
Abstract
Hemorrhagic shock is characterized by both macrovascular hemodynamic abnormalities (decreased venous return, decreased cardiac output and systemic hypotension) and alterations of the microcirculation. The microcirculation is a critical component of the cardiovascular system, which regulates flow to the tissues. Several studies have shown a significant decrease in microvascular blood flow in various organs during the acute phase of hemorrhagic shock and resuscitation [1–4]. Persistence of these microvascular alterations is believed to be a contributing factor to the development of organ dysfunction. The pathogenesis of the microvascular alterations involves both vascular and cellular components. In current clinical practice, guidelines for resuscitation are provided by monitoring macrocirculatory variables, such as arterial blood pressure, heart rate, and cardiac output.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Hemorrhagic Shock
- Fluid Resuscitation
- Microvascular Blood Flow
- Postcapillary Venule
- Functional Capillary Density
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Hemorrhagic shock is characterized by both macrovascular hemodynamic abnormalities (decreased venous return, decreased cardiac output and systemic hypotension) and alterations of the microcirculation. The microcirculation is a critical component of the cardiovascular system, which regulates flow to the tissues. Several studies have shown a significant decrease in microvascular blood flow in various organs during the acute phase of hemorrhagic shock and resuscitation [1–4]. Persistence of these microvascular alterations is believed to be a contributing factor to the development of organ dysfunction. The pathogenesis of the microvascular alterations involves both vascular and cellular components. In current clinical practice, guidelines for resuscitation are provided by monitoring macrocirculatory variables, such as arterial blood pressure, heart rate, and cardiac output. However, whether these resuscitation procedures are effective in restoring organ microcirculation remains to be elucidated. The purpose of this review is to focus on the mechanisms involved in the development of microvascular alterations in hemorrhagic shock and to discuss the potential therapeutic implications for resuscitation in hemorrhagic shock.
Cardiovascular Response in Hemorrhagic Shock
In the acute phase of hemorrhage, macrovascular and microvascular responses rapidly act to compensate for the loss of blood volume and to limit tissue hypoxia.
Macrovascular Response
The macrovascular compensatory mechanism involves the autonomic nervous system. Decreases in venous return and arterial pressure lead to unloading of cardiopulmonary and arterial baroreceptors inducing a decrease in the activation of the vasomotor inhibitory center in the brainstem, which leads to activation of the sympathetic center and inhibition of vagal activity (sinoatrial node). The increased activity of the sympathetic nerves produces an increase in heart rate, cardiac contractility and arterial and venous tone with an activation of the renin-angiotensin-aldosterone system. The magnitude of the compensatory vasoconstriction that follows is the net result of the interaction of the effects of norepinephrine (from the peripheral nerves) and epinephrine (from the adrenal medulla) on the peripheral vascular adrenoreceptors, and nonadrenergic mechanisms (i. e., angiotensin and vasopressin). Arterial vasoconstriction rapidly decreases non-vital organ blood flow (musculocutaneous, splanchnic and renal blood flow) to maintain perfusion pressure and blood flow to vital organs (the heart and the brain). It is important to keep in mind that the sympathetic stimulation exerts both arterial and venous α-adrenergic stimulation [5]. Indeed, in addition to its arterial vasoconstricting action, the sympathetic stimulation induces venoconstriction (specially in splanchnic circulation) enhancing a shift of splanchnic blood volume to the systemic circulation [6]. This venous adrenergic stimulation may recruit blood from the venous unstressed volume helping to maintain venous return and cardiac output [6].
Microvascular Response
The microcirculation regulates the distribution of blood flow throughout individual organs to provide adequate oxygen delivery (DO2) for the oxygen demands of every cell within an organ. In order to achieve this, the microcirculation responds to changes in metabolic demand by limiting blood flow in microvascular units with low oxygen demand and increasing blood flow in microvascular units with high oxygen demand. This microvascular heterogeneity of blood flow is an essential property of normal microcirculatory perfusion to provide adequate DO2 for the tissue. During hemorrhagic shock, in addition to the macrovascular distribution of arterial blood flow at the expense of non-vital organs, blood flow is redistributed within the capillary networks of each organ according to arteriolar and capillary resistances, rheologic factors and oxygen demand. Increase in arteriolar and capillary resistances associated with unfavorable rheologic factors takes blood flow away from microvascular units with low oxygen demands and non-essential cell functions. The microvascular units where the blood flow is reduced most severely might adjust their function and their energy utilization to prevent hypoxia. This down-regulation of cellular metabolism is called conformance or hibernation [7]. The possible involvement of such a mechanism may limit tissue hypoxia. However, the observed increase in lactate level during the acute phase of hemorrhagic shock indicates the limits of this adaptative metabolic down-regulation.
The microvascular response to the decrease in DO2 in microvascular units with high oxygen demand involves several compensatory mechanisms to increase oxygen extraction and maintain tissue oxygenation. Two major mechanisms have been proposed to account for the local oxygen delivery regulation: Regulation of arteriolar tone and control of the functional surface area for oxygen exchange.
Regulation of the Arteriolar Tone
Arterioles dilate in response to decreased tissue PO2 to increase perfusion and DO2. The arteriolar tone is the net result of the interaction of the autonomic nervous system, vasoactive substances in the blood (catecholamines, angiotensin and vasopressin) and local regulation of arteriolar tone. Local regulation of arteriolar tone is a crucial factor in microvascular regulation to match oxygen supply to oxygen demand. Several mechanisms contribute to the local regulation of arteriolar tone, including response to intraluminal pressure (myogenic response), shear stress on the endothelial cells (shear-dependent response), and tissue metabolite concentrations (metabolic response).
The vascular myogenic response refers to the intrinsic ability of a blood vessel to constrict to an increase in intraluminal pressure or dilate to a decrease in intraluminal pressure. The vasodilation induced by shear stress (nitric oxide [NO]-dependent mechanism) is dependent on endothelial sensing/transduction of the shear induced by blood flow. The proposed mechanosensors on the luminal surface of the endothelium include components of the glycocalyx (glycoproteins and proteoglycans), stretch-activated ion channels, cytoskeletal rearrangements and cell-cell and cell-extracellular matrix connections [8].
The metabolic response allows the vascular tone to adapt to cellular oxygen demand. During hemorrhagic shock, the decrease in DO2 limits the production of adenosine 5′ triphosphate (ATP) and adenosine 5′ diphosphate (ADP) accumulates because its stock is not completely rephosphorylated with a resulting accumulation of ADP and its degradation products (adenosine 5′ monophosphate [AMP], adenosine). Furthermore, glycolysis is activated with a production of lactate and hydrogen ion. Adenosine, lactate and hydrogen ions are arteriolar vasodilators and contribute to the close link between metabolite production and tissue oxygenation. CO2 is also a powerful vasodilator, which accumulates when there is an increase in cellular metabolism or reduced clearance of CO2 during tissue hypoperfusion.
Finally, an increasingly important role in the regulation of the microvascular tone and in the matching of oxygen supply to oxygen demand is being attributed to the red blood cell (RBC) and the hemoglobin molecule. Ellsworth et al. [9, 10] suggest that the RBC behaves as a mobile oxygen-sensor and controls vascular tone by means of release of ATP. ATP is released from erythrocytes in response to mechanical deformation of the membrane, to exposure to low PO2 associated with a decrease in the hemoglobin oxygen saturation within erythrocytes, and to receptor-mediated activation of erythrocyte membrane-bound ß-adrenergic receptors or prostacyclin receptors. The erythrocyte-derived ATP can then interact with endothelial purinergic receptors, inducing release of vasodilator mediators. This vasodilation is conducted in a retrograde fashion, resulting in increased blood flow (oxygen supply) to areas of increased oxygen demand. Certainly this concept still needs to be confirmed, but it is an attractive track to explain the microvascular response to oxygen demand. Other mechanisms involving the erythrocyte in the regulation of the vascular tone have been proposed. Stamler et al. proposed that the erythrocyte could regulate DO2 through the transport of NO in a protected form as S-nitrosothiol (SNO) [11, 12]. This vasorelaxant moiety is released by hemoglobin when the hemoglobin oxygen saturation falls in response to an increase in local oxygen demand. Finally, another hypothesis has been proposed in which deoxyhemoglobin would function as a nitrite reductase to transform nitrite (NO2 −) into NO with resulting vasodilatory action [13, 14]. The possible involvement of other oxygen sensors, such as cytochrome oxidase or NADPH oxidase, is interesting, but requires further work to establish their respective contribution.
Control of Functional Surface Area for Oxygen Exchange
To maintain tissue oxygenation despite the decrease in DO2, there is an immediate need to extract more oxygen from the incoming blood. Oxygen extraction depends on the incoming blood flow (convective oxygen transport determined mainly by arteriolar tone) and on the functional surface area for oxygen exchange (diffusive oxygen transport) related to the number of RBCs and the number of perfused capillaries. Thus, oxygen extraction is facilitated by a high capillary density which increases the surface area for oxygen exchange and reduces capillary-to-mitochondrial diffusion distances (Fig. 1).


Schematic representation of the microvascular response to a decrease in oxygen delivery in microvascular units with high oxygen demands
This high capillary density can be achieved by recruiting capillaries (i. e., initiation of RBC flux in previously non-flowing capillaries). However, in some microvascular beds, such as skeletal muscle or myocardium microcirculation, it has been reported that most capillaries may sustain RBC flux at rest and capillary recruitment does not appear to be requisite for the increase in oxygen extraction [15, 16]. So, in these microcirculations, the oxygen extraction is mainly dependent on the distribution of RBC within the previously flowing capillaries. Recruitment is then more a longitudinal recruitment (along the vessel) than a recruitment of new capillaries [16]. Capillary resistance and rheologic factors (blood viscosity and RBC deformability) determine the RBC distribution within the capillary bed [17, 18]. These factors play a crucial role in determining capillary homogeneity and functional capillary density, especially at low flow states [19]. During the acute phase of hemorrhagic shock, the decrease in capillary pressure will cause increased net fluid absorption with fluid shift from the interstitium to the vascular compartment helping to restore blood volume. This effect associated with hemodilution during the resuscitation phase could theoretically decrease blood viscosity and contribute to decrease the heterogeneity of RBC distribution. However, during the late phase of hemorrhagic shock, the inflammatory process can induce fluid leakage with fluid shift to the interstitial compartment resulting in an increase in viscosity and heterogeneity of RBC distribution [20, 21].
Alterations of the Microcirculation in Hemorrhagic Shock
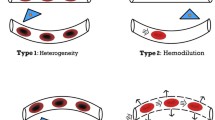
Despite efficient microvascular adaptative mechanisms, when the blood loss is severe enough, alterations of microcirculatory blood flow and tissue oxygenation have been described in various experimental models of hemorrhagic shock. Progressive decrease in cardiac output and oxygen delivery induces a progressive decrease in capillary blood flow, RBC velocities and functional capillary density with an increase in flow heterogeneity [4, 22]. These microvascular alterations are more pronounced in non-vital organ microcirculations (splanchnic, renal and musculocutaneous microcirculations) and in microvascular units with low oxygen demand and non-essential cell functions. Increase in RBC aggregation causing blood flow slowing with intermittent/no flow capillaries and ‘plasmatic’ capillaries contribute to the increased heterogeneity (Fig. 2) [2]. There is evidence that hemorrhagic shock impairs RBC deformability and causes RBC cellular damage [23, 24]. These microvascular alterations are associated with a decrease in microvascular PO2 through convective arteriovenous shunting, direct diffusion of oxygen from arterioles to venules lying in close proximity to each other, and functional shunting of disadvantaged microcirculatory units [25, 26].


Illustration of potential mechanisms involved in the development of microvascular alterations in hemorrhagic shock
In septic shock, alteration of microcirculatory blood flow is a major pathophysiological feature. Microvascular density and microvascular blood flow are both reduced in association with an increased heterogeneous perfusion in septic patients [27, 28]. Moreover, the degree of microvascular impairment has a prognostic value since it worsens in non-surviving septic patients compared to those who ultimately overcome their septic episode [29]. Early systemic hemodynamic resuscitation of septic patients may improve the time-course of microcirculatory dysfunction and eventually patient outcome. However, even when systemic hemodynamic alterations seem to be reversed with fluid resuscitation and vasoactive agents, significant alterations in the microcirculation may persist and participate in the development of multiple organ failure. Therefore in septic shock, relationships between macrovascular hemodynamic and microcirculatory changes during resuscitation are complex (global hemodynamics do not necessarily reflect regional microcirculation) with a critical disorder of the microcirculation. Nakajima et al. [30] compared microvascular perfusion in intestinal villi in mouse models of septic shock and hemorrhagic shock. These authors demonstrated that, after one hour at the same level of hypotension (mean arterial pressure [MAP] ≈ 40 mm Hg), mucosal perfusion disorders were considerably more pronounced in endotoxin-induced hypotension than in hemorrhagic hypotension. RBC velocity was maintained in hemorrhagic shock but not during endotoxic shock. During the initial phase of hemorrhagic shock, the microvasculature was still able to regulate microvascular perfusion, but during sepsis the regulatory response was impaired. Similar results were achieved by Fang et al. [3], who found that impaired buccal capillary blood flow in septic animals (cecal ligation and perforation) was more severe than that in hemorrhagic animals with the same level of hypotension. In addition, major findings reported by these authors were that the impaired buccal capillary blood flow was similar in both types of shock for the same reduction in cardiac index and that significantly improved global hemodynamics after fluid resuscitation did not effectively improve the buccal capillary blood flow in septic shock, in contrast to the hemorrhagic shock condition during which buccal capillary blood flow was significantly improved. Therefore, microvascular alterations are closely related to macrocirculatory variables (especially cardiac index and DO2) during hemorrhage and resuscitation [3, 31]. Hence, fluid resuscitation, associated with blood transfusion, is the major therapy to improve the microcirculation in hemorrhagic shock. In this respect, Legrand et al. [26] reported that correcting arterial blood pressure and cardiac index with only fluid resuscitation was not a guarantee for providing sufficient oxygen and correct shock-induced microcirculatory hypoxia. Only transfusion of blood associated with fluid resuscitation was able to improve tissue oxygenation [26]. Blood transfusion has to be considered early during the management of hemorrhagic shock to improve microvascular DO2 and match oxygen supply to oxygen demand.
Even if a considerable part of the microvascular alterations during hemorrhagic shock are closely related to a decrease in DO2, other factors could be involved in addition to hemodynamic factors; these include changes in the microvascular endothelium, leukocyte adherence to venules, interstitial and endothelial edema, RBC alterations, and coagulation activation (Fig. 2). During the resuscitation phase of hemorrhagic shock, neutrophil rolling and adherence to postcapillary venules is enhanced with leukocyte plugging and resulting increase in vascular resistances [1]. In addition, generation of reactive oxygen species (ROS) precedes leukocyte adherence following hemorrhagic shock and promotes endothelial dysfunction with an increase in microvascular permeability and tissue edema, which may lead to alterations in oxygen diffusion [32]. Machiedo et al. [24] reported that transfusion of RBCs from trauma/hemorrhagic shock rats into naïve rats led to impaired microcirculatory flow to several important organs, including the lungs, spleen, ileum, and cecum, as well as deleterious systemic hemodynamic effects with reduced cardiac output. The role of these factors tends to increase with the degree of inflammation. Presumably, when the inflammatory response is marked (e. g., severe or sustained hemorrhagic shock, traumatic hemorrhagic shock or associated hypoxemia), sepsis-like microvascular alterations with persistent alterations may be observed despite adequate macrovascular resuscitation and associated with organ dysfunction. This possibility is supported by studies documenting that microcirculatory alterations partly persist after resuscitation despite correction of macrovascular parameters. For example, persistent decreased microcirculatory PO2 values have been reported despite an adequate macrovascular resuscitation [25, 26].
It should be stressed that observations about alterations of the microcirculation in hemorrhagic shock are mainly derived from experimental models during the acute phase of resuscitation. Thus, assessing microvascular alterations in hemorrhagic shock patients seems urgent.
Implications for Resuscitation of Patients with Hemorrhagic Shock
Objectives of Fluid Resuscitation
In hemorrhagic shock, the therapeutic priority is to stop the bleeding as quickly as possible. A critical element in the resuscitation of patients with hemorrhagic shock is to prevent a potential increase of bleeding by being too aggressive. Indeed, fluid resuscitation may promote coagulopathy by diluting coagulation factors and favoring hypothermia. Moreover, an excessive arterial pressure can favor bleeding by preventing clot formation. Two concepts have emerged in recent years: The concept of ‘low volume resuscitation’ and that of ‘hypotensive resuscitation’. Often these two concepts are merged. Indeed, the fluid resuscitation strategy and the blood pressure target are two associated elements during hemorrhagic shock resuscitation. Several experimental studies have suggested that limited administration of fluids with a low blood-pressure level as an endpoint may decrease bleeding without the associated increased risk of death, if lasting for a short period of time [33, 34]. Recently, Li et al. reported that a target resuscitation pressure of 50–60 mm Hg was the ideal blood pressure for uncontrolled hemorrhagic shock in rats [35]. Ninety minutes of permissive hypotension is the tolerance limit and 120 min of hypotensive resuscitation can cause a significant alteration in mitochondrial function and severe organ damage and should be avoided [35]. Therefore, the initial objective is to control bleeding as soon as possible and to maintain a minimal arterial pressure to limit tissue hypoxia, inflammation and organ dysfunction. European guidelines for the management of the bleeding trauma patient recommend a target systolic blood pressure of 80 to 100 mm Hg in the acute phase of hemorrhagic shock until major bleeding has been stopped [36]. However, the optimal level of blood pressure during resuscitation of patients with hemorrhagic shock is still debated and we need more experimental and clinical studies to evaluate the consequences of hypotensive resuscitation on the microcirculation and tissue oxygenation. It appears crucial to develop bedside devices to assess microcirculatory perfusion or tissue oxygenation to better titrate the hemodynamic resuscitation strategy during hemorrhagic shock.
It is important to remember that restoration of the microcirculation implies not only restoration of blood volume to enhance organ perfusion, but also restoration of the functional capillary density. In this respect, it is crucial to test the influence of the currently used therapies (i. e., fluids, vasopressors and transfusion) on functional capillary density.
Fluid Resuscitation and Vasoactive Agents
Hypertonic saline (HTS) has been proposed as an interesting tool to improve the microcirculation in trauma hemorrhagic shock. It has been reported in experimental studies that resuscitation with HTS improves intestinal perfusion associated with selective arteriolar vasodilation of distal premucosal arterioles (A3), decreases interstitial and endothelial edema and prevents leukocyte adhesion to postcapillary venules and hemorrhagic shock-induced inflammation [1, 37]. However, despite these microvascular beneficial effects, fluid resuscitation with HTS failed to improve outcomes in trauma patients with hemorrhagic shock in recent studies [38, 39]. Moreover, there was a higher mortality rate in patients receiving HTS who did not receive any blood transfusion in the first 24 hours. To explain this effect, the authors evoked the possibility that out-of-hospital administration of HTS could mask the signs of hypovolemia and delay the diagnosis of hemorrhagic shock.
In the context of restoring functional capillary density, a new approach to fluid resuscitation is based on fluid with high viscosity in order to increase plasma viscosity and wall shear stress with NO production causing microcirculatory vasodilation with resulting capillary recruitment. Cabrales et al. suggested that hemorrhagic shock resuscitation (hamster window model) with polyethylene glycol (PEG)-conjugated bovine serum albumin provides early and long-term sustained systemic and microvascular recovery compared to hydroxyethyl starch (HES) [40]. Recently in the same model, Villela et al. reported that increasing blood and plasma viscosities during hemorrhage resuscitation with increased viscosity Ringer’s lactate (addition of 0.3 % alginate) significantly improved arteriolar diameter and venular flow and maintained functional capillary density [41]. It is obvious that this concept has to be confirmed but thinking about the rheological properties of fluids could generate new ways of improving the microcirculation.
Vasopressor agents may be transiently required in hemorrhagic shock to maintain tissue perfusion in the presence of life-threatening hypotension, even when fluid expansion is in progress and hypovolemia has not yet been corrected. The microvascular effects of vasopressors in hemorrhagic shock are still under debate. More works will be required to establish the net microvascular effect of vasopressors during resuscitation of hemorrhagic shock patients. It is conceivable that correction of hypotension by vasopressors may improve microvascular perfusion by increasing the driving pressure of capillary beds [42, 43].
Transfusion
As previously mentioned, early administration of RBCs is a priority to maintain arterial DO2 and to restore effective microcirculation and tissue oxygenation [25, 26]. An increasing body of data suggests that RBC plays a crucial role as an oxygen sensor in the regulation of microvascular tone and in the matching of oxygen supply to oxygen demand. Therefore, blood transfusion may improve microvascular DO2 not only as an oxygen carrier but also as an oxygen sensor that improves functional capillary density by interfering with local microvascular control [44, 45]. This finding is important because a better understanding of the role of the RBC could change transfusion strategy and influence recommended optimal hemoglobin levels in hemorrhagic shock; moreover, it may be relevant to assess the impact of the quality of transfused RBCs on the microcirculation [46].
Conclusion
In hemorrhagic shock, a considerable part of the microvascular alterations are closely related to macrocirculatory variables (especially DO2). Other factors could be involved in addition to macrohemodynamic factors. These include changes in the microvascular endothelium, leukocyte adherence to postcapillary venules, interstitial and endothelial edema, RBC deformability alterations, and coagulation activation. Restoration of the microcirculation after hemorrhagic shock has to focus on improvement in the functional capillary density within the organ. In this respect, it is crucial to test the influence of currently used therapies in hemorrhagic shock (i. e., fluids, vasopressors and transfusion) on functional capillary density. It is important to develop bedside devices to assess microcirculatory perfusion and tissue oxygenation to better titrate the hemodynamic resuscitation strategy during hemorrhagic shock. Understanding the impact of currently used therapies in hemorrhagic shock is the first step toward developing interventions that target microcirculatory perfusion.
References
Pascual JL, Ferri LE, Seely AJ et al (2002) Hypertonic saline resuscitation of hemorrhagic shock diminishes neutrophil rolling and adherence to endothelium and reduces in vivo vascular leakage. Ann Surg 236:634–642
Sordia T, Tatarishvili J, Varazashvili M, McHedlishvili G (2004) Hemorheological disorders in the microcirculation following hemorrhage. Clin Hemorheol Microcirc 30:461–462
Fang X, Tang W, Sun S, Huang L, Chang YT, Castillo C, Weil MH (2006) Comparison of buccal microcirculation between septic and hemorrhagic shock. Crit Care Med 34(Suppl):S447–S453
Dubin A, Pozo MO, Ferrara G et al (2009) Systemic and microcirculatory responses to progressive hemorrhage. Intensive Care Med 35:556–564
Imai Y, Satoh K, Taira N (1978) Role of the peripheral vasculature in changes in venous return caused by isoproterenol, norepinephrine, and methoxamine in anesthetized dogs. Circ Res 43:553–561
Gelman S, Mushlin PS (2004) Catecholamine-induced changes in the splanchnic circulation affecting systemic hemodynamics. Anesthesiology 100:434–439
Schumacker PT (1998) Oxygen supply dependency in critical illness: an evolving understanding. Intensive Care Med 24:97–99
Dahl KN, Kalinowski A, Pekkan K (2010) Mechanobiology and the microcirculation: cellular, nuclear and fluid mechanics. Microcirculation 17:179–191
Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH, Sprague RS (2009) Erythrocytes: Oxygen sensors and modulators of vascular tone. Physiology 24:107–116
Sprague RS, Bowles EA, Achilleus D, Ellsworth ML (2010) Erythrocytes as controllers of perfusion distribution in the microvasculature of skeletal muscle. Acta Physiologica 202:285–292
Jia L, Bonaventura C, Bonaventura J, Stamler JS (1996) S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 380:221–226
Stamler JS, Jia L, Eu JP et al (1997) Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276:2034–2037
Cosby K, Partovi KS, Crawford JH et al (2003) Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 9:1498–1505
Gladwin MT, Crawford JH, Patel RP (2004) The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radic Biol Med 36:707–717
Kindig CA, Richardson TE, Poole DC (2002) Skeletal muscle capillary hemodynamics from rest to contractions: implications for oxygen transfer. J Appl Physiol 92:2513–2520
Poole DC, Copp SW, Hirai DM, Musch TI (2011) Dynamics of muscle microcirculatory and blood-myocyte O2 flux during contractions. Acta Physiologica 202:293–310
Bateman RM, Sharpe MD, Ellis CG (2003) Bench-to-bedside review: Microvascular dysfunction in sepsis – hemodynamics, oxygen transport, and nitric oxide. Crit Care 7:359
Ellis CG, Jagger J, Sharpe M (2005) The microcirculation as a functional system. Crit Care 9(Suppl 4):S3
Groom AC, Ellis CG, Wrigley SJ, Potter RF (1995) Capillary network morphology and capillary flow. Int J Microcirc Clin Exp 15:223–230
Funk W, Baldinger V (1995) Microcirculatory perfusion during volume therapy. A comparative study using crystalloid or colloid in awake animals. Anesthesiology 82:975–982
Hoffmann JN, Vollmar B, Laschke MW, Inthorn D, Schildberg FW, Menger MD (2002) Hydroxyethyl starch (130 kD), but not crystalloid volume support, improves microcirculation during normotensive endotoxemia. Anesthesiology 97:460–470
Vajda K, Szabo A, Boros M (2004) Heterogeneous microcirculation in the rat small intestine during hemorrhagic shock: Quantification of the effects of hypertonic-hyperoncotic resuscitation. Eur Surg Res 36:338–344
Zaets SB, Berezina TL, Morgan C et al (2003) Effect of trauma-hemorrhagic shock on red blood cell deformability and shape. Shock 19:268–273
Machiedo GW, Zaets SB, Berezina TL et al (2009) Trauma-hemorrhagic shock-induced red blood cell damage leads to decreased microcirculatory blood flow. Crit Care Med 37:1000–1010
Ince C, Sinaasappel M (1999) Microcirculatory oxygenation and shunting in sepsis and shock. Crit Care Med 27:1369–1377
Legrand M, Mik EG, Balestra GM et al (2010) Fluid resuscitation does not improve renal oxygenation during hemorrhagic shock in rats. Anesthesiology 112:119–127
De Backer D, Creteur J, Preiser JC, Dubois MJ, Vincent JL (2002) Microvascular blood flow is altered in patients with sepsis. Am J Respir Crit Care Med 166:98–104
Trzeciak S, Dellinger RP, Parrillo JE et al (2007) Early microcirculatory perfusion derangements in patients with severe sepsis and septic shock: relationship to hemodynamics, oxygen transport, and survival. Ann Emerg Med 49:88–98
Sakr Y, Dubois MJ, De Backer D, Creteur J, Vincent JL (2004) Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit Care Med 32:1825–1831
Nakajima Y, Baudry N, Duranteau J, Vicaut E (2001) Microcirculation in intestinal villi: a comparison between hemorrhagic and endotoxin shock. Am J Respir Crit Care Med 164:1526–1530
van Iterson M, Bezemer R, Heger M, Siegemund M, Ince C (2012) Microcirculation follows macrocirculation in heart and gut in the acute phase of hemorrhagic shock and isovolemic autologous whole blood resuscitation in pigs. Transfusion 52:1552–1559
Childs EW, Udobi KF, Wood JG, Hunter FA, Smalley DM, Cheung LY (2002) In vivo visualization of reactive oxidants and leukocyte-endothelial adherence following hemorrhagic shock. Shock 18:423–427
Capone AC, Safar P, Stezoski W, Tisherman S, Peitzman AB (1995) Improved outcome with fluid restriction in treatment of uncontrolled hemorrhagic shock. J Am Coll Surg 180:49–56
Kowalenko T, Stern S, Dronen S, Wang X (1992) Improved outcome with hypotensive resuscitation of uncontrolled hemorrhagic shock in a swine model. J Trauma 33:349–353
Li T, Zhu Y, Hu Y et al (2011) Ideal permissive hypotension to resuscitate uncontrolled hemorrhagic shock and the tolerance time in rats. Anesthesiology 114:111–119
Rossaint R, Bouillon B, Cerny V et al (2010) Management of bleeding following major trauma: an updated European guideline. Crit Care 14:R52
el Zakaria R, Tsakadze NL, Garrison RN (2006) Hypertonic saline resuscitation improves intestinal microcirculation in a rat model of hemorrhagic shock. Surgery 140:579–587
Bulger EM, Jurkovich GJ, Nathens AB et al (2008) Hypertonic resuscitation of hypovolemic shock after blunt trauma: a randomized controlled trial. Arch Surg 143:139–148
Bulger EM, May S, Kerby JD et al (2011) Out-of-hospital hypertonic resuscitation after traumatic hypovolemic shock. Ann Surg 253:431–441
Cabrales P, Intaglietta M, Tsai AG (2005) Increased plasma viscosity sustains microcirculation after resuscitation from hemorrhagic shock and continuous bleeding. Shock 23:549–555
Villela NR, Tsai AG, Cabrales P, Intaglietta M (2011) Improved resuscitation from hemorrhagic shock with Ringer’s lactate with increased viscosity in the hamster window chamber model. J Trauma 71:418–424
Deruddre S, Cheisson G, Mazoit JX, Vicaut E, Benhamou D, Duranteau J (2007) Renal arterial resistance in septic shock: effects of increasing mean arterial pressure with norepinephrine on the renal resistive index assessed with Doppler ultrasonography. Intensive Care Med 33:1557–1562
Georger JF, Hamzaoui O, Chaari A, Maizel J, Richard C, Teboul JL (2010) Restoring arterial pressure with norepinephrine improves muscle tissue oxygenation assessed by near-infrared spectroscopy in severely hypotensive septic patients. Intensive Care Med 36:1882–1889
Sakr Y, Chierego M, Piagnerelli M et al (2007) Microvascular response to red blood cell transfusion in patients with severe sepsis. Crit Care Med 35:1639–1644
Yuruk K, Almac E, Bezemer R, Goedhart P, de Mol B, Ince C (2011) Blood transfusions recruit the microcirculation during cardiac surgery. Transfusion 51:961–967
Raat NJH, Ince C (2007) Oxygenating the microcirculation: the perspective from blood transfusion and blood storage. Vox Sang 93:12–18
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Harrois, A., Tanaka, S., Duranteau, J. (2013). The Microcirculation in Hemorrhagic Shock. In: Vincent, JL. (eds) Annual Update in Intensive Care and Emergency Medicine 2013. Annual Update in Intensive Care and Emergency Medicine. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-35109-9_22
Download citation
DOI: https://doi.org/10.1007/978-3-642-35109-9_22
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-35108-2
Online ISBN: 978-3-642-35109-9
eBook Packages: MedicineMedicine (R0)