with mature cystic teratoma (57–80 %), although in 15–42 % of cases [411, 412], tumors are larger and not associated with teratoma. Neoplasms have a heterogeneous morphology, with various neuroendocrine differentiations of embryonal fore-, mid-, and hindgut, often coexisting in the same neoplasm [305]. Each predominant histologic subtype presents a relatively characteristic clinicopathologic profile. On a histological basis alone, they are similar to carcinoids metastatic to the ovary, from which they should be differentiated, since they have a different prognosis.
Histologically, the following carcinoid patterns are present in the ovary:
-
(a)
The most frequent insular patterns of midgut type.
-
(b)
Trabecular patterns of fore- and hindgut type that have in some series a similar frequency to insular carcinoids [411] but in others [28] are considered less common than strumal carcinoid.
-
(c)
Rare mucinous carcinoids, similar to appendiceal adenocarcinoids (goblet cell carcinoids).
-
(d)
A combination of thyroid tissue with trabecular or insular variants: strumal carcinoid. This peculiar tumor is exclusive to the ovary, not found in the testis or in any other GCT location and different from medullary carcinoma of the thyroid.
-
(e)
Metastatic carcinoids from the gastrointestinal tract differ from primary carcinoids by their monomorphic pattern; the absence of accompanying teratoid tissues; their bilateral, multinodular ovarian involvement; and frequent vascular and peritoneal invasion. Clinically, the age distribution of patients with ovarian carcinoid is similar to that of those with secondary neoplasms originating in mature teratomas, and, in most cases, symptoms are nonspecific. Rare functioning cases may present with Cushing’s and hyperinsulinism syndromes secondary to ectopic ACTH or insulin secretion by ovarian carcinoid [103, 386] as well as an association with other neuroendocrine tumors in multiple endocrine neoplasia I syndrome [360].
Up to a third of cases are associated with carcinoid syndrome [413], which is more frequent in patients with large insular carcinoid tumors and/or metastatic disease [411]. In the absence of hepatic involvement, carcinoid syndrome may occur by a direct release of vasoactive substances into the systemic circulation bypassing the liver. Its symptoms include flushes, diarrhea, wheezing, cramping, pruritus, etc. Late-onset, right-sided congestive heart failure related to tricuspid fibrosis can occur, which in rare cases may resolve after tumor removal [414]. Massive medial hypertrophy of the ovarian drainage vein has also been reported [415]. Carcinoid syndrome occurs more frequently in association with insular, midgut-type carcinoids, less so with strumal carcinoid, where it may be associated with constipation [416] and rarely, if ever, with trabecular [411] and mucinous carcinoids [28].
Macroscopically , tumors are unilateral, although they may be associated with MCT or mucinous cystadenoma in the contralateral ovary. Often, carcinoids are found as nodular growths in a teratoma (Fig. 6.24a) or, less frequently, as uniform solid masses. They are only rarely discovered as incidental microscopic findings in a MCT. On cut section, tissue is firm and tan or yellow; mucinous microcysts may be present. An association with both benign and malignant ovarian mucinous tumors [417] has been reported.


Ovarian carcinoids. Characteristic gross appearance of carcinoid originating from teratoma (a). Tumor is tan colored. Insular pattern (b) with gland-like spaces. Insular carcinoid with prominent granular cytoplasm at the periphery (c), expressing chromogranin (d). Trabecular carcinoid (e). Adenocarcinoid with compact granular areas coexisting with chords of goblet cells (f). Vascular invasion in the lung (g) in a patient with ovarian mucinous carcinoid. Trabecular carcinoid with colonic-type mucinous glands (h). Strumal carcinoid (i) with thyroid tissue (left), trabecular carcinoid (center), and appendiceal-type mucinous epithelium (bottom right). Strumal carcinoid: trabecular areas coexist with abortive follicles (j)
Microscopically , insular carcinoid is a midgut carcinoid constituted by anastomosing sheets and nests of uniform, non-mitotic regular cells with salt and pepper nuclei arranged in gland-like or acinar formations (Fig. 6.24b) that may contain proteinaceous or calcified material. A granular, chromogranin positive, cytoplasm is usually evident in most cells and is prominent at the periphery (Figs. 6.24c, d). Luteinized stromal cells are often present in the pushing margins. Their differential diagnosis includes adult granulosa, Sertoli-Leydig cell, and Brenner tumors.
Trabecular carcinoids are arranged in bland uni-or bicellular ribbons of cylindrical cells with clear cytoplasm (Fig. 6.24e) in a background of a dense stroma. Granules are not prominent. Sertoli-Leydig cell tumors with a trabecular pattern should be considered in the differential diagnosis.
Primary mucinous (goblet cell) carcinoid is composed of small glands, cords, or nests populated by varying proportions of goblet and granular cells. Mucin pools are present in the stroma. When not associated with other mature teratoid components, they are indistinguishable from metastases from an appendiceal mucinous carcinoid [418]. A histologic spectrum, ranging from well-differentiated to atypical crowded or microcystic glands to openly malignant goblet cell carcinoma (Fig. 6.24f) with angioinvasive features, is found (Fig. 6.24g) [419]. Mucinous glands lined by goblet cells (Fig. 6.24h) are often focally present in various types of ovarian carcinoid and should not be considered as mucinous carcinoid [28].
Strumal carcinoid shows an intimate admixture of thyroid colloid-containing follicles and trabecular carcinoid (Fig. 6.24i, j), although insular carcinoid nests and, more commonly, mucinous glands with interspersed neuroendocrine cells can also be present. Notable differences with medullary carcinoma of the thyroid include the fact that strumal carcinoid is almost invariably benign, rarely secretes calcitonin, or has an amyloid stroma. In the rare event of metastases, it may do so as carcinoid [258] and exceptionally as follicular carcinoma [420]. The frequent mucinous component can be appendiceal type and may be associated with pseudomyxoma peritonei [305].
Rare primary neuroendocrine carcinomas of the ovary are usually of large cell type and are not associated with teratoma. Their germ cell origin is questionable. They should be differentiated from small cell pulmonary type neuroendocrine carcinomas that originate from MCT [380].
6.3.3.6 Immunohistochemistry
Immunohistochemically, each histologic variant of ovarian carcinoid has a relatively characteristic ultrastructure and neuroendocrine immunophenotype. Insular carcinoid shows pleomorphic neurosecretory granules, which, in contrast, are rounded and uniform in trabecular carcinoid. Strumal carcinoid may show a hybrid (amphicrine) phenotype displaying thyroidal and neuroendocrine features [421] including the expression of TTF-1 in follicular areas. All carcinoids express chromogranin-A, synaptophysin, and CD56 in varying amounts, as well as, specifically, neuropeptides such as YY, calcitonin, substance P, insulin, etc. but rarely have any hormonally functioning activity. Prostate-specific acid phosphatase may be expressed by trabecular strumal carcinoids. CDX2 positivity has been considered a discriminating factor between metastases from gastrointestinal and primary carcinoids, being overexpressed in metastases and only weakly positive in ovarian, teratoma-associated primaries [412].
6.4 Mixed Germ and Sex Cord-Stromal Cell Tumors
Simultaneous growth of primitive sex cord and germ cells occurs in two well-defined histo- and clinicopathologic entities: gonadoblastoma and mixed germ cell-sex cord-stromal tumor (MGC-SCST).
6.4.1 Gonadoblastoma (GB)
GB [422] is a benign lesion that is, however, a precursor of primitive GCT. It characteristically involves gonads of patients with DSD which genetically contain the gonadoblastoma locus on the Y-chromosome (GBY), responsible for the testis-specific protein Y-encoded-1 (TSPY-1) [423]. In these patients, mutations or deletions of genes involved in the development of functional Sertoli cells disrupt their regulation of germ cell maturation, contributing to the maintenance of pluripotency features in the germ cells with a corresponding marker expression, being identical to testicular GCNIS [424]. Consequently, these germ cells may give rise to primitive GCT, more often dysgerminomas and yolk sac tumors, as well as other type II GCT such as EC and mixed GCT.
Since GB almost invariably occurs in Y-chromosome-containing cells, it is not stricto sensu an ovarian (female) tumor. GB also represents a curious crossroads between genetic malformation and neoplasia. Its genetic and clinicopathologic features are also considered in Chaps. 3, 7, and 10.
GB is diagnosed in all age groups with an average age of 18 years and may occur in identical twins. Many present in virilized, phenotypic females [425], frequently with Swyer syndrome. The bilateral gonadal lesions are often an incidental finding in amenorrheic patients presenting microcalcifications [426]. In phenotypic females, they occur in either streak or unknown nature gonads. They are often microscopic unless they are overgrown by primitive GCT, which may present in either gonad.
Microscopically, the characteristic appearance is that of well-delineated insular aggregates of intimately admixed germ and immature sex cord cells arranged in pseudofollicular spaces filled with an eosinophilic, hyaline material that often becomes calcified (Fig. 6.25a, b). The clear germ cell component presents features identical to GCNIS, expressing pluripotentiality markers such as OCT4 (Fig. 6.25c) and SALL4, which contrasts with their negativity in the otherwise α-inhibin-positive sex cord cells. In children, microscopic foci of cribriform, microfollicular structures with eosinophilic contents (Fig. 6.25d) resembling sex cord tumor with annular tubules, but lacking germ cells, have been called gonadoblastoma-like structures [427]. They merely represent examples of abnormal folliculogenesis that involute with sexual maturity.

Gonadoblastoma. (a) Partly calcified nodules of gonadoblastoma in a streak gonad. (b) Higher magnification of gonadoblastoma with large, atypical germ cells positive for OCT4 (c) coexisting with small sex cord cells. (d) Gonadoblastoma-like structures resembling sex cord tumor with annular tubules in a 15 year old girl
Initial overgrowth by dysgerminoma can be seen in discrete areas and is usually revealed by the presence of lymphocytic infiltrates peripheral to GB.
6.4.2 Unclassified Mixed Germ Cell-Sex Cord-Stromal Tumor (UMGC-SCST)
This exceptionally rare tumor was initially reported in the French literature [428] and later in English by Talerman, who coined its current nomenclature [429]. Due to the few reported cases, there is no clear profile of age, distribution, or clinical presentation. UMGC-SCST have been considered in both reviews [430–432] and in a few well-documented case reports [433–437].
Although this tumor occurs in both female and male gonads, it is much more frequent in the ovary. Female patients lack any DSD features and have a normal 46,XX karyotype, some of them being able to reach term pregnancies [431]. It is diagnosed due to the presence of an abdominal mass in girls younger than 10 years, but may also involve any age and only rarely presents with isosexual precocity [434].
Macroscopically, tumors are large and unilateral [431] and only occasionally, bilateral [438].
Histologically, ovarian UMGC-SCST grow in a predominantly solid pattern (Figs. 6.26a, b), although tubular/cribriform, trabecular (Fig. 6.26c), or haphazard, often mixed, arrangements can be found. All patterns show an admixture of sex cord cells with scanty cytoplasm and rounded elongated nuclei that coexist with large, atypical, clear germ cells of variable size, some presenting mitoses [432] and granular (lead shot) chromatin reminiscent of spermatocytic tumors (Fig. 6.26d). Coexistence with gonadoblastoma areas has been reported [439, 440].

Mixed germ cell-sex cord-stromal tumor in a young girl (a), exhibiting a diffusely mixed population (b) of clear germ cells and sex cord elements. This trabecular pattern reminiscent of embryonic sex cords (c) occurred in a 24-year-old phenotypic female. Higher magnification of solid area in the same case (d). Abundant germ cells with nuclei exhibiting a “lead shot”·chromatin coexist with smaller sex cord cells with fusiform nuclei
Retiform [435, 436] and heterologous [437] areas are occasionally present. Similar features are found in Sertoli-Leydig cell tumors which, in their retiform variant, are primitive tumors of sex cord lineage that mirror early ovarian organogenesis, showing sex cord blastema and its connection with the primitive rete cords [441]. It is therefore not surprising that the earliest descriptions of UMGC-SCST in the French literature used the term Pflügeromes [442] due to their resemblance to the trabecular embryonic sex cords described by Pflüger [443].
Immunohistochemically, their germ cells express pluripotentiality markers such as OCT4 and SALL4 [430, 432] as well as other germ cell markers such as PLAP and CD117. 12p amplification does not usually occur in the germ cell component, being present only in isolated instances [430]. Sex cord components characteristically express α-inhibin.
Both pluripotential germ cell elements and immature sex cord components are able to metastasize [434] and recur in the abdominal cavity [433]. Only rare cases give rise to malignant germ cell tumors [444].
6.5 Germ Cell Tumors Originating from Somatic Müllerian Neoplasms (Type VI GCT)
This group of rare, type VI GCT includes secondary malignant GCT growth patterns arising from somatic-cell-derived tumors. They likely originate from tumor stem cells behaving as induced pluripotential cells (iPSC) (see Chap. 3) and occur in association with neoplasms of Müllerian derivation, including endometriosis, and are not related to chemotherapy. The GCT patterns reproduced are exclusively non-dysgerminomas and frequently, glandular YST. They occur predominantly in pre-and postmenopausal patients, reaching a peak in the seventh decade of life. Often they are diagnosed in advanced clinical stages and behave aggressively. Similar tumors occur with a lesser frequency in the testis (see Chap. 7) as well as in extragonadal locations such as the uterus, head and neck region [445], stomach [446], urinary tract [109], etc. (see Chaps. 11 and 12).
In the WHO classification of female genital tract tumors [447], they are not considered a discrete entity. However, although currently very unusual tumors, they mainly involve elderly women, an age group that is increasing in numbers due the progressive aging of the population in developed countries. This, coupled with their difficult diagnosis and poor prognosis, would merit considering them as a separate category in tumor classifications of ovarian and uterine tumors. In the literature, most publications are case reports with only few relevant series [77, 78].
Recent data from histopathologic-based registries from the European Union (see Chap. 2), the United Kingdom [22], and the United States [23] reveal an unusual GCT incidence peak at 65–75 that corresponds to GCT arising from somatic Müllerian neoplasms (GCT-SMN). Although they are not referred to as such in the various registries, it is possible that many such cases pass unrecognized due to their rarity and difficult diagnosis, often complicated by the malignant GCT overgrowth effacing the original somatic tumor [78]. European and US population-based statistics predict that almost 30 % of the population will be over 65 by 2035 and the population pyramid inverted by 2080, with a substantial increase of women over the age of 85 [448, 449]. This would mean that, in the not too distant future, age-related tumors such as these and type II endometrial cancers will experiment a remarkable increase in incidence.
Historically, GCT-SMN were described for the first time in the female genital tract associated with endometriosis [450] and, later, with endometrioid adenocarcinomas and carcinosarcomas of both ovary and endometrium [77, 451, 452] and much less frequently with serous [453] and mucinous carcinomas [454].
We have studied 25 cases of ovarian and uterine GCT-SMN arising from both endometrioid and clear cell carcinomas [133, 455]. Clinically, their ages ranged from 30 to 94 with the majority of cases in the sixth and seventh decades of life (Fig. 6.27). They presented with the usual nonspecific symptoms of ovarian and uterine tumors. At the time of surgery, they were often found in FIGO stages II–IV. Survival data indicate both poor prognosis and response to treatment [77]. A r.

Age distribution in 25 cases of somatic Müllerian malignancies of ovary and uterus associated with germ cell tumor
Histologically, most tumors show diffuse intermingling of Müllerian somatic tumors with non-dysgerminomatous GCT.
The main histologic categories of GCT-SMN correspond to:
-
(a)
Endometrioid neoplasms associated with YST
-
(b)
Clear cell carcinoma (CCC) associated with YST
-
(c)
Endometrioid or CCC associated with YST and other GCT patterns
6.5.1 Endometrioid Neoplasms Associated with YST
These somatically derived YST [456] represent the most numerous category of GCT-SMN. We have analyzed a series of 15 cases [455] corresponding to women aged between 30 and 80 years. Of these, most (11/15) were in the fifth and sixth decades of life. Tumors were in an advanced stage in over half of cases. Mortality was high with a poor response to chemotherapy. Macroscopically, the tumors were bulky with an average size of 15 cm and only once were bilateral. A third of cases were associated with endometriosis or endometrioid cystoadenofibromas. The endometrioid adenocarcinoma constituted the main neoplastic component and was well differentiated with morular formation in four cases, moderately differentiated in six and the remaining five were carcinosarcomas. A recently published series [456] provides a similar a clinicopathologic profile. The YST element is characteristically heterogeneous, showing small foci of classic microcystic YST (Fig. 6.28a, b) in half of cases, coexisting with areas of glandular pattern that were the predominant or the exclusive pattern in three cases (Fig. 6.28c). The cylindrical cell lining of glandular spaces of both endometrioid tumor and glandular YST can be very similar, and, often, apical and basal vacuolation of the YST intestinal-type glands is the only reliable histologic discriminating feature (Fig. 6.28c). For this reason, immunohistochemistry is necessary to identify the differential diagnostic phenotypes of each component. The endometrioid component expresses EMA (Fig. 6.29a), CK7 [132], and estrogen receptors [77] as well as PAX8 (Fig. 6.29b) [457]. In contrast, glandular YST can be focally positive for CK7 and EMA but expresses diffusely SALL4 and villin (Fig. 6.29c, d) as well as focal, patchy AFP and GPC3. Intestinal differentiation in YST reveals a strong CDX2 and HepPar-1 expression. Immunohistochemical differential diagnostic data are shown in Table 6.6.

Interphase of well-differentiated endometrioid adenocarcinoma with microcystic (a) and hepatoid (b) YST. Glandular YST (c) resembles endometrioid adenocarcinoma except for the diffuse basal vacuolation and periglandular rarefaction

Coexistence of glandular YST with endometrioid adenocarcinoma. (a, b) EMA and PAX8 are only expressed in the endometrioid component, while only glandular YST expresses SALL4 (c) and villin (d)
6.5.2 Clear Cell Carcinoma (CCC) Associated with YST
We have analyzed twelve cases of this unusual association from several European consultation materials [133]. They involved a similar group than endometrioid tumors, ranging from 37 to 94 years, with an average of 62. They were also smaller in size, ranging from 2 to 20 cm in diameter, average 12 cm. Endometriosis was present in all but two cases and 50 % were in advanced clinical stages. Histologically, all corresponded to characteristic tubulopapillary patterns of clear cell carcinomas (CCC) except one case of borderline clear cell adenofibroma. The YST component was almost invariably of glandular type, being predominant in over half of cases. Clinically, eight patients died 16 months to three years after diagnosis. Four are alive and well with short follow ups of up to 4 years.
Since the initial descriptions of YST [458], CCC has been the main differential diagnosis with YST. Since glandular YST is the most frequently associated pattern with CCC [133], the difficulty in its differentiation from CCC lies in the fact that glandular YST may have a papillary growth pattern and extensive vacuolation, so closely resembling CCC that it can be taken for a hybrid phenotype (Fig. 6.30a). Histologically, glandular YST coexists and merges with CCC (Fig. 6.30b) but differs from it in their more regular nuclei, polarized vacuoles in apical or subnuclear position, subepithelial stromal rarefaction, and occasional presence of goblet cells and hyaline globules. These features should differentiate it from the usually complex CCC histology [459]. For these reasons, immunohistochemistry is necessary for differential diagnosis and should not be based on the expression of a single antibody [112, 460], since some otherwise “characteristic markers” can be expressed by both components. Therefore, diagnostic accuracy is improved by contrasting wider immunohistochemical panels for CCC [459] and YST [96]. Before these comprehensive panels were available, the differentiation between YST and CCC was problematic; thus it is difficult to know if previously reported cases of CCC with AFP expression were either pure CCCs with ectopic AFP secretion, papillary glandular YST resembling CCC exhibiting AFP expression, or an admixture of both [461–464].

(a) YST resembling clear cell carcinoma (CCC). (b) Interphase between CCC (left) and glandular YST (right). HNF1-ß (c) is expressed by both glandular YST (top) and CCC (bottom). EMA (d) can show a strong expression in glandular papillary YST. However, Napsin A (e) is only expressed in CCC, while villin (f) only stains glandular YST
Neither AFP nor GPC3 [460] fully discriminates between CCC and YST and the same is true for other traditional markers of CCC and YST: HNF1-β, a well-known marker for most CCC [465], is also expressed in YST [133, 464] (Fig. 6.30c), which limits its usefulness in differentiating these patterns. The absence of expression of CK7 and EMA [466] has also been considered characteristic of YST. However, it can also be focally present in glandular YST (Fig. 6.30d), where this expression may represent gastropancreatic differentiation [133].
PAX8 and Napsin A are more specific antibodies (Fig. 6.30e) expressed in CCC [467–469] but not in YST patterns even the glandular ones [133].
SALL4 is an excellent marker of cells retaining stemness, and consequently it is expressed in YST of both classic and glandular type where it represents, as in carcinoma of the stomach, fetal gut differentiation [470]. It is, however, negative in CCC [119]. Villin, although considered a relatively nonspecific antibody, is constantly expressed in YST (Fig. 6.30f) but not in CCC [96]. Finally, HepPar-1 is frequently focally positive in YST and invariably absent in CCC.
As a summary (Table 6.6), CCC and glandular YST may coexpress HNF1-β, CK7, EMA, and GPC3. Identification of CCC areas is based on expression of PAX8 and NapsinA, while YST is best identified by its AFP, SALL4, villin, and, eventually, focal HepPar-1 expression.
6.5.3 Endometrioid or CCC Associated with YST and Other Polydifferentiated GCT Patterns
This rare association occurs in neoplasms of the uterus and ovary as a combination of endometrioid or CCC, with glandular YST and other GCT growth patterns such as choriocarcinoma, mature teratoma, immature neural structures similar to those found in IT, as well as mimics of embryonal carcinoma and polyembryoma. It is possible that, in the past, some tumors in this category were reported as immature teratoma in extraovarian locations such as the uterus [471, 472]. Only isolated well-documented cases [473] have been reported in the literature.
We have studied five cases, associated with endometrioid carcinoma and CCC [474]. All had a glandular YST component. Coexistence of glandular YST and CCC was observed in some cysts which presented a biphasic lining of both clear cell and endoderm. Other components included, in three cases, an abundance of both neural blastema and neuroepithelial pigmented tubules of the type seen in immature teratoma (Fig. 6.31), one of them differentiating a glioblastoma multiforme pattern. In two instances, there were patterns resembling areas of polyembryoma with embryoids expressing a characteristic ecto- and endodermal immunophenotype (Fig. 6.32). Another included a choriocarcinoma and yet another, mature components such as squamous and respiratory epithelia, striated muscle and cartilage of the type seen on mature teratoma, but lacking a dermoid appearance. No areas of dysgerminoma were present.

Pigmented neuroepithelium in a mixed CCC-glandular YST

Embryoid body in an endometrioid carcinoma associated with embryonal carcinoma-like patterns. The area corresponding to endodermal differentiation expresses AFP and only focally HepPar-1. GPC3 stains both ecto- and endodermal areas, while SOX2 is only positive in the ectoderm. SALL4 stains all immature areas
Immunohistochemically, all GCT components (glandular YST, neural components, choriocarcinoma, and embryoids) show a characteristic phenotype. Some areas may have an incomplete EC immunophenotype expressing OCT4, but weak and focal SOX2 and CD30. These incomplete EC features partly resemble type II testicular-type tumors and would exemplify the developmental capacities of iPSC. The existence in these tumors of cells retaining a high degree of stemness [452] is confirmed by the constant presence of foci of cells expressing OCT4 (see Fig. 3.34) [474] (Fig. 6.33), similar to iPSC. OCT4 expression has been reported in high-grade immature teratomas [187], but not in Müllerian-derived GCT.

Uterine carcinosarcoma associated with YST. Atypical glandular areas have a patchy expression of OCT4
Tumor stem cells have been demonstrated in neoplasms classically associated with polydifferentiations such as uterine carcinosarcomas [475], endometrioid carcinoma [476, 477], endometriosis [478], and CCC [479]. An intriguing paper [480] has demonstrated that the ovarian surface epithelium in the menopause is able to produce stem cells capable of giving rise to embryoid body-, oocyte-, and blastocyst-like structures but not teratomas when transplanted to mice. This could be exemplified by a recent case of mature uterine teratoma where the comparison of DNA profile of normal uterine tissue and a uterine teratoma suggested an origin from a pluripotential stem cell of the uterus [481].
As a summary, somatic-derived malignant stem cells behaving as an induced pluripotential stem cell (iPSC) can give rise to a malignant stem cell tumor with features of embryonal carcinoma, even with an organoid arrangement into embryoids. They may also differentiate into endodermal structures of YST and trophoblast as well as embryonal mesenchymal and neural structures present in teratoid carcinosarcoma. It is also speculated that rare “neometaplastic” monophyletic neoplasm such as extrarenal Wilms, extra-axial ependymomas, hepatoid tumors, and even intestinal mucinous tumors may also represent differentiations from induced pluripotential stem cells.
6.6 Extraovarian Germ Cell Tumors of the Female Genital Tract
Extraovarian GCT in the female genital tract are rare. Their true incidence is, as in other extragonadal teratomas, difficult to know, since most reports are clinically oriented and few have a well-documented histopathologic description. Likewise, the loose usage of the term teratoma in the older literature to describe some polydifferentiated neoplasms may have included some cases of mixed Müllerian tumors. Additionally, in the uterus, cervix, and perhaps the fallopian tube, iatrogenic fetal implants [482] may display various mature tissues embedded in the endometrium [483, 484] that can be erroneously diagnosed as mature teratoma.
Fallopian tube GCT are possibly the most common GCT in female genital tract outside the ovary and are also type IV tumors with a parthenogenetic origin. Most represent incidental findings and are small mature teratomas that tend to adopt either a solid [485] (Figs. 6.34a, b) or cystic [486, 487] appearance. There are, however, some reported cases of immature teratomas [488].

A small solid mature teratoma in the lumen of a fallopian tube (a) is composed of fibroadipose tissue with some sebaceous glands (b)
Uterine and cervical GCT are extremely rare lesions (see Chap. 2) and are often polypoid, mature teratomas [489–491] even showing the presence of thyroid tissue [492]. It is possible that cases reported as immature teratomas or those with malignant transformation [493, 472] may represent examples of type VI tumors of somatic Müllerian tumors associated with the polydifferentiated GCT patterns, as described above. In the cervix, both mature [494, 495] and immature [496] teratomas have been reported, as well as rare cases of YST [497, 498].
Vulvovaginal GCT include primary YST [499–501] that may correspond to type I GCT in childhood as well as rare immature teratoma [502].
Bibliography
Marsden HB, Birch JM, Swindell R. Germ cell tumours of childhood: a review of 137 cases. J Clin Pathol. 1981;34(8):879–83.
Katsube Y, Berg JW, Silverberg SG. Epidemiologic pathology of ovarian tumors: a histopathologic review of primary ovarian neoplasms diagnosed in the Denver Standard Metropolitan Statistical Area, 1 July-31 December 1969 and 1 July-31 December 1979. Int J Gynecol Pathol. 1982;1(1):3–16.
Lo Curto M, D’Angelo P, Cecchetto G, Klersy C, Dall’Igna P, Federico A, et al. Mature and immature teratomas: results of the first paediatric Italian study. Pediatr Surg Int. 2007;23(4):315–22. doi:10.1007/s00383-007-1890-1.
Quirk JT, Natarajan N, Mettlin CJ. Age-specific ovarian cancer incidence rate patterns in the United States. Gynecol Oncol 2005;99(1):248–50. doi:S0090-8258(05)00506-8 [pii] 10.1016/j.ygyno.2005.06.052.
Moller H, Evans H. Epidemiology of gonadal germ cell cancer in males and females. APMIS 2003;111(1):43–6; discussion 6–8. doi:apm1110107 [pii].
Lee ST, Choi MH, Lee EJ, Gong SP, Jang M, Park SH et al. Establishment of autologous embryonic stem cells derived from preantral follicle culture and oocyte parthenogenesis. Fertil Steril 2008;90(5):1910–20. doi:S0015-0282(07)00207-5 [pii] 10.1016/j.fertnstert.2007.01.099.
Oliveira FG, Dozortsev D, Diamond MP, Fracasso A, Abdelmassih S, Abdelmassih V et al. Evidence of parthenogenetic origin of ovarian teratoma: case report. Hum Reprod 2004;19(8):1867–70. doi:10.1093/humrep/deh345. deh345 [pii].
Weiss JR, Burgess JR, Kaplan KJ. Fetiform teratoma (homunculus). Arch Pathol Lab Med 2006;130(10):1552–6. doi:CR51040 [pii] 10.1043/1543-2165(2006)130[1552:FTH]2.0.CO;2.
Sheikine Y, Genega E, Melamed J, Lee P, Reuter VE, Ye H. Molecular genetics of testicular germ cell tumors. Am J Cancer Res. 2012;2(2):153–67.
Goodman S, Zhang L, Cheng L, Jiang Z. Differential expression of IMP3 between male and female mature teratomas–immunohistochemical evidence of malignant nature. Histopathology. 2014;65(4):483–9. doi:10.1111/his.12409.
Sigismondi C, Scollo P, Ferrandina G, Candiani M, Angioli R, Vigano R, et al. Management of bilateral malignant ovarian germ cell tumors: a MITO-9 Retrospective Study. Int J Gynecol Cancer. 2015;25(2):203–7. doi:10.1097/IGC.0000000000000358.
Horton Z, Schlatter M, Schultz S. Pediatric germ cell tumors. Surg Oncol. 2007;16(3) doi:10.1016/j.suronc.2007.07.005.
Gobbi D, Fascetti Leon F, Aquino A, Melchionda F, Lima M. Metachronous bilateral ovarian teratoma: a germ-line familial disorder and review of surgical management options. J Pediatr Adolesc Gynecol 2013;26(5):e105–7. doi:10.1016/j.jpag.2013.02.006. S1083-3188(13)00102-2 [pii].
Galani E, Alamanis C, Dimopoulos MA. Familial female and male germ cell cancer. A new syndrome? Gynecol Oncol 2005;96(1):254–5. doi:S0090-8258(04)00802-9 [pii] 10.1016/j.ygyno.2004.09.047.
Giambartolomei C, Mueller CM, Greene MH, Korde LA. A mini-review of familial ovarian germ cell tumors: an additional manifestation of the familial testicular germ cell tumor syndrome. Cancer Epidemiol 2009;33(1):31–6. doi:10.1016/j.canep.2009.04.015. S1877-7821(09)00038-1 [pii].
Stevens LC, Varnum DS. The development of teratomas from parthenogenetically activated ovarian mouse eggs. Dev Biol. 1974;37(2):369–80. doi:0012-1606(74)90155-9 [pii].
Prat J, Cao D, Carinelli SG, Nogales FF, Vang R, Zaloudek CJ. Germ cell tumours. In:WHO classification of tumours of female reproductive organs. 4th ed. Lyon: International Agency for Research on Cancer; 2014. p. 57–62.
Nogales FF, Dulcey I, Preda O. Germ cell tumors of the ovary: an update. Arch Pathol Lab Med. 2014;138(3):351–62. doi:10.5858/arpa.2012-0547-RA.
Prat J, Cao D, Carinelli SG, Nogales FF, Vang R, Zaloudek CJ. World health organization classification of tumours In: Kurman RY, Young RH. editor. Tumours of the female genital organs. 2nd ed. Lyon: IARCPress; 2014.
Smith HO, Berwick M, Verschraegen CF, Wiggins C, Lansing L, Muller CY et al. Incidence and survival rates for female malignant germ cell tumors. Obstet Gynecol. 2006;107(5):1075–85. doi:107/5/1075 [pii] 10.1097/01.AOG.0000216004.22588.ce.
Quirk JT, Natarajan N. Ovarian cancer incidence in the United States, 1992–1999. Gynecol Oncol 2005;97(2):519–23. doi: S0090-8258(05)00123-X [pii] 10.1016/j.ygyno.2005.02.007.
Arora RS, Alston RD, Eden TO, Geraci M, Birch JM. Comparative incidence patterns and trends of gonadal and extragonadal germ cell tumors in England, 1979 to 2003. Cancer. 2012;118(17):4290–7. doi:10.1002/cncr.27403.
Stang A, Trabert B, Wentzensen N, Cook MB, Rusner C, Oosterhuis JW, et al. Gonadal and extragonadal germ cell tumours in the United States, 1973–2007. Int J Androl. 2012;35(4):616–25. doi:10.1111/j.1365-2605.2011.01245.x.
Pierce GB, Speers WC. Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer Res. 1988;48(8):1996–2004.
Nogales FF. Haines & Taylor obstetrical and gynaecological pathology. In: Fox H, Wells M, editors. Haines & Taylor obstetrical and gynaecological pathology. 5th ed. Edinburgh: Churchill Livingstone; 2003. p. 771–820.
Ulbright TM. Germ cell tumors of the gonads: a selective review emphasizing problems in differential diagnosis, newly appreciated, and controversial issues. Mod Pathol 2005;18 Suppl 2:S61–79. doi:3800310 [pii] 10.1038/modpathol.3800310.
Prat JC, D, Carinelli SG, Nogales FF, Vang R, Zaloudek CJ, International Agency for Research on Cancer., World Health Organization. WHO classification of tumours of female reproductive organs. World Health Organization classification of tumours. 4th ed. Lyon: International Agency for Research on Cancer; 2014. p. 57–62.
Oliva H, Young RH. Germ cell tumours of the ovary: selected topics. Diagnostic Histopathol. 2014;20(9):364–375.
Pectasides D, Pectasides E, Kassanos D. Germ cell tumors of the ovary. Cancer Treat Rev 2008;34(5):427–41. doi:10.1016/j.ctrv.2008.02.002. S0305-7372(08)00025-X [pii].
Lee KH, Lee IH, Kim BG, Nam JH, Kim WK, Kang SB et al. Clinicopathologic characteristics of malignant germ cell tumors in the ovaries of Korean women: a Korean Gynecologic Oncology Group Study. Int J Gynecol Cancer 2009;19(1):84–7. doi:10.1111/IGC.0b013e31819914c5. 00009577-200901000-00016 [pii].
Vazquez I, Rustin GJ. Current controversies in the management of germ cell ovarian tumours. Curr Opin Oncol 2013;25(5):539–45. doi:10.1097/01.cco.0000432609.39293.77. 00001622-201309000-00014 [pii].
Gershenson DM. Treatment of ovarian cancer in young women. Clin Obstet Gynecol. 2012;55(1):65–74. doi:10.1097/GRF.0b013e318248045b. 00003081-201203000-00008 [pii].
Gershenson DM. Management of ovarian germ cell tumors. J Clin Oncol. 2007;25(20):2938–43. doi:25/20/2938 [pii] 10.1200/JCO.2007.10.8738.
Solheim O, Gershenson DM, Trope CG, Rokkones E, Sun CC, Weedon-Fekjaer H et al. Prognostic factors in malignant ovarian germ cell tumours (The Surveillance, Epidemiology and End Results experience 1978–2010). Eur J Cancer 2014;50(11):1942–50. doi:10.1016/j.ejca.2014.03.288. S0959-8049(14)00573-5 [pii].
Mangili G, Sigismondi C, Lorusso D, Cormio G, Scollo P, Vigano R et al. Is surgical restaging indicated in apparent stage IA pure ovarian dysgerminoma? The MITO group retrospective experience. Gynecol Oncol 2011;121(2):280–4. doi:10.1016/j.ygyno.2011.01.006. S0090-8258(11)00018-7 [pii].
Gershenson DM, Miller AM, Champion VL, Monahan PO, Zhao Q, Cella D et al. Reproductive and sexual function after platinum-based chemotherapy in long-term ovarian germ cell tumor survivors: a Gynecologic Oncology Group Study. J Clin Oncol. 2007;25(19):2792–7. doi:25/19/2792 [pii] 10.1200/JCO.2006.08.4590.
Solheim O, Trope CG, Rokkones E, Kaern J, Paulsen T, Salvesen HB et al. Fertility and gonadal function after adjuvant therapy in women diagnosed with a malignant ovarian germ cell tumor (MOGCT) during the “cisplatin era”. Gynecol Oncol 2015;136(2):224–9. doi:10.1016/j.ygyno.2014.12.010 S0090-8258(14)01565-0 [pii].
Cossu-Rocca P, Zhang S, Roth LM, Eble JN, Zheng W, Karim FW et al. Chromosome 12p abnormalities in dysgerminoma of the ovary: a FISH analysis. Mod Pathol 2006;19(4):611–5. doi:3800576 [pii] 10.1038/modpathol.3800576.
Cheng L, Roth LM, Zhang S, Wang M, Morton MJ, Zheng W, et al. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer. 2011;117(10):2096–103. doi:10.1002/cncr.25794.
Meyer R. Ovarialtumoren und Geschlechtlichkeit. Ein Beitrag zur funktionellen Betrachtung der Geschwülste: I.“Disgerminome” beider Geschlechter bei Störung in der Entwicklung der Keimdrüsen, II. Granulosazelltumoren mit “Verweiblichung,” III. Arrhenoblastome mit “Vermännlichung”. Klin Wochenschr. 1930;9:2237–40.
Stewart CJ, Baker E, Beaton C, Crook M, Peverall J, Wallace S. Detection of Y-chromosome in gonadal tumours using fluorescence in situ hybridization: diagnostic value in intersex conditions including older patients with clinically unsuspected androgen insensitivity syndrome. Histopathology 2008;52(2):175–82. doi:10.1111/j.1365-2559.2007.02927.x. HIS2927 [pii].
Boyes DA, Pankratz E, Walliford BW, White GW, Fairey RN. Experience with dysgerminomas at the Cancer Control Agency of British Columbia. Gynecol Oncol. 1978;6(2):123–9.
Linton EB. Dysgerminoma of the ovary. J Reprod Med. 1981;26(5):255–60.
Kraggerud SM, Szymanska J, Abeler VM, Kaern J, Eknaes M, Heim S, et al. DNA copy number changes in malignant ovarian germ cell tumors. Cancer Res. 2000;60(11):3025–30.
Zhao S, Kato N, Endoh Y, Jin Z, Ajioka Y, Motoyama T. Ovarian gonadoblastoma with mixed germ cell tumor in a woman with 46, XX karyotype and successful pregnancies. Pathol Int. 2000;50(4):332–5. doi:pin1041 [pii]
Koo YJ, Chun YK, Kwon YS, Lee IH, Kim TJ, Lee KH, et al. Ovarian gonadoblastoma with dysgermi-noma in a woman with 46XX karyotype. Pathol Int. 2011;61(3):171–3. doi:10.1111/j.1440-1827.2010.02636.x.
Tanaka Y, Fujiwara K, Yamauchi H, Mikami Y, Kohno I. Pregnancy in a woman with a Y chromosome after removal of an ovarian dysgerminoma. Gynecol Oncol 2000;79(3):519–21. doi:10.1006/gyno.2000.6004. S0090-8258(00)96004-9 [pii].
Paliwal P, Sharma A, Birla S, Kriplani A, Khadgawat R. Identification of novel SRY mutations and SF1 (NR5A1) changes in patients with pure gonadal dysgenesis and 46, XY karyotype. Mol Hum Reprod 2011;17(6):372–8. doi:10.1093/molehr/gar002 gar002 [pii].
Rozenholc A, Abdulcadir J, Pelte MF, Petignat P. A pelvic mass on ultrasonography and high human chorionic gonadotropin level: not always an ectopic pregnancy. BMJ Case Rep. 2012;2012. doi:10.1136/bcr.01.2012.5577 bcr0120125577 [pii] bcr.01.2012.5577 [pii].
Brettell JR, Miles PA, Herrera G, Greenberg H. Dysgerminoma with syncytiotrophoblastic giant cells presenting as a hydatidiform mole. Gynecol Oncol. 1984;18(3):393–401.
Nourani M, Manera RB. Pediatric ovarian dysgerminoma presenting with hypercalcemia and chronic constipation: a case report. J Pediatr Hematol Oncol. 2013;35(7):e272–3. doi:10.1097/MPH.0b013e31829bcaf1.
Rotman M, Dorfmann H, de Seze S, Kahn MF. Coexistence d’un lupus erythemateux disseminé et d’un seminome de l’ovarie. Guérison apparente rapide du lupus après ablation de la tumeur. Recul de 7 ans. Nouv Press Med. 1972;1(13):853–7.
Yeh KE, Marcus PS, Fong TL. Paraneoplastic cholestasis associated with ovarian dysgerminoma. Obstet Gynecol. 2015;126(2):431–4. doi:10.1097/AOG.0000000000000699.
Buskirk SJ, Schray MF, Podratz KC, Lee RA, Stanhope CR, Gaffey TA, et al. Ovarian dysgerminoma: a retrospective analysis of results of treatment, sites of treatment failure, and radiosensitivity. Mayo Clin Proc. 1987;62(12):1149–57.
De Palo G, Lattuada A, Kenda R, Musumeci R, Zanini M, Pilotti S, et al. Germ cell tumors of the ovary: the experience of the National Cancer Institute of Milan. I. Dysgerminoma. Int J Radiat Oncol Biol Phys. 1987;13(6):853–60. doi:0360-3016(87)90099-X [pii].
Schweyer S, Soruri A, Baumhoer D, Peters J, Cattaruzza M, Radzun HJ, et al. Expression of CXC chemokine IP-10 in testicular germ cell tumours. J Pathol. 2002;197(1):89–97. doi:10.1002/path.1094.
Kato N, Kurotaki H, Uchigasaki S, Fukase M, Kurose A. Ovarian clear cell carcinoma with plasma cell-rich inflammatory stroma: a clear cell carcinoma subgroup with distinct clinicopathological features. Histopathology. 2015; doi:10.1111/his.12783.
Sellami-Dhouib R, Nasfi A, Mejri NT, Doghri R, Charfi L, Sassi S, et al. Ovarian ALK+ diffuse large B-cell lymphoma: a case report and a review of the literature. Int J Gynecol Pathol. 2013;32(5):471–5. doi:10.1097/PGP.0b013e31826cbd6e.
Alvarado-Cabrero I, Valencia-Cedillo R, Mohs-Alfaro M, De Anda-Gonzalez J. Ovarian dysgerminoma associated with fibrosarcoma: a case report. Int J Gynecol Pathol. 2011;30(5):466–9. doi:10.1097/PGP.0b013e318217139e.
Iczkowski KA, Butler SL, Shanks JH, Hossain D, Schall A, Meiers I et al. Trials of new germ cell immunohistochemical stains in 93 extragonadal and metastatic germ cell tumors. Hum Pathol 2008;39(2):275–81. doi:S0046-8177(07)00357-7 [pii] 10.1016/j.humpath.2007.07.002.
Idrees M, Saxena R, Cheng L, Ulbright TM, Badve S. Podoplanin, a novel marker for seminoma: A comparison study evaluating immunohistochemical expression of podoplanin and OCT3/4. Ann Diagn Pathol 2010;14(5):331–6. doi:10.1016/j.anndiagpath.2010.05.008 S1092-9134(10)00095-X [pii].
Chang MC, Vargas SO, Hornick JL, Hirsch MS, Crum CP, Nucci MR. Embryonic stem cell transcription factors and D2-40 (podoplanin) as diagnostic immunohistochemical markers in ovarian germ cell tumors. Int J Gynecol Pathol. 2009;28(4):347–55. doi:10.1097/PGP.0b013e318195da86.
Rijlaarsdam MA, van Herk HA, Gillis AJ, Stoop H, Jenster G, Martens J et al. Specific detection of OCT3/4 isoform A/B/B1 expression in solid (germ cell) tumours and cell lines: confirmation of OCT3/4 specificity for germ cell tumours. Br J Cancer 2011;105(6):854–63. doi:10.1038/bjc.2011.270 bjc2011270 [pii].
Cossu-Rocca P, Jones TD, Roth LM, Eble JN, Zheng WX, Karim FWA, et al. Cytokeratin and CD30 expression in dysgerminoma. Hum Pathol. 2006;37(8):1015–21. doi:10.1016/j.humpath.2006.02.018.
Battifora H, Sheibani K, Tubbs RR, Kopinski MI, Sun TT. Antikeratin antibodies in tumor diagnosis. Distinction between seminoma and embryonal carcinoma. Cancer. 1984;54(5):843–8.
Parkash V, Carcangiu ML. Transformation of ovarian dysgerminoma to yolk sac tumor: evidence for a histogenetic continuum. Mod Pathol. 1995;8(8):881–7.
Tatekawa Y, Kemmotsu H, Mouri T, Joe K, Ohkawa H. A case of pediatric ovarian dysgerminoma associated with high serum levels and positive immunohistochemical staining of neuron-specific enolase. J Pediatr Surg. 2004;39(9):1437–9. doi:S0022346804003628 [pii].
Nogales FF, Ruiz Avila I, Concha A, del Moral E. Immature endodermal teratoma of the ovary: embryologic correlations and immunohistochemistry. Hum Pathol. 1993;24(4):364–70.
Young RH. A brief history of the pathology of the gonads. Mod Pathol. 2005;18:S3–S17. doi:10.1038/modpathol.3800305.
Young RH. The yolk sac tumor: reflections on a remarkable neoplasm and two of the many intrigued by It-Gunnar Teilum and Aleksander Talerman-and the bond it formed between them. Int J Surg Pathol 2014;22(8):677–87. doi:10.1177/1066896914558265 1066896914558265 [pii].
Nogales FF, Preda O, Nicolae A. Yolk sac tumours revisited. A review of their many faces and names. Histopathology. 2012;60(7):1023–33. doi:10.1111/j.1365-2559.2011.03889.x.
Nogales FF, Talerman A, Kubik-Huch RA, Tavassoli FA, Devousssoux-Shisheboran M. Germ cell tumours In: Tavassoli FA, P Devilee, Eds. Tumours of the breast and female genital tract. Lyon: IARC Press; 2003. p. 163–75.
Mosbech CH, Rechnitzer C, Brok JS, Rajpert-De Meyts E, Hoei-Hansen CE. Recent advances in understanding the etiology and pathogenesis of pediatric germ cell tumors. J Pediatr Hematol Oncol. 2014;36(4):263–70. doi:10.1097/MPH.0000000000000125.
Simon RA, Laughlin TS, Nuccie B, Wang N, Rothberg PG, Wang X. A 46 XY phenotypic female adolescent with bilateral gonadal tumors consisting of five different components. Int J Gynecol Pathol 2008;27(3):407–11. doi:10.1097/PGP.0b013e31815d05d4 00004347-200807000-00013 [pii].
Langley FA, Govan AD, Anderson MC, Gowing NF, Woodcock AS, Harilal KR. Yolk sac and allied tumours of the ovary. Histopathology. 1981;5(4):389–401.
Cooper C, Cooper M, Carter J, Russell P. Gonadoblastoma progressing to dysgerminoma in a 55-year-old woman with normal karyotype. Pathology 2007;39(2):284–5. doi:773354847 [pii] 10.1080/00313020701230708.
Nogales FF, Bergeron C, Carvia RE, Alvaro T, Fulwood HR. Ovarian endometrioid tumors with yolk sac tumor component, an unusual form of ovarian neoplasm. Analysis of six cases. Am J Surg Pathol. 1996;20(9):1056–66.
Roth LM, Talerman A, Levy T, Sukmanov O, Czernobilsky B. Ovarian yolk sac tumors in older women arising from epithelial ovarian tumors or with no detectable epithelial component. Int J Gynecol Pathol. 2011;30(5):442–51. doi:10.1097/PGP.0b013e3182164386.
Elhosseiny A, Gonzalez O, Hedjazi M, Pillari V, Howard R. Endodermal sinus tumor and benign cystic teratoma of the ovary occurring simultaneously. N Y State J Med. 1989;89(4):233–4.
Kurman RJ, Norris HJ. Endodermal sinus tumor of the ovary: a clinical and pathologic analysis of 71 cases. Cancer. 1976;38(6):2404–19.
Arima N, Tanimoto A, Hayashi R, Hamada T, Sasaguri Y. Ovarian yolk sac tumor with virilization during pregnancy: Immunohistochemical demonstration of Leydig cells as functioning stroma. Pathol Int. 2000;50(6):520–5. doi:10.1046/j.1440-1827.2000.01073.x.
Pecorelli S, Sartori E, Favalli G, Ugazio AG, Gastaldi A. Ataxia-telangiectasia and endodermal sinus tumor of the ovary: report of a case. Gynecol Oncol. 1988;29(2):240–4. doi:0090-8258(88)90219-3 [pii].
Billmire DF, Cullen JW, Rescorla FJ, Davis M, Schlatter MG, Olson TA et al. Surveillance after initial surgery for pediatric and adolescent girls with stage I ovarian germ cell tumors: report from the Children’s Oncology Group. J Clin Oncol 2014;32(5):465–70. doi:10.1200/JCO.2013.51.1006 JCO.2013.51.1006 [pii].
Mooney EE, Nogales FF, Tavassoli FA. Hepatocytic differentiation in retiform Sertoli-Leydig cell tumors: distinguishing a heterologous element from Leydig cells. Hum Pathol. 1999;30(6):611–7.
Williams SD, Einhorn LH. Brain metastases in disseminated germinal neoplasms: incidence and clinical course. Cancer. 1979;44(4):1514–6.
Clement PB, Young RH, Scully RE. Endometrioid-like variant of ovarian yolk sac tumor. A clinicopathological analysis of eight cases. Am J Surg Pathol. 1987;11(10):767–78.
Nogales Jr FF, Matilla A, Nogales O, galera-Davidson HL. Yolk sac tumors with pure and mixed polyvesicular vitelline patterns. Hum Pathol. 1978;9(5):553–66.
Young RH, Ulbright TM, Policarpio-Nicolas MLC. Yolk sac tumor with a prominent polyvesicular vitelline pattern a report of three cases. Am J Surg Pathol. 2013;37(3):393–8. doi:10.1097/Pas.0b013e31827dcc2b.
Vos A, Oosterhuis JW, de Jong B, Castedo SM, Hollema H, Buist J, et al. Karyotyping and DNA flow cytometry of metastatic ovarian yolk sac tumor. Cancer Genet Cytogenet. 1990;44(2):223–8. doi:0165-4608(90)90051-B [pii].
Vandeputte M, Sobis H. Experimental rat model for human yolk sac tumor. Eur J Cancer Clin Oncol. 1988;24(3):551–8.
Ulbright TM, Roth LM, Brodhecker CA. Yolk sac differentiation in germ cell tumors. A morphologic study of 50 cases with emphasis on hepatic, enteric, and parietal yolk sac features. Am J Surg Pathol. 1986;10(3):151–64.
Damjanov I, Amenta PS, Zarghami F. Transformation of an AFP-positive yolk sac carcinoma into an AFP-negative neoplasm. Evidence for in vivo cloning of the human parietal yolk sac carcinoma. Cancer. 1984;53(9):1902–7.
Nogales-Fernandez F, Silverberg SG, Bloustein PA, Martinez-Hernandez A, Pierce GB. Yolk sac carcinoma (endodermal sinus tumor): ultrastructure and histogenesis of gonadal and extragonadal tumors in comparison with normal human yolk sac. Cancer. 1977;39(4):1462–74.
Kao CS, Idrees MT, Young RH, Ulbright TM. Solid pattern yolk sac tumor: a morphologic and immunohistochemical study of 52 cases. Am J Surg Pathol. 2012;36(3):360–7. doi:10.1097/PAS.0b013e31823c510b.
Kao CS, Idrees MT, Young RH, Ulbright TM. Solid pattern yolk sac tumor: a morphologic and immunohistochemical study of 52 cases. Am J Surg Pathol. 2012;36(3):360–7.
Nogales FF, Quinonez E, Lopez-Marin L, Dulcey I, Preda O. A diagnostic immunohistochemical panel for yolk sac (primitive endodermal) tumours based on an immunohistochemical comparison with the human yolk sac. Histopathology. 2014;65(1):51–9. doi:10.1111/his.12373.
Heifetz SA, Cushing B, Giller R, Shuster JJ, Stolar CJ, Vinocur CD, et al. Immature teratomas in children: pathologic considerations: a report from the combined Pediatric Oncology Group/Children’s Cancer Group. Am J Surg Pathol. 1998;22(9):1115–24.
Prat J, Bhan AK, Dickersin GR, Robboy SJ, Scully RE. Hepatoid yolk sac tumor of the ovary (endodermal sinus tumor with hepatoid differentiation): a light microscopic, ultrastructural and immunohistochemical study of seven cases. Cancer. 1982;50(11):2355–68.
Salazar HK, Tobon, A. et al. Endodermal cell derivatives in embryonal carcinoma of the ovary: an electron microscopy study of two cases. Am J Pathol. 1974;74:108a.(abstr).
Michael H, Ulbright TM, Brodhecker CA. The pluripotential nature of the mesenchyme-like component of yolk sac tumor. Arch Pathol Lab Med. 1989;113(10):1115–9.
Nogales FF, Buritica C, Regauer S, Gonzalez T. Mucinous carcinoid as an unusual manifestation of endodermal differentiation in ovarian yolk sac tumors. Am J Surg Pathol. 2005;29(9):1247–51. doi:00000478-200509000-00017 [pii].
Dickersin GR, Oliva E, Young RH. Endometrioid-like variant of ovarian yolk sac tumor with foci of carcinoid: an ultrastructural study. Ultrastruct Pathol. 1995;19(5):421–9.
Battocchio M, Zatelli MC, Chiarelli S, Trento M, Ambrosio MR, Pasquali C, et al. Ovarian tumors secreting insulin. Endocrine. 2015;49(3):611–9. doi:10.1007/s12020-015-0605-y.
Mann JR, Gray ES, Thornton C, Raafat F, Robinson K, Collins GS, et al. Mature and immature extracranial teratomas in children: The UK Children’s Cancer Study Group experience. J Clin Oncol. 2008;26(21):3590–7. doi:10.1200/jco.2008.16.0622.
Nogales FF. Unpublished observations.
Fujii K, Yamashita Y, Yamamoto T, Takahashi K, Hashimoto K, Miyata T et al. Ovarian mucinous tumors arising from mature cystic teratomas–a molecular genetic approach for understanding the cellular origin. Hum Pathol 2014;45(4):717–24. doi:10.1016/j.humpath.2013.10.031 S0046-8177(13)00478-4 [pii].
Nogales FF, Dulcey I. The secondary human yolk sac has an immunophenotype indicative of both hepatic and intestinal differentiation. Int J Dev Biol. 2012;56(9):755–760. doi:10.1387/ijdb.120080fn 120080fn [pii].
Xue D, Peng Y, Wang F, Allan RW, Cao D. RNA-binding protein LIN28 is a sensitive marker of ovarian primitive germ cell tumours. Histopathology. 2011;59(3):452–9. doi:10.1111/j.1365-2559.2011.03949.x.
Preda O, Dema A, Iacob M, Goyenaga P, Dulcey I, Aneiros Fernandez J, et al. Urothelial carcinoma of the renal pelvis with simultaneous trophoblastic and malignant clear cell endodermal-type differentiation. Virchows Arch. 2012;460(3):353–6. doi:10.1007/s00428-012-1211-5.
Zynger DL, McCallum JC, Luan C, Chou PM, Yang XJ. Glypican 3 has a higher sensitivity than alpha-fetoprotein for testicular and ovarian yolk sac tumour: immunohistochemical investigation with analysis of histological growth patterns. Histopathology 2010;56(6):750–7. doi:10.1111/j.1365-2559.2010.03553.x HIS3553 [pii].
Preda O, Nicolae A, Aneiros-Fernandez J, Borda A, Nogales FF. Glypican 3 is a sensitive, but not a specific, marker for the diagnosis of yolk sac tumours. Histopathology 2011;58(2):312–4; author reply 4–5. doi:10.1111/j.1365-2559.2010.03735.x.
Esheba GE, Pate LL, Longacre TA. Oncofetal protein glypican-3 distinguishes yolk sac tumor from clear cell carcinoma of the ovary. Am J Surg Pathol. 2008;32(4):600–7. doi:10.1097/PAS.0b013e31815a565a.
Pitman MB, Triratanachat S, Young RH, Oliva E. Hepatocyte paraffin 1 antibody does not distinguish primary ovarian tumors with hepatoid differentiation from metastatic hepatocellular carcinoma. Int J Gynecol Pathol. 2004;23(1):58–64. doi:10.1097/01.pgp.0000101141.31270.a1.
Gulluoglu MG, Karayigit E, Ozden I, Kapran Y, Dizdaroglu F. Does HepPar-1 immunoexpression have a role in differential diagnosis of periampullary cancer? Pathology. 2008;40(1):35–41. doi:10.1080/00313020701716391.
Pelosi G, Petrella F, Sandri MT, Spaggiari L, Galetta D, Viale G. A primary pure yolk sac tumor of the lung exhibiting CDX-2 immunoreactivity and increased serum levels of alkaline phosphatase intestinal isoenzyme. Int J Surg Pathol. 2006;14(3):247–51. doi:14/3/247 [pii] 1177/1066896906290657.
Bing Z, Pasha T, Tomaszewski JE, Zhang P. CDX2 expression in yolk sac component of testicular germ cell tumors. Int J Surg Pathol 2009;17(5):373–7. doi:10.1177/1066896909338598 1066896909338598 [pii].
Wong HH, Chu P. Immunohistochemical features of the gastrointestinal tract tumors. J Gastrointest Oncol. 2012;3(3):262–84. doi:10.3978/j.issn.2078-6891.2012.019.
Cao D, Li J, Guo CC, Allan RW, Humphrey PA. SALL4 is a novel diagnostic marker for testicular germ cell tumors. Am J Surg Pathol. 2009;33(7):1065–77. doi:10.1097/PAS.0b013e3181a13eef.
Cao D, Guo S, Allan RW, Molberg KH, Peng Y. SALL4 is a novel sensitive and specific marker of ovarian primitive germ cell tumors and is particularly useful in distinguishing yolk sac tumor from clear cell carcinoma. Am J Surg Pathol. 2009;33(6):894–904. doi:10.1097/PAS.0b013e318198177d.
Wang F, Liu A, Peng Y, Rakheja D, Wei L, Xue D, et al. Diagnostic utility of SALL4 in extragonadal yolk sac tumors: an immunohistochemical study of 59 cases with comparison to placental-like alkaline phosphatase, alpha-fetoprotein, and glypican-3. Am J Surg Pathol. 2009;33(10):1529–39. doi:10.1097/PAS.0b013e3181ad25d5.
Khalifa MA, Gersell DJ, Hansen CH, Lage JM. Hepatic (hepatocellular) adenoma of the placenta: a study of four cases. Int J Gynecol Pathol. 1998;17(3):241–4.
Matsuyama TA, Kushima M, Yamochi-Onizuka T, Ota H. Placental yolk sac tumor with divergent endodermal differentiation. Int J Gynecol Pathol. 2004;23(4):398–402. doi:00004347-200410000-00014 [pii].
West JA, Viswanathan SR, Yabuuchi A, Cunniff K, Takeuchi A, Park IH et al. A role for Lin28 in primordial germ-cell development and germ-cell malignancy. Nature 2009;460(7257):909–13. doi:10.1038/nature08210 nature08210 [pii].
Cao D, Allan RW, Cheng L, Peng Y, Guo CC, Dahiya N et al. RNA-binding protein LIN28 is a marker for testicular germ cell tumors. Hum Pathol 2011;42(5):710–8. doi:10.1016/j.humpath.2010.09.007 S0046-8177(10)00364-3 [pii].
Cao D, Liu A, Wang F, Allan RW, Mei K, Peng Y, et al. RNA-binding protein LIN28 is a marker for primary extragonadal germ cell tumors: an immunohistochemical study of 131 cases. Mod Pathol. 2011;24(2):288–96. doi:10.1038/modpathol.2010.195.
Nogales FF, Dulcey I, Preda O. Issues in Gynecologic Pathology. Germ Cell Tumors of the Ovary. An Update. Arch Pathol Lab Med. 2014.
Miettinen M, McCue PA, Sarlomo-Rikala M, Rys J, Czapiewski P, Wazny K, et al. GATA3: a multispecific but potentially useful marker in surgical pathology: a systematic analysis of 2500 epithelial and nonepithelial tumors. Am J Surg Pathol. 2014;38(1):13–22. doi:10.1097/PAS.0b013e3182a0218f.
Schuldt M, Rubio A, Preda O, Nogales FF. GATA3 expression is present in primitive patterns of Yolk Sac Tumors but is not expressed by differentiated variants. Histopathology. 2015; doi:10.1111/his.12776.
Iacobelli JF, Charles AK, Crook M, Stewart CJ. NUT protein immunoreactivity in ovarian germ cell tumours. Pathology 2015;47(2):118–122. doi:10.1097/PAT.0000000000000208 01268031-201502000-00005 [pii].
Damato S, Haldar K, McCluggage WG. Primary endometrial yolk sac tumor with endodermal-intestinal differentiation masquerading as metastatic colorectal adenocarcinoma. Int J Gynecol Pathol. 2015; doi:10.1097/PGP.0000000000000236.
Retamero JA, Schuldt M, Nogales FF. Yolk sac tumors in postmenopausal patients arising in endometrioid adenocarcinoma. AJSP Rev Rep. 2016;21:189–94.
Ramalingam P, Malpica A, Silva EG, Gershenson DM, Liu JL, Deavers MT. The use of cytokeratin 7 and EMA in differentiating ovarian yolk sac tumors from endometrioid and clear cell carcinomas. Am J Surg Pathol. 2004;28(11):1499–505.
Nogales FF, Schuldt M, Cruz BN, Oosterhuis JW, Kaur B, Matias-Guiu X et al. Germ cell tumor growth patterns originated from clear cell carcinoma of the ovary and endometrium. Comparative immunohistochemical study proposing a putative origin from neoplastic stem cells of somatic tumors. In preparation 2017.
De Backer A, Madern GC, Pieters R, Haentjens P, Hakvoort-Cammel FG, Oosterhuis JW, et al. Influence of tumor site and histology on long-term survival in 193 children with extracranial germ cell tumors. Eur J Pediatr Surg. 2008;18(1):1–6. doi:10.1055/s-2007-989399.
Topuz S, Iyibozkurt AC, Akhan SE, Keskin N, Yavuz E, Salihoglu Y, et al. Malignant germ cell tumors of the ovary: a review of 41 cases and risk factors for recurrence. Eur J Gynaecol Oncol. 2008;29(6):635–7.
Moch H, Humphrey P, Ubright TM, Reuter VE. WHO classification of tumours of the urinary system and male genital organs, World health organization classification of tumours. 4th ed. Lyon: IARC; 2016.
Hennes E, Zahn S, Lopes LF, Schonberger S, Leuschner I, Gobel U, et al. Molecular genetic analysis of bilateral ovarian germ cell tumors. Klin Padiatr. 2012;224(6):359–65. doi:10.1055/s-0032-1327606.
Kleinsmith LJ, Pierce Jr GB. Multipotentiality of single embryonal carcinoma cells. Cancer Res. 1964;24:1544–51.
Malecki M, Anderson M, Beauchaine M, Seo S, Tombokan X, Malecki R. TRA-1-60, SSEA-4, Oct4A, Nanog clones of pluripotent stem cells in the embryonal carcinomas of the ovaries. J Stem Cell Res Ther. 2012;2(5) doi:130 [pii].
Cheng L, Zhang SB, Talerman A, Roth LM. Morphologic, immunohistochemical, and fluorescence in situ hybridization study of ovarian embryonal carcinoma with comparison to solid variant of yolk sac tumor and immature teratoma. Hum Pathol. 2010;41(5):716–23. doi:10.1016/j.humpath.2009.10.016.
Prat J, Bhan AK, Dickersin GR, Robboy SJ, Scully RE. Hypothesis: when is a seminoma not a seminoma? J Clin Pathol. 1981;34(11):1308–9.
Kurman RJ, Norris HJ. Embryonal carcinoma of the ovary: a clinicopathologic entity distinct from endodermal sinus tumor resembling embryonal carcinoma of the adult testis. Cancer. 1976;38(6):2420–33.
Young RH, Stall JN, Sevestre H. The polyembryoma: one of the most intriguing human neoplasms, with comments on the investigator who brought it to light, Albert Peyron. Int J Gynecol Pathol. 2016;35(2):93–105. doi:10.1097/PGP.0000000000000282.
Ulbright TM, Young RH. Tumors of the tesis and adjacent structures. Atlas of tumor pathology (4th series). American Registry of Pathology: Silver Spring; 2013.
Poulos C, Cheng L, Zhang S, Gersell DJ, Ulbright TM. Analysis of ovarian teratomas for isochromosome 12p: evidence supporting a dual histogenetic pathway for teratomatous elements. Mod Pathol 2006;19(6):766–71. doi:3800596 [pii] 10.1038/modpathol.3800596.
Wang D, Hu Y, He Y, Xie C, Yin R. Pure ovarian choriocarinoma mimicking ectopic pregnancy in true hermaphroditism. Acta Obstet Gynecol Scand 2009;88(7):850–2. doi:10.1080/00016340902902883 910379889 [pii].
Joneborg U, Papadogiannakis N, Lindell G, Marions L. Choriocarcinoma following ovarian hydatidiform mole: a case report. J Reprod Med. 2011;56(11–12):511–4.
Sehn JK, Kuroki LM, Hopeman MM, Longman RE, McNicholas CP, Huettner PC. Ovarian complete hydatidiform mole: case study with molecular analysis and review of the literature. Hum Pathol 2013;44(12):2861–4. doi:10.1016/j.humpath.2013.07.027 S0046-8177(13)00316-X [pii].
Gardner JK, Konner JA. Germ cell and sex cord stromal tumors: clinical introduction. In: Soslow RA, Tornos C, Eds. Diagnostic Pathology of Ovarian Tumors. New York: Springer; 2011. p. 145–53.
Cho NH, Kim YT, Lee JH, Song C, Cho SW, Cho SH, et al. Diagnostic challenge of fetal ontogeny and its application on the ovarian teratomas. Int J Gynecol Pathol. 2005;24(2):173–82. doi:00004347-200504000-00010 [pii].
Gutierrez-Aranda I, Ramos-Mejia V, Bueno C, Munoz-Lopez M, Real PJ, Macia A, et al. Human induced pluripotent stem cells develop teratoma more efficiently and faster than human embryonic stem cells regardless the site of injection. Stem Cells. 2010;28(9):1568–70. doi:10.1002/stem.471.
Doss BJ, Jacques SM, Qureshi F, Chang CH, Christensen CW, Morris RT et al. Immature teratomas of the genital tract in older women. Gynecol Oncol 1999;73(3):433–8. doi:S0090-8258(99)95345-3 [pii] 10.1006/gyno.1999.5345.
Nogales FF. Germ cell tumours of the ovary. In: Fox H, Wells M, editors. Haines & Taylor obstetrical and gynaecological pathology. 5th ed. Edinburgh: Churchill Livingstone; 2003. p. 771–820.
Dabner M, McCluggage WG, Bundell C, Carr A, Leung Y, Sharma R, et al. Ovarian teratoma associated with anti-N-methyl D-aspartate receptor encephalitis: a report of 5 cases documenting prominent intratumoral lymphoid infiltrates. Int J Gynecol Pathol. 2012;31(5):429–37. doi:10.1097/PGP.0b013e31824a1de2.
Nogales Jr FF, Favara BE, Major FJ, Silverberg SG. Immature teratoma of the ovary with a neural component (“solid” teratoma). A clinicopathologic study of 20 cases. Hum Pathol. 1976;7(6):625–42.
Marina NM, Cushing B, Giller R, Cohen L, Lauer SJ, Ablin A, et al. Complete surgical excision is effective treatment for children with immature teratomas with or without malignant elements: A Pediatric Oncology Group/Children’s Cancer Group Intergroup Study. J Clin Oncol. 1999;17(7):2137–43.
Norris HJ, Zirkin HJ, Benson WL. Immature (malignant) teratoma of the ovary: a clinical and pathologic study of 58 cases. Cancer. 1976;37(5):2359–72.
Yanai H, Matsuura H, Kawasaki M, Takada Y, Tabuchi Y, Yoshino T. Immature teratoma of the ovary with a minor rhabdomyosarcomatous component and fatal rhabdomyosarcomatous metastases: the first case in a child. Int J Gynecol Pathol. 2002;21(1):82–5.
Byun JC, Choi IJ, Han MS, Lee SC, Roh MS, Cha MS. Soft tissue metastasis of an immature teratoma of the ovary. J Obstet Gynaecol Res. 2011;37(11):1689–93. doi:10.1111/j.1447-0756.2011.01553.x.
Kurata A, Hirano K, Nagane M, Fujioka Y. Immature teratoma of the ovary with distant metastases: favorable prognosis and insights into chemotherapeutic retroconversion. Int J Gynecol Pathol. 2010;29(5):438–44. doi:10.1097/PGP.0b013e3181cef16b.
Merard R, Ganesan R, Hirschowitz L. Growing teratoma syndrome: a report of 2 cases and review of the literature. Int J Gynecol Pathol 2015;34(5):465–72. doi:10.1097/PGP.0000000000000180. 00004347-201509000-00009 [pii].
Nogales FF, Aguilar D. Florid vascular proliferation in grade 0 glial implants from ovarian immature teratoma. Int J Gynecol Pathol. 2002;21(3):305–7.
O’Connor DM, Norris HJ. The influence of grade on the outcome of stage I ovarian immature (malignant) teratomas and the reproducibility of grading. Int J Gynecol Pathol. 1994;13(4):283–9.
King ME, Micha JP, Allen SL, Mouradian JA, Chaganti RS. Immature teratoma of the ovary with predominant malignant retinal anlage component. A parthenogenically derived tumor. Am J Surg Pathol. 1985;9(3):221–31.
Liang L, Olar A, Niu N, Jiang Y, Cheng W, Bian XW, et al. Primary glial and neuronal tumors of the ovary or peritoneum: a clinicopathologic study of 11 cases. Am J Surg Pathol. 2016; doi:10.1097/PAS.0000000000000635.
Euscher ED, Deavers MT, Lopez-Terrada D, Lazar AJ, Silva EG, Malpica A. Uterine tumors with neuroectodermal differentiation: a series of 17 cases and review of the literature. Am J Surg Pathol 2008;32(2):219–28. doi:10.1097/PAS.0b013e318093e421 00000478-200802000-00006 [pii].
Clinkard DJ, Khalifa M, Osborned RJ, Bouffet E. Successful management of medulloblastoma arising in an immature ovarian teratoma in pregnancy. Gynecol Oncol 2011;120(2):311–2. doi:10.1016/j.ygyno.2010.10.022 S0090-8258(10)00768-7 [pii].
Morency EG, Lerner D, Garcia R, Kalir T. High-grade sarcoma masquerading as growing teratoma syndrome after resection of ovarian immature teratoma: report of a case. Int J Gynecol Pathol. 2012;31(3):276–9. doi:10.1097/PGP.0b013e31823ef912.
Malagon HD, Valdez AM, Moran CA, Suster S. Germ cell tumors with sarcomatous components: a clinicopathologic and immunohistochemical study of 46 cases. Am J Surg Pathol 2007;31(9):1356–62. doi:10.1097/PAS.0b013e318033c7c4 00000478-200709000-00008 [pii].
Mengshol SC, Demars LR, Schned AR. Gliomatosis peritonei and teratomatous implant with carcinomatous transformation presenting 54 years following oophorectomy for dermoid cyst. Gynecol Oncol. 2004;92(1):353–6.
DiSaia PJ, Saltz A, Kagan AR, Morrow CP. Chemotherapeutic retroconversion of immature teratoma of the ovary. Obstet Gynecol. 1977;49(3):346–50.
Logothetis CJ, Samuels ML, Trindade A, Johnson DE. The growing teratoma syndrome. Cancer. 1982;50(8):1629–35.
Kato N, Uchigasaki S, Fukase M. How does secondary neoplasm arise from mature teratomas in growing teratoma syndrome of the ovary? A report of two cases. Pathol Int. 2013;63(12):607–10. doi:10.1111/pin.12112.
Djordjevic B, Euscher ED, Malpica A. Growing teratoma syndrome of the ovary: review of literature and first report of a carcinoid tumor arising in a growing teratoma of the ovary. Am J Surg Pathol 2007;31(12):1913–8. doi:10.1097/PAS.0b013e318073cf44 00000478-200712000-00017 [pii].
Nogales FF, Preda O, Dulcey I. Gliomatosis peritonei as a natural experiment in tissue differentiation. Int J Dev Biol 2012;56(10–12):969–74. doi:10.1387/ijdb.120172fn 120172fn [pii].
Wu XH, Han LY, Xu X, Li ZT. Recurrent immature teratoma of the ovary: a case report of radical secondary cytoreduction with replacement of the aortic bifurcation. Gynecol Oncol. 2004;95(3):746–9. doi:10.1016/j.ygyno.2004.08.007.
Yoon NR, Lee JW, Kim BG, Bae DS, Sohn I, Sung CO, et al. Gliomatosis peritonei is associated with frequent recurrence, but does not affect overall survival in patients with ovarian immature teratoma. Virchows Arch. 2012;461(3):299–304. doi:10.1007/s00428-012-1285-0.
Gonzalez-Campora R, Nogales Jr FF, Davidson HG, Mendez JA. Case report: ultrastructure of mature neurogenic implants from ovarian immature teratoma. Histopathology. 1979;3(3):233–40.
Webman R, Talishinskiy T, Raetz E, Lala S, Tomita S. Spontaneous regression of thoracic and extraperitoneal glial implants in child with gliomatosis peritonei after resection of ovarian teratoma. J Pediatr Hematol Oncol. 2014; doi:10.1097/MPH.0000000000000230.
Webman R, Talishinskiy T, Raetz E, Lala S, Tomita S. Spontaneous regression of thoracic and extraperitoneal glial implants in child with gliomatosis peritonei after resection of ovarian teratoma. J Pediatr Hematol Oncol. 2015;37(3):230–1. doi:10.1097/MPH.0000000000000230.
Liang L, Zhang Y, Malpica A, Ramalingam P, Euscher ED, Fuller GN, et al. Gliomatosis peritonei: a clinicopathologic and immunohistochemical study of 21 cases. Mod Pathol. 2015;28(12):1613–20. doi:10.1038/modpathol.2015.116.
Kim NR, Lim S, Jeong J, Cho HY. Peritoneal and nodal gliomatosis with endometriosis, accompanied with ovarian immature teratoma: a case study and literature review. Korean J Pathol. 2013;47(6):587–91. doi:10.4132/KoreanJPathol.2013.47.6.587.
Lobotesis K, U-King-Im JM, Cross JJ, Gillard JH, Antoun NM. Gliomatosis peritonei associated with a ventriculo-peritoneal shunt. Clin Radiol 2009;64(1):95–9. doi:10.1016/j.crad.2008.08.015 S0009-9260(08)00374-7 [pii].
Ferguson AW, Katabuchi H, Ronnett BM, Cho KR. Glial implants in gliomatosis peritonei arise from normal tissue, not from the associated teratoma. Am J Pathol. 2001;159(1):51–5. doi:10.1016/s0002-9440(10)61672-0.
Kwan MY, Kalle W, Lau GT, Chan JK. Is gliomatosis peritonei derived from the associated ovarian teratoma? Hum Pathol. 2004;35(6):685–8. doi:S004681770400108X [pii].
Alexander M, Cope N, Renninson J, Hong A, Simpson RH, Hirschowitz L. Relationship between endometriosis, endometrioid adenocarcinoma, gliomatosis peritonei, and carcinoid tumor in a patient with recurrent ovarian teratoma. Int J Gynecol Pathol. 2011;30(2):151–7. doi:10.1097/PGP.0b013e3181f6bcd4.
Abiko K, Mandai M, Hamanishi J, Matsumura N, Baba T, Horiuchi A et al. Oct4 expression in immature teratoma of the ovary: relevance to histologic grade and degree of differentiation. Am J Surg Pathol 2010;34(12):1842–8. doi:10.1097/PAS.0b013e3181fcd707 00000478-201012000-00012 [pii].
Linder D, McCaw BK, Hecht F. Parthenogenic origin of benign ovarian teratomas. N Engl J Med. 1975;292(2):63–6. doi:10.1056/NEJM197501092920202.
Dahl N, Gustavson KH, Rune C, Gustavsson I, Pettersson U. Benign ovarian teratomas. An analysis of their cellular origin. Cancer Genet Cytogenet. 1990;46(1):115–23.
Besser MJ, Posey DM. Cystic teratoma in a supernumerary ovary of the greater omentum. A case report. J Reprod Med. 1992;37(2):189–93.
Nezhat C, Kotikela S, Mann A, Hajhosseini B, Veeraswamy A, Lewis M. Familial cystic teratomas: four case reports and review of the literature. J Minim Invasive Gynecol 2010;17(6):782–6. doi:10.1016/j.jmig.2010.06.006 S1553-4650(10)00333-X [pii].
Brown Jr EH. Identical twins with twisted benign cystic teratoma of the ovary. Am J Obstet Gynecol. 1979;134(8):879–80. doi:0002-9378(79)90860-3 [pii].
Saba L, Guerriero S, Sulcis R, Virgilio B, Melis G, Mallarini G. Mature and immature ovarian teratomas: CT, US and MR imaging characteristics. Eur J Radiol 2009;72(3):454–63. doi:10.1016/j.ejrad.2008.07.044 S0720-048X(08)00457-9 [pii].
Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25–36. doi:10.1002/ana.21050.
Dalmau J, Rosenfeld MR. Update on paraneoplastic neurologic disorders. Commun Oncol. 2010;7(5):219–24.
Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005;58(4):594–604. doi:10.1002/ana.20614.
Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr Res 2014. doi:S0920-9964(14)00546-5 [pii] 10.1016/j.schres.2014.10.007.
Dulcey I, Cespedes MU, Ballesteros JL, Preda O, Aneiros-Fernandez J, Clavero PA et al. Necrotic mature ovarian teratoma associated with anti-N-methyl-D-aspartate receptor encephalitis. Pathol Res Pract 2012;208(8):497–500. doi:10.1016/j.prp.2012.05.018 S0344-0338(12)00156-2 [pii].
Templeman CL, Fallat ME, Lam AM, Perlman SE, Hertweck SP, O’Connor DM. Managing mature cystic teratomas of the ovary. Obstet Gynecol Surv. 2000;55(12):738–45.
Kim MJ, Kim NY, Lee DY, Yoon BK, Choi D. Clinical characteristics of ovarian teratoma: age-focused retrospective analysis of 580 cases. Am J Obstet Gynecol. 2011;205(1):32 e1–4. doi:10.1016/j.ajog.2011.02.044 S0002-9378(11)00234-1 [pii].
Peitsidou A, Peitsidis P, Goumalatsos N, Papaspyrou R, Mitropoulou G, Georgoulias N. Diagnosis of an autoamputated ovary with dermoid cyst during a Cesarean section. Fertil Steril. 2009;91(4):1294 e9–12. doi:10.1016/j.fertnstert.2008.12.029 S0015-0282(08)04725-0 [pii].
Kusaka M, Mikuni M. Ectopic ovary: a case of autoamputated ovary with mature cystic teratoma into the cul-de-sac. J Obstet Gynaecol Res. 2007;33(3):368–70. doi:JOG538 [pii] 10.1111/j.1447-0756.2007.00538.x.
Siegel MJ, McAlister WH, Shackelford GD. Radiographic findings in ovarian teratomas in children. AJR Am J Roentgenol. 1978;131(4):613–5. doi:10.2214/ajr.131.4.613.
Shetty NS, Vallabhaneni S, Patil A, Babu MM, Baig A. Unreported location and presentation for a parasitic ovarian dermoid cyst in an indirect inguinal hernia. Hernia. 2013;17(2):263–5. doi:10.1007/s10029-011-0876-z.
Sinha R, Sundaram M, Lakhotia S. Multiple intraabdominal parasitic cystic teratomas. J Minim Invasive Gynecol 2009;16(6):789–91. doi:10.1016/j.jmig.2009.08.003 S1553-4650(09)00443-9 [pii].
Takeda A, Imoto S, Mori M, Yamada J, Nakamura H. Early abdominal pregnancy complicated by parasitic dermoid cyst: diagnosis by diffusion-weighted magnetic resonance imaging and management by laparoendoscopic single-site surgery. J Minim Invasive Gynecol 2012;19(5):647–50. doi:10.1016/j.jmig.2012.03.024 S1553-4650(12)00127-6 [pii].
Cucinella G, Granese R, Venezia R, Mangione D, Calagna G, Perino A. Parasitic dermoid cyst coexisting with absence of an adnexa. Acta Obstet Gynecol Scand. 2011;90(6):677–8. doi:10.1111/j.1600-0412.2011.01125.x.
Walid MS, Boddy MG. Bilateral dermoid cysts of the ovary in a pregnant woman: case report and review of the literature. Arch Gynecol Obstet. 2009;279(2):105–8. doi:10.1007/s00404-008-0695-3.
Feltingoff M, Heller DS, Bleiweiss IJ. Ovarian pregnancy with ipsilateral mature cystic teratoma of the ovary: case report. Mt Sinai J Med. 1992;59(1):82–4.
Nogales FF, Fernandez-Sanz J, Fulwood H, Matilla A, Galera-Davidson H. Estudio clínico-patológico de 288 teratomas quísticos de ovario. Patologia (Madrid). 1979;16:12–25.
Goh W, Bohrer J, Zalud I. Management of the adnexal mass in pregnancy. Curr Opin Obstet Gynecol 2014;26(2):49–53. doi:10.1097/GCO.0000000000000048 00001703-201404000-00002 [pii].
Tariel O, Huissoud C, Rudigoz RC, Dubernard G. [Presumed benign ovarian tumors during pregnancy]. J Gynecol Obstet Biol Reprod (Paris). 2013;42(8):842–55. doi:10.1016/j.jgyn.2013.09.038 S0368-2315(13)00284-6 [pii].
Erdemoglu M, Kuyumcuoglu U, Kale A. Pregnancy and adnexal torsion: analysis of 20 cases. Clin Exp Obstet Gynecol. 2010;37(3):224–5.
Ulstein M, Rana G, Yangzom K, Gurung R. Prolapse of an ovarian tumor during labor. Acta Obstet Gynecol Scand. 1987;66(8):721–2.
Nader R, Thubert T, Deffieux X, de Laveaucoupet J, Ssi-Yan-Kai G. Delivery induced intraperitoneal rupture of a cystic ovarian teratoma and associated chronic chemical peritonitis. Case Rep Radiol. 2014;2014:189409. doi:10.1155/2014/189409.
Yoshikata R, Yamamoto T, Kobayashi M, Ota H. Immunohistochemical characteristics of mature ovarian cystic teratomas in patients with postoperative recurrence. Int J Gynecol Pathol. 2006;25(1):95–100. doi:00004347-200601000-00014 [pii].
Anteby EY, Ron M, Revel A, Shimonovitz S, Ariel I, Hurwitz A. Germ cell tumors of the ovary arising after dermoid cyst resection: a long-term follow-up study. Obstet Gynecol. 1994;83(4):605–8.
Chapron C, Dubuisson JB, Samouh N, Foulot H, Aubriot FX, Amsquer Y, et al. Treatment of ovarian dermoid cysts. Place and modalities of operative laparoscopy. Surg Endosc. 1994;8(9):1092–5.
Arora DS, Haldane S. Carcinosarcoma arising in a dermoid cyst of the ovary. J Clin Pathol. 1996;49(6):519–21.
Amerigo J, Nogales Jr FF, Fernandez-Sanz J, Oliva H, Velasco A. Squamous cell neoplasms arising from ovarian benign cystic teratoma. Gynecol Oncol. 1979;8(3):277–83.
Pansky M, Shade D, Moskovitch M, Halperin R, Ben-Ami I, Smorgick N. Inadvertent rupture of benign cystic teratoma does not impair future fertility. Am J Obstet Gynecol. 2010;203(5):442 e1–4. doi:10.1016/j.ajog.2010.06.062 S0002-9378(10)00842-2 [pii].
Pantoja E, Noy MA, Axtmayer RW, Colon FE, Pelegrina I. Ovarian dermoids and their complications. Comprehensive historical review. Obstet Gynecol Surv. 1975;30(1):1–20.
Stuart GC, Smith JP. Ruptured benign cystic teratomas mimicking gynecologic malignancy. Gynecol Oncol. 1983;16(1):139–43.
Gendre J, Sebban-Rozot C, Regent D, Ranchoup Y, Ridereau-Zins C, Vullierme MP et al. [Peritoneal parasitic teratoma and chemical dermoid peritonitis]. J Radiol 2011;92(5):382–92. doi:10.1016/j.jradio.2011.03.005 S0221-0363(11)00117-X [pii].
Maiti S, Fatima Z, Anjum ZK, Hopkins RE. Ruptured ovarian cystic teratoma in pregnancy with diffuse peritoneal reaction mimicking advanced ovarian malignancy: a case report. J Med Case Rep 2008;2:203. doi:10.1186/1752-1947-2-203 1752-1947-2-203 [pii].
Sasaki H, Nagasako K, Harada M, Kobayashi S, Uetake K. Benign cystic teratoma of the ovary with rupture into the rectum: report of a unique rectal tumor. Dis Colon Rectum. 1979;22(4):248–51.
Suzuki M, Fukasawa M, Hara T. Dermoid cyst with thyroid follicle perforated into bladder and ileum: a case report. Nihon Hinyokika Gakkai Zasshi. 1999;90(5):586–9.
von-Walter AR, Nelken RS. Benign cystic ovarian teratoma with a fistula into the small and large bowel. Obstet Gynecol 2012;119(2 Pt 2):434–6. doi:10.1097/AOG.0b013e3182319185 00006250-201202001-00007 [pii].
Sardesai S, Raghoji V, Dabade R, Shaikh H. Benign cytic teratoma of ovary perforating into the urinary bladder: a rare case. J Obstet Gynaecol India 2012;62(Suppl 1):54–5. doi:10.1007/s13224-013-0354-x 354 [pii].
Candela G, Di Libero L, Varriale S, Manetta F, Napolitano S, Scetta G, et al. Hemoperitoneum caused by the rupture of a giant ovarian teratoma in a 9-year-old female. Case report and literature review. Ann Ital Chir. 2009;80(2):141–4.
Fukushima M, Sharpe L, Okagaki T. Peritoneal melanosis secondary to a benign dermoid cyst of the ovary: a case report with ultrastructural study. Int J Gynecol Pathol. 1984;2(4):403–9.
Jaworski RC, Boadle R, Greg J, Cocks P. Peritoneal “melanosis” associated with a ruptured ovarian dermoid cyst: report of a case with electron-probe energy dispersive X-ray analysis. Int J Gynecol Pathol. 2001;20(4):386–9.
Brelje MC, Garcia-Bunuel R. Salmonella infection in an ovarian dermoid cyst in pregnancy: report of a case. Obstet Gynecol. 1964;24:279–80.
Matsubayashi T, Hamajima T, Asano K, Mizukami A, Seguchi M, Kohno C, et al. Salmonella infection of an ovarian dermoid cyst. Pediatr Int. 2001;43(2):164–5. doi:ped1351 [pii].
Salehpour S, Akbari Sene A. Super infection of an ovarian dermoid cyst with actinomyces in an infertile woman. Int J Fertil Steril. 2013;7(2):134–7.
Melato M, Muuse MM, Hussein AM, Falconieri G. Schistosomiasis in a cystic teratoma of the ovary. Clin Exp Obstet Gynecol. 1987;14(1):57–9.
Sane SY, Patel CV. A filarial worm in the wall of a cystic teratoma of the ovary–(a case report). J Postgrad Med. 1989;35(4):217–8.
Cobo F, Pereira A, Nomdedeu B, Gallart T, Ordi J, Torne A, et al. Ovarian dermoid cyst-associated autoimmune hemolytic anemia: a case report with emphasis on pathogenic mechanisms. Am J Clin Pathol. 1996;105(5):567–71.
Payne D, Muss HB, Homesley HD, Jobson VW, Baird FG. Autoimmune hemolytic anemia and ovarian dermoid cysts: case report and review of the literature. Cancer. 1981;48(3):721–4.
Ibarra M, Chou P, Pachman LM. Ovarian teratoma mimicking features of juvenile dermatomyositis in a child. Pediatrics 2011;128(5):e1293–6. doi:10.1542/peds.2010-2115 peds.2010-2115 [pii].
Wiese W, Alansari H, Tranchida P, Madrid FF. Paraneoplastic polyarthritis in an ovarian teratoma. J Rheumatol. 2004;31(9):1854–7. doi:0315162X-31-1854 [pii].
Deen R, de Silva A, Wijesuriya R. Twisted benign ovarian teratoma presenting with pain and generalized pruritus: a case report. J Med Case Rep 2013;7(1):130. doi:10.1186/1752-1947-7-130 1752-1947-7-130 [pii].
Di Stefano F, Siriruttanapruk S, Di Gioacchino M. Exercise-induced urticarial vasculitis as a paraneoplastic manifestation of cystic teratoma. Rheumatology (Oxford). 2003;42(11):1418–9. doi:10.1093/rheumatology/keg346 42/11/1418 [pii].
Frawley KJ, Calvo-Garcia MA, Krueger DA, McMasters RL. ‘Benign’ ovarian teratoma and N-methyl-D-aspartate receptor (NMDAR) encephalitis in a child. Pediatr Radiol. 2012;42(1):120–3. doi:10.1007/s00247-011-2111-6.
McCarthy A, Dineen J, McKenna P, Keogan M, Sheehan J, Lynch T, et al. Anti-NMDA receptor encephalitis with associated catatonia during pregnancy. J Neurol. 2012;259(12):2632–5. doi:10.1007/s00415-012-6561-z.
Lupi I, Fessehatsion R, Manca A, Cossu-Rocca P, Martino E, Macchia E. Hashimoto’s thyroiditis in a benign cystic teratoma of the ovary: case report and literature review. Gynecol Endocrinol. 2012;28(1):39–42. doi:10.3109/09513590.2011.579659.
Amareen VN, Haddad FH, Al-Kaisi NS. Hypothyroidism due to Hashimoto thyroiditis post struma ovarii excision. Saudi Med J. 2004;25(7):948–50. doi:20030587’ [pii].
Elms AF, Carlan SJ, Rich AE, Cerezo L. Ovarian tumor-derived ectopic hyperprolactinemia. Pituitary. 2012;15(4):552–5. doi:10.1007/s11102-011-0366-4.
Klonoff DC, Kahn DG, Rosenzweig W, Wilson CB. Hyperprolactinemia in a patient with a pituitary and an ovarian dermoid tumor: case report. Neurosurgery. 1990;26(2):335–9.
Takeuchi K, Murata K, Funaki K, Kitazawa S, Kitazawa R. Immunohistochemical detection of parathyroid hormone-related protein in a squamous cell carcinoma arising from mature cystic teratoma causing humoral hypercalcemia of malignancy. Gynecol Oncol 2000;79(3):504–7. doi:10.1006/gyno.2000.5945 S0090-8258(00)95945-6 [pii].
Gunasekaran S, Kopecka E, Maung KH, England RJ. Struma ovarii and the thyroid surgeon. J Laryngol Otol 2012;126(8):858–60. doi:10.1017/S0022215112000904 S0022215112000904 [pii].
Deffieux X, Thubert T, Huchon C, Demoulin G, Rivain AL, Faivre E et al. [Complications of presumed benign ovarian tumors]. J Gynecol Obstet Biol Reprod (Paris). 2013;42(8):816–32. doi:10.1016/j.jgyn.2013.09.036 S0368-2315(13)00282-2 [pii].
Axiotis CA, Lippes HA, Merino MJ, deLanerolle NC, Stewart AF, Kinder B. Corticotroph cell pituitary adenoma within an ovarian teratoma. A new cause of Cushing’s syndrome. Am J Surg Pathol. 1987;11(3):218–24.
Tsujimoto T, Takaichi M, Endo H, Yasuda K, Kishimoto M, Noto H, et al. A patient with diabetes and breast cancer in whom virilization was caused by a testosterone-producing mature cystic teratoma containing a Brenner tumor. e. 2011;341(1):74–7. doi:10.1097/MAJ.0b013e3181f54e59.
Hoffman JG, Strickland JL, Yin J. Virilizing ovarian dermoid cyst with leydig cells. J Pediatr Adolesc Gynecol 2009;22(3):e39–40. doi:10.1016/j.jpag.2008.05.012 S1083-3188(08)00194-0 [pii].
Lopez-Beltran A, Calanas AS, Jimena P, Escudero AL, Campello TR, Munoz-Torres M, et al. Virilizing mature ovarian cystic teratomas. Virchows Arch. 1997;431(2):149–51.
Rutgers JL, Scully RE. Functioning ovarian tumors with peripheral steroid cell proliferation: a report of twenty-four cases. Int J Gynecol Pathol. 1986;5(4):319–37.
Robboy SJ, Scully RE. Strumal carcinoid of the ovary: an analysis of 50 cases of a distinctive tumor composed of thyroid tissue and carcinoid. Cancer. 1980;46(9):2019–34.
Ober WB. Solid ovarian teratoma with struma ovarii, theca lutein reaction, and endometrial hyperplasia. Report of a case. J Obstet Gynaecol Br Emp. 1960;67:451–4.
Murdoch W, Sadoski J, Rosin FC. Multisystem manifestations of benign ovarian teratomas. J Am Board Fam Med. 2014;27(3):421–3. doi:10.3122/jabfm.2014.03.130221 27/3/421 [pii].
Sahin AA, Ro JY, Chen J, Ayala AG. Spindle cell nodule and peptic ulcer arising in a fully developed gastric wall in a mature cystic teratoma. Arch Pathol Lab Med. 1990;114(5):529–31.
Tiltman AJ, Glass DR, Duffield MS. Peptic ulceration in a mature cystic teratoma of the ovary. Histopathology. 1995;26(4):375–7.
Kirova YM, Feuilhade F, de Baecque-Fontaine C, Le Bourgeois JP. Metastasis of a breast carcinoma in a mature teratoma of the ovary. Eur J Gynaecol Oncol. 1999;20(3):223–5.
Damewood M, Rosenshein NB, Woodruff JD. Multiple benign cystic teratomas of the ovary. Report of two cases and review of the literature. Diagn Gynecol Obstet. 1982;4(3):243–5.
Monk BE, Isaacs AJ, Bayliss R. Giant ovarian dermoid masked by obesity. Postgrad Med J. 1980;56(660):748–9.
Canda AE, Astarcioglu H, Obuz F, Canda MS. Cystic ovarian teratoma with intracystic floating globules. Abdom Imaging. 2005;30(3):369–71. doi:10.1007/s00261-004-0266-4.
Devoize L, Collangettes D, Le Bouedec G, Mishellany F, Orliaguet T, Dallel R et al. Giant mature ovarian cystic teratoma including more than 300 teeth. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008;105(3):e76–9. doi:10.1016/j.tripleo.2007.10.001 S1079-2104(07)00819-0 [pii].
Chavan SS, Yenni VV. Mandible like structure with fourteen teeth in a benign cystic teratoma. Indian J Pathol Microbiol. 2009;52(4):595–6. doi:10.4103/0377-4929.56145 IndianJPatholMicrobiol_2009_52_4_595_56145 [pii].
Ingale Y, Shankar AA, Routray S, Agrawal M, Kadam A, Patil T. Ectopic teeth in ovarian teratoma: a rare appearance. Case Rep Dent. 2013;2013:970464. doi:10.1155/2013/970464.
Willis RA. The spread of tumours in the human body. 3rd ed. London: Butterworths; 1973.
Kuno N, Kadomatsu K, Nakamura M, Miwa-Fukuchi T, Hirabayashi N, Ishizuka T. Mature ovarian cystic teratoma with a highly differentiated homunculus: a case report. Birth Defects Res A Clin Mol Teratol. 2004;70(1):40–6. doi:10.1002/bdra.10133.
Abbott TM, Hermann Jr WJ, Scully RE. Ovarian fetiform teratoma (homunculus) in a 9-year-old girl. Int J Gynecol Pathol. 1984;2(4):392–402.
Joutel N, Pirot N, Pop I, Khoutech K, Bisson V, Cingotti M et al. Homunculus: fetiform teratoma. Gynecol Obstet Fertil 2012;40(2):121–3. doi:10.1016/j.gyobfe.2011.07.046 S1297-9589(11)00273-6 [pii].
Weldon-Linne CM, Rushovich AM. Benign ovarian cystic teratomas with homunculi. Obstet Gynecol. 1983;61(3 Suppl):88S–94S.
Di Donato N, Zanello M, Corti B, Seracchioli R, Paradisi R. Incidental diagnosis of ectopic adenohypophyseal tissue in an ovarian dermoid cyst: a case report. Eur J Obstet Gynecol Reprod Biol. 2014;183:203–4. doi:10.1016/j.ejogrb.2014.10.044. S0301-2115(14)00568-5 [pii].
McKeel Jr DW, Askin FB. Ectopic hypophyseal hormonal cells in benign cystic teratoma of the ovary. Light microscopic histochemical dye staining and immunoperoxidase cytochemistry. Arch Pathol Lab Med. 1978;102(3):122–8.
Khan N, Sen Ray P, Hakim S, Ziyauddin F. Retinal tissue in mature cystic teratoma of ovary presenting with full-term pregnancy. BMJ Case Rep. 2014;2014. doi:10.1136/bcr-2013-202914. bcr2013202914 [pii] bcr-2013-202914 [pii].
Mercur L, Farkas TG, Anker PM, Brill R, Mund ML. Developing eye in an ovarian teratoma. Arch Ophthalmol. 1976;94(4):597–8.
Oi RH, Dobbs M. Lactating breast tissue in benign cystic teratoma. Am J Obstet Gynecol. 1978;130(6):729–31. doi:0002-9378(78)90342-3 [pii].
Ulirsch RC, Goldman RL. An unusual teratoma of the ovary: neurogenic cyst with lactating breast tissue. Obstet Gynecol. 1982;60(3):400–2.
Muretto P. The relationship of Langerhans cells to melanocytes and Schwann cells in mature cystic teratomas of the ovary. Int J Surg Pathol. 2007;15(3):266–71. doi:15/3/266 [pii] 10.1177/1066896907302227.
Nogales Jr FF, Ortega I, Rivera F, Armas JR. Metanephrogenic tissue in immature ovarian teratoma. Am J Surg Pathol. 1980;4(3):297–9.
Lopez JI, de Santamaria IS, Claros IJ, Garmendia M, Gaafar A, Bilbao FJ. Prostatic remnants in mature cystic teratoma of the ovary. Ann Diagn Pathol 2008;12(5):378–80. doi:10.1016/j.anndiagpath.2007.04.005 S1092-9134(07)00054-8 [pii].
Nogales FF, Vergara E, Medina MT. Prostate in ovarian mature cystic teratoma. Histopathology. 1995;26(4):373–5.
Melniker LA, Slavutin LJ. Prostatic tissue in a benign cystic teratoma of the ovary. Diagn Gynecol Obstet. 1980;2(2):139–45.
Shelekhova KV, Hejda V, Kazakov DV, Michal M. Mature cystic teratoma of the ovary with male accessory sexual glands including seminal vesicles, prostatic tissue, and bulbo-urethral glands: a case report. Virchows Arch. 2008;452(1):109–11. doi:10.1007/s00428-007-0546-9.
Glavina-Durdov M, Forempoher G. Thymus tissue in ovarian cystic teratoma: possible clues to its histogenesis. Pathol Res Pract 2000;196(11):795–7; discussion 8–9. doi:S0344-0338(00)80116-8 [pii] 10.1016/S0344-0338(00)80116-8.
Agarwal K, Agarwal C, Agarwal S, Chaddha R. Mature teratoma with loops of colon: a case report. Indian J Pathol Microbiol. 2007;50(2):405–7.
Woodfield B, Katz DA, Cantrell CJ, Bogard PJ. A benign cystic teratoma with gastrointestinal tract development. Am J Clin Pathol. 1985;83(2):236–40.
Zaragoza Vargas PE, Dickter S, Baquera Heredia J, Ortiz Hidalgo C. Gastric wall in benign cystic teratoma of the ovary. Ginecol Obstet Mex. 2007;75(1):43–5.
Agaimy A, Lindner M, Wuensch PH. Interstitial cells of Cajal (ICC) in mature cystic teratoma of the ovary. Histopathology 2006;48(2):208–9. doi:HIS2211 [pii] 10.1111/j.1365-2559.2005.02211.x.
Agaimy A, Wunsch PH. Coexistence of interstitial cell of Cajal hyperplasia and microcarcinoidosis in a mature cystic teratoma of the ovary. Histopathology 2008;52(2):260–2. doi:HIS2910 [pii] 10.1111/j.1365-2559.2007.02910.x.
Marucci G, Collina G. Mature cystic teratoma of the ovary with a small ganglioneuroma. Pathologica. 2006;98(1):41–3.
Ishida M, Iwai M, Yoshida K, Kagotani A, Okabe H. Well-differentiated cerebellum within a mature cystic teratoma of the ovary. Int J Clin Exp Pathol. 2014;7(3):1255–7.
Dominguez-Rosado I, Michel J, Ortiz-Hidalgo C. Ovarian mature cystic teratoma exhibiting well-differentiated cerebellum in a 14-year-old female. Pediatr Dev Pathol. 2005;8(2):247–9. doi:10.1007/s10024-004-7083-1.
MacSween RN. Foetal cerebellar tissue in an ovarian teratoma. J Pathol Bacteriol. 1968;96(2):513–4. doi:10.1002/path.1700960235.
Nogales FF, Aguilar D. Neural tissue in human teratomas. In:The biology of teratomas. Clifton: Human Press; 1983.
PN V. Spontaneous myospherulosis developing at contact with a mature cystic dysembryoma of the ovary. Pathogenetic discussion apropos of a case diagnosed by laparoscopic biopsy. J Gynecol Obstet Biol Reprod (Paris). 1988;17(1):15–8.
Hirakawa T, Tsuneyoshi M, Enjoji M. Squamous cell carcinoma arising in mature cystic teratoma of the ovary. Clinicopathologic and topographic analysis. Am J Surg Pathol. 1989;13(5):397–405.
McKenney JK, Soslow RA, Longacre TA. Ovarian mature teratomas with mucinous epithelial neoplasms: morphologic heterogeneity and association with pseudomyxoma peritonei. Am J Surg Pathol. 2008;32(5):645–55. doi:10.1097/PAS.0b013e31815b486d.
Choi EJ, Koo YJ, Jeon JH, Kim TJ, Lee KH, Lim KT. Clinical experience in ovarian squamous cell carcinoma arising from mature cystic teratoma: A rare entity. Obstet Gynecol Sci. 2014;57(4):274–80. doi:10.5468/ogs.2014.57.4.274.
Park JY, Kim DY, Kim JH, Kim YM, Kim YT, Nam JH. Malignant transformation of mature cystic teratoma of the ovary: experience at a single institution. Eur J Obstet Gynecol Reprod Biol 2008;141(2):173–8. doi:10.1016/j.ejogrb.2008.07.032 S0301-2115(08)00298-4 [pii].
Park JY, Song JS, Choi G, Kim JH, Nam JH. Pure primary squamous cell carcinoma of the ovary: a report of two cases and review of the literature. Int J Gynecol Pathol 2010;29(4):328–34. doi:10.1097/PGP.0b013e3181c6d965 00004347-201007000-00005 [pii].
Vang R, Gown AM, Zhao C, Barry TS, Isacson C, Richardson MS et al. Ovarian mucinous tumors associated with mature cystic teratomas: morphologic and immunohistochemical analysis identifies a subset of potential teratomatous origin that shares features of lower gastrointestinal tract mucinous tumors more commonly encountered as secondary tumors in the ovary. Am J Surg Pathol 2007;31(6):854–69. doi:10.1097/PAS.0b013e31802efb45 00000478-200706000-00005 [pii].
Quinonez E, Schuldt M, Retamero JA, Nogales FF. Ovarian strumal carcinoid containing appendiceal-type mucinous tumor patterns presenting as pseudomyxoma peritonei. Int J Gynecol Pathol. 2015;34(3):293–7. doi:10.1097/PGP.0000000000000138.
Nakatsuka S, Wakimoto T, Ozaki K, Nagano T, Kimura H, Nakajo K, et al. Mucinous borderline-like tumor of the gastrointestinal type arising from mature cystic teratoma of the ovary and its immunohistochemical cytokeratin and mucin phenotype. J Obstet Gynaecol Res. 2012;38(2):471–5. doi:10.1111/j.1447-0756.2011.01729.x.
McCluggage WG, Bissonnette JP, Young RH. Primary malignant melanoma of the ovary: a report of 9 definite or probable cases with emphasis on their morphologic diversity and mimicry of other primary and secondary ovarian neoplasms. Int J Gynecol Pathol 2006;25(4):321–9. doi:10.1097/01.pgp.0000215301.39900.07 00004347-200610000-00002 [pii].
Hurwitz JL, Fenton A, McCluggage WG, McKenna S. Squamous cell carcinoma arising in a dermoid cyst of the ovary: a case series. BJOG 2007;114(10):1283–7. doi:BJO1478 [pii] 10.1111/j.1471-0528.2007.01478.x.
Pantoja E, Rodriguez-Ibanez I, Axtmayer RW, Noy MA, Pelegrina I. Complications of dermoid tumors of the ovary. Obstet Gynecol. 1975;45(1):89–94.
Marcial-Rojas RA, Medina R. Cystic teratomas of the ovary; a clinical and pathological analysis of two hundred sixty-eight tumors. AMA Arch Pathol. 1958;66(4):577–89.
Klionsky BL, Nickens OJ, Amortegui AJ. Squamous cell carcinoma in situ arising in adult cystic teratoma of the ovary. Arch Pathol. 1972;93(2):161–3.
Nogales FF, Luna MA, Vergara E, O’Valle F, Lopez-Cantarero M. Small-cell epidermoid carcinoma with melanocyte colonization arising in a mature ovarian cystic teratoma. Arch Pathol Lab Med. 1995;119(6):551–4.
Karateke A, Gurbuz A, Kir G, Haliloglu B, Kabaca C, Devranoglu B et al. Mucoepidermoid variant of adenosquamous carcinoma arising in ovarian dermoid cyst: a case report and review of the literature. Int J Gynecol Cancer 2006;16 Suppl 1:379–84. doi:IJG233 [pii] 10.1111/j.1525-1438.2006.00233.x.
Iwasa A, Oda Y, Kaneki E, Ohishi Y, Kurihara S, Yamada T et al. Squamous cell carcinoma arising in mature cystic teratoma of the ovary: an immunohistochemical analysis of its tumorigenesis. Histopathology 2007;51(1):98–104. doi:HIS2727 [pii] 10.1111/j.1365-2559.2007.02727.x.
Fox H. Bronchial carcinoma in an ovarian cystic teratoma (dermoid). J Clin Pathol. 1965;18:164–6.
Yahata T, Kawasaki T, Serikawa T, Suzuki M, Tanaka K. Adenocarcinoma arising from respiratory ciliated epithelium in benign cystic teratoma of the ovary: a case report with analyzes of the CT, MRI, and pathological findings. J Obstet Gynaecol Res. 2008;34(3):408–12. doi:10.1111/j.1447-0756.2008.00784.x JOG784 [pii].
Hershkovitz D, Vlodavsky E, Simon E, Ben-Izhak O. KRAS mutation positive mucinous adenocarcinoma originating in mature ovarian teratoma: Case report and review of literature. Pathol Int. 2013;63(12):611–4. doi:10.1111/pin.12123.
Fujiwara K, Ginzan S, Silverberg SG. Mature cystic teratomas of the ovary with intestinal wall structures harboring intestinal- type epithelial neoplasms. Gynecol Oncol 1995;56(1):97–101. doi:S0090-8258(85)71017-7 [pii] 10.1006/gyno.1995.1017.
Clark ME, Will MD. Intestinal-type adenocarcinoma arising in a mature cystic teratoma of the ovary. Int J Gynecol Pathol. 2016; doi:10.1097/PGP.0000000000000258.
Halabi M, Oliva E, Mazal PR, Breitenecker G, Young RH. Prostatic tissue in mature cystic teratomas of the ovary: a report of four cases, including one with features of prostatic adenocarcinoma, and cytogenetic studies. Int J Gynecol Pathol. 2002;21(3):261–7.
Stanhiser J, Mahdi H, Rosa G, Harper H, Shepard D, Rose PG, et al. Prostate-type adenocarcinoma in mature cystic ovarian teratoma. Int J Gynecol Pathol. 2016;35(2):185–90. doi:10.1097/PGP.0000000000000217.
Wheeler L, Westhoff GL, O’Keefe MC, Kong CS, Karam A. Adenocarcinoma with breast/adnexal and upper gastrointestinal differentiation arising in an ovarian mature cystic teratoma: a case report and review of the literature. Int J Gynecol Pathol. 2016;35(1):72–7. doi:10.1097/PGP.0000000000000218.
Monteagudo C, Torres JV, Llombart-Bosch A. Extramammary Paget’s disease arising in a mature cystic teratoma of the ovary. Histopathology. 1999;35(6):582–4. doi:his833d [pii].
Shimizu S, Kobayashi H, Suchi T, Torii Y, Narita K, Aoki S. Extramammary Paget’s disease arising in mature cystic teratoma of the ovary. Am J Surg Pathol. 1991;15(10):1002–6.
Randall BJ, Ritchie C, Hutchison RS. Paget’s disease and invasive undifferentiated carcinoma occurring in a mature cystic teratoma of the ovary. Histopathology. 1991;18(5):469–70.
Shintaku M, Nakagawa E, Tsubura A. Extramammary Paget’s disease arising in a mature cystic teratoma of the ovary: immunohistochemical expression of androgen receptor. Pathol Int. 2011;61(8):498–500. doi:10.1111/j.1440-1827.2011.02692.x.
Hyun HS, Mun ST. Primary malignant melanoma arising in a cystic teratoma. Obstet Gynecol Sci. 2013;56(3):201–4. doi:10.5468/ogs.2013.56.3.201.
Lee J, Magpayo J, Panchari C, Peng SK. Primary malignant melanoma of the ovary arising in a cystic teratoma; case report and review of the literature. Dermatol Online J. 2014;20(10). doi:13030/qt0tr2b2z9 [pii].
Godoy HE, Kesterson JP, Kasznica JM, Lele S. Malignant melanoma arising in mature cystic teratoma of the ovary. Gynecol Oncol Case Rep 2011;2(1):14–5. doi:10.1016/j.gynor.2011.10.007 S2211-338X(11)00016-0 [pii].
Boughton RS, Hughmanick S, Marin-Padilla M. Malignant melanoma arising in an ovarian cystic teratoma in pregnancy. J Am Acad Dermatol. 1987;17(5 Pt 2):871–5.
Mathios AJ, McCausland AM. Basal cell carcinoma originating in a benign cystic teratoma of the ovary. Obstet Gynecol. 1973;42(6):892–6.
Alfsen GC, Strom EH. Pilomatrixoma of the ovary: a rare variant of mature teratoma. Histopathology. 1998;32(2):182–3.
An HJ, Jung YH, Yoon HK, Jung SJ. Sebaceous carcinoma arising in mature cystic teratoma of ovary. Korean J Pathol. 2013;47(4):383–7. doi:10.4132/KoreanJPathol.2013.47.4.383.
Chumas JC, Scully RE. Sebaceous tumors arising in ovarian dermoid cysts. Int J Gynecol Pathol. 1991;10(4):356–63.
Yasui W, Kameda T, Harada D, Nakatani H, Hata J, Ito H, et al. A case of sweat gland carcinoma arising in a mature cystic teratoma of the ovary. Gan No Rinsho. 1987;33(7):875–80.
Morimitsu Y, Nakashima O, Nakashima Y, Kojiro M, Shimokobe T. Apocrine adenocarcinoma arising in cystic teratoma of the ovary. Arch Pathol Lab Med. 1993;117(6):647–9.
Kudo M. The nature of “blue nevus” in cystic teratomas of the ovary. An ultrastructural evidence for Schwann cell origin. Acta Pathol Jpn. 1985;35(3):693–8.
Kuroda N, Hirano K, Inui Y, Yamasaki Y, Toi M, Nakayama H, et al. Compound melanocytic nevus arising in a mature cystic teratoma of the ovary. Pathol Int. 2001;51(11):902–4. doi:1288 [pii]
Tsang P, Berman L, Kasznica J. Adnexal tumor and a pigmented nevoid lesion in a benign cystic ovarian teratoma. Arch Pathol Lab Med. 1993;117(8):846–7.
Amortegui AJ, Kanbour AI. Compound nevus in a cystic teratoma of the ovary. Arch Pathol Lab Med. 1981;105(2):115–6.
Kruger S, Schmidt H, Kupker W, Rath FW, Feller AC. Fibrosarcoma associated with a benign cystic teratoma of the ovary. Gynecol Oncol 2002;84(1):150–4. doi:10.1006/gyno.2001.6408 S009082580196408X [pii].
Stamp GW, McConnell EM. Malignancy arising in cystic ovarian teratomas. A report of 24 cases. Br J Obstet Gynaecol. 1983;90(7):671–5.
Kelley RR, Scully RE. Cancer developing in dermoid cysts of the ovary. A report of 8 cases, including a carcinoid and a leiomyosarcoma. Cancer. 1961;14:989–1000.
Allam-Nandyala P, Bui MM, Caracciolo JT, Hakam A. Squamous cell carcinoma and osteosarcoma arising from a dermoid cyst–a case report and review of literature. Int J Clin Exp Pathol. 2010;3(3):313–8.
Ngwalle KE, Hirakawa T, Tsuneyoshi M, Enjoji M. Osteosarcoma arising in a benign dermoid cyst of the ovary. Gynecol Oncol. 1990;37(1):143–7.
Yasunaga M, Saito T, Eto T, Okadome M, Ariyoshi K, Nishiyama K, et al. Dedifferentiated chondrosarcoma arising in a mature cystic teratoma of the ovary: a case report and review of the literature. Int J Gynecol Pathol. 2011;30(4):391–4. doi:10.1097/PGP.0b013e318211f56e.
Climie AR, Heath LP. Malignant degeneration of benign cystic teratomas of the ovary. Review of the literature and report of a chondrosarcoma and carcinoid tumor. Cancer. 1968;22(4):824–32.
Contreras AL, Malpica A. Angiosarcoma arising in mature cystic teratoma of the ovary: a case report and review of the literature. Int J Gynecol Pathol 2009;28(5):453–7. doi:10.1097/PGP.0b013e31819d4574 00004347-200909000-00006 [pii].
den Bakker MA, Ansink AC, Ewing-Graham PC. “Cutaneous-type” angiosarcoma arising in a mature cystic teratoma of the ovary. J Clin Pathol. 2006;59(6):658–60. doi:59/6/658 [pii] 10.1136/jcp.2005.029751.
Kefeli M, Kandemir B, Akpolat I, Yildirim A, Kokcu A. Rhabdomyosarcoma arising in a mature cystic teratoma with contralateral serous carcinoma: case report and review of the literature. Int J Gynecol Pathol. 2009;28(4):372–5. doi:10.1097/PGP.0b013e3181929269.
Ueda G, Sato Y, Yamasaki M, Inoue M, Hiramatsu K, Tanaka Y, et al. Malignant fibrous histiocytoma arising in a benign cystic teratoma of the ovary. Gynecol Oncol. 1977;5(3):313–22. doi:0090-8258(77)90041-5 [pii].
Richardson G, Robertson DI, O’Connor ME, Nation JG, Stuart GC. Malignant transformation occurring in mature cystic teratomas of the ovary. Can J Surg. 1990;33(6):499–503.
Adams H, Went P, Tolnay M, Sartorius G, Singer G. Meningothelial meningioma in a mature cystic teratoma of the ovary. Pathologe. 2007;28(4):278–80. doi:10.1007/s00292-006-0828-z.
Takeshima Y, Kaneko M, Furonaka O, Jeet AV, Inai K. Meningioma in mature cystic teratoma of the ovary. Pathol Int 2004;54(7):543–8. doi:10.1111/j.1440-1827.2004.01662.x PIN1662 [pii].
Elliot VJ, Shaw EC, Walker M, Jaynes E, Theaker JM. Ovarian paraganglioma arising from mature cystic teratoma. Int J Gynecol Pathol. 2012;31(6):545–6. doi:10.1097/PGP.0b013e31824d3564.
Mahdavi A, Silberberg B, Malviya VK, Braunstein AH, Shapiro J. Gangliocytic paraganglioma arising from mature cystic teratoma of the ovary. Gynecol Oncol. 2003;90(2):482–5. doi:S0090825803002725 [pii].
den Boon J, van Dijk CM, Helfferich M, Peterse HL. Glioblastoma multiforme in a dermoid cyst of the ovary. A case report. Eur J Gynaecol Oncol. 1999;20(3):187–8.
Bjersing L, Cajander S, Rogo K, Ottosson UB, Stendahl U. Glioblastoma multiform in a dermoid cyst of the ovary. Eur J Gynaecol Oncol. 1989;10(6):389–92.
Yadav A, Lellouch-Tubiana A, Fournet JC, Quazza JE, Kalifa C, Sainte-Rose C, et al. Glioblastoma multiforme in a mature ovarian teratoma with recurring brain tumours. Histopathology. 1999;35(2):170–3. doi:his695 [pii].
Spaulding R, Alatassi H, Stewart Metzinger D, Moghadamfalahi M. Ependymoma and carcinoid tumor associated with ovarian mature cystic teratoma in a patient with multiple endocrine neoplasia I. Case Rep Obstet Gynecol. 2014;2014:712657. doi:10.1155/2014/712657.
Stolnicu S, Furtado A, Sanches A, Nicolae A, Preda O, Hincu M et al. Ovarian ependymomas of extra-axial type or central immunophenotypes. Hum Pathol 2011;42(3):403–8. doi:10.1016/j.humpath.2010.07.017 S0046-8177(10)00311-4 [pii].
Ud Din N, Memon A, Aftab K, Ahmad Z, Ahmed R, Hassan S. Oligodendroglioma arising in the glial component of ovarian teratomas: a series of six cases and review of literature. J Clin Pathol 2012;65(7):631–4. doi:10.1136/jclinpath-2012-200714 jclinpath-2012-200714 [pii].
Opris I, Ducrotoy V, Bossut J, Lamy A, Sabourin JC. Oligodendroglioma arising in an ovarian mature cystic teratoma. Int J Gynecol Pathol. 2009;28(4):367–71. doi:10.1097/PGP.0b013e318196c4c0.
Skopelitou A, Mitselou A, Michail M, Mitselos V, Stefanou D. Pilocytic astrocytoma arising in a dermoid cyst of the ovary: a case presentation. Virchows Arch. 2002;440(1):105–6.
Wey SL, Chen CK, Chen TC, Chen TY. Desmoplastic medulloblastoma arising from an ovarian teratoma: a case report and review of the literature. Int J Surg Pathol 2013;21(4):427–31. doi:10.1177/1066896912464048 1066896912464048 [pii].
Olah KS, Needham PG, Jones B. Multiple neuroectodermal tumors arising in a mature cystic teratoma of the ovary. Gynecol Oncol. 1989;34(2):222–5. doi:0090-8258(89)90147-9 [pii].
Block M, Gilbert E, Davis C. Metastatic neuroblastoma arising in an ovarian teratoma with long-term survival. Case report and review of the literature. Cancer. 1984;54(3):590–5.
Reid HA, van der Walt JD, Fox H. Neuroblastoma arising in a mature cystic teratoma of the ovary. J Clin Pathol. 1983;36(1):68–73.
Kleinman GM, Young RH, Scully RE. Primary neuroectodermal tumors of the ovary. A report of 25 cases. Am J Surg Pathol. 1993;17(8):764–78.
Aguirre P, Scully RE. Malignant neuroectodermal tumor of the ovary, a distinctive form of monodermal teratoma: report of five cases. Am J Surg Pathol. 1982;6(4):283–92.
Boor PJ, Schoene WC. Fetal cerebellar tissue associated with a primitive neuro-epithelial tumor in an ovarian teratoma. Can J Neurol Sci. 1975;2(2):139–41.
Hirschowitz L, Ansari A, Cahill DJ, Bamford DS, Love S. Central neurocytoma arising within a mature cystic teratoma of the ovary. Int J Gynecol Pathol. 1997;16(2):176–9.
von Gunten M, Burger H, Vajtai I. Choroid plexus papilloma developing in a dermoid cyst of ovary. Histopathology 2006;49(2):204–5. doi:HIS2383 [pii] 10.1111/j.1365-2559.2006.02383.x.
Marquette S, Amant F, Vergote I, Moerman P. Pseudomyxoma peritonei associated with a mucinous ovarian tumor arising from a mature cystic teratoma. A case report. Int J Gynecol Pathol 2006;25(4):340–3. doi:10.1097/01.pgp.0000209571.16340.20 00004347-200610000-00005 [pii].
Ronnett BM, Seidman JD. Mucinous tumors arising in ovarian mature cystic teratomas: relationship to the clinical syndrome of pseudomyxoma peritonei. Am J Surg Pathol. 2003;27(5):650–7.
Valli R, Froio E, Alvarez de Celis MI, Mandato VD, Piana S. Diffuse large B-cell lymphoma occurring in an ovarian cystic teratoma: expanding the spectrum of large B-cell lymphoma associated with chronic inflammation. Hum Pathol 2014;45(12):2507–11. doi:10.1016/j.humpath.2014.09.002 S0046-8177(14)00368-2 [pii].
Gandhi N, Soomro IN, O’Connor S, Sovani V. Primary lymphoma arising in a mature cystic teratoma of the ovary. Histopathology. 2012;61(6):1238–40. doi:10.1111/j.1365-2559.2012.04343.x.
Damiani S. Pulmonary papillary adenoma-like tumour arising in ovarian teratoma. Virchows Arch. 2004;445(1):96–7. doi:10.1007/s00428-004-1022-4.
Ikota H, Kaneko K, Takahashi S, Kawarai M, Tanaka Y, Yokoo H, et al. Malignant transformation of ovarian mature cystic teratoma with a predominant pulmonary type small cell carcinoma component. Pathol Int. 2012;62(4):276–80. doi:10.1111/j.1440-1827.2012.02804.x.
Rubio A, Schuldt M, Chamorro C, Crespo-Lora V, Nogales FF. Ovarian small cell carcinoma of pulmonary type arising in mature cystic teratomas with metastases to the contralateral ovary. Int J Surg Pathol 2015;23(5):388–92. doi:10.1177/1066896915586254 1066896915586254 [pii].
Miyamoto M, Takano M, Goto T, Kato M, Sasaki N, Furuya K. Large cell neuroendocrine carcinoma arising in mature cystic teratoma: a case report and review of the literature. Eur J Gynaecol Oncol. 2012;33(4):414–8.
Chenevert J, Bessette P, Plante M, Tetu B, Dube V. Mixed ovarian large cell neuroendocrine carcinoma, mucinous adenocarcinoma, and teratoma: a report of two cases and review of the literature. Pathol Res Pract 2009;205(9):657–61. doi:10.1016/j.prp.2009.01.013 S0344-0338(09)00040-5 [pii].
Choi YD, Lee JS, Choi C, Park CS, Nam JH. Ovarian neuroendocrine carcinoma, non-small cell type, associated with serous carcinoma. Gynecol Oncol 2007;104(3):747–52. doi:S0090-8258(06)00900-0 [pii] 10.1016/j.ygyno.2006.11.008.
Ting WH, Hsiao SM, Lin HH, Wei MC. Primary carcinoid tumor of the ovary arising in a mature cystic teratoma: a case report. Eur J Gynaecol Oncol. 2014;35(1):100–2.
Guney N, Sayilgan T, Derin D, Ozcan D. Primary carcinoid tumor arising in a mature cystic teratoma of the ovary: a case report. Eur J Gynaecol Oncol. 2009;30(2):223–5.
Huang B, Wu X, Zhou Q, Hu Y, Zhao H, Zhu H, et al. Cushing’s syndrome secondary to ectopic ACTH secretion from carcinoid tumor within an ovarian mature teratoma: a case report and review of the literature. Gynecol Endocrinol. 2014;30(3):192–6. doi:10.3109/09513590.2013.871518.
Hsu CY, Yang CF, Chen WY, Chiang H. Adenosquamous carcinoma and schneiderian papilloma-like lesion in a mature cystic teratoma of the ovary: a case report. Zhonghua Yi Xue Za Zhi (Taipei). 1996;57(5):375–9.
Las Heras F, Pritzker KP, Colgan TJ. Chordoma arising in a mature cystic teratoma of the ovary: a case report. Pathol Res Pract 2007;203(6):467–71. doi:S0344-0338(07)00031-3 [pii] 10.1016/j.prp.2006.12.010.
Silver SA, Tavassoli FA. Glomus tumor arising in a mature teratoma of the ovary: report of a case simulating a metastasis from cervical squamous carcinoma. Arch Pathol Lab Med. 2000;124(9):1373–5. doi:10.1043/0003-9985(2000)124<1373:GTAIAM>2.0.CO;2.
Genadry R, Parmley T, Woodruff JD. Secondary malignancies in benign cystic teratomas. Gynecol Oncol. 1979;8(2):246–51. doi:0090-8258(79)90032-5 [pii].
Hanada M, Tsujimura T, Shimizu H. Multiple malignancies (squamous cell carcinoma and sarcoma) arising in a dermoid cyst of the ovary. Acta Pathol Jpn. 1981;31(4):681–8.
Leroy C, Gontier MF, Gontier F, Minh HN, Smadja A. Malignant dermoid cyst of the ovary with double tumor cell population, revealed by an umbilical metastasis. Ann Pathol. 1992;12(3):178–82.
Terada T, Tateoka K. Ovarian cystic tumor composed of Brenner tumor and struma ovarii. Int J Clin Exp Pathol. 2012;5(3):274–7.
Nogales FF. Primary chondrosarcoma of the ovary. Histopathology. 1982;6:376.
Nogales Jr FF, Silverberg SG. Epidermoid cysts of the ovary: a report of five cases with histogenetic considerations and ultrastructural findings. Am J Obstet Gynecol. 1976;124(5):523–8.
Isaac MA, Vijayalakshmi S, Madhu CS, Bosincu L, Nogales FF. Pure cystic nephroblastoma of the ovary with a review of extrarenal Wilms’ tumors. Hum Pathol. 2000;31(6):761–4. doi:10.1053/hupa.2000.7627.
Stolnicu S, Preda O, Dohan M, Puscasiu L, Garcia-Galvis OF, Nogales FF. Pseudoglandular hepatoid differentiation in endometrioid carcinoma of the ovary simulates oxyphilic cell change. Int J Gynecol Pathol. 2008;27(4):521–5. doi:10.1097/PGP.0b013e318178e566.
Nogales FF, Concha A, Plata C, Ruiz-Avila I. Granulosa cell tumor of the ovary with diffuse true hepatic differentiation simulating stromal luteinization. Am J Surg Pathol. 1993;17(1):85–90.
Devaney K, Snyder R, Norris HJ, Tavassoli FA. Proliferative and histologically malignant struma ovarii: a clinicopathologic study of 54 cases. Int J Gynecol Pathol. 1993;12(4):333–43.
Jin C, Dong R, Bu H, Yuan M, Zhang Y, Kong B. Coexistence of benign struma ovarii, pseudo-Meigs’ syndrome and elevated serum CA 125: case report and review of the literature. Oncol Lett 2015;9(4):1739–42. doi:10.3892/ol.2015.2927 ol-09-04-1739 [pii].
Outwater EK, Siegelman ES, Hunt JL. Ovarian teratomas: tumor types and imaging characteristics. Radiographics. 2001;21(2):475–90. doi:10.1148/radiographics.21.2.g01mr09475.
Moon S, Waxman M. Mixed ovarian tumor composed of Brenner and thyroid elements. Cancer. 1976;38(5):1997–2001.
Yoshida M, Obayashi C, Tachibana M, Minami R. Coexisting Brenner tumor and struma ovarii in the right ovary: case report and review of the literature. Pathol Int 2004;54(10):793–7. doi:PIN1757 [pii] 10.1111/j.1440-1827.2004.01757.x.
Shaco-Levy R, Peng RY, Snyder MJ, Osmond GW, Veras E, Bean SM, et al. Malignant struma ovarii: a blinded study of 86 cases assessing which histologic features correlate with aggressive clinical behavior. Arch Pathol Lab Med. 2012;136(2):172–8. doi:10.5858/arpa.2011-0092-OA.
Garg K, Soslow RA, Rivera M, Tuttle MR, Ghossein RA. Histologically bland “extremely well differentiated” thyroid carcinomas arising in struma ovarii can recur and metastasize. Int J Gynecol Pathol 2009;28(3):222–30. doi:10.1097/PGP.0b013e31818a2b99 00004347-200905000-00003 [pii].
Robboy SJ, Shaco-Levy R, Peng RY, Snyder MJ, Donahue J, Bentley RC et al. Malignant struma ovarii: an analysis of 88 cases, including 27 with extraovarian spread. Int J Gynecol Pathol 2009;28(5):405–22. doi:10.1097/PGP.0b013e3181a27777 00004347-200909000-00001 [pii].
Wolff EF, Hughes M, Merino MJ, Reynolds JC, Davis JL, Cochran CS, et al. Expression of benign and malignant thyroid tissue in ovarian teratomas and the importance of multimodal management as illustrated by a BRAF-positive follicular variant of papillary thyroid cancer. Thyroid. 2010;20(9):981–7. doi:10.1089/thy.2009.0458.
Boutross-Tadross O, Saleh R, Asa SL. Follicular variant papillary thyroid carcinoma arising in struma ovarii. Endocr Pathol. 2007;18(3):182–6. doi:10.1007/s12022-007-0022-8.
Schmidt J, Derr V, Heinrich MC, Crum CP, Fletcher JA, Corless CL et al. BRAF in papillary thyroid carcinoma of ovary (struma ovarii). Am J Surg Pathol 2007;31(9):1337–43. doi:10.1097/PAS.0b013e31802f5404 00000478-200709000-00005 [pii].
Reed NS, Gomez-Garcia E, Gallardo-Rincon D, Barrette B, Baumann K, Friedlander M et al. Gynecologic Cancer InterGroup (GCIG) consensus review for carcinoid tumors of the ovary. Int J Gynecol Cancer 2014;24(9 Suppl 3):S35–41. doi:10.1097/IGC.0000000000000265 00009577-201411003-00009 [pii].
Soga J, Osaka M, Yakuwa Y. Carcinoids of the ovary: an analysis of 329 reported cases. J Exp Clin Cancer Res. 2000;19(3):271–80.
Desouki MM, Lioyd J, Xu H, Cao D, Barner R, Zhao C. CDX2 may be a useful marker to distinguish primary ovarian carcinoid from gastrointestinal metastatic carcinoids to the ovary. Hum Pathol. 2013;44(11):2536–41. doi:10.1016/j.humpath.2013.06.014.
Davis KP, Hartmann LK, Keeney GL, Shapiro H. Primary ovarian carcinoid tumors. Gynecol Oncol 1996;61(2):259–65. doi:S0090-8258(96)90136-5 [pii] 10.1006/gyno.1996.0136.
Goldman T, Adamson K, Yang E. Resolution of right-sided heart failure symptoms after resection of a primary ovarian carcinoid tumor. Tex Heart Inst J. 2014;41(5):533–6. doi:10.14503/THIJ-13-3314.
Dessauvagie BF, Lai PH, Oost E, Thomas A, Stewart CJ. Medial hypertrophy of the ovarian vein: a novel type of vascular pathology associated with a primary ovarian carcinoid tumor. Int J Gynecol Pathol. 2015;34(1):36–9. doi:10.1097/PGP.0000000000000114.
Muller KE, Tafe LJ, Gonzalez JL, West LA, Schned AR. Ovarian strumal carcinoid producing peptide YY associated with severe constipation: a case report and review of the literature. Int J Gynecol Pathol. 2015;34(1):30–5. doi:10.1097/PGP.0000000000000117.
Robboy SJ. Insular carcinoid of ovary associated with malignant mucinous tumors. Cancer. 1984;54(10):2273–6.
Mandai M, Konishi I, Tsuruta Y, Suginami N, Kusakari T, Iwasaki T, et al. Krukenberg tumor from an occult appendiceal adenocarcinoid: a case report and review of the literature. Eur J Obstet Gynecol Reprod Biol. 2001;97(1):90–5.
Baker PM, Oliva E, Young RH, Talerman A, Scully RE. Ovarian mucinous carcinoids including some with a carcinomatous component: a report of 17 cases. Am J Surg Pathol. 2001;25(5):557–68.
Armes JE, Ostor AG. A case of malignant strumal carcinoid. Gynecol Oncol 1993;51(3):419–23. doi:S0090-8258(83)71316-8 [pii] 10.1006/gyno.1993.1316.
Kimura N, Sasano N, Namiki T. Evidence of hybrid cell of thyroid follicular cell and carcinoid cell in strumal carcinoid. Int J Gynecol Pathol. 1986;5(3):269–77.
Scully RE. Gonadoblastoma; a gonadal tumor related to the dysgerminoma (seminoma) and capable of sex-hormone production. Cancer. 1953;6(3):455–63.
Hertel JD, Huettner PC, Dehner LP, Pfeifer JD. The chromosome Y-linked testis-specific protein locus TSPY1 is characteristically present in gonadoblastoma. Hum Pathol. 2010;41(11):1544–9. doi:10.1016/j.humpath.2010.04.007.
Ulbright TM, Young RH. Gonadoblastoma and selected other aspects of gonadal pathology in young patients with disorders of sex development. Semin Diagn Pathol 2014;31(5):427–40. doi:10.1053/j.semdp.2014.07.001 S0740-2570(14)00064-1 [pii].
Scully RE. Gonadoblastoma. A review of 74 cases. Cancer. 1970;25(6):1340–56.
Schiller PJ, Tagatz GE. Gonadal calcification. Indication for gonadectomy in gonadal dysgenesis. Obstet Gynecol. 1976;48(4):486–90.
Manivel JC, Dehner LP, Burke B. Ovarian tumorlike structures, biovular follicles, and binucleated oocytes in children: their frequency and possible pathologic significance. Pediatr Pathol. 1988;8(3):283–92.
Serment H, Laffargue P, Piana L, Blanc B. Tumeurs endocrines de l’ovaire chez l’enfant. Rev Fr Gynecol Obstet. 1970;65(9):465–77.
Talerman A. A distinctive gonadal neoplasm related to gonadoblastoma. Cancer. 1972;30(5):1219–24.
Michal M, Vanecek T, Sima R, Mukensnabl P, Hes O, Kazakov DV, et al. Mixed germ cell sex cord-stromal tumors of the testis and ovary. Morphological, immunohistochemical, and molecular genetic study of seven cases. Virchows Arch. 2006;448(5):612–22. doi:10.1007/s00428-006-0155-z.
Talerman A, Roth LM. Recent advances in the pathology and classification of gonadal neoplasms composed of germ cells and sex cord derivatives. Int J Gynecol Pathol. 2007;26(3):313–21. doi:10.1097/01.pgp.0000250148.52215.ce.
Roth LM, Cheng L. Expression of transcription factors and nuclear receptors in mixed germ cell-sex cord stromal tumor and related tumors of the gonads. Int J Gynecol Pathol. 2015; doi:10.1097/PGP.0000000000000192.
Arroyo JG, Harris W, Laden SA. Recurrent mixed germ cell-sex cord-stromal tumor of the ovary in an adult. Int J Gynecol Pathol. 1998;17(3):281–3.
Lacson AG, Gillis DA, Shawwa A. Malignant mixed germ-cell-sex cord-stromal tumors of the ovary associated with isosexual precocious puberty. Cancer. 1988;61(10):2122–33.
Tokuoka S, Aoki Y, Hayashi Y, Yokoyama T, Ishii T. A mixed germ cell-sex cord-stromal tumor of the ovary with retiform tubular structure: a case report. Int J Gynecol Pathol. 1985;4(2):161–70.
Tavassoli FA. A combined germ cell–gonadal stromal–epithelial tumor of the ovary. Am J Surg Pathol. 1983;7(1):73–84.
Zuntova A, Motlik K, Horejsi J, Eckschlager T. Mixed germ cell-sex cord stromal tumor with heterologous structures. Int J Gynecol Pathol. 1992;11(3):227–33.
Jacobsen GK, Braendstrup O, Talerman A. Bilateral mixed germ cell sex-cord stroma tumour in a young adult woman. Case Rep APMIS Suppl. 1991;23:132–7.
Bhathena D, Haning Jr RV, Shapiro S, Hafez GR. Coexistence of a gonadoblastoma and mixed germ cell-sex cord stroma tumor. Pathol Res Pract. 1985;180(2):203–8. doi:10.1016/S0344-0338(85)80171-0.
Colafranceschi M, Massi D. Gonadoblastoma with coexistent features of mixed germ cell-sex cord stroma tumor: a case report. Tumori. 1995;81(3):215–8.
Mooney EE, Nogales FF, Bergeron C, Tavassoli FA. Retiform Sertoli-Leydig cell tumours: clinical, morphological and immunohistochemical findings. Histopathology. 2002;41(2):110–7.
Cabanne F. Gonadoblastomes et tumeurs de l ’ebauche gonadique. Ann Anat Pathol (Paris). 1971;16(4):387–404.
Pflüger E. Ueber die Bewegungen der Ovarien. Archiv für Anatomie, Physiologie und wissenschaftliche Medicin. 1859;S:30–2.
Pang S, Zhang L, Shi Y, Liu Y. Unclassified mixed germ cell-sex cord-stromal tumor with multiple malignant cellular elements in a young woman: a case report and review of the literature. Int J Clin Exp Pathol. 2014;7(8):5259–66.
Shanmugaratnam K, Kunaratnam N, Chia KB, Chiang GS, Sinniah R. Teratoid carcinosarcoma of the paranasal sinuses. Pathology. 1983;15(4):413–9.
Kinjo T, Taniguchi H, Kushima R, Sekine S, Oda I, Saka M, et al. Histologic and immunohistochemical analyses of alpha-fetoprotein–producing cancer of the stomach. Am J Surg Pathol. 2012;36(1):56–65. doi:10.1097/PAS.0b013e31823aafec.
Kurman RJ, Carcangiu ML, Herrington CS, Young RH. WHO classification of tumours of female reproductive organs. 4th ed. Lyon: International Agency for Research on Cancer; 2014.
http://ec.europa.eu/eurostat/statistics-explained/index.php/Population_structure_and_ageing. Accessed: December 2016.
Praxis Strategy Group Analysis of U.S. Census Population Projections, https://www.census.gov/population/projections/data/national/2014.html.
Lankerani MR, Aubrey RW, Reid JD. Endometriosis of the colon with mixed “germ cell” tumor. Am J Clin Pathol. 1982;78(4):555–9.
Rutgers JL, Young RH, Scully RE. Ovarian yolk sac tumor arising from an endometrioid carcinoma. Hum Pathol. 1987;18(12):1296–9.
OF G-G, Cabrera-Ozoria C, Fernandez JA, Stolnicu S, Nogales FF. Malignant mullerian mixed tumor of the ovary associated with yolk sac tumor, neuroepithelial and trophoblastic differentiation (teratoid carcinosarcoma). Int J Gynecol Pathol. 2008;27(4):515–20. doi:10.1097/PGP.0b013e31817b06c7.
Varia M, McCluggage WG, Oommen R. High grade serous carcinoma of the ovary with a yolk sac tumour component in a postmenopausal woman: report of an extremely rare phenomenon. J Clin Pathol. 2012;65(9):853–4. doi:10.1136/jclinpath-2011-200606.
Lopez JM, Malpica A, Deavers MT, Ayala AG. Ovarian yolk sac tumor associated with endometrioid carcinoma and mucinous cystadenoma of the ovary. Ann Diagn Pathol. 2003;7(5):300–5. doi:S1092913403000819 [pii].
Retamero JA, Schuldt M, Nogales FF. Yolk Sac tumours arising from Endometrioid carcinoma of the ovary and endometrium. A review of 14 cases. AJSP Reviews Reports. 2016;(in press).
McNamee T, Damato S, McCluggage WG. Yolk sac tumours of the female genital tract in older adults derive commonly from somatic epithelial neoplasms: somatically derived yolk sac tumours. Histopathology. 2016;69(5):739–51. doi:10.1111/his.13021.
Yemelyanova A, Gown AM, Wu LS, Holmes BJ, Ronnett BM, Vang R. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33(5):492–9. doi:10.1097/PGP.0b013e3182a54afa.
Teilum G. Endodermal sinus tumors of the ovary and testis. Comparative morphogenesis of the so-called mesoephroma ovarii (Schiller) and extraembryonic (yolk sac-allantoic) structures of the rat’s placenta. Cancer. 1959;12:1092–105.
Offman SL, Longacre TA. Clear cell carcinoma of the female genital tract (not everything is as clear as it seems). Adv Anat Pathol. 2012;19(5):296–312. doi:10.1097/PAP.0b013e31826663b1.
Maeda D, Ota S, Takazawa Y, Aburatani H, Nakagawa S, Yano T, et al. Glypican-3 expression in clear cell adenocarcinoma of the ovary. Mod Pathol. 2009;22(6):824–32. doi:10.1038/modpathol.2009.40.
Ballotta MR, Bianchini E, Borghi L, Fortini RM. Clear cell carcinoma simulating the “endometrioid-like variant” of yolk sac tumor. Pathologica. 1995;87(1):87–90.
Cetin A, Bahat Z, Cilesiz P, Demirbag N, Yavuz E. Ovarian clear cell adenocarcinoma producing alpha-fetoprotein: case report. Eur J Gynaecol Oncol. 2007;28(3):241–4.
El-Bahrawy M. Alpha-fetoprotein-producing non-germ cell tumours of the female genital tract. Eur J Cancer. 2010;46(8):1317–22. doi:10.1016/j.ejca.2010.01.028.
Meguro S, Yasuda M. alpha-Fetoprotein-producing ovarian tumor in a postmenopausal woman with germ cell differentiation. Ann Diagn Pathol. 2013;17(1):140–4. doi:10.1016/j.anndiagpath.2011.07.010.
Hoang LN, Han G, McConechy M, Lau S, Chow C, Gilks CB, et al. Immunohistochemical characterization of prototypical endometrial clear cell carcinoma–diagnostic utility of HNF-1beta and oestrogen receptor. Histopathology. 2014;64(4):585–96. doi:10.1111/his.12286.
Ramalingam P, Malpica A, Silva EG, Liu JL, Gershenson DM, Deavers MT. The use of cytokeratin 7 in differentiating yolk sac tumors from endometrioid and clear cell carcinomas of the ovary. Lab Investig. 2003;83(1):207A–A.
Iwamoto M, Nakatani Y, Fugo K, Kishimoto T, Kiyokawa T. Napsin A is frequently expressed in clear cell carcinoma of the ovary and endometrium. Hum Pathol. 2015;46(7):957–62. doi:10.1016/j.humpath.2015.03.008.
Nonaka D, Chiriboga L, Soslow RA. Expression of pax8 as a useful marker in distinguishing ovarian carcinomas from mammary carcinomas. Am J Surg Pathol. 2008;32(10):1566–71. doi:10.1097/PAS.0b013e31816d71ad.
Schaner ME, Ross DT, Ciaravino G, Sorlie T, Troyanskaya O, Diehn M, et al. Gene expression patterns in ovarian carcinomas. Mol Biol Cell. 2003;14(11):4376–86. doi:10.1091/mbc.E03-05-0279.
Ushiku T, Shinozaki A, Shibahara J, Iwasaki Y, Tateishi Y, Funata N, et al. SALL4 represents fetal gut differentiation of gastric cancer, and is diagnostically useful in distinguishing hepatoid gastric carcinoma from hepatocellular carcinoma. Am J Surg Pathol. 2010;34(4):533–40. doi:10.1097/PAS.0b013e3181d1dcdd.
Iwanaga S, Shimada A, Hasuo Y, Yoh S, Miyajima S, Nishimura H, et al. Immature teratoma of the uterine fundus. Kurume Med J. 1993;40(3):153–8.
Ansah-Boateng Y, Wells M, Poole DR. Coexistent immature teratoma of the uterus and endometrial adenocarcinoma complicated by gliomatosis peritonei. Gynecol Oncol. 1985;21(1):106–10.
Yu XJ, Zhang L, Liu ZP, Shi YQ, Liu YX. Ovarian malignant mixed germ cell tumor with clear cell carcinoma in a postmenopausal woman. Int J Clin Exp Pathol. 2014;7(12):8996–9001.
Nogales FF, Schuldt M, Prat J. Germ cell tumour patterns occurring in somatic malignacies of the female genital tract. 2016.
Choijamts B, Jimi S, Kondo T, Naganuma Y, Matsumoto T, Kuroki M, et al. CD133+ cancer stem cell-like cells derived from uterine carcinosarcoma (malignant mixed Mullerian tumor). Stem Cells. 2011;29(10):1485–95. doi:10.1002/stem.711.
Rutella S, Bonanno G, Procoli A, Mariotti A, Corallo M, Prisco MG, et al. Cells with characteristics of cancer stem/progenitor cells express the CD133 antigen in human endometrial tumors. Clin Cancer Res. 2009;15(13):4299–311. doi:10.1158/1078-0432.CCR-08-1883.
Chang SJ, Wang TY, Tsai CY, Hu TF, Chang MD, Wang HW. Increased epithelial stem cell traits in advanced endometrial endometrioid carcinoma. BMC Genomics. 2009;10:613. doi:10.1186/1471-2164-10-613.
Gotte M, Wolf M, Staebler A, Buchweitz O, Kelsch R, Schuring AN, et al. Increased expression of the adult stem cell marker Musashi-1 in endometriosis and endometrial carcinoma. J Pathol. 2008;215(3):317–29. doi:10.1002/path.2364.
Mizuno T, Suzuki N, Makino H, Furui T, Morii E, Aoki H, et al. Cancer stem-like cells of ovarian clear cell carcinoma are enriched in the ALDH-high population associated with an accelerated scavenging system in reactive oxygen species. Gynecol Oncol. 2015;137(2):299–305. doi:10.1016/j.ygyno.2014.12.005.
Virant-Klun I, Rozman P, Cvjeticanin B, Vrtacnik-Bokal E, Novakovic S, Rulicke T, et al. Parthenogenetic embryo-like structures in the human ovarian surface epithelium cell culture in postmenopausal women with no naturally present follicles and oocytes. Stem Cells Dev. 2009;18(1):137–49. doi:10.1089/scd.2007.0238.
Wang WC, Lee MS, Ko JL, Lai YC. Origin of uterine teratoma differs from that of ovarian teratoma: a case of uterine mature cystic teratoma. Int J Gynecol Pathol 2011;30(6):544–8. doi:10.1097/PGP.0b013e31821c3205 00004347-201111000-00006 [pii].
Siddon A, Hui P. Glial heterotopia of the uterine cervix: DNA genotyping confirmation of its fetal origin. Int J Gynecol Pathol. 2010;29(4):394–7. doi:10.1097/PGP.0b013e3181c5a7e8.
Newton 3rd CW, Abell MR. Iatrogenic fetal implants. Obstet Gynecol. 1972;40(5):686–91.
Russell P, de Costa C, Yeoh G. Fetal glial allograft in the endometrium: case report of a recurrent pseudo-tumor. Pathology. 1993;25(3):247–9.
Chao TJ, Chao J, Kuan LJ, Li YT, Kuo TC, Chang YC, et al. Mature solid teratoma of the fallopian tube associated with uterine leiomyomas. J Chinese Med Assoc. 2008;71(8):425–7. doi:10.1016/s1726-4901(08)70095-9.
Kutteh WH, Albert T. Mature cystic teratoma of the fallopian tube associated with an ectopic pregnancy. Obstet Gynecol. 1991;78(5 Pt 2):984–6.
Hurd Jr JK. Benign cystic teratoma of the fallopian tube. Obstet Gynecol. 1978;52(3):362–4.
Frost RG, Roongpisuthipong A, Cheek BH, Majmudar BN. Immature teratoma of the fallopian tube. A case report. J Reprod Med. 1989;34(1):62–4.
Papadia A, Rutigliani M, Gerbaldo D, Fulcheri E, Ragni N. Mature cystic teratoma of the uterus presenting as an endometrial polyp. Ultrasound Obstet Gynecol. 2007;29(4):477–8. doi:10.1002/uog.3969.
Takahashi O, Shibata S, Hatazawa J, Takisawa J, Sato H, Ota H, et al. Mature cystic teratoma of the uterine corpus. Acta Obstet Gynecol Scand. 1998;77(9):936–8.
Kamgobe E, Massinde A, Matovelo D, Ndaboine E, Rambau P, Chaula T. Uterine myometrial mature teratoma presenting as a uterine mass: a review of literature. BMC Clin Pathol. 2016;16:5. doi:10.1186/s12907-016-0026-8.
Akai M, Isoda H, Sawada S, Matsuo I, Kanzaki H, Sakaida N, et al. A case of struma uteri. AJR Am J Roentgenol. 2005;185(1):216–8. doi:10.2214/ajr.185.1.01850216.
Newsom-Davis T, Poulter D, Gray R, Ameen M, Lindsay I, Papanikolaou K et al. Case report: malignant teratoma of the uterine corpus. BMC Cancer 2009;9:195. doi:10.1186/1471-2407-9-195 1471-2407-9-195 [pii].
Lim SC, Kim YS, Lee YH, Lee MS, Lim JY. Mature teratoma of the uterine cervix with lymphoid hyperplasia. Pathol Int. 2003;53(5):327–31. doi:1468 [pii].
Iwanaga S, Ishii H, Nagano H, Shimizu M, Nishida T, Yakushiji M. Mature cystic teratoma of the uterine cervix. Asia Oceania J Obstet Gynaecol. 1990;16(4):363–6.
Panesar NK, Sidhu JS. Uterine cervical teratoma with divergent neuroepithelial differentiation and development of an oligodendroglioma: report of a case and review of the literature. Ann Diagn Pathol. 2007;11(4):293–6. doi:10.1016/j.anndiagpath.2006.03.019.
Spatz A, Bouron D, Pautier P, Castaigne D, Duvillard P. Primary yolk sac tumor of the endometrium: a case report and review of the literature. Gynecol Oncol 1998;70(2):285–8. doi:S0090-8258(98)95036-3 [pii] 10.1006/gyno.1998.5036.
Wang C, Li G, Xi L, Gu M, Ma D. Primary yolk sac tumor of the endometrium. Int J Gynaecol Obstet 2011;114(3):291–3. doi:10.1016/j.ijgo.2011.03.020 S0020-7292(11)00259-1 [pii].
Mardi K, Gupta N, Bindra R. Primary yolk sac tumor of cervix and vagina in an adult female: a rare case report. Indian J Cancer. 2011;48(4):515–6. doi:10.4103/0019-509X.92249 IndianJournalofCancer_2011_48_4_515_92249 [pii].
Mochizuki K, Obatake M, Taura Y, Inamura Y, Takatsuki M, Nagayasu T, et al. Yolk sac tumor of the vulva: a case report with recurrence after long-term follow-up. Pediatr Surg Int. 2012;28(9):931–4. doi:10.1007/s00383-012-3153-z.
Flanagan CW, Parker JR, Mannel RS, Min KW, Kida M. Primary endodermal sinus tumor of the vulva: a case report and review of the literature. Gynecol Oncol 1997;66(3):515–8. doi:S0090-8258(97)94759-4 [pii] 10.1006/gyno.1997.4759.
Mamoon N, Mushtaq S, Akhter N, Aslam A, Chaudary A, Rashid M. Immature teratoma of the vulva with an inguinal lymph node metastasis: report of a case and review of literature. Int J Gynecol Pathol 2010;29(2):197–200. doi:10.1097/PGP.0b013e3181b8e73e 00004347-201003000-00015 [pii].
Acknowledgments
The authors are grateful to the following pathologists for kindly providing the cases illustrating the following figures:
Figure 6-3B. Dr. Isabel Alvarado-Cabrero, Hospital Siglo XXI, Mexico DF, Mexico
Figure 6-6B. Dr. Carmen Tornos, Stony Brook University, NY. USA
Figure 6-6R. Dr. Silvia Chiarelli, Università degli Studi di Padova. Italy
Figure 6-6S. Dr. Harry Hollema, University of Groningen. The Netherlands
Figure 6-20A. Dr. Miguel Marin-Padilla, Geisel School of Medicine at Dartmouth, Hanover, NH, USA
Figure 6-23B. Dr. Lara Alberte, CHUVI Vigo, Spain
Figure 6-30. Dr. Baljeet Kaur. Hammersmith Hospital, London, UK
Figure6-31. Dr. Maria J Pareja.Complejo Hospitalario Universitario de Huelva, Spain
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer-Verlag GmbH Germany
About this chapter
Cite this chapter
Nogales, F.F., Schuldt, M. (2017). Germ Cell Tumors of the Female Genital Tract. In: Nogales, F., Jimenez, R. (eds) Pathology and Biology of Human Germ Cell Tumors. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-53775-6_6
Download citation
DOI: https://doi.org/10.1007/978-3-662-53775-6_6
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-53773-2
Online ISBN: 978-3-662-53775-6
eBook Packages: MedicineMedicine (R0)