
Abstract
Autoimmune diseases, a group of dozens of distinct disorders that target every organ system, affect as much as 10 % of the US population and are increasing in prevalence worldwide. The mechanisms which underlie these diverse diseases are not understood. Autoimmune disease has a strong genetic component, and nearly 80 % of its victims are women, suggesting a strong hormonal contribution. Environmental agents (e.g., lupus-like syndromes induced by pharmaceuticals) can also be a factor. Since the risk of autoimmune disease and levels of circulating antibodies rise with age, a progressive deterioration of the immune system (termed immunosenescence) is implicated.
As data on the origin of autoimmune disease begin to come together from diverse disciplines, it is becoming clear that a set of shared proteins common to several immune pathways are involved in multiple diseases and that the respective contributions from genetics, sex hormones, environmental insults, and immunosenescence are put in play by epigenetic mechanisms that control gene expression. Since those changes are reversible, epigenetics presents a promising new approach for autoimmune disease therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Systemic Lupus Erythematosus
- Autoimmune Disease
- Celiac Disease
- Experimental Autoimmune Encephalomyelitis
- Systemic Lupus Erythematosus Patient
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The immune system prevents an immune response to the body’s own tissues, generating immune tolerance with regard to self-antigens during T-cell generation in the thymus and B-cell generation in the bone marrow. Mechanisms of tolerance to sequestered or otherwise aberrant self-antigens exist in the periphery as well [1].
Autoimmune disease (AID), nonetheless, exists and is increasing in prevalence worldwide [2]. A group of at least 80 distinct syndromes, these are mostly of relatively rare complaints that together comprise a wide range of genetically complex diseases that afflict as much as 10 % of the population [3]. Autoimmune disorders are defined by a dysregulation of immune function that potentiates a breakdown in immune tolerance. The breakdown in immune tolerance in turn permits an overexpression of autoantibodies and autoreactive T cells [3] that will, unchecked, eventually result in inflammation and tissue destruction [4]. Autoimmune diseases target a variety of tissue types but share certain characteristics, foremost being the fact that they affect far more women than men (Tables 24.1a, Part 1 and 24.1b Part 2).
The mechanisms which underlie this diverse group of diseases are still poorly understood. Many autoimmune diseases display both genetic predisposition and familial associations, firmly establishing a genetic foundation for their eventual development [2,41].
Most also have obvious environmental triggers as well; a wide variety of environmental insults including infectious agents, pharmaceuticals, dietary factors, smoking, chemicals, and pollutants are shown to increase the risk [2,41].
In addition, since autoimmune disease affects predominantly women (80 % of autoimmune disease sufferers are female) [13], with the activity of disease fluctuating in parallel with estrogen levels, sex steroids are widely proposed as a key component of autoimmune pathology [42].
Finally, as both the levels of circulating of autoantibodies (in association with a general dysregulation of immune function) and the risk of autoimmune disease are observed to increase as humans age [43], it is probable that immunosenescence also plays a role.
Although the precise pathways through which a genetic predisposition to autoimmune disease becomes a full-blown autoimmune disease (through the influence of endogenous hormones, environmental insults, and the natural decay of the aging immune system) are still unknown [3], progress suggests that lifelong epigenetic changes to deoxyribonucleic acid (DNA) are a primary means to gene regulation. Because of this, autoimmune diseases, potentially devastating conditions with complex etiology and paradoxical nature, are better understood.
2 The Genetic Underpinnings of Autoimmune Disease
The existence of a genetic platform that confers susceptibility to or protection against autoimmune disease has been long suspected. Only a handful of these diseases display strict Mendelian inheritance [44]. Certain autoimmune diseases, nonetheless, consistently occur at statistically improbable levels in certain families [45].
Apart from Mendelian inheritance, familial aggregation is modest [44]. For example, despite the fact that a family history of systemic lupus erythematosus (SLE) increases the risk 25-fold, only 2 % of an AID patient’s close relatives actually develop the disease [46]. Thus, the manner in which the genome confers protection or susceptibility through modulation of immune tolerance is not solidly identified, and patients in which a genetic profile has been firmly linked to the development of disease are a substantial minority [47]. This situation arises because many of the identified genetic associations have odds ratios (typically 1.1–1.5) that are too small to meaningfully increase individual risk [44].
The genetic contribution to autoimmunity is “extraordinarily complex” [44]. Refinements in methodologies in molecular biology, however, are beginning to unlock the genetic basis of autoimmunity. Culpable genes in the pathogenesis of autoimmune diseases were first pursued through candidate gene association studies, then by linkage analysis in affected families, and then through genome-wide association scans [44] (Table 24.2). Genome-wide association studies, built on the completion of the Human Genome and the haplotype map (HapMap) projects, have particularly accelerated solid associations between disease and specific genes, courtesy of their ability to scan the entire genome for polymorphisms implicated in disease [44].
More than 200 genetic loci have now been documented as contributing to various autoimmune diseases [4]. Some genetic variants, that is, human leukocyte antigens (HLAs), have been reliably associated with several different autoimmune diseases, including type I diabetes mellitus (T1DM), rheumatoid arthritis (RA), and SLE [3]; the signal transducer and activator of transcription 4 (STAT4) (a transcription factor gene) haplotype is associated with an increased risk of both RA and SLE [21]. Through these genetic techniques, multiple potential pathways for the origin of autoimmunity have emerged. These are primarily related to innate immunity, cytokine signaling, or lymphocyte activation [3], and include molecules like intracellular tyrosine phosphatases, tumor necrosis factor(s), intracellular pattern recognition receptors, nuclear factor kappa-light-chain-enhancer (NF-Кβ) and other transcription factors, cytokines (and their receptors), cell-surface receptors, signaling molecules, enzymes, autoantigens, and major histocompatibility complex (MHC) profiles [44].
Intriguingly, the loci identified are often shown to be common to a number of autoimmune diseases [4], and the presence of an autoimmune disease in one individual increases the risk in close family members to a variety of others. For example, analysis of variants of the protein tyrosine phosphatase (PTPN) gene in Swedish patients with autoimmune diseases such as RA, T1DM, or Crohn’s disease (N = 172,242) found that the relative risk of several autoimmune diseases increased in offspring when the parent suffered from an existing autoimmune disease [45] (Table 24.3). Pairwise analyses demonstrated a shared familial risk for RA, SLE, T1DM, ankylosing spondylitis (AS), Crohn’s disease, celiac disease, and ulcerative colitis (UC) (see Table 24.3).
A genome-wide association study looked at single-nucleotide polymorphisms (SNPs) outside the MHC and identified a total of 107 SNPs that were associated with an increased risk for at least one of seven individual autoimmune diseases: celiac disease, Crohn’s disease, multiple sclerosis (MS), psoriasis, RA, SLE, and T1DM. Again, a substantial proportion (44 %) of the SNPs identified were also implicated in more than one disease [48]. The implicated SNPs clustered near deoxyribonucleic acid (DNA) sequences that encoded proteins also implicated in the same subsets of diseases [49].
Such proteins, with genes located in proximity to genes implicated in the increased risk for multiple autoimmune diseases, may represent an underlying shared mechanism that constitutes the physiological basis for disease risk. This is a theory supported by the observation that autoimmune patients frequently suffer from more than one autoimmune disorder at a time or from different autoimmune diseases during different stages of their lives [3].
Genome-wide association studies ostensibly reveal a set of genes common to many autoimmune diseases which represent little increased risk on their own [44]. Multiple polymorphisms, however, each carry a slight risk on their own, but together create a lower threshold for the development of an autoimmune event [3]. It is also possible that in rare instances, copy number [44] or somatic mutations de novo can create a genetic platform for autoimmune disease [50,51]. It is reported that somatic mutation can, in adults, generate B cells with autoreactive antigen receptors [52].
Concordance rates among monozygotic (MZ) twins, although consistently higher than those in dizygotic (DZ) twins, are low (see Table 24.1b, Part 2). This observation and the observation that in animal studies the gender bias characteristic of autoimmune disease was revealed to be strain specific suggest an interaction between sex chromosomes and the background genes [36]. It makes it clear that AID requires more than just a genetic foundation. Despite the fact that genome-wide association studies have identified multiple significant associations with specific genetic loci, concordance rates for MZ twins are still below 50 % (with a few exceptions), a statistic that suggests additional complementary mechanisms in the genesis of AID [47].
3 Contributions of Gender to Autoimmune Disease
The most striking feature in the diverse group of disorders that make up AID, arguably, is their definitive predominance for females [53] (see Tables 24.1a, Part 1 and 24.1b Part 2). Autoimmune disease is one of the top ten causes of death in women under the age of 65 [54], the second highest cause of chronic illness, and the top cause of morbidity in women in the United States [55]. Suspicion with regard to the physiological basis for the dramatic surfeit of female AID patients has rested primarily on the female sex hormone estrogen [56], with lots of evidence to support it [46]. The role of estrogen in immune function as well as autoimmune disease is supported by a substantial body of evidence [42].
3.1 Estrogen
The immune response is heightened in women as compared to men [57]. Both cellular and humoral immune responses are more vigorous in women than men, making women more resistant to infection, but also more subject to autoimmune diseases [41]. Women typically respond to infection (natural and by vaccination) as well as trauma with the increased antibody production characteristic of a T-helper 2 cell (Th2) immune response, while men respond with a T-helper 1 cell (Th1) response, characterized predominantly by inflammation [58]. Estrogen’s effect on T-helper cells appears to be estrogen dependent. Low doses of estrogen stimulate Th1 response and higher doses, Th2 [59], a phenomenon also driven by increasing levels of circulating estrogen of pregnancy. Such a shift is also observed during pregnancy [60]. The Th2 response, wherein estrogen stimulates Th lymphocytes to secrete type II cytokines, thereby promotes synthesis of antibodies [61].
Estrogen drastically reduces the size of the bone marrow cavity and induces significant atrophy of the thymus, sites where deletions of autoreactive cells occur. Estrogens are known to stimulate lymphopoiesis outside of marrow (promoting auto reactivity by bypassing the normal selection process) [62], so B cells may develop at alternative sites (liver and spleen) where less stringent selection is occurring. Mice treated with endogenous estrogen have extensive hemopoietic centers in the liver and spleen with B-cell activation including autoantibody-rousing cells in the spleen and liver [42].
Estrogen, a principal regulator of proinflammatory molecules [52], is known to exert numerous effects on individual immune parameters (Table 24.4).
Numerous intrinsic (e.g., pregnancy, menopause, disease states) and extrinsic (e.g., oral contraceptives, hormone replacement therapy [HRT]) factors influence serum estrogen levels. The course of autoimmune diseases typically fluctuates in parallel to changes in estrogen levels [46]; the role that sex hormones play is clearly evidenced by dramatic differences in prevalence related to circulating levels of estrogen over the female life span [42].
Autoimmune hepatitis (AIH), for example, often ameliorates during pregnancy, with an increase in first-time diagnoses in the postpartum period [75,76]. The severity of MS as well as RA decreases during pregnancy, particularly in the third trimester when hormone levels are highest [46].
SLE varies over the course of the menstrual cycle, with flares much more likely during peak estrogen periods such as the high estrogen levels of pregnancy [52]. Similarly, in vitro fertilization procedures and drugs that induce ovulation also induce SLE flares or the onset of SLE symptoms [52]. New diagnoses and flares of existing SLE are rare in the postmenopausal period [52].
Treatment of human peripheral blood mononuclear cells (PBMCs) from SLE patients enhanced total immunoglobulin G (IgG) production as well as anti-double-stranded DNA (dsDNA) autoantibody levels; PBMCs from healthy individuals, however, did not; the disparity proved to be interleukin (IL)-10 dependent [77]. Testosterone significantly inhibited IgM and IgG production by PBMC [78].
Similar estrogen effects were observed in animal models [46]. Animal studies confirm that hormonal manipulations can influence disease expression [53]. In a murine lupus model of experimental autoimmune encephalomyelitis (EAE) in which male mice were castrated, castration produced SLE with disease parameters very similar to that in female mice, while ovariectomized females produced disease parameters similar to male mice of the same strain (NZBxNZW) F1 [46]. Estradiol protected the female mice from EAE.
In other animal studies, five different genes, all regulated by either estradiol or dihydrotestosterone, contributed to autoimmunity (in vivo mouse model with lupus) [79].
The importance of the hormonal component in autoimmunity was illuminated by an experiment in mice, which isolated the chromosomal from the hormonal component by moving the testis-determining gene SRY from the Y chromosome to an autosomal chromosome [80]. This approach revealed that although the XY genotype alone stimulated autoantibody proliferation, androgens exerted an overriding suppressive effect [81]. Male sex hormones appear to mask the otherwise immunostimulatory influence of the XY genotype.
In contrast, however, estradiol increases susceptibility to experimental myasthenia gravis, experimental autoimmune uveoretinitis, and lupus [5]. The effects of estrogen on female-predominant AIDs, however, are not generalizable—some are abrogated by testosterone and some are amplified [82]. Gender differences in autoimmunity are at least partially mediated by regulation at the level of the estrogen receptor.
3.2 Estrogen Receptors
Nuclear receptors are factors that bind to DNA and act to regulate gene expression at the transcription level (thus known also as transcription factors) and include receptors for estrogens and androgens [52]. Estrogen (17-β estradiol), which is the principal estrogen in circulation during the reproductive years, acts as a ligand for two different nuclear receptor proteins, thus forming two functionally distinct estrogen receptors, estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ), which exist in both homo- and heterodimers.
Estrogen receptors translocate to the nucleus where they bind to estrogen-responsive elements (EREs) in gene promoters and thus act as an on/off switch for gene transcription. The estrogen/estrogen-receptor (E/ER) complex may also interact with other transcription factors such as NF-Κβ, which is capable of binding to non-ERE sites in other promoters [83].
Both types of estrogen receptors are found all over the body, including the ovary, womb, breast, bone, and immune cells, T and B cells, dendritic cells, neutrophils, macrophages, natural killer (NK) cells, thymic stroma cells, bone marrow, and endothelial cells [83].
Although both ERα and ERβ both act to modulate gene expression of estrogen-responsive genes, they modulate that expression in very different ways [52]. ERα deficiency in a murine model of lupus, for example, resulted in disease abrogation and prolonged survival, while ERβ deficiency had minimal effect [84].
Estrogen receptors also occur in two places, with distinctive purposes. Membrane-associated ERs amplify signal transduction cascades, where nuclear ones induce gene transcription. Mature lymphocytes express membrane-associated ERs which are activated by estrogen to invoke increased calcium flex in antigen-activated cells [83].
The effects of estrogen-receptor regulation of immunity and their implications for autoimmunity are many. In one study, estrogen-receptor blockers blocked estrogen-receptor activation in lupus T cells but not in healthy controls [85]. Decreased ERα in macrophages resulted in the stimulation of a cluster of differentiation 4 (CD4) cells [82]. Estradiol (E2), acting via ERα, increased proinflammatory cytokine expression, enhanced proliferative and interferon alpha (INF-α) production by CD4+T cells [82], and differential expression of ER-α and ER-β observed in SLE and RA [83].
The presence of two ERs, occurring in two different places, adds another layer to hormonal regulation of immune processes since the same estrogen produces different effects at one receptor versus another [41], giving the body the ability to fine-tune immune response.
Estrogen, then, is believed to be involved in the regulation of a wide variety of immune cells and a key player in the dysregulation of immunity that leads to both autoimmune and autoinflammatory diseases. Paradoxically, however, comparisons of estrogen levels in female patients with different autoimmune diseases (including AID patients) showed no difference between patients and healthy controls, an indication that estrogen is just part of a more complex etiology [78,86]. In fact, estrogen is not the only component of female gender with the potential to contribute to autoimmunity.
3.3 Microchimerism
Fetomaternal microchimerism is the presence, after pregnancy and delivery, of foreign fetal cells in the mother. These include hemapoietic progenitor cells with the capacity to induce autoimmunity and which have been implicated in autoimmune disease, in particular, Graves’ disease [87]. It is known that fetal cells are found in peripheral blood of nearly all women during pregnancy, including hematopoietic progenitor cells with the potential to become both effectors and targets of immune processes, and that maternal DNA can be detected by polymerase chain reaction (PCR) in a majority of cord blood samples [87].
Female subjects with autoimmune thyroid disease (Graves and Hashimoto’s) are often shown to have microchimeric fetal cells in their thyroid glands [87]. In addition, women with autoimmune thyroid disease with sons had higher prevalence of male cells in their thyroid than women with sons but healthy thyroids [87].
Sixty percent of women with Graves’ disease with onset during reproductive years developed the disease in the postpartum period [87]. Also supporting microchimerism as the etiological basis of Graves’ disease is a large study that found a significant increase in thyroid peroxidase autoantibody (TPO) associated with increase in parity [88].
3.4 X Chromosome
Interestingly, in a murine model in which the authors artificially produced XX, XY mice without alteration of actual gonads, the XX genotype was associated with greater severity of EAE and pristane-induced lupus than XY [46], demonstrating that the sex chromosomes beyond the influence of the sex hormones themselves, contribute to autoimmunity.
The X chromosome itself is implicated in the genesis of autoimmunity, with the presence of two copies of the X chromosome in women held responsible for the observed female predominance in AID. Aberrant, congenital X-chromosome doses, as in Klinefelter’s syndrome (XXY) in men, which greatly increases the risk for female-predominant autoimmune disease, underlines the X chromosome’s importance [15]. Gene expression in X and Y, however, is not well understood. The fact that the X chromosome itself contains 1,000 unique genes, including genes associated with systemic sclerosis, autoimmune thyroid disease, and SLE [89], in and of itself infers a substantial role of the X chromosome in the regulation of autoimmunity.
In women, one X chromosome is habitually but randomly inactivated to preserve equal gene expression in men and women. However, the inactivated X is not completely inactivated, meaning that some X-associated genes are overexpressed in women [46] as compared to males. Partial inactivation of X, then, may contribute to gender disparity in AID. The mouse strain BXSB, with a translocation mutation in which part of the X chromosome has been transferred to the Y, develops lupus-like syndrome with high frequency in males, shown to involve overexpression of Toll-like receptors (TLR)-7, shown to be a key player in lupus development [46].
In addition, although the choice of which X chromosome to inactivate is random, certain cell types are preferentially inactivated or maintained, resulting in “skewed inactivation” that acts to modulate gene expression. A high percentage of women with systemic sclerosis display skewed X inactivation [46]; skewed X-chromosome inactivation is also implicated in scleroderma in association with reduced regulatory T cell (Treg) activity [83].
Finally, there is sometimes spontaneous loss of the X chromosome with age, observed in increasing frequency with age in blood cells of patients with systemic sclerosis and autoimmune thyroid disease [46]. The origin and the effect of this spontaneous loss are unclear.
Beyond issues of gender created by the gene complement of sex chromosomes and hormones that the complement stimulates, environmental interactions contribute to autoimmunity as well.
4 The Contribution of Environmental Agents in Autoimmune Disease
The fact that monozygotic pairs of female twins (with identical genetic and hormonal constitutions) can attribute only 20 % of their phenotypic variance to their shared genetic polymorphisms implicates environmental exposures in the development of autoimmune disease [3]. These exposures are numerous and contribute to a variety of autoimmune diseases. Geographic clusterings of specific diseases are observed, with wide disparities in prevalence in different parts of the world. Even in the confines of the US, multiple sclerosis has dramatic geographical specificity, with a prevalence of 61/100,000 in the West, compared to only 3/100,000 in the South [33].
Autoimmunity is associated with seemingly improbable environmental factors like season of birth or weather patterns [3].
Many environmental factors, both intrinsic and extrinsic, are associated with the development of autoimmune disorders (Table 24.5). The internal biological environment (e.g., endogenous hormones and aging) obviously contributes to autoimmune disease; extrinsic insults, including environmental agents like ultraviolet (UV) light, chemical or other occupational exposures, pollutants, or health habits like smoking or alcohol consumption, are now recognized as important components of autoimmune disease as well. Environmental factors may initiate immunity through nonspecific activation of resting T cells, modification or release of previously sequestered proteins, cross-reactivity between virus and self-protein (molecular mimicry), and modulation of gene expression [5]. Viruses and other infectious agents are also potential environmental inducers of AID as well, although, since viral infection can occur years before the actual onset of a particular AID, definitive associations are difficult to establish [5].
Potential contributions to autoimmunity from the environment are virtually endless and differ widely with occupation, place of residence, hobbies, and medical history. Environmental interactions, like diet, occupation, exposure to environmental chemicals, radiation, UV light, pathogenic organisms, and medications, can also differ by gender [36] and contribute to gender disparities in AID prevalence [99]. Interestingly, the lupus-like syndrome produced by the drug procainamide has a male to female ratio of 2:1, while in asymptomatic patients, the ratio is 5:1, implying an as yet unresolved interaction between gender determinants and susceptibility to an environmental exposure, procainamide, in the development of lupus [36].
Environmental insults may increase with age, including cortisol levels, sleep dysregulation, decrease in physical activity, increase in oxidative stress, or nutritional deficiencies [100].
5 Immunosenescence and Autoimmune Disease
The immune system undergoes extensive remodeling as humans age, characterized by a progressive deterioration in the ability to mount an effective immune response. However, an unexplained paradox is a simultaneous increase in autoantibody production [43]. The clinical consequences of this remodeling include a risk of neoplasia and infectious disease, as well as autoimmune disease [43]. Immunosenescence is a subject of increasing interest in medicine, as life expectancy, especially in developed countries, increases without a parallel improvement in health in old age [101].
The immune system, a complex and exquisitely regulated collection of interdependent molecular pathways of both innate and adaptive defense, begins to undergo dysregulation of cell homeostasis. This causes multiple changes in immune function which increase susceptibility to the development of autoimmune diseases [43] (Table 24.6). Old age is characterized by the rising incidence of autoimmunity [103] and an associated increase in both levels of circulating antibodies and numbers of specific antibodies in circulation. Persistent high antigen levels activate memory cells, and costimulatory T cells become less susceptible to downregulation [117].
Although immunosenescence affects all cell types and results in deterioration of both humoral and cellular immunity and multiple cell types, age-associated cumulative effects on T-cell function are the most dramatic and most detrimental [118]. With age, there is a decrease in thymic epithelial space and thymic cellularity, called thymic involution; in humans, the increase in perivascular thymic space is replaced progressively with fat [101]. Humoral immunity as well is severely compromised in the aged as a result of decreased production of long-term Ig-producing B cells and the loss of immunoglobulin diversity and affinities [101].
Although B-cell numbers do not change significantly, B cells do exhibit a decreased response to primary antigenic stimulation [119]. B-cell response (immunoglobulins produced) becomes more random [119] with decreased affinity of IgG produced for a given antigen [119], and IL-15 stimulates proliferation of memory T cells. IL-15 levels nearly double in healthy adults 95 years or older (3.05 pg/mL) as compared to both older adults (60–89, 1.94 pg/mL) and midlife adults (30–59, 1.73 pg/mL) [120]. With an increasing number of memory B cells, the number of autoantibodies also increases with age [121].
Antinuclear antibody (ANA) levels remain constant until age 60 and then increase rapidly. Over the age of 70, more than a third of otherwise healthy senior citizens exhibit high levels of circulating antibody.
Autoantibodies rise because the efficiency of physical barriers is reduced, with higher exposure to pathogens and novel exposure to previously sequestered self-antigens [117]. Infections can trigger immune-mediated inflammatory disease, either by cross-reactivity or by interfering with signaling processes regulating immune response [122]. Increasing autoantibodies in old age may also be the result of reactivation of self-reactive memory B cells originally generated in childhood but reactivated [119].
Defective clearance of cellular debris can also result in prolonged exposure to autoantigens in higher concentrations than normal with subsequent activation of lymphocytes [103]. An increased basal level of inflammatory activity may result from increased production of proinflammatory cytokines such as IL6, tumor necrosis factor (TNF) alpha, and free radicals [123].
A large percentage of older people have relatively high titers of autoantibodies due to higher exposure to exogenous factors such as polypharmia or multiple infections, with cumulative exposure to antibody specificities, supported by a study in Cameroonians 60 years and older [124]. Autoantibodies (AABs) were observed at a rate similar to US averages (49 %) but with a markedly different pattern which implied a strong contribution from extrinsic factors, putatively the widespread presence of chronic infection [124]. In a US study which evaluated Medicare files for the prevalence of AIDs, the risk increased 41 % with a prior infection-related medical visit and by 90 % with a prior (pathogen-free) transfusion [125].
Immunosenescence does not affect men and women equally. Men (and postmenopausal women), for example, have reduced T-cell immunity as compared to premenopausal women [126]. Furthermore, different forms of estrogen are produced at different points of the female life span. During the reproductive years, the primary circulating form of estrogen is estradiol (E2), produced in the ovaries; after menopause, it is estrone (E1), which is produced primarily by adipose tissues. Estrone is the primary form of circulating estrogen in men at all ages. Estradiol binds both estrogen receptors (α and β) at equal affinity. Estrone however, has a fivefold higher affinity for ERα than for ERβ [127].
6 Genes, Estrogen, Environment, and Aging: Hints of a Nexus in SLE
Human lupus is a systemic autoimmune disease which primarily affects women, characterized by the formation of autoantibodies to nuclear antigens like DNA, with varied pathogenicity, for example, deposition of immune complexes in the kidneys or in the skin [128].
SLE, one of the more common autoimmune diseases and therefore one of the most studied, is nonetheless a disease whose pathogenesis is still somewhat murky. Dysregulation of immune function seems to be global, as multiple genes appear to more or less simultaneously escape control [52]. In a study which analyzed SLE sera, 30 disparate proteins (cytokines, chemokines, growth factors, and soluble receptors) were observed to be outside the range observed in normal controls [52], highlighting the multifactorial etiology of AIDs like SLE, a complexity that has made definitive etiologies elusive. Over the last several years, however, the multidisciplinary profile of SLE has come together that moves toward integration of the disparate influences on SLE for a more comprehensive understanding.
6.1 The Genetic Foundation of SLE
SLE has solid evidence of a genetic foundation. Multiple genetic loci are identified with risk [128]; HLA variant HLA DR3-DQ2.5-C4AQ0 is strongly associated with SLE (odds ratio [OR] 2.8, 95 % confidence interval [CI] 1.7–4.5) [129]. No single gene or group of structural gene defects, however, has emerged as a defining genetic factor [74]. In addition, there is minimal concordance between MZ twins, implying an existence for other complementary components as well [128].
6.2 The Contribution of Estrogen to SLE
Estrogen is abnormally metabolized in SLE, with an increase in the production of 16-hydroxyestrone and estriol metabolites which putatively lead to a chronic hyperestrogenic state [41]. Estrogen is clearly an important component of disease course, which fluctuates in close parallel to estrogen levels [130].
In addition, when a cohort of 238,308 women was evaluated prospectively, risk factors for SLE showed a strongly positive association with cumulative estrogen exposure. Menarche earlier than age 10 (relative risk [RR] 2.1, 95 % CI 1.4–3.2), the use of oral contraceptives (RR 1.5, 95 % CI 1.1–2.1), and the use of HRT (RR 1.9, 95 % CI 1.2–3.1) in menopause all raise the risk significantly [131].
Several etiological pathways have been implicated in estrogen’s effects. One strong candidate is the strong association of SLE with an observed demethylation of CD40 ligand on CD40+T cells [132]. This demethylation, when present on the inactive X chromosome, results in the overexpression of CD40 ligand on CD4+ cells, which in turn induces the production of autoantibodies [85].
SLE is characterized by the largest number of detectable autoantibody specificities among the autoimmune diseases, which could result from T-cell escape from normal regulation [52]; autoantibody secretion could result from abnormal T-cell regulation. Estrogen-dependent T-cell stimulation, therefore, provides a specific pathway between hormone activation of the T cell, increased T-cell interactions with B cells, and the subsequent overexpression of autoantibodies characteristic of SLE [52].
Expression of the T-cell activator calcineurin is increased when estrogen is cultured with SLE T cells but not with T cells from normal women. This is an upregulation of T-cell function that appears to be estrogen driven since estradiol, bound to the ER, evokes a direct increase in calcineurin expression in T cells from female lupus patients. This does not happen in males, implying a disease-related alteration of the ER specific to women [133]. Estrogen stimulation of calcineurin expression is dose dependent [130]. Thus, estrogen stimulation of calcineurin expression may be what upregulates T-cell regulation, adding to the existing genetic-, environmental-, and immunosenescence-burdened threshold for disease.
ERα also plays a role in SLE disease activity. ERα promotes lupus by inducing interferon (IFN) and cytokines [134]. A deficit in ERα attenuates lupus in NZB/NZW mice disruption of ERα in female NZB/NZW, delays onset of glomerulonephritis, increases survival, and delays the production of autoantibodies; ERα deficiency in male mice increases survival and decreases anti-DNA antibodies [134].
A decrease in SLE disease activity also correlates with abrogation of interferon alpha/beta (INF-αβ) signaling, implicating INF receptors in SLE pathogenesis [135]. Increased levels of estradiol could seemingly be assumed to cause dose-dependent effects across the immune system. Estrogen levels in autoimmune diseases like SLE, however, do not differ dramatically from normal controls. Clearly, then, estrogen does not act in a vacuum.
6.3 The Role of Environment in SLE
SLE, in addition to strong evidence for both genetic and sex-hormone components to the disease state, is the autoimmune disease most strongly associated with a specific environmental component. Numerous lupus-like syndromes are widely recognized to result from exposure to over 100 different pharmaceuticals (most commonly procainamide and hydralazine). Drug-induced lupus syndromes, in fact, permitted the use of mouse models in targeted investigation into pathogenic mechanisms, for example, the realization that 5-azacytidine (known to inhibit DNA methylation) causes CD4+T cells to become autoreactive with the ability to respond to antigen-presenting cells (APCs) directly (without exposure to antigen). Intriguingly, the effects were reversible when the drug was removed [128]. Bacterial and viral infections are also believed to be environmental triggers [136]. Ultraviolet light is also known to cause lupus flares [128].
6.4 The Contribution of Immunosenescence to SLE
Defects in apoptosis mechanism as well as impaired clearance of apoptotic cells have also been implicated in SLE pathogenesis. Impaired clearance creates a progressive accumulation of autoantigens with an increasing likelihood of an autoimmune response [137]. Regulation of apoptosis is also deranged with increased age, with impaired clearance of cellular debris [138], a factor which could augment effects produced by estrogen.
As the pathogenic basis for SLE is further revealed, we begin to glimpse a variety of known, implicated, and potential mechanisms, from disparate influences but which have plausible interdependent actions with plausible ability to create a genesis of autoimmunity. The genetic platform of susceptibility is built upon, brick by brick, by the multiple potential influences of the immune dysregulation characteristic of increasing age, the chromosomal and hormonal contributions of gender, and a lifetime of environmental insults. Combined, they push the immune system gradually toward immunodysregulation until the threshold is crossed to overt SLE [139]. The question is how that happens.
7 NF-КΒ: Insights from One Protein’s Role in Gene Regulation
NF-Кβ is a family of transcription factors ubiquitous in the cytoplasm of immune cells, which clearly plays a fundamental role in immunoregulation (Table 24.7). NF-Кβ proteins occur as dimers and its distribution can differ between tissues. Nuclear factor-Кβ1 (aka p50) and RelA (aka p65) heterodimers are expressed ubiquitously; nuclear factor-Кβ2 (aka p52), RelB, and c-Rel are expressed only in lymphoid cells and tissues. The intercellular balance between different nuclear factor-Кβ dimers determines which complex will bind target DNA sequences. NF-Кβ is activated by specific factors like lipopolysaccharide (LPS), TNF-α, and IL-1 as well as by nonspecific factors like UV radiation or oxidative stress, which leads to dissociation of nuclear factor-Кβ from a binding protein inhibitory Кβ (IКβ), allowing NF-Кβ to enter the nucleus [1]. In some normal cells, such as B cells, some T cells, Sertoli cells, and some neurons, NF-Кβ is constitutively located in the nucleus [140].
Nearly countless genes, many of which are implicated in pathways to AID (e.g., inflammatory molecules, apoptosis inhibitors, growth factors, proteins for viral replication, and self-regulatory proteins for NF-Кβ actions) appear to either alter NF-Кβ regulation or to be altered by NF-Кβ (Table 24.8). Not surprisingly, NF-Кβ is often proposed as an agent in the pathogenesis of autoimmune disease.
NF-Кβ appears to encourage autoimmune disease in several ways. It increases tolerance to self-antigens by acting on APCs and thymocytes in such a way that negative selection is deranged, causing increased survival of autoreactive T cells in the peripheral circulation and an enhanced susceptibility to environmental insults with the potential to trigger autoimmune disease [1].
NF-Кβ also initiates the inflammatory response when activated by bacterial or viral interaction with TLRs on macrophages, dendritic cells, and other cells that effect innate immunity [1]. NF-Кβ, in turn, activates transcription for genes encoding inflammation-related cytokines, chemokines, and other molecules required for the migration of inflammatory and phagocytic cells into areas of infection or injury [1]. TLRs in the endoplasmic reticulum, endosomes, and lysosomes also recognize bacterial and nuclear DNA and, through NF-Кβ, induce autoimmune reactions [153]. Inflammation, in turn, causes tissue breakdown and the erosion of peripheral tolerance [1].
NF-Кβ also promotes stimulation of an immune response in the absence of antigen due to dysregulation of apoptosis, as many of NF-Кβ target genes are also NF-Кβ inducers, creating a continual feedback loop that can drive an inflammatory response explosively even in the dearth or absence of antigenic stimuli [1]. Reduction of NF-Кβ activity, in fact, promotes the survival of a hyperactivated autoreactive T cells [13].
Functional control of apoptosis is integral for both the development and selection of both T and B cells. NF-Кβ regulates apoptosis in many cell types; whether apoptosis is induced or suppressed depends on the specific type of cell, the specific inducer, and the relative level of intracellular levels of subunits [1].
7.1 NF-Кβ and the Genetic Predilection for Autoimmune Disease
NF-Кβ builds upon genetically conferred risk by acting to inappropriately induce gene products. NF-Кβ, once activated, is able to enter the nucleus. There, it binds rapidly to DNA, stimulating the expression of target genes, including many cytokines, chemokines, and other factors involved in immune-cell signaling (see Table 24.8). Monocytes and B cells appear to produce express active NF-Кβ; T cells, however, require proteasome-dependent activation [13].
7.2 NF-Кβ and Sex Hormones
Estrogens, interestingly, are known to regulate NF-Кβ activity. 17-β estradiol raises NF-Кβ levels as well as the levels of TNF-α that it induces in a dose-dependent fashion, causing enough overexpression of relevant cytokines to potentially cause autoimmune disease in the physiological context [13]. High concentrations of E2 modulates NF-КB signaling and affects T-cell survival [82]; estrogen regulates inflammation [154]. Estrogen acts to regulate inflammation through posttranslational modification of STAT-1 and NF-Кβ proteins [74].
NF-Кβ interacts directly with estrogen, binding to both ERs, and forming both homo- and heterodimers. When bound, ERs translocate to the nucleus where they bind to EREs in gene promoters, thereby directly controlling gene transcription. The E/ER complex may also interact with other transcription factors such as additional NF-Кβ, capable of binding to transcription factors that are not estrogen related.
Estrogen interactions with NF-Кβ intriguingly also appear to drive gender specificity of autoimmune disease. A recent report revealed that peroxisome proliferator-activated receptor (PPARα) mediates inflammatory responses by activating NF-Кβ and thus inducing production of INF-γ and TNF downregulation of Th2 cytokines. Men are less prone to develop some autoimmune diseases because CD4+T cells express higher levels of PPARα [155].
7.3 NF-Кβ and Environment and Aging
Cigarette smoke, an environmental factor associated with numerous autoimmune diseases, is known to induce the release of TNF, TNF alpha receptors, IL-1, IL-6, IL-8, and granulocyte macrophage colony-stimulating factors (GM-CSF) and decrease IL-10 and IFN-gamma [33]. Many of these cytokines and other factors are inducers of the NF-Кβ pathway. Numerous other chemical agents and environmental threats are implicated in the modulation of the actions (see Table 24.8).
The NF-Кβ functions are implicated in the processes of immunosenescence itself [156]. In another positive feedback loop, the progressive deterioration of immune functions regulated by multiple cytokines and other regulatory factors that are known inducers are targets of NF-Кβ actions, it is probable that immunosenescence augments NF-Κβ contributions to autoimmune disease. Suppression of NF-Кβ activity is a common denominator in at least T1DM, SLE, MS, Crohn’s disease, and RA.
NF-Кβ, then, provides an example of a transcription factor, one of many that provide a link between the genetic background of the individual and the influences of aging, estrogen, and environmental interactions on that genetic predisposition, driving genetic risk toward the onset of autoimmune disease [1] (Table 24.9).
8 Epigenetics: The Missing Link in an Integrated Theory of Autoimmune Disease
Epigenetic actions are cell specific, and these actions represent stable changes to DNA that act to regulate gene expression, but do not cause mutation. Epigenetic changes do not alter DNA sequence, but instead control gene expression [47]. These epigenetic changes effect real-time control of homeostasis in the body and maintain the normal function of every cell and its metabolism.
Epigenetic mechanisms confer “phenotypic plasticity” to the genotypic platform by giving the body, at the cellular level, an ability to respond to both internal and external environmental cues [165]. Cells, for example, monitor inventories of necessary compounds and are able to modulate transcription of appropriate genes through epigenetic mechanisms [166]. Epigenetic modifications of DNA are abundant in every cell, changes which are stable because they are heritable during cell division [165].
Epigenetic changes are involved in normal development as well as in disease. Epigenetic variation over time depends on genotype, environment, sex hormone interactions, and undoubtedly other undetermined stochastic factors [80]. Such epigenetic changes provide a ready explanation for discordance in MZ twins with regard to epigenetic diseases that clearly have a strong genetic foundation [94], literally serving as the physiological link between the genome and the genesis of disease. Phenotypic differences in identical twins, caused by epigenetic changes, increase steadily with age (“epigenetic drift”), driven by regular environmental insult [47].
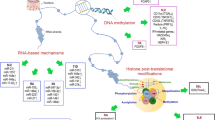
Epigenetic changes are effected by three primary mechanisms as follows: (1) methylation or demethylation of DNA, particularly in promoter sequences of genes; (2) histone modifications, which effect steric changes to chromatin that act to regulate gene transcription; and (3) micro ribonucleic acid (miRNA) particles, typically 22 nucleotides in length, which bind to and degrade complementary messenger RNA (mRNA) after transcription, therefore preventing translation [47] (Table 24.10). Monozygotic twins, with age, show increasing variance in total DNA methylation as well as in acetylation of histone H3K9, with older sets of twins acquiring significantly more epigenetic variance than younger sets [165].
8.1 Mechanisms of Epigenetic Change
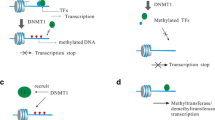
8.1.1 DNA Methylation
Inherited patterns of DNA methylation are largely wiped away shortly after an embryo is fertilized, with a rapid eradication of the paternal genome patterns but only partial demethylation of maternal DNA. Genome-wide remethylation occurs in the blastocyst stage and tissue-specific remethylation follows later [80]. DNA methylation enzymes DNMT1, DNMT3a, and DNMT3b enact these changes during fetal development; they reproduce epigenetic markings after cell division as well as maintain or copy methylation marks after DNA replication that remethylate in the blastocyte stage [80].
DNA methylation occurs mainly in DNA sequences in which cytosine residues precede guanine residues to yield 5-methylcytosine (5-meC). This resulting dinucleotide is called C-phosphate-G (CpG). Many areas of the human genome are rich in CpGs; CpG-rich areas are known as CpG islands. CpG islands typically occur in gene promoter regions and are usually unmethylated in normal cells [80].
8.1.2 Histone Modifications
Chromatin is made up of four core histones, wrapped in 147 base pairs (bp) of DNA. Four epigenetic modifications to DNA in conjunction with histones (typically what is called the “histone tail,” a stretch of DNA that protrudes from the histone) are commonly observed: acetylation, methylation, phosphorylation, and ubiquitination [80]. Histone methylation is controlled by histone methyltransferases and demethylases [166].
DNA methylation and histone modification are coupled through both DNA methyltransferases (DNMTs) and different families of methylated DNA binding proteins that associate with histone-modifying enzymes in multiprotein complexes; both regulate transcription in the nucleus and affect nuclear architecture that also acts to regulate gene expression, therefore producing multiple levels of regulation of gene expression [137].
The third more recent epigenetic effectors are miRNAs, typically 22-nucleotide noncoding RNAs that control gene expression at the posttranscriptional level by binding (through partial sequence homology) to the three prime untranslated region (3′UTR) of RNAs, producing translation inhibition or degradation of miRNAs [74].
MicroRNA acting posttranslationally provides a mechanism for quantitative regulation of genes (rather than off/on signals at the genome level) and acts to fine-tune cellular responses to environmental influences [167], thereby regulating immune-cell development as well as maintaining immune homeostasis and immune tolerance [74].
8.2 Epigenetic Contributions to Autoimmunity and Gender
ER function is regulated by epigenetic mechanisms. ER occurs in nearly every immune-cell type and is distributed at the cellular membrane, cytoplasm, nucleus, and mitochondria [168].
ER-α is epigenetically regulated; T cells from SLE patients have decreased total genomic methylation compared with age-matched controls [52].
Nuclear ERs provide DNA-dependent regulation of gene expression mediated through the histone-acetylation enzyme histone acetyltransferase (HAT). The binding of HATs to the ER produces acetylation of local histones, which in turn cause steric changes that facilitate gene transcription by permitting transcription factor binding with promoter regions of estrogen-responsive genes.
Dysregulated mRNA expression has also been observed to be estrogen driven in association with autoimmune disease [169].
DNA methylation is also involved in genomic imprinting (the preferential gender-specific silencing of genetic material, for example, the paternal X chromosome in mammals) and X-chromosome inactivation [80].
It is also reported that differentiation of T-helper cells in the direction of either the Th1 or Th2 pathways is epigenetically regulated—Th1 cells have a demethylated IFN-γ promoter, with repressive epigenetic histone modifications at the IL-4 to IL-13 locus, while Th2 cells carry the opposite profile [170].
8.2.1 Aging
Global DNA methylation decreases with age [80], simultaneously coupled with hypermethylation specific to CpG island gene promoters or ribosomal DNA clusters [80], changes that almost certainly contribute to immunosenescence [80].
Several papers have reported age-associated changes in global and specific histone profiles, as well as in several histone-modifying enzymes [171].
The biological basis of aging is a progressive decline in the ability to adapt to the environment due to loss of normal gene regulation precipitated by changes in gene promoters or gene silencing regions [165]. Age-associated changes to DNA methylation patterns or histones and their regulatory enzymes certainly increase the risk for dysregulation of gene expression, including the myriad of genes related to immunoregulatory molecules.
8.2.2 Environment
An expansive list of environmental factors are now known to directly or indirectly induce epigenetic changes that, through changes in gene expression, modulate immune-cell function. Epigenetics then provides molecular mechanisms that explain the environmental effects on the development of autoimmune disorders [3]. The fact that many autoimmune disease are striking greater numbers of individuals, in populations whose background of genetic, gender, and aging can be assumed to be relatively stable, is an indication of the importance of environmental interactions in autoimmune disease. Genetically identical laboratory animals raised under identical conditions similarly evidence environmental importance and also evidence epigenetic drift [80].
One of the first and most interesting examples of the power of epigenetics was the Dutch Hunger Winter. A severe famine at the end of World War II affecting a specific area of the Netherlands suggests that famine exposure at a critical period in utero can lead to adverse phenotypes. In this group, there was an increased risk of coronary artery disease (CAD) specific for exposure to famine early in gestation. A similar influence of the environment has been shown in agouti mice, in which epigenetic changes related to diet determine coat color; foods rich in methyl donors change the coat color in offspring, a modification produced by altered DNA methylation [47,172].
Many other environmental interactions effect epigenetic changes that contribute to disease (see Table 24.5).
8.2.3 Systemic Lupus Erythematosus
Lupus, one of the most investigated of autoimmune diseases, is arguably also the most well characterized in terms of epigenetic etiology [128]. DNA methylation patterns have been associated with immune dysregulation in SLE. A study which looked at genome-wide DNA methylation patterns in a cohort of MZ twins discordant for SLE found DNA methylation differences in genes relevant to SLE pathogenesis, with a decrease in total methylation content [173]. Human SLE patients have reduced total deoxymethylcytosine content and decreased levels of DNMT1, an enzyme which drives the methylation of cytosine residues; demethylated T cells stimulated antibody production by autologous B cells [47]. Drug-induced lupus is predictably induced by procainamide and hydralazine (methylation inhibitors) [47] and is the mechanism found with other drugs as well.
Global acetylation of H3 and H4 histones was observed in active SLE CD4+ cells, a change negatively correlated with disease activity.
Estrogen and the female chromosome complement are known to contribute to female predisposition to lupus (in a mouse model) through both hormonal influence and epigenetic effects on the X chromosome [174].
Epigenetic processes contribute to dysregulation of both B- and T-cell function [47,165,175]. Numerous associations of lupus with miRNA changes have been observed. In human patients, PBMC displayed abnormal miRNA expression patterns in PBMC as compared to controls [74], although the association of these abnormal patterns with overt dysregulation was not consistent, a conundrum which may involve sex-hormone levels or other environmental influences [167]. Specific miRNAs have recently been identified to influence T-cell sensitivity and selection [167] as well as the IFN-gamma pathway; the underexpression of miR-146a in lupus patients effects alterations in type I IFN pathways via key signaling proteins. Models have also observed disrupted miRNA patterns in lupus-affected mice [74].
Epigenetic changes are now being identified as the root of nearly every autoimmune disease. The accumulation of epigenetic changes that disrupt immune function and contribute to a breakdown in immune tolerance through DNA methylation, histone modification, and miRNA binding creates autoimmune disease through aberrant regulation of gene expression.
Epigenetics is then the missing link in the chain of events that produces autoimmune disease from the starting point of modest genetic risk. That risk, acted on by a lifetime of hormone changes, environmental interactions, and inexorable aging, eventually results in a full-blown autoimmune disease. Epigenetics, reversible but acting at the DNA level, defines mechanisms that can explain the environmental effects on the development of autoimmune disorders [3].
9 Summary
Although autoimmunity is affected largely by autoantibodies, the development of autoimmune disease requires an aberrant yet sophisticated interplay of a multitude of immunoactive genes, resulting after years of random boosts in risk from genes, estrogen, environmental insults, and the inexorable process of aging, in a point at which immune tolerance and the onset of autoimmune disease occurs. Up until the last few years, the manner in which the process unfolds within the body, in which both set internal parameters and variable external risks act in concert to bring about disease, has been somewhat mysterious.
Epigenetics is a new frontier in understanding the etiology of complex disease, providing a firm footing for etiology theories by defining mechanisms by which environmental conditions, both internal and external, are acted upon to create disease. Moreover, since epigenetic changes are reversible, they provide great potential for therapies in autoimmune diseases. Further research will provide better understanding of epigenetic processes. With targeted intervention, a cure for autoimmune disease may be on the horizon.
References
Kuryłowicz A, Nauman J. The role of nuclear factor-kappaB in the development of autoimmune diseases: a link between genes and environment. Acta Biochim Pol. 2008;55(4):629–47.
The Autoimmune Diseases Coordinating Committee. Progress in autoimmune disease research. Report to Congress. NIH publication 05-5140. 2005.
Javierre BM, Hernando H, Ballestar E. Environmental triggers and epigenetic deregulation in autoimmune disease. Discov Med. 2011;12(67):535–45.
Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med. 2011;365(17):1612–23.
Béland K, Lapierre P, Alvarez F. Influence of genes, sex, age and environment on the onset of autoimmune hepatitis. World J Gastroenterol. 2009;15(9):1025–34.
Kurata JH, Kantor-Fish S, Frankl H, Godby P, Vadheim CM. Crohn’s disease among ethnic groups in a large health maintenance organization. Gastroenterology. 1992;102(6):1940–8.
Reinshagen M, Loeliger C, Kuehnl P, Weiss U, Manfras BJ, Adler G, et al. HLA class II gene frequencies in Crohn’s disease: a population based analysis in Germany. Gut. 1996;38(4):538–42.
Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43(11):1066–73.
Shapira Y, Agmon-Levin N, Shoenfeld Y. Defining and analyzing geoepidemiology and human autoimmunity. J Autoimmun. 2010;34(3):J168–77.
Brix TH, Knudsen GPS, Kristiansen M, Kyvik KO, Orstavik KH, Hegedüs L. High frequency of skewed X-chromosome inactivation in females with autoimmune thyroid disease: a possible explanation for the female predisposition to thyroid autoimmunity. J Clin Endocrinol Metab. 2005;90(11):5949–53.
Djilali-Saiah I, Larger E, Harfouch-Hammoud E, Timsit J, Clerc J, Bertin E, et al. No major role for the CTLA-4 gene in the association of autoimmune thyroid disease with IDDM. Diabetes. 1998;47(1):125–7.
Gough SCL, Simmonds MJ. The HLA region and autoimmune disease: associations and mechanisms of action. Curr Genomics. 2007;8(7):453–65.
Dale E, Davis M, Faustman DL. A role for transcription factor NF-kappaB in autoimmunity: possible interactions of genes, sex, and the immune response. Adv Physiol Educ. 2006;30(4):152–8.
Fukazawa T, Yanagawa T, Kikuchi S, Yabe I, Sasaki H, Hamada T, et al. CTLA-4 gene polymorphism may modulate disease in Japanese multiple sclerosis patients. J Neurol Sci. 1999;171(1):49–55.
Invernizzi P, Pasini S, Selmi C, Gershwin ME, Podda M. Female predominance and X chromosome defects in autoimmune diseases. J Autoimmun. 2009;33(1):12–6.
Miozzo M, Selmi C, Gentilin B, Grati FR, Sirchia S, Oertelt S, et al. Preferential X chromosome loss but random inactivation characterize primary biliary cirrhosis. Hepatology. 2007;46(2):456–62.
Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med. 2009;360(24):2544–55.
Nigam P, Singh D, Matreja VS, Saxena HN. Psoriatic arthritis: a clinico-radiological study. J Dermatol. 1980;7(1):55–9.
Chandran V, Raychaudhuri SP. Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis. J Autoimmun. 2010;34(3):J314–21.
Chabchoub G, Uz E, Maalej A, Mustafa CA, Rebai A, Mnif M, et al. Analysis of skewed X-chromosome inactivation in females with rheumatoid arthritis and autoimmune thyroid diseases. Arthritis Res Ther. 2009;11(4):R106.
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357(10):977–86.
Ozbalkan Z, Bagişlar S, Kiraz S, Akyerli CB, Ozer HTE, Yavuz S, et al. Skewed X chromosome inactivation in blood cells of women with scleroderma. Arthritis Rheum. 2005;52(5):1564–70.
Gladman DD, Kung TN, Siannis F, Pellett F, Farewell VT, Lee P. HLA markers for susceptibility and expression in scleroderma. J Rheumatol. 2005;32(8):1481–7.
Agarwal SK, Reveille JD. The genetics of scleroderma (systemic sclerosis). Curr Opin Rheumatol. 2010;22(2):133–8.
Bizzarro A, Valentini G, Di Martino G, DaPonte A, De Bellis A, Iacono G. Influence of testosterone therapy on clinical and immunological features of autoimmune diseases associated with Klinefelter’s syndrome. J Clin Endocrinol Metab. 1987;64(1):32–6.
Harley JB, Reichlin M, Arnett FC, Alexander EL, Bias WB, Provost TT. Gene interaction at HLA-DQ enhances autoantibody production in primary Sjögren’s syndrome. Science. 1986;232(4754):1145–7.
Desai-Mehta A, Lu L, Ramsey-Goldman R, Datta SK. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest. 1996;97(9):2063–73.
Chung SA, Taylor KE, Graham RR, Nititham J, Lee AT, Ortmann WA, et al. Differential genetic associations for systemic lupus erythematosus based on anti-dsDNA autoantibody production. PLoS Genet. 2011;7(3):e1001323.
Barker JM. Clinical review: type 1 diabetes-associated autoimmunity: natural history, genetic associations, and screening. J Clin Endocrinol Metab. 2006;91(4):1210–7.
Thompson AI, Lees CW. Genetics of ulcerative colitis. Inflamm Bowel Dis. 2011;17(3):831–48.
Teitelbaum JE, Perez-Atayde AR, Cohen M, Bousvaros A, Jonas MM. Minocycline-related autoimmune hepatitis: case series and literature review. Arch Pediatr Adolesc Med. 1998;152(11):1132–6.
Alla V, Abraham J, Siddiqui J, Raina D, Wu GY, Chalasani NP, et al. Autoimmune hepatitis triggered by statins. J Clin Gastroenterol. 2006;40(8):757–61.
Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun. 2010;34(3):J258–65.
Kappelman MD, Rifas-Shiman SL, Kleinman K, Ollendorf D, Bousvaros A, Grand RJ, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clin Gastroenterol Hepatol. 2007;5(12):1424–9.
Brooks WH, Le Dantec C, Pers J, Youinou P, Renaudineau Y. Epigenetics and autoimmunity. J Autoimmun. 2010;34(3):J207–19.
Pollard KM. Gender differences in autoimmunity associated with exposure to environmental factors. J Autoimmun. 2012;38(2–3):J177–86.
Orton S, Wald L, Confavreux C, Vukusic S, Krohn JP, Ramagopalan SV, et al. Association of UV radiation with multiple sclerosis prevalence and sex ratio in France. Neurology. 2011;76(5):425–31.
Ascherio A, Munch M. Epstein-Barr virus and multiple sclerosis. Epidemiology. 2000;11(2):220–4.
Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127(2):485–92.
Ogrendik M. Does periodontopathic bacterial infection contribute to the etiopathogenesis of the autoimmune disease rheumatoid arthritis? Discov Med. 2012;13(72):349–55.
González DA, Díaz BB, Rodríguez Pérez MDC, Hernández AG, Chico BND, de León AC. Sex hormones and autoimmunity. Immunol Lett. 2010;133(1):6–13.
Ahmed SA, Hissong BD, Verthelyi D, Donner K, Becker K, Karpuzoglu-Sahin E. Gender and risk of autoimmune diseases: possible role of estrogenic compounds. Environ Health Perspect. 1999;107 Suppl 5:681–6.
Ramos-Casals M, García-Carrasco M, Brito MP, López-Soto A, Font J. Autoimmunity and geriatrics: clinical significance of autoimmune manifestations in the elderly. Lupus. 2003;12(5):341–55.
Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annu Rev Immunol. 2009;27:363–91.
Hemminki K, Li X, Sundquist K, Sundquist J. Shared familial aggregation of susceptibility to autoimmune diseases. Arthritis Rheum. 2009;60(9):2845–7.
Rubtsov AV, Rubtsova K, Kappler JW, Marrack P. Genetic and hormonal factors in female-biased autoimmunity. Autoimmun Rev. 2010;9(7):494–8.
Meda F, Folci M, Baccarelli A, Selmi C. The epigenetics of autoimmunity. Cell Mol Immunol. 2011;8(3):226–36.
Cotsapas C, Voight BF, Rossin E, Lage K, Neale BM, Wallace C, et al. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet. 2011;7(8):e1002254.
Gervin K, Vigeland MD, Mattingsdal M, Hammerø M, Nygård H, Olsen AO, et al. DNA methylation and gene expression changes in monozygotic twins discordant for psoriasis: identification of epigenetically dysregulated genes. PLoS Genet. 2012;8(1):e1002454.
Manheimer-Lory AJ, Zandman-Goddard G, Davidson A, Aranow C, Diamond B. Lupus-specific antibodies reveal an altered pattern of somatic mutation. J Clin Invest. 1997;100(10):2538–46.
Davidson A, Manheimer-Lory A, Aranow C, Peterson R, Hannigan N, Diamond B. Molecular characterization of a somatically mutated anti-DNA antibody bearing two systemic lupus erythematosus-related idiotypes. J Clin Invest. 1990;85(5):1401–9.
Rider V, Abdou NI. Hormones: epigenetic contributors to gender-based autoimmunity. In: Zouali M, editor. The epigenetics of autoimmune diseases. Chichester: Wiley; 2009. p. 309–26.
Olsen NJ, Kovacs WJ. Effects of androgens on T and B lymphocyte development. Immunol Res. 2001;23(2–3):281–8.
Walsh SJ, Rau LM. Autoimmune diseases: a leading cause of death among young and middle-aged women in the United States. Am J Public Health. 2000;90(9):1463–6.
IOM (Institute of Medicine). Women’s health research: progress, pitfalls, and promise. Washington, DC: The National Academies Press; 2010.
Gameiro C, Romao F. Changes in the immune system during menopause and aging. Front Biosci (Elite Ed). 2010;2:1299–303.
Papenfuss TL, Whitacre CC. Sex hormones, pregnancy, and immune function. In: Pfaff DW, Arnold AP, Etgen AM, Fahrbach SE, Rubin RT, editors. Hormones, brain, and behavior. San Diego: Academic; 2009. p. 367–76.
Fairweather D, Frisancho-Kiss S, Rose NR. Sex differences in autoimmune disease from a pathological perspective. Am J Pathol. 2008;173(3):600–9.
Delpy L, Douin-Echinard V, Garidou L, Bruand C, Saoudi A, Guéry J. Estrogen enhances susceptibility to experimental autoimmune myasthenia gravis by promoting type 1-polarized immune responses. J Immunol. 2005;175(8):5050–7.
Marzi M, Vigano A, Trabattoni D, Villa ML, Salvaggio A, Clerici E, et al. Characterization of type 1 and type 2 cytokine production profile in physiologic and pathologic human pregnancy. Clin Exp Immunol. 1996;106(1):127–33.
Beagley KW, Gockel CM. Regulation of innate and adaptive immunity by the female sex hormones oestradiol and progesterone. FEMS Immunol Med Microbiol. 2003;38(1):13–22.
Grimaldi CM, Michael DJ, Diamond B. Cutting edge: expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J Immunol. 2001;167(4):1886–90.
Kanda N, Tamaki K. Estrogen enhances immunoglobulin production by human PBMCs. J Allergy Clin Immunol. 1999;103(2 Pt 1):282–8.
Nalbandian G, Kovats S. Estrogen, immunity & autoimmune disease. Curr Med Chem Immun Endoc Metab Agents. 2005;5:85–91.
Tanriverdi F, Silveira LFG, MacColl GS, Bouloux PMG. The hypothalamic-pituitary-gonadal axis: immune function and autoimmunity. J Endocrinol. 2003;176(3):293–304.
Tai P, Wang J, Jin H, Song X, Yan J, Kang Y, et al. Induction of regulatory T cells by physiological level estrogen. J Cell Physiol. 2008;214(2):456–64.
Thongngarm T, Jenkins JK, Ndebele K, McMurray RW. Estrogen and progesterone modulate monocyte cell cycle progression and apoptosis. Am J Reprod Immunol. 2003;49(3):129–38.
O’Connor M, Motivala SJ, Valladares EM, Olmstead R, Irwin MR. Sex differences in monocyte expression of IL-6: role of autonomic mechanisms. Am J Physiol Regul Integr Comp Physiol. 2007;293(1):R145–51.
Lamon-Fava S, Posfai B, Schaefer EJ. Effect of hormonal replacement therapy on C-reactive protein and cell-adhesion molecules in postmenopausal women. Am J Cardiol. 2003;91(2):252–4.
Miller AP, Feng W, Xing D, Weathington NM, Blalock JE, Chen Y, et al. Estrogen modulates inflammatory mediator expression and neutrophil chemotaxis in injured arteries. Circulation. 2004;110(12):1664–9.
Bynoe MS, Grimaldi CM, Diamond B. Estrogen up-regulates Bcl-2 and blocks tolerance induction of naive B cells. Proc Natl Acad Sci U S A. 2000;97(6):2703–8.
Srivastava S, Weitzmann MN, Cenci S, Ross FP, Adler S, Pacifici R. Estrogen decreases TNF gene expression by blocking JNK activity and the resulting production of c-Jun and JunD. J Clin Invest. 1999;104(4):503–13.
Koh KK, Ahn JY, Jin DK, Yoon B, Kim HS, Kim DS, et al. Effects of continuous combined hormone replacement therapy on inflammation in hypertensive and/or overweight postmenopausal women. Arterioscler Thromb Vasc Biol. 2002;22(9):1459–64.
Dai R, Zhang Y, Khan D, Heid B, Caudell D, Crasta O, et al. Identification of a common lupus disease-associated microRNA expression pattern in three different murine models of lupus. PLoS One. 2010;5(12):e14302.
Buchel E, Van Steenbergen W, Nevens F, Fevery J. Improvement of autoimmune hepatitis during pregnancy followed by flare-up after delivery. Am J Gastroenterol. 2002;97(12):3160–5.
Samuel D, Riordan S, Strasser S, Kurtovic J, Singh-Grewel I, Koorey D. Severe autoimmune hepatitis first presenting in the early post partum period. Clin Gastroenterol Hepatol. 2004;2(7):622–4.
Kanda N, Tsuchida T, Tamaki K. Estrogen enhancement of anti-double-stranded DNA antibody and immunoglobulin G production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. 1999;42(2):328–37.
Kanda N, Tsuchida T, Tamaki K. Testosterone inhibits immunoglobulin production by human peripheral blood mononuclear cells. Clin Exp Immunol. 1996;106(2):410–5.
Gubbels Bupp MR, Jørgensen TN, Kotzin BL. Identification of candidate genes that influence sex hormone-dependent disease phenotypes in mouse lupus. Genes Immun. 2008;9(1):47–56.
Aguilera O, Fernández AF, Muñoz A, Fraga MF. Epigenetics and environment: a complex relationship. J Appl Physiol. 2010;109(1):243–51.
Palaszynski KM, Smith DL, Kamrava S, Burgoyne PS, Arnold AP, Voskuhl RR. A yin-yang effect between sex chromosome complement and sex hormones on the immune response. Endocrinology. 2005;146(8):3280–5.
Abdou N, Rider V. Gender differences in autoimmune diseases: immune mechanisms and clinical applications. In: Legato MJ, editor. Principles of gender-specific medicine. London: Academic; 2009. p. 585–91.
Pennell LM, Galligan CL, Fish EN. Sex affects immunity. J Autoimmun. 2012;38(2–3):J282–91.
Cunningham M, Gilkeson G. Estrogen receptors in immunity and autoimmunity. Clin Rev Allergy Immunol. 2011;40(1):66–73.
Rider V, Jones S, Evans M, Bassiri H, Afsar Z, Abdou NI. Estrogen increases CD40 ligand expression in T cells from women with systemic lupus erythematosus. J Rheumatol. 2001;28(12):2644–9.
Abdou NI, Rider V, Greenwell C, Li X, Kimler BF. Fulvestrant (Faslodex), an estrogen selective receptor downregulator, in therapy of women with systemic lupus erythematosus. Clinical, serologic, bone density, and T cell activation marker studies: a double-blind placebo-controlled trial. J Rheumatol. 2008;35(5):797.
Galofré JC. Microchimerism in Graves’ disease. J Thyroid Res. 2012;2012:724382.
Greer LG, Casey BM, Halvorson LM, Spong CY, McIntire DD, Cunningham FG. Antithyroid antibodies and parity: further evidence for microchimerism in autoimmune thyroid disease. Am J Obstet Gynecol. 2011;205(5):471.e1–4.
Zandman-Goddard G, Peeva E, Shoenfeld Y. Gender and autoimmunity. Autoimmun Rev. 2007;6(6):366–72.
Tremolizzo L, Carboni G, Ruzicka WB, Mitchell CP, Sugaya I, Tueting P, et al. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc Natl Acad Sci U S A. 2002;99(26):17095–100.
Kim K, Friso S, Choi S. DNA methylation, an epigenetic mechanism connecting folate to healthy embryonic development and aging. J Nutr Biochem. 2009;20(12):917–26.
Shaw GM, Carmichael SL, Yang W, Selvin S, Schaffer DM. Periconceptional dietary intake of choline and betaine and neural tube defects in offspring. Am J Epidemiol. 2004;160(2):102–9.
Choi S, Friso S. Epigenetics: a new bridge between nutrition and health. Adv Nutr. 2010;1(1):8–16.
MacFarlane AJ, Strom A, Scott FW. Epigenetics: deciphering how environmental factors may modify autoimmune type 1 diabetes. Mamm Genome. 2009;20(9–10):624–32.
Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308(5727):1466–9.
Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179(7):572–8.
Fernández-Ruiz J, Gómez M, Hernández M, de Miguel R, Ramos JA. Cannabinoids and gene expression during brain development. Neurotox Res. 2004;6(5):389–401.
Roth TL, Zoladz PR, Sweatt JD, Diamond DM. Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. J Psychiatr Res. 2011;45(7):919–26.
Borchers AT, Gershwin ME. Sociological differences between women and men: implications for autoimmunity. Autoimmun Rev. 2012;11(6–7):A413–21.
Larbi A, Franceschi C, Mazzatti D, Solana R, Wikby A, Pawelec G. Aging of the immune system as a prognostic factor for human longevity. Physiology (Bethesda). 2008;23:64–74.
Aw D, Silva AB, Palmer DB. Immunosenescence: emerging challenges for an ageing population. Immunology. 2007;120(4):435–46.
Allman D, Miller JP. B cell development and receptor diversity during aging. Curr Opin Immunol. 2005;17(5):463–7.
Hasler P, Zouali M. Immune receptor signaling, aging, and autoimmunity. Cell Immunol. 2005;233(2):102–8.
Huppert FA, Solomou W, O’Connor S, Morgan K, Sussams P, Brayne C. Aging and lymphocyte subpopulations: whole-blood analysis of immune markers in a large population sample of healthy elderly individuals. Exp Gerontol. 1998;33(6):593–600.
Franceschi C, Cossarizza A. Introduction: the reshaping of the immune system with age. Int Rev Immunol. 1995;12(1):1–4.
Utsuyama M, Hirokawa K, Kurashima C, Fukayama M, Inamatsu T, Suzuki K, et al. Differential age-change in the numbers of CD4+CD45RA+ and CD4+CD29+ T cell subsets in human peripheral blood. Mech Ageing Dev. 1992;63(1):57–68.
Johnson SA, Cambier JC. Ageing, autoimmunity and arthritis: senescence of the B cell compartment—implications for humoral immunity. Arthritis Res Ther. 2004;6(4):131–9.
Frasca D, Riley RL, Blomberg BB. Humoral immune response and B-cell functions including immunoglobulin class switch are downregulated in aged mice and humans. Semin Immunol. 2005;17(5):378–84.
Whisler RL, Liu BQ, Newhouse YG, Walters JD, Breckenridge MB, Grants IS. Signal transduction in human B cells during aging: alterations in stimulus-induced phosphorylations of tyrosine and serine/threonine substrates and in cytosolic calcium responsiveness. Lymphokine Cytokine Res. 1991;10(6):463–73.
Paganelli R, Quinti I, Fagiolo U, Cossarizza A, Ortolani C, Guerra E, et al. Changes in circulating B cells and immunoglobulin classes and subclasses in a healthy aged population. Clin Exp Immunol. 1992;90(2):351–4.
Giglio T, Imro MA, Filaci G, Scudeletti M, Puppo F, De Cecco L, et al. Immune cell circulating subsets are affected by gonadal function. Life Sci. 1994;54(18):1305–12.
Fann M, Chiu WK, Wood 3rd WH, Levine BL, Becker KG, Weng N. Gene expression characteristics of CD28null memory phenotype CD8+ T cells and its implication in T-cell aging. Immunol Rev. 2005;205:190–206.
Mo R, Chen J, Han Y, Bueno-Cannizares C, Misek DE, Lescure PA, et al. T cell chemokine receptor expression in aging. J Immunol. 2003;170(2):895–904.
Weng N. Aging of the immune system: how much can the adaptive immune system adapt? Immunity. 2006;24(5):495–9.
Tarazona R, DelaRosa O, Alonso C, Ostos B, Espejo J, Peña J, et al. Increased expression of NK cell markers on T lymphocytes in aging and chronic activation of the immune system reflects the accumulation of effector/senescent T cells. Mech Ageing Dev. 2000;121(1–3):77–88.
Ershler WB, Sun WH, Binkley N, Gravenstein S, Volk MJ, Kamoske G, et al. Interleukin-6 and aging: blood levels and mononuclear cell production increase with advancing age and in vitro production is modifiable by dietary restriction. Lymphokine Cytokine Res. 1993;12(4):225–30.
Farage MA, Miller KW, Maibach HI. The effects of menopause on autoimmune diseases. Expert Rev Obstet Gynecol. 2012;7(6):557–71.
Ku LT, Gercel-Taylor C, Nakajima ST, Taylor DD. Alterations of T cell activation signalling and cytokine production by postmenopausal estrogen levels. Immun Ageing. 2009;6:1.
Stacy S, Krolick KA, Infante AJ, Kraig E. Immunological memory and late onset autoimmunity. Mech Ageing Dev. 2002;123(8):975–85.
Gangemi S, Basile G, Monti D, Merendino RA, Di Pasquale G, Bisignano U, et al. Age-related modifications in circulating IL-15 levels in humans. Mediators Inflamm. 2005;2005(4):245–7.
Fairweather D, Rose N. Immunopathogenesis of autoimmune diseases. In: Luebke R, House RV, Kimber I, editors. Immunotoxicology and immunopharmacology. Boca Raton: CRC Press; 2007. p. 423–36.
Hasler P, Zouali M. Subversion of B lymphocyte signaling by infectious agents. Genes Immun. 2003;4(2):95–103.
Gameiro CM, Romão F, Castelo-Branco C. Menopause and aging: changes in the immune system–a review. Maturitas. 2010;67(4):316–20.
Njemini R, Meyers I, Demanet C, Smitz J, Sosso M, Mets T. The prevalence of autoantibodies in an elderly sub-Saharan African population. Clin Exp Immunol. 2002;127(1):99–106.
Rogers MAM, Levine DA, Blumberg N, Fisher GG, Kabeto M, Langa KM. Antigenic challenge in the etiology of autoimmune disease in women. J Autoimmun. 2012;38(2–3):J97–102.
Pietschmann P, Gollob E, Brosch S, Hahn P, Kudlacek S, Willheim M, et al. The effect of age and gender on cytokine production by human peripheral blood mononuclear cells and markers of bone metabolism. Exp Gerontol. 2003;38(10):1119–27.
Zhu BT, Han G, Shim J, Wen Y, Jiang X. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology. 2006;147(9):4132–50.
Strickland FM, Richardson BC. Epigenetics in human autoimmunity. Epigenetics in autoimmunity—DNA methylation in systemic lupus erythematosus and beyond. Autoimmunity. 2008;41(4):278–86.
Jönsen A, Bengtsson AA, Sturfelt G, Truedsson L. Analysis of HLA DR, HLA DQ, C4A, FcgammaRIIa, FcgammaRIIIa, MBL, and IL-1Ra allelic variants in Caucasian systemic lupus erythematosus patients suggests an effect of the combined FcgammaRIIa R/R and IL-1Ra 2/2 genotypes on disease susceptibility. Arthritis Res Ther. 2004;6(6):R557–62.
Rider V, Foster RT, Evans M, Suenaga R, Abdou NI. Gender differences in autoimmune diseases: estrogen increases calcineurin expression in systemic lupus erythematosus. Clin Immunol Immunopathol. 1998;89(2):171–80.
Costenbader KH, Feskanich D, Stampfer MJ, Karlson EW. Reproductive and menopausal factors and risk of systemic lupus erythematosus in women. Arthritis Rheum. 2007;56(4):1251–62.
Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol. 2007;179(9):6352–8.
Rider V, Jones SR, Evans M, Abdou NI. Molecular mechanisms involved in the estrogen-dependent regulation of calcineurin in systemic lupus erythematosus T cells. Clin Immunol. 2000;95(2):124–34.
Bynoté KK, Hackenberg JM, Korach KS, Lubahn DB, Lane PH, Gould KA. Estrogen receptor-alpha deficiency attenuates autoimmune disease in (NZB x NZW)F1 mice. Genes Immun. 2008;9(2):137–52.
Jørgensen TN, Roper E, Thurman JM, Marrack P, Kotzin BL. Type I interferon signaling is involved in the spontaneous development of lupus-like disease in B6.Nba2 and (B6.Nba2 x NZW)F(1) mice. Genes Immun. 2007;8(8):653–62.
New discoveries on environmental triggers of lupus. http://lupusresearchinstitute.org/news/discoveries/080528.
Ballestar E, Esteller M, Richardson BC. The epigenetic face of systemic lupus erythematosus. J Immunol. 2006;176(12):7143–7.
Aprahamian T, Takemura Y, Goukassian D, Walsh K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol. 2008;152(3):448–55.
Grimaldi CM, Hill L, Xu X, Peeva E, Diamond B. Hormonal modulation of B cell development and repertoire selection. Mol Immunol. 2005;42(7):811–20.
Gilmore T. NK-kB Transcription factors. http://www.bu.edu/nf-kb/.
Knecht H, Berger C, Rothenberger S, Odermatt BF, Brousset P. The role of Epstein-Barr virus in neoplastic transformation. Oncology. 2001;60(4):289–302.
Seppänen M, Vihko KK. Activation of transcription factor NF-kappaB by growth inhibitory cytokines in vulvar carcinoma cells. Immunol Lett. 2000;74(2):103–9.
Ramdass B, Maliekal TT, Lakshmi S, Rehman M, Rema P, Nair P, et al. Coexpression of Notch1 and NF-kappaB signaling pathway components in human cervical cancer progression. Gynecol Oncol. 2007;104(2):352–61.
Peña AS, Peñate M. Genetic susceptibility and regulation of inflammation in Crohn’s disease. Relationship with the innate immune system. Rev Esp Enferm Dig. 2002;94(6):351–60.
Staudt LM. The molecular and cellular origins of Hodgkin’s disease. J Exp Med. 2000;191(2):207–12.
Atreya I, Atreya R, Neurath MF. NF-kappaB in inflammatory bowel disease. J Intern Med. 2008;263(6):591–6.
Gilmore TD. Multiple myeloma: lusting for NF-kappaB. Cancer Cell. 2007;12(2):95–7.
Satoh J, Illes Z, Peterfalvi A, Tabunoki H, Rozsa C, Yamamura T. Aberrant transcriptional regulatory network in T cells of multiple sclerosis. Neurosci Lett. 2007;422(1):30–3.
Huang S, Robinson JB, Deguzman A, Bucana CD, Fidler IJ. Blockade of nuclear factor-kappaB signaling inhibits angiogenesis and tumorigenicity of human ovarian cancer cells by suppressing expression of vascular endothelial growth factor and interleukin 8. Cancer Res. 2000;60(19):5334–9.
Greetham D, Ellis CD, Mewar D, Fearon U, Ultaigh SN, Veale DJ, et al. Functional characterization of NF-kappaB inhibitor-like protein 1 (NFkappaBIL1), a candidate susceptibility gene for rheumatoid arthritis. Hum Mol Genet. 2007;16(24):3027–36.
Oikonomidou O, Vlachoyiannopoulos PG, Kominakis A, Kalofoutis A, Moutsopoulos HM, Moutsatsou P. Glucocorticoid receptor, nuclear factor kappaB, activator protein-1 and C-jun N-terminal kinase in systemic lupus erythematosus patients. Neuroimmunomodulation. 2006;13(4):194–204.
Eldor R, Yeffet A, Baum K, Doviner V, Amar D, Ben-Neriah Y, et al. Conditional and specific NF-kappaB blockade protects pancreatic beta cells from diabetogenic agents. Proc Natl Acad Sci U S A. 2006;103(13):5072–7.
Hurst J, von Landenberg P. Toll-like receptors and autoimmunity. Autoimmun Rev. 2008;7(3):204–8.
Dai R, Phillips RA, Karpuzoglu E, Khan D, Ahmed SA. Estrogen regulates transcription factors STAT-1 and NF-kappaB to promote inducible nitric oxide synthase and inflammatory responses. J Immunol. 2009;183(11):6998–7005.
Dunn SE, Ousman SS, Sobel RA, Zuniga L, Baranzini SE, Youssef S, et al. Peroxisome proliferator-activated receptor (PPAR)alpha expression in T cells mediates gender differences in development of T cell-mediated autoimmunity. J Exp Med. 2007;204(2):321–30.
Csiszar A, Wang M, Lakatta EG, Ungvari Z. Inflammation and endothelial dysfunction during aging: role of NF-kappaB. J Appl Physiol. 2008;105(4):1333–41.
Levine A, Shamir R, Wine E, Weiss B, Karban A, Shaoul RR, et al. TNF promoter polymorphisms and modulation of growth retardation and disease severity in pediatric Crohn’s disease. Am J Gastroenterol. 2005;100(7):1598–604.
Hayashi T, Faustman D. NOD mice are defective in proteasome production and activation of NF-kappaB. Mol Cell Biol. 1999;19(12):8646–59.
Gale EA, Gillespie KM. Diabetes and gender. Diabetologia. 2001;44(1):3–15.
van Heel DA, Udalova IA, De Silva AP, McGovern DP, Kinouchi Y, Hull J, et al. Inflammatory bowel disease is associated with a TNF polymorphism that affects an interaction between the OCT1 and NF(-kappa)B transcription factors. Hum Mol Genet. 2002;11(11):1281–9.
Miterski B, Böhringer S, Klein W, Sindern E, Haupts M, Schimrigk S, et al. Inhibitors in the NFkappaB cascade comprise prime candidate genes predisposing to multiple sclerosis, especially in selected combinations. Genes Immun. 2002;3(4):211–9.
Okamoto T, Tsuchiya A. NF-kappaB as a therapeutic target of rheumatoid arthritis. Nihon Rinsho Meneki Gakkai Kaishi. 2007;30(5):383–9. Japanese.
Krause S, Kuckelkorn U, Dörner T, Burmester G, Feist E, Kloetzel P. Immunoproteasome subunit LMP2 expression is deregulated in Sjogren’s syndrome but not in other autoimmune disorders. Ann Rheum Dis. 2006;65(8):1021–7.
Kammer GM, Tsokos GC. Abnormal T lymphocyte signal transduction in systemic lupus erythematosus. Curr Dir Autoimmun. 2002;5:131–50.
Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–40.
Luo J, Kuo MH. Linking nutrient metabolism to epigenetics. Cell Sci Rev. 2009;6:49–54.
Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60(4):1065–75.
Leader JE, Wang C, Popov VM, Fu M, Pestell RG. Epigenetics and the estrogen receptor. Ann N Y Acad Sci. 2006;1089:73–87.
Dai R, Ahmed SA. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl Res. 2011;157(4):163–79.
Baguet A, Bix M. Chromatin landscape dynamics of the Il4-Il13 locus during T helper 1 and 2 development. Proc Natl Acad Sci U S A. 2004;101(31):11410–5.
Calvanese V, Lara E, Kahn A, Fraga MF. The role of epigenetics in aging and age-related diseases. Ageing Res Rev. 2009;8(4):268–76.
Wolff GL, Kodell RL, Moore SR, Cooney CA. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998;12(11):949–57.
Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20(2):170–9.
Strickland FM, Hewagama A, Lu Q, Wu A, Hinderer R, Webb R, et al. Environmental exposure, estrogen and two X chromosomes are required for disease development in an epigenetic model of lupus. J Autoimmun. 2012;38(2–3):J135–43.
Korganow A, Knapp A, Nehme-Schuster H, Soulas-Sprauel P, Poindron V, Pasquali J, et al. Peripheral B cell abnormalities in patients with systemic lupus erythematosus in quiescent phase: decreased memory B cells and membrane CD19 expression. J Autoimmun. 2010;34(4):426–34.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Farage, M.A., Miller, K.W., Lahita, R.G. (2015). Genes, Hormones, Immunosenescence, and Environmental Agents: Toward an Integrated View of the Genesis of Autoimmune Disease. In: Farage, M., Miller, K., Fugate Woods, N., Maibach, H. (eds) Skin, Mucosa and Menopause. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-44080-3_24
Download citation
DOI: https://doi.org/10.1007/978-3-662-44080-3_24
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-44079-7
Online ISBN: 978-3-662-44080-3
eBook Packages: MedicineMedicine (R0)