
Abstract
Chronic inflammation resulting from infections, altered metabolism, inflammatory diseases or other environmental factors can be a major contributor to the development of several types of cancer. In fact around 20% of all cancers are linked to some form of inflammation. Evidence gathered from genetic, epidemiological and molecular pathological studies suggest that inflammation plays a crucial role at various stages of prostatic carcinogenesis and tumor progression. These include initiation, promotion, malignant conversion, invasion, and metastasis. Detailed basic and clinical research in these areas, focused towards understanding the etiology of prostatic inflammation, as well as the exact roles that various signaling pathways play in promoting tumor growth, is critical for understanding this complex process. The information gained would be useful in developing novel therapeutic strategies such as molecular targeting of inflammatory mediators and immunotherapy-based approaches.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Prostate cancer
- Inflammation
- Oxidative stress
- Innate immune system
- Adaptive immune system
- Macrophages
- Lymphocytes
- Cytokines
- Chemokines
Abbreviations
- AML:
-
Acute myeloid leukemia
- ARE:
-
Antioxidant response element
- Bcl:
-
B-cell lymphoma
- BMI1:
-
B lymphoma Mo-MLV insertion region 1 homolog
- BMP7:
-
Bone morphogenetic protein 7
- BPH:
-
Benign prostatic hyperplasia
- BRCA1:
-
Breast cancer 1
- CCL2:
-
C-C motif chemokine ligand 2
- CD:
-
Cluster of differentiation
- CDKN1B:
-
Cyclin dependent kinase inhibitor 1B
- CMV:
-
Cytomegalovirus
- COX2:
-
Cyclooxygenase-2
- CSC:
-
Cancer stem cells
- CSF-1R:
-
Colony stimulating factor 1 receptor
- CXCL12:
-
C-X-C motif chemokine ligand 12
- DAMP:
-
Danger-associated molecular patterns
- DHT:
-
Dihydrotestosterone
- DNA:
-
Deoxyribonucleic acid
- DNMT1:
-
DNA methyltransferase 1
- DPI :
-
Diphenyleneiodonium
- EBV:
-
Epstein-Barr virus
- EMT:
-
Epithelial to mesenchymal transformation
- EZH2:
-
Enhancer of zeste homology 2
- GATA1:
-
GATA sequence binding factor 1
- GST:
-
Glutathione-S-transferase
- GSTP1:
-
Glutathione-S-transferase Pi 1
- HCA:
-
Heterocyclic amines
- HCC:
-
Hepatocellular carcinoma
- Hh:
-
Hedgehog
- HMG-CoA:
-
3-hydroxy-3-methylglutaryl-coenzyme A
- HNE:
-
4-hydroxy-2-nonenal
- HPV:
-
Human papillomavirus
- HSV2:
-
Herpes simplex virus 2
- IFN:
-
Interferon
- IL:
-
Interleukin
- JAK:
-
Janus kinase
- LNCaP:
-
Prostate adenocarcinoma cell line
- MAPK:
-
Mitogen activated protein kinase
- mCRPC:
-
Metastatic castration-resistant prostate cancer
- M-CSF:
-
Macrophage colony stimulating factor
- MDSC:
-
Myeloid-derived suppressor cells
- MIC1:
-
Macrophage inhibitory cytokine 1
- miR:
-
MicroRNA
- MLH1:
-
MutL homolog 1
- MMPs:
-
Matrix metalloproteinases
- MSR1:
-
Macrophage scavenger receptor 1
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NF-κB:
-
Nuclear factor kappa B subunit
- NKX3.1:
-
NK3 transcription factor related, locus 1
- NO:
-
Nitric oxide
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- NSAID:
-
Nonsteroidal anti-inflammatory drug
- PAMP:
-
Pathogen-associated molecular patterns
- PC3:
-
Prostate CRPC cell line
- PCa:
-
Prostate cancer
- PI3K:
-
Phosphoinositide 3-kinase
- PIA:
-
Proliferative inflammatory atrophy
- PIN:
-
Prostatic intraepithelial neoplasia
- PRR:
-
Pattern recognition receptors
- PSA:
-
Prostate specific antigen
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxyen species
- RUNX3:
-
Runt related transcription factor 3
- SMAD:
-
Small body size mothers against decapentaplegic
- STAT3:
-
Signal transducer and activator of transcription 3
- TAMs:
-
Tumor associated macrophages
- TCR:
-
T-cell receptors
- TGF:
-
Transforming growth factor
- TLR:
-
Toll like receptors
- TNF:
-
Tumor necrosis factor
- TRAMP:
-
Transgenic adenocarcinoma mouse prostate
- US:
-
United States
- VEGF:
-
Vascular endothelial growth factor
3.1 Introduction
Inflammation is a physiological process that is initiated upon exposure to various infections or tissue injury. The inflammatory processes leads to a cascade of chemical events targeted towards eradication of pathogens, clearing tissue and cellular debris, regeneration of the epithelium and remodeling of the stroma. However, if this highly regulated process remains unchecked, or normal healthy tissue integrity is not restored and the inflammatory response persists, it results in significant cellular and genomic damage. The sustained inflammation generates a multitude of various reactive nitrogen and oxygen species, cytokines, chemokines, and growth factors. The persistent high level of these factors potentially leads to uncontrolled cellular proliferation and enhanced genomic instability. Genomic instability (e.g. activation of oncogenes and/or loss of tumor suppressors) coupled with unchecked cell proliferation due to presence of growth factors increases the risk of developing several types of malignancies, including prostate cancer (PCa) [1,2,3].
PCa is a leading public health concern that places a significant burden on health-care systems worldwide [4]. PCa risk factors include family history, old age and ethnicity. Nearly 3 million men in the US currently live with the disease and approximately 14% of men will be diagnosed with PCa in their lifetime. This year alone in the US over 26,000 patients will die from PCa [4]. In Europe, there are approximately 346,000 new PCa cases and 87,000 deaths per year [5].
The longstanding observation and epidemiological link between inflammation and cancer has been recognized since the dawn of modern medicine when the German physician Rudolf Virchow in 1863 first described leucocyte infiltration and their distribution in neoplastic tissues. He further proposed that these “lymphoreticular infiltrates” in the tumor perhaps were remnants of chronic inflammation sites [6].
Given that less than 10% of all cancers are caused by germline mutations, there is great emphasis placed on understanding the underlying mechanisms that cause the vast majorities of human tumors; that is, those that initiate from acquired somatic mutations and detrimental environmental factors [7]. There exists a strong association between chronic inflammatory diseases such as gastritis, hepatitis, prostatitis or colitis and the increased risk of developing carcinomas in the afflicted organ. In fact, out of the nearly 600,000 thousand people who will die this year from hepatocellular carcinoma (HCC), 90% of the cases present with some form of hepatic injury and inflammation [8]. For men diagnosed with prostatitis around 18% of them will develop prostate cancer. Moreover, men who show signs of chronic inflammation in non-cancerous prostate tissue have nearly twice the risk of actually developing prostate cancer later on than those without inflammation [9]. This association is even stronger for those who ultimately develop high-grade disease (Gleason score ≥ 7) [10]. Overall approximately 20% of diagnosed adult cancers have been attributed to chronic inflammatory diseases [11].
3.1.1 Prostatic Inflammation and Cancer
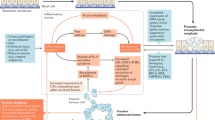
A recent resurgence of interest into the tumor-promoting effects of the inflammatory microenvironment has been led by the abundance of clinical, molecular, histo-pathological and epidemiological-based evidence connecting prostate cancer and inflammation concurrence [12]. PCa development is mediated in part by hereditary components, but particularly in regards to inflammation-induced disease, also by environmental exposures such as infectious agents and dietary carcinogens. This is evidenced by the apparent increase in prostate cancer risk among men from geographic areas with low prostate cancer incidence (Southeast and East Asia) who immigrate to western countries [13]. While the exact sources of prostatic inflammation are still being investigated the environmental exposures that induce or increase the risk include (see also Fig. 3.1):
-
1)
Infectious microorganisms such as E. coli, Propionibacterium acnes and others associated with the intraprostatic reflux of urine and sexually transmitted diseases (Neisseria gonorrhoeae) and prostatitis [14, 15]. Viruses such as Human papillomavirus (HPV), herpes simplex virus 2 (HSV2) and cytomegalovirus (CMV) can also infect the prostate [16, 17]. However, their frequency of infection and role in inflammation-induced carcinoma is largely unknown.
-
2)
Noxious dietary elements and lifestyle-risk factors, including exposure to heterocyclic amines (HCAs) produced from cooking meat at high temperatures, estrogen, and obesity.
-
3)
Treatment for reproductive ailments or tumor-induced inflammation [18].
-
4)
Urine reflux causing chemical irritation with metabolites such as uric acid can lead to chronic inflammation within the prostate [19]. It can synergize with infection to further aggravate chronic inflammation.
-
5)
Sperm seen in prostate tissue have been associated with PIA and inflammation. However, frequent ejaculation could potentially flush out urinary carcinogens and has been linked with decrease in prostate cancer incidence [20].
-
6)
Hereditary inflammation-related genes such as macrophage inhibitory cytokine-1 (MIC1), Toll like receptors (TLRs), and interleukin receptor −1 antagonist (IL-1RN).

Various intrinsic and extrinsic factors can increase the risk of prostatic inflammation. See text for details
Inflammation Based Therapeutics
Interestingly, studies have also found significant correlation between intake of anti-inflammatory compounds and reduced prostate cancer risk. In one study the anti-inflammatory phytoestrogens, genistein and daidzein, found in soy and green tea were associated with reduced risk of PCa [12], perhaps through modification of glutathione S-transferase P (GTSP1) and ephrin B2 (EphB2) promoter regions [12]. Furthermore, the use of aspirin and non-steroidal anti-inflammatory drugs (NSAIDs) has been associated with reduced risk of PCa, possibly through inhibition of the cyclooxygenase 2 (COX-2) enzyme [12, 21]. COX-2 is an inflammation related protein that facilitates the production of prostaglandins, which in turn promotes neoangiogenesis and cell migration, and reduces apoptosis [22]. Finally, the use of statins (prescribed primarily for lowering cholesterol) has been correlated with reduced risk of advanced and aggressive PCa by inhibiting 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) [23, 24] (Fig. 3.1).
3.1.2 Proliferative Inflammatory Atrophy (PIA)
Proliferative inflammatory atrophy (PIA) are lesions within the prostate that are atrophic and are associated with increased acute or chronic inflammatory cell infiltration [25]. Some atrophic lesions show increased number of epithelial cells but are devoid of inflammatory cells. Morphological studies have shown association between the presence of these lesions and prostatic intraepithelial neoplasia (PIN) and carcinoma [25, 26]. Remarkably, these lesions are mostly found in the peripheral zone of the prostate where PCa most commonly occurs [27]. In fact, no cancerous lesions have been reported in the central zone of the prostate [28]. Although some evidence of molecular changes in PIA has been observed (such as GSTP1 hypermethylation), no studies have shown clonal genetic alterations in PIA [29]. Other genes such as NKX3.1 and CDKN1B, which are downregulated or lost in PIN and PCa, have also been shown to be downregulated in PIA [25, 30]. Indeed, targeted disruption of these genes in mouse models results in the development of PIN or invasive PCa [31].
3.1.3 Role of Innate and Adaptive Immunity
Inflammation is a process that involves the interplay between innate and adaptive immune responses following infection or injury and have a powerful influence on the development of the tumor microenvironment by producing a wide variety of pro-inflammatory oxidizing species, cytokines and chemokines, which results in carcinogenesis by promoting growth, angiogenesis, differentiation, survival and migration of tumor cells [32].
3.1.3.1 Immune Cells: Do they Help or Harm?
Although immune cells are important mediators of PCa progression, the incomplete phenotypic characterization of these cellular infiltrates combined with conflicting clinical evidence represents a gap of knowledge critical to understanding the intricate roles immune cells play in the tumor microenvironment [18]. One recent study analyzing lymphocyte infiltration in tumor tissues observed that both high and low levels of CD3+ cells (T-cell co-receptors) were correlated with reduced PSA recurrence-free survival [33]. Inclusion of other T-cell subtype markers (such as CD4+, CD8+, etc.) would have perhaps helped in parsing this discrepancy [18]. In normal prostates and prostates with benign prostatic hyperplasia (BPH), inflammatory cells have been shown to be comprised of CD3+ T cells (~70–80%) and B cells CD20+ (~15–20%), along with a high number of macrophages. Whereas normal prostates contain a greater fraction of CD8+ T-cells, inflammatory lesions are enriched in the CD4+ T cells subtype. Moreover, most of the observed T cells were alpha beta while less than 1% were gamma delta TCR positive T-cells. Additionally, 40% of the T cells present in inflammatory lesions were memory T-cells [34,35,36]. Regulatory T-cells (Tregs) are potent suppressors of anti-tumor adaptive immune responses, and higher amounts of CD4+, CD25+, and Foxp3+ Treg cells are found in the tumors and blood of prostate cancer patients with clinically localized disease [37]. With this in mind, a thorough analysis of various T-cell subsets in the context of various grades and stages of prostate cancer would shed immense new light on understanding the effects of immune cell presence in the tumor microenvironment.
Tumor-associated macrophages (TAMs) are another class of cells that are strongly associated with disease progression [38], yet only one study was able to determine the M2 macrophage subtype responsible for this phenomenon [39], which may explain the fact that despite the several studies documenting the pro-tumorigenic properties of TAMs only a handful of studies were able to directly correlate TAM infiltration with disease recurrence [33].
Future work aimed at completely characterizing the inflammatory cells within the tumor microenvironment will be essential in developing novel immunotherapies and identifying immune-based prognostic indicators.
3.1.3.2 Innate Immunity and Prostate Cancer
The innate immune system is the rapid-acting ‘first line of defense’ against pathogens and is comprised of several types of cells including granulocytes (i.e. neutrophils, basophils and eosinophils), dendritic cells, natural killer cells, macrophages, and mast cells. Mast cells and macrophages are probably the two most extensively studied innate immune cells in prostate cancer.
Mast cells are able to produce both pro- and anti-tumorigenic cytokines and as such are dynamic and effective regulators of the interactions within the tumor microenvironment. Their function is dependent on their environment, and probably varies in different types and stages of cancer [38]. For example some studies of PCa patients have noted high densities of intra-tumor infiltrated mast cells associating with overall lower Gleason-grade tissue and better prognosis [40, 41], yet in a separate study of PCa patients, low counts of mast cells were linked to lower Gleason scores and longer progression-free survival times [42]. These conflicting observations are better understood when considering that mast cells are capable of producing an immense number of distinct regulatory cytokines and effector molecules, including serotonin, heparin and proteases [43]. More evidence however is needed to support their roles in prostate carcinogenesis.
Macrophages represent another major class of immune cells that have been studied for their use in evaluating disease prognosis in PCa. Significantly greater levels of macrophage colony-stimulating factor (M-CSF) and colony-stimulating factor-1 receptor (CSF-1R) have been observed in tumor and stromal cells near primary tumors of patients with metastatic disease compared to those without metastases [44]. Macrophages may also promote prostate cancer invasion by secreting proteases (notably cathepsin K and cathepsin S) that breakdown the extracellular matrix. In a study aimed at identifying proteins associated with tumor progression, cathepsin S was found to be upregulated in poorly-differentiated and metastatic tumors taken from TRAMP (transgenic adenocarcinoma mouse prostate) mice models of PCa as well as in high Gleason grade tumors from patients [45]. Another recent study demonstrated that tumor growth in bones was impaired in cathepsin K-deficient mice injected intra-tibially with PC3 cells (a PCa cell line model of castration-resistant disease) [46].
3.1.3.3 Adaptive Immunity and Prostate Cancer
Similarly to cells of the innate immune system, T and B lymphocytes of the adaptive immune system are known to have paradoxical roles in carcinogenesis, especially in chronically inflamed tumors and lesions that typically are associated with prolonged interactions with adaptive immune cells [38]. T-lymphocytes in particular have been frequently examined for their use as prognostic markers and in general have been associated with good prognosis as they may act to illicit an anti-tumor response [38]. The study of T-cells is hindered as they are normally differentiated by their cytokine secretions, which is difficult to analyze by immunohistochemistry, and more advanced approaches using flow cytometry to separate immune cells from prostate cells can be difficult to accomplish [38]. However, using flow cytometry for the phenotypic analysis of T-cells using serial needle aspirates of peripheral prostate tissue is certainly possible [47].
Although they are present in the tumor microenvironment [33], much less is known about the role of B-lymphocytes in prostate cancer. They have been reported to promote the progression of castration-resistant cancer cells by activating STAT3 and the proto-oncogene BMI1 [48].
3.2 Mechanisms of Inflammation Induced Carcinogenesis
Inflammation Genes in Prostate Cancer
Several hereditary prostate cancer risk studies have revealed the potential involvement of inflammation-related genes as risk factors for hereditary prostate cancer. Linkage studies in families with prostate cancer have identified an E265X mutation in the innate immune response gene RNAse L (which is located on chromosome 1q and is involved in interferon (IFN) signaling) as a PCa-susceptibility gene in a family of European descent, and a M1I mutation in the same region in a family of African descent [49]. Another gene identified in families with PCa and associated with increased susceptibility is located on chromosome 8p and encodes a macrophage specific scavenger receptor (MSR1) [50]. Other inflammatory genes identified in Swedish case-control cohort studies which are associated with risk of developing PCa include MIC1, Toll like receptors (TLRs) and interleukin receptor −1 antagonist (IL-1RN) [51,52,53].
The inflammatory state can initiate or promote neoplastic progression if it is able to transform cells in the local environment into the full malignant phenotype. Phenotypic hallmarks include tissue remodeling, angiogenesis and metastasis. A tumor microenvironment rich in sustained proinflammatory cytokines and inflammatory cells can induce or promote neoplastic and malignant progression in several ways:
-
1)
Generating reactive oxygen and nitrogen species that inflict cellular, epigenetic, and genetic alterations and damage
-
2)
Sustaining the inflammatory tumor-microenvironment and recruiting additional leukocytes that promote angiogenesis, proliferation, vascular and tissue growth and remodeling
-
3)
Elaborating the cytokine and chemokine network that promotes cell replication, differentiation and inhibition of apoptosis
-
4)
Each mechanism has unique and significant contributions to carcinogenesis, and has been described in detail in the sections below.
3.2.1 Reactive Oxygen and Nitrogen Species (ROS and RNS) Generation
In response to infections, inflammatory cells (usually neutrophils and macrophages) synthesize a variety of toxic compounds designed to eradicate microorganisms. These compounds are reactive oxygen and reactive nitrogen species (ROS and RNS, respectively) and include hydrogen peroxide (H2O2), the hydroxyl radical (OH•), nitric oxide (NO), organic peroxides, singlet oxygen and the superoxide anion (O2•-) [54, 55]. Under normal metabolic conditions the majority of the free radicals, ROS and RNS produced are byproducts of aerobic cellular respiration, generated in the intracellular milieu by mitochondria. However in response to pathogens, neutrophils and macrophages produce these compounds via extracellular membrane-bound enzyme complexes known as NADPH oxidases (i.e. NOX enzymes) and release ROS rapidly into the tumor microenvironment in an ‘oxidative burst’ [54]. During chronic inflammation there exists an imbalance between the amount of oxidizing agents produced and the host’s ability to process them. The enzymes (e.g. superoxide dismutase, catalase, peroxiredoxin, thioredoxin glutathione reductase and glutathione S-transferase) and antioxidant molecules (e.g. glutathione, flavonoids and vitamins A, C and E) responsible for detoxifying the environment become overwhelmed, leading to a state of continued oxidative stress [56]. The buildup of these highly reactive compounds leads to significant mutagenic and genome-destabilizing DNA lesions; in fact there are over 100 oxidized DNA products currently known [57]. Some of these damages can either arrest or promote transcription, bring about point mutations, induce replication errors, and inhibit DNA repair [58]. One type of point mutation (a G to T transversion) has been observed in both Ras [59] and p53 genes [60] in multiple cancers, indicating that ROS and RNS may directly activate or inactivate proto-oncogenes and tumor suppressor genes, respectively.
Similar to mutations, epigenetic alterations can also contribute to carcinogenesis. The presence of reactive oxidizing species in the inflamed microenvironment has also been associated with epigenetic damage through aberrant DNA methylation and histone modifications [61]. Generally hypermethylation in the promoter regions of genes blocks transcription thereby regulating genetic expression within a cell. Exactly how ROS and RNS lead to increased DNA methylation is still unclear, however one proposed mechanism suggests that 5-halogenated cytosines formed from ROS can prevent DNA methyltransferases (i.e. DNMT1) from distinguishing methylated from halogenated cytosines leading to altered methylation [62]. Hypermethylation of putative tumor suppressor genes, such as RUNX3 in esophageal cancer [63] and GATA-4 and GATA-5 in colorectal and gastric cancers [64] leads to complete deactivation of their downstream targets. Moreover, DNA repair genes such as MLH1 and BRCA1 are also targets of hypermethylation, whereby their silencing results in accumulation of further genetic damage leading to the development of the malignant phenotype [65].
Lipids and proteins are also highly susceptible to damage from free radicals. Peroxidation of lipids generates lipid radicals and aldehydes, such as 4-hydroxy-2-nonenal (HNE), a well-characterized molecule known to affect the function of proteins involved in signaling pathways [66]. Oxidative damage to proteins can alter their structure and stability and commonly involves the carbonylation or nitrosylation of amino acid side chains. But the most severe and permanent damage results from disulfide bond–mediated protein cross-linkages or the formation of bulky protein aggregates [66]. Oxidizing species therefore pose a significant threat to the maintenance of structural integrity of both the cell membrane and many proteins involved in cell signaling and essential enzymatic pathways. Ultimately, the sum of these damages is important for the first step in carcinogenesis, the initiation stage, where normal cells acquire the right amount and type of mutations to help them survive and rapidly proliferate.
Deregulation of the transcription factor erythroid 2p45 (NF-E2)-related factor 2 (Nrf2) can also potentially contribute to ROS accumulation. Nrf2 is known to mediate the expression of several important antioxidant enzymes by interacting with the antioxidant-response element (ARE) promoter region of these genes. In fact, the expression of Nrf2 has been shown to be significantly downregulated in prostate tumors [67]. The loss of Nrf2 results in the suppression of glutathione-S-transferase (GST) expression (a target gene of Nrf2), leading to ROS accumulation and DNA damage in Nrf2-deficient cells [67]. GST itself is also prone to mutations, and its somatic silencing has been observed in nearly all cases of prostate cancer examined by Nelson and colleagues [68]. In fact, of all the genes known to be aberrantly methylated in PCa, GST is the most frequently methylated, with its methylation status positively correlating with both Gleason grade and tumor volume [69].
A significant portion of ROS come directly from the NOX family of enzymes [70]. Ectopic expression of the NOX1 isoform has been found to enhance the growth and tumorigenicity of prostate epithelial cells. Moreover, tumors that express NOX1 also overexpress VEGF and VEGF receptors, thereby vascularizing previously-dormant tumors and enabling their growth [71]. Conversely, downregulation of NOX5 leads to dramatic growth inhibition and treatment with the NOX inhibitor diphenylene iodonium (DPI) caused cells to undergo apoptosis [72].
Age and Testosterone
Age is a risk factor for PCa development (median age at diagnosis is 65) suggesting that changes in cellular metabolism might initiate the onset of PCa [70]. Advancing age has been associated with the increased risk of developing metabolic abnormalities that impairs a cell’s ability to detoxify ROS leading to the development of PCa [70].
Steroid hormones (i.e. testosterone and dihydrotestosterone [DHT]) are critical for the proper maintenance and functioning of the prostate and have long been thought to regulate redox homeostasis within the tissue. Studies in rats have shown that castration induced the expression of NOX enzymes and reduced the expression of superoxide dismutase 2, glutathione peroxidase 1, thioredoxin, and peroxiredoxin 5 [73] Furthermore, replacement of testosterone levels decreased the NOX expression and restored the above-mentioned antioxidant enzymes to normal levels. Other studies have demonstrated that prostate cancer cells stimulated by androgens experience increased oxidative stress [74, 75]. While circulating levels of androgens can influence the production of ROS, exactly how their presence leads to redox imbalance is unclear.
3.2.2 The Inflammatory Tumor–Microenvironment
As mentioned previously, the innate immune system is the rapid-acting non-specific ‘first line of defense’ of the body and is comprised of cells that express on their surface Toll-like receptors (TLRs) that can recognize structurally conserved molecular domains found on microbes [76]. The activation of TLRs leads to a multitude of intracellular events including the activation NF-κB signaling pathways resulting in increased production of proinflammatory cytokines, chemokines and increased synthesis of nitric oxide (NO) [76]. The cytokines produced by innate immune cells alert the immune system to the presence of pathogens and promote the differentiation and activation of B and T lymphocytes, which are the major adaptive immune cells that are committed to the recognition of specific antigens [76].
Over time the accumulation of somatic mutations and other damage resulting from oxidative stress alters the growth and migration of epithelial cells. These epithelial cells, along with tumor cells, produce various cytokines and chemokines to attract leukocytes (i.e. dendritic cells, eosinophils, lymphocytes, macrophages, mast cells and neutrophils) to the affected area [77]. While these immune cells are all known to contribute to tumor angiogenesis, invasion, metastasis and proliferation [78], tumor-associated macrophages (TAMs) in particular have been associated with poor prognosis in several cancers and contribute to carcinogenesis in multiple ways [77, 79]. TAMs release interleukins and prostaglandins to suppress anti-tumor responses, and work to vascularize the tumor by releasing angiogenic factors such as endothelin-2 [80] and vascular endothelial growth factor (VEGF) [77]. TAMs can also stimulate tumor cell migration and proliferation by releasing several types of epidermal growth factors. Furthermore TAMs synthesize proteases such as cathepsins, matrix metalloproteinases (MMPs, i.e. MMP-2 and MMP-9) and urokinase-type plasminogen activator (uPA), which breakdown the basement membrane of cells and allows for the remodeling of the stromal matrix thereby promoting tumor cell invasion and metastasis [81]. Mast cells and neutrophils release many of the same, or similar, growth factors and proteases as macrophages and are therefore thought to contribute significantly to both angiogenesis and metastasis [77]. Moreover, tumor cells express vital pro-inflammatory transcription factors (e.g. STAT3, NFκB) [82]. These transcription factors in turn induce the production of key cytokines (IL-6 and TNF), chemokines (CCL-2 and CXCL12) and inflammatory enzymes (COX-2), thereby leading to a complex inflammatory microenvironment surrounding the tumor and infiltrated immune cells. Autocrine and paracrine cytokine signaling within the tumor microenvironment may lead to constitutive altered signaling leading to cancer-related inflammation which influences cell survival, proliferation, angiogenesis, invasion and metastasis, and immune suppressor phenotype [83]. Thus, the immune cells in the tumor microenvironment are able to directly transform the milieu into one that benefits the growth of tumor cells, by vascularizing and remodeling tumor tissues to firmly entrench them into the local environment, provide nutrients, and allow for the invasion of tumor cells into distant parts of the body (Fig. 3.3).
3.2.2.1 Inflammasomes
Inflammasomes are protein complexes assembled during heightened inflammation by cells of the innate immune system. A characteristic feature of inflammasomes is the presence of pattern recognition receptors (PRRs) [such as Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD) receptors (NLRs)] which recognize pathogen-associated molecular patterns (PAMP) or danger-associated molecular patterns (DAMP) [84]. These complexes regulate caspase-1, which promotes an inflammatory response through activation and secretion of IL-1β and IL18 and induction of pyroptosis, an immune-regulated form of programmed cell death [85, 86]. Once activated by caspase-1, IL-18 induces IFN-γ production in NK cells and T-cells. This, in turn, enables anti-pathogen responses by macrophages including the production of reactive oxygen and nitrogen species [87, 88]. Unregulated expression of IL-1 has been indicated in malignancies and CD4+ T-cell production [89, 90]. CD4+ T cells produce IL-17, which in conjunction with IL-23, progresses skin carcinogenesis [90]. On the other hand, lack of IL-1 signaling has been shown to inhibit tumorigenesis by increasing myeloid-derived suppressor cell (MDSC) infiltration [91]. Also, IL-18-mediated IFN-γ production can also limit carcinogenesis, as seen in murine colorectal cancer [88, 92]. Finally, radiotherapy and chemotherapy can promote inflammasome activity, which in turn may stimulate antitumor immune responses [93]. Since inflammasomes play a role in both the protection and progression of cancer further research is needed to parse out the roles of various members of the complex and the downstream pathways affected in order to harness their protective capacities for therapeutic purposes.
3.2.2.2 microRNAs and Inflammation
MicroRNAs are non-coding RNA molecules usually 19–24 nucleotides long that form hairpin-like structrues and play an instrumental role in post-transcriptional regulation, either through the degradation of mRNA or by blocking translation thereby affecting cellular processes such as cell growth, angiogenesis, immune response and survival [94,95,96]. Inflammation mediated production of reactive oxygen species can cause genomic instability and production of aberrant miRNAs [97]. Additionally, NF-κB and other transcription factors can regulate the expression of genes that code for miRNAs [98]. Aberrant regulation of certain miRNAs can cause an increase in oncogene expression, or suppression of tumor suppressors or both, Fig. 3.2. IL-6 produced by immune cells stimulates miR-21 (a microRNA highly expressed in inflammatory diseases and responsible for carcinogenesis) through NF-κB [99]. Incidentally, inhibition of miR-21 results in tumor regression in xenograft mouse models [100]. In breast cancer, Let-7 miRNAs regulate the level of IL6 and its inhibition through NF-κB constitutes a positive feedback loop ultimately resulting in further IL6 production [101]. miR-155, another oncogene, is highly expressed in inflammatory conditions such as H. pylori and EBV infections and inhibits TP53-induced nuclear protein 1 (TP53INP1), a pro-apoptotic gene, leading to increased tumor cell survival [102, 103]. On the other hand, miR-663 behaves as a tumor-suppressor and its loss in gastric cancer increases oncogenesis [104]. miR-146 also shows tumor suppressor properties and its overexpression results in reduction in the levels of IL6 and IL8 [105]. In prostate cancer, miR-146 has been identified to have tumor suppressor properties through its inhibitory effect on Rac1 [106]. Also, overexpression of miR-101 results in the inhibition of prostate cancer cell growth [107]. In metastatic prostate cancer, loss of miR-101 results in up-regulation of EZH2, an E-cadherin silencer [108]. Since miRNAs exhibit distinct expression patterns in drug-resistant cancers, they may be used to differentiate between drug-sensitive and insensitive malignancies [109, 110]. Although microRNAs constitute a small fraction of the genome, growing evidence suggests that they play a substantial role in inflammation related-cancers and may hold diagnostic, prognostic, and therapeutic value.

Simplified schematic showing that miRNAs can be ‘oncogenic’ or ‘tumor suppressive’, based on whether they are lost (resulting in increased expression of oncogenes) or are overexpressed (resulting in the suppression of tumor suppressors), respectively
3.2.3 The Network of Cytokines and Chemokines (Fig. 3.2)
The complex system of chemokine and cytokine signaling between stromal, tumor, and immune cells are involved in promoting cell replication and survival within the inflammatory environment. Cytokines can be classified as proteins, peptides, or glycoproteins that are secreted or are membrane-bound and regulate the differentiation and activation of immune cells. These include interleukins, growth factors, TNF-α and colony-stimulating factors [111].
Several ILs are known to associate with the diseased prostate such as IL-1, IL-4, IL-5, IL-6, IL-8, IL-10, IL-13, IL-17, IL-23, TGF-β and TNF-α [112, 113]. While some cytokines (i.e. IL-1 and IL-4) are primarily associated with the development of benign prostatic hyperplasia (BPH) [114, 115] others such as IL-6, IL-8, TGF-β and TNF-α are known to directly contribute to carcinogenesis. Perhaps one of the best-studied pro-inflammatory cytokines in cancer, IL-6 was first discovered to enhance the proliferation of intestinal epithelial cells and was elevated in the serum of colon cancer patients [77]. Patients with PCa display high levels of IL-6 and its soluble receptor in the circulating plasma [116]. A crosstalk between IL-6 and androgen receptor activation has also been observed [117]. Remarkably, an androgen-sensitive PCa cell line (LNCaP) that was continuously exposed to IL-6 in vitro developed neuroendocrine features [118] and has been thought to be a factor driving the neuroendocrine phenotype in prostate tumors [119].
Incidentally mutations in Ras and TP53 also lead to increased production of IL-6 [120, 121]. IL-6 is also a potent activator of members of the Janus kinase (JAK) family of tyrosine kinases, which in turn further activate transcription factors known as signal transducers and activators of transcription (STATs), especially STAT3 [122]. STAT3 is constitutively active in many cancers and promotes cell proliferation by upregulating the expression of cyclins, the proto-oncogene c-Myc, and anti-apoptotic genes such as Bcl-2, Bcl-XL and survivin [113]. Besides activating the STAT3 pathway, depending on the cellular context IL-6 can also signal through the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3 kinase (PI3K) compensatory signaling pathways that are upregulated in castration-resistant PCa cell lines [123].
From a therapeutic standpoint, Siltuximab, a chimeric humanized antibody, has been shown to have high specificity and affinity for binding to IL-6 [124]. In in vitro studies, the antibody sensitized PCa cancer cell lines to cis-diamminedichloroplatinum- and etoposide- mediated cytotoxicity [125]. This antibody has been shown to be safe for combination therapy with docetaxel in a Phase I study of mCRPC patients where 62% patients showed reduction in serum PSA [126].
Tumor necrosis factor (TNF) is another major cytokine involved in systemic inflammation, especially during the early events of carcinogenesis by recruiting inflammatory cytokines, growth factors, and epithelial adhesion factors to damaged tissue [77]. The process of angiogenesis is supported by TNF via the induction of various angiogenic factors (e.g. VEGF, basic fibroblast growth factor) and enzymes (thymidine phosphorylase) [127]. TNF is also a major inducer of nuclear factor-κB (NF-κB), a transcription factor that upregulates many of the same pro-replication, pro-survival genes as STAT3. NF-κB is a dimer formed by Rel family proteins (i.e. RelA/p65, RelB, c-Rel, NF-κB1/p50, and NF-κB2/p52) and is held in an inactive conformation in the cytoplasm by inhibitory IκB proteins. Upon activation by external stimuli, such as TNF, the dimer is released and it enters the nucleus where it binds to the promoter of NF-κB-responsive genes [128, 129]. In PCa, the NF-κB pathway is dysregulated resulting in the progression to the androgen-independent state that ultimately leads to lethal CRPC. Constitutive NF-κB activation has been reported in prostate tumors [130] and the active, nuclear-localized NF-κB has been observed in organ-confined prostate tumors, but not in benign tissues, suggesting that constitutive NF-κB activation may also be an important early event in prostate carcinogenesis [131] (Fig. 3.3).

Simplified schematic of the various processes involved in the progression of inflammation-initiated tumorigenesis
3.2.3.1 EMT-Linking Inflammation and Cancer
Growing evidence suggests that the tumor microenvironment transmits inflammatory signals that enhance the metastatic capacity of cancer through the activation of a developmental process known as epithelial to mesenchymal transition (EMT). As mentioned previously, immune cells including DCs, TAMs, NK cells, regulatory T cells, neutrophils, B cells and MDSCs constitute a considerable proportion of the tumor microenvironment and behave as mediators of inflammation-induced EMT. For EMT to occur, cells must have the capacity to undergo this process irrespective of their oncogenic content and have sufficient signals that promote EMT induction [132]. Moreover studies have demonstrated that epithelial cells can undergo various degrees of EMT when induced with TGF-β1 resulting in the activation of SMAD transcription factors that then stimulates EMT proteins such as Snail, Zeb, and AP-1 [133, 134]. Additionally, Wnt signaling sensitizes cells to TGF-β induced EMT by inhibiting GSK3-β, which inactivates Snail, Zeb, and β-catenin via phosphorylation [135]. TGF-β also interacts with BMP7, a promoter of epithelial cell differentiation that impedes metastasis in prostate and breast cancer [136]. Additionally, oncogenic pathways such as Ras, Notch, and Hedgehog, have been shown to be involved in stemness through the induction of EMT by TGF-β [137,138,139,140].
Within the tumor microenvironment, TAMs, MDSCs, and Tregs produce TGF-β1, exacerbating cancer to a more aggressive and invasive state [141, 142]. Reciprocally, tumor cells produce TGF-β1 for immune cell recruitment and polarization; these cells collectively form a tumor-permissive microenvironment that drives EMT [132]. In prostate cancer, IGF-1 induction by TGF-β1 has been linked to EMT [143]. The NF-κB pathway, a Snail-1 stabilizer, is activated by TNF-α produced by TAMs and enhances TGF-β induced EMT [144, 145]. Although inflammation induced EMT is a well-established phenomenon; little is known about the migratory behavior of disseminated tumor cells. In fact, tumor cells undergoing EMT may co-migrate with macrophages by acquiring immune cell properties, mainly chemotaxis [146].
3.2.3.2 Stem Cell Theory
Tumors are composed of an array of cell types with differential tumorigenic capacities. Cancer stem cells (CSCs) are a subpopulation of cells within the tumor that have been implicated as key drivers of carcinogenesis. The defining characteristics of CSCs are their ability to differentiate into cells that reinforce the oncogenic phenotypes of the tumor, as well as their ability to self-renew and sustain tumor growth [147, 148]. Since inflammatory signals have been shown to induce processes that regulate normal stem cells, they may also play a critical role in the initiation and maintenance of CSCs [149]. NFκ-B signaling, activated by TNF-a, has been indicated in neural stem cell proliferation and inhibition of differentiation [150]. IL-6 can supplement the self-renewal of hematopoietic stem cells [151]. Additionally, low levels of oxygen and ROS enhance the self-renewal capacity of normal stem cells. Deregulation of pathways that maintain levels of oxygen and ROS can impair stem-cell function [152,153,154,155,156,157]. Inflammatory cytokines produce high levels of ROS, and interestingly, cancer stem cells in acute myeloid leukemia (AML) have higher level of ROS compared to normal stem cells [158]. It is possible that high ROS levels may lead to deregulated stem-cell mechanisms—perhaps through genomic instability—that give rise to cancer stem cells. This discrepancy between normal stem cells and CSCs may hold significant therapeutic value [159,160,161]. Several studies have suggested a strong relationship between CSCs and EMT through the well-defined TGF-β pathway [162]. Sustained expression of Snail by TGF-β treatment on breast cancer cell lines leads to loss of E-cadherin and a phenotype synonymous to breast CSCs [163,164,165]. HIF-1α activation in hypoxic conditions—a CSC-friendly environment—can promote EMT through Twist-1 expression [166, 167]. Furthermore, developmental pathways such as Wnt and Hedgehog (Hh) have been linked to self-renewal and CSCs [168, 169]. Thus, inflammation may enhance tumorigenic potential and drive disease progression through the initiation, and subsequent utilization, of stem cell properties. Discovery of CSC-specific markers may supplement current diagnostic and prognostic tools for disease detection, monitoring and potentially be of therapeutic value.
3.3 Conclusions
There exists an association between the incidence of chronic inflammation and the ability of the inflammatory microenvironment to initiate or promote prostate carcinogenesis. However, the molecular details surrounding inflammation and prostate cancer are still far from being completely understood. This is particularly important in the case of the immune cells surrounding the tumor. There exists a fine balance between their protective versus aggravating role, and more work is needed to specifically identify the subpopulations of the various immune cells that contribute to each of the scenarios. Certainly, elucidation of the molecular and immunobiological mechanisms linking inflammation and PCa will be beneficial to the development of novel therapies and prognostic markers to treat and detect inflammation-associated malignancies of the prostate.
References
Albini A, Sporn MB (2007) The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer 7(2)
Gonzalgo ML, Isaacs WB (2003) Molecular pathways to prostate cancer. J Urol 170(6 Pt 1):2444–2452
Pihan GA et al (2003) Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res 63(6):1398–1404
Savage L (2007) Unreported VA data may affect SEER research, Cancer surveillance, and statistics gathering. JNCI J. Natl. Cancer Inst 99(23):1744–1752
Ferlay J et al (2006) Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol 18(3):581–592
Virchow R (1881) An address on the value of pathological experiments. Br Med J 2(1075):198–203
Elinav E et al (2013) Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 13(11):759–771
Bishayee A (2014) The Inflammation and Liver Cancer, in Advances in Experimental Medicine and Biology. Springer Nature, pp 401–435
Sfanos KS, Isaacs WB, De Marzo AM (2013) Infections and inflammation in prostate cancer. Am J Clin Exp Urol 1(1):3–11
Gurel B et al (2014) Chronic inflammation in benign prostate tissue is associated with high-grade prostate Cancer in the placebo arm of the prostate Cancer prevention trial. Cancer Epidemiol Biomark Prev 23(5):847–856
Mantovani A et al (2008) Cancer-related inflammation. Nature 454(7203):436–444
Sfanos KS, De Marzo AM (2011) Prostate cancer and inflammation: the evidence. Histopathology 60(1):199–215
Lee J et al (2007) Cancer incidence among Korean-American immigrants in the United States and native Koreans in South Korea. Cancer Control 14(1):78–85
Pelouze PS (1935) Obscure pseudomembranous Trigonitis: Trigonitis Areata Alba. Ann Surg 101(1):594–598
Bushman W (2000) In: Lepor H (ed) Etiology of prostate. Prostatic diseases. W B Saunders Company, Philadelphia, pp 550–557
Zambrano A et al (2002) Detection of human polyomaviruses and papillomaviruses in prostatic tissue reveals the prostate as a habitat for multiple viral infections. Prostate 53(4):263–276
Samanta M et al (2003) High prevalence of human cytomegalovirus in prostatic intraepithelial neoplasia and prostatic carcinoma. J Urol 170(3):998–1002
Strasner A, Karin M (2015) Immune infiltration and prostate Cancer. Front Oncol 5
Persson BE, Ronquist G (1996) Evidence for a mechanistic association between nonbacterial prostatitis and levels of urate and creatinine in expressed prostatic secretion. J Urol 155(3):958–960
Leitzmann MF et al (2004) Ejaculation frequency and subsequent risk of prostate cancer. JAMA 291(13):1578–1586
Vidal AC et al (2014) Aspirin, NSAIDs, and risk of prostate cancer: Results from the REDUCE study. Clin Cancer Res 21(4):756–762
Masferrer JL et al (2000) Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res 60(5):1306–1311
Boudreau DM, Yu O, Johnson J (2010) Statin use and cancer risk: a comprehensive review. Expert Opin Drug Saf 9(4):603–621
Blake GJ, Ridker PM (2000) Are statins anti-inflammatory? Curr Control Trials Cardiovasc Med 1(3):161–165
De Marzo AM et al (1999) Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am J Pathol 155(6):1985–1992
Montironi R, Mazzucchelli R, Scarpelli M (2002) Precancerous lesions and conditions of the prostate: from morphological and biological characterization to chemoprevention. Ann N Y Acad Sci 963:169–184
Rich AR (1979) Classics in oncology. On the frequency of occurrence of occult carcinoma of the prostate: Arnold rice Rich, M.D., Journal of urology 33:3, 1935. CA Cancer J Clin 29(2):115–119
McNeal JE et al (1988) Zonal distribution of prostatic adenocarcinoma. Correlation with histologic pattern and direction of spread. Am J Surg Pathol 12(12):897–906
Nakayama M et al (2003) Hypermethylation of the human glutathione S-transferase-pi gene (GSTP1) CpG island is present in a subset of proliferative inflammatory atrophy lesions but not in normal or hyperplastic epithelium of the prostate: a detailed study using laser-capture microdissection. Am J Pathol 163(3):923–933
Bethel CR et al (2006) Decreased NKX3.1 protein expression in focal prostatic atrophy, prostatic intraepithelial neoplasia, and adenocarcinoma: association with Gleason score and chromosome 8p deletion. Cancer Res 66(22):10683–10690
Abate-Shen C, Shen MM (2002) Mouse models of prostate carcinogenesis. Trends Genet 18(5):S1–S5
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420(6917):860–867
Flammiger A et al (2012) Intratumoral T but not B lymphocytes are related to clinical outcome in prostate cancer. APMIS 120(11):901–908
Steiner GE et al (2002) The picture of the prostatic lymphokine network is becoming increasingly complex. Rev Urol 4(4):171–177
Steiner GE et al (2003) Expression and function of pro-inflammatory interleukin IL-17 and IL-17 receptor in normal, benign hyperplastic, and malignant prostate. Prostate 56(3):171–182
Steiner GE et al (2003) Cytokine expression pattern in benign prostatic hyperplasia infiltrating T cells and impact of lymphocytic infiltration on cytokine mRNA profile in prostatic tissue. Lab Investig 83(8):1131–1146
Miller AM et al (2006) CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol 177(10):7398–7405
Sfanos KS, Hempel HA, De Marzo AM (2014) The role of inflammation in prostate cancer, in advances in experimental medicine and biology. Springer Nature, pp 153–181
Lanciotti M et al (2014) The role of M1 and M2 macrophages in prostate Cancer in relation to extracapsular tumor extension and biochemical recurrence after radical prostatectomy. Biomed Res Int 2014:1–6
Johansson A et al (2010) Mast cells are novel independent prognostic markers in prostate Cancer and represent a target for therapy. Am J Pathol 177(2):1031–1041
Fleischmann A et al (2009) Immunological microenvironment in prostate cancer: high mast cell densities are associated with favorable tumor characteristics and good prognosis. Prostate 69(9):976–981
Nonomura N et al (2007) Decreased number of mast cells infiltrating into needle biopsy specimens leads to a better prognosis of prostate cancer. Br J Cancer 97:952–956
Khazaie K et al (2011) The significant role of mast cells in cancer. Cancer Metastasis Rev 30(1):45–60
Richardsen E et al (2008) The prognostic impact of M-CSF, CSF-1 receptor, CD68 and CD3 in prostatic carcinoma. Histopathology 53(1):30–38
Lindahl C et al (2009) Increased levels of macrophage-secreted cathepsin S during prostate cancer progression in TRAMP mice and patients. Cancer Genomics Proteomics 6(3):149–159
Herroon MK et al (2012) Macrophage cathepsin K promotes prostate tumor progression in bone. Oncogene 32(12):1580–1593
Sfanos KS et al (2008) Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res 14(11):3254–3261
Ammirante M et al (2010) B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 464(7286):302–305
Smith JR et al (1996) Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science 274(5291):1371–1374
Xu J et al (2002) Germline mutations and sequence variants of the macrophage scavenger receptor 1 gene are associated with prostate cancer risk. Nat Genet 32(2):321–325
Zheng SL et al (2004) Sequence variants of toll-like receptor 4 are associated with prostate cancer risk: results from the CAncer prostate in Sweden study. Cancer Res 64(8):2918–2922
Lindmark F et al (2004) H6D polymorphism in macrophage-inhibitory cytokine-1 gene associated with prostate cancer. J Natl Cancer Inst 96(16):1248–1254
Lindmark F et al (2005) Interleukin-1 receptor antagonist haplotype associated with prostate cancer risk. Br J Cancer 93(4):493–497
Khanna RD et al (2014) Inflammation, free radical damage, oxidative stress and cancer. Microinflammation 1:109. https://doi.org/10.4172/2381-8727.1000109
Klaunig JE, Kamendulis LM, Hocevar BA (2009) Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol 38(1):96–109
Liou G-Y, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44(5):479–496
Federico A et al (2007) Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer 121(11):2381–2386
Maynard S et al (2008) Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 30(1):2–10
Bos JL (1988) The ras gene family and human carcinogenesis. Mutation Research/Reviews in Genetic Toxicology 195(3):255–271
Takahashi T et al (1989) p53: a frequent target for genetic abnormalities in lung cancer. Science 246(4929):491–494
Franco R et al (2008) Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett 266(1):6–11
Valinluck V, Sowers LC (2007) Endogenous cytosine damage products Alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res 67(3):946–950
Long C et al (2007) Promoter Hypermethylation of the RUNX3 gene in esophageal squamous cell carcinoma. Cancer Investig 25(8):685–690
Akiyama Y et al (2003) GATA-4 and GATA-5 transcription factor genes and potential downstream antitumor target genes are epigenetically silenced in colorectal and gastric Cancer. Mol Cell Biol 23(23):8429–8439
Sharma S, Kelly TK, Jones PA (2009) Epigenetics in cancer. Carcinogenesis 31(1):27–36
Trachootham D et al (2008) Redox regulation of cell survival. Antioxid Redox Signal 10(8):1343–1374
Frohlich DA et al (2008) The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 27(31):4353–4362
Nelson WG et al (2004) The role of inflammation in the pathogenesis of prostate cancer. J Urol 172(5):S6–S12
Zhou M et al (2004) Quantitative GSTP1 methylation levels correlate with Gleason grade and tumor volume in prostate needle biopsies. J Urol 171(6):2195–2198
Khandrika L et al (2009) Oxidative stress in prostate cancer. Cancer Lett 282(2):125–136
Arbiser JL et al (2002) Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc Natl Acad Sci 99(2):715–720
Brar SS et al (2003) NOX5 NAD(P)H oxidase regulates growth and apoptosis in DU 145 prostate cancer cells. AJP: Cell Physiol 285(2):C353–C369
Tam NNC et al (2003) Androgenic regulation of oxidative stress in the rat prostate. Am J Pathol 163(6):2513–2522
Pathak S et al (2008) Androgen manipulation alters oxidative DNA adduct levels in androgen-sensitive prostate cancer cells grown in vitro and in vivo. Cancer Lett 261(1):74–83
Miyake H et al (2004) Oxidative DNA damage in patients with prostate cancer and its response to treatment. J Urol 171(4):1533–1536
de Visser KE, Coussens LM (2005) The interplay between innate and adaptive immunity regulates cancer development. Cancer Immunol Immunother 54(11):1143–1152
Lu H (2006) Inflammation, a key event in cancer development. Mol Cancer Res 4(4):221–233
Coussens LM, Werb Z (2001) Inflammatory cells and Cancer. J Exp Med 193(6):F23–F26
Lin EY, Pollard JW (2004) Role of infiltrated leucocytes in tumour growth and spread. Br J Cancer 90(11):2053–2058
Grimshaw MJ, Wilson JL, Balkwill FR (2002) Endothelin-2 is a macrophage chemoattractant: implications for macrophage distribution in tumors. Eur J Immunol 32(9):2393–2400
Pollard JW (2004) Opinion: tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4(1):71–78
Grivennikov SI, Karin M (2010) Inflammation and oncogenesis: a vicious connection. Curr Opin Genet Dev 20(1):65–71
Crusz SM, Balkwill FR (2015) Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol 12(10):584–596
Medzhitov R (2009) Approaching the asymptote: 20 years later. Immunity 30(6):766–775
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10(2):417–426
Chen KW et al (2014) In: Hiraku YU, Kawanishi SO, Oshima H (eds) Inflammasomes and inflammation, in cancer and inflammation mechanisms : chemical, biological, and clinical aspects, p 1 online resource (409 pages)
Gu Y et al (1997) Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science 275(5297):206–209
Schroder K et al (2004) Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75(2):163–189
Scheede-Bergdahl C et al (2012) Is IL-6 the best pro-inflammatory biomarker of clinical outcomes of cancer cachexia? Clin Nutr 31(1):85–88
Langowski JL et al (2006) IL-23 promotes tumour incidence and growth. Nature 442(7101):461–465
Bunt SK et al (2007) Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 67(20):10019–10026
Zaki MH et al (2010) IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol 185(8):4912–4920
Ghiringhelli F et al (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15(10):1170–1178
Gong Z et al (2014) In: Hiraku YU, Kawanishi SO, Oshima H (eds) MicroRNAs and Inflammation-related cancer, in cancer and inflammation mechanisms : chemical, biological, and clinical aspects, p 1 online resource (409 pages)
Sonkoly E, Pivarcsi A (2009) microRNAs in inflammation. Int Rev Immunol 28(6):535–561
Schetter AJ, Heegaard NH, Harris CC (2010) Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 31(1):37–49
Karin M, Greten FR (2005) NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 5(10):749–759
Murdoch C et al (2008) The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 8(8):618–631
Loffler D et al (2007) Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 110(4):1330–1333
Si ML et al (2007) miR-21-mediated tumor growth. Oncogene 26(19):2799–2803
Iliopoulos D, Hirsch HA, Struhl K (2009) An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139(4):693–706
Motsch N et al (2007) Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) induces the expression of the cellular microRNA miR-146a. RNA Biol 4(3):131–137
Gironella M et al (2007) Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc Natl Acad Sci U S A 104(41):16170–16175
Pan J et al (2010) Tumor-suppressive mir-663 gene induces mitotic catastrophe growth arrest in human gastric cancer cells. Oncol Rep 24(1):105–112
Bhaumik D et al (2009) MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY) 1(4):402–411
Sun Q et al (2014) miR-146a functions as a tumor suppressor in prostate cancer by targeting Rac1. Prostate 74(16):1613–1621
Hao Y et al (2011) Enforced expression of miR-101 inhibits prostate cancer cell growth by modulating the COX-2 pathway in vivo. Cancer Prev Res (Phila) 4(7):1073–1083
Varambally S et al (2008) Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322(5908):1695–1699
Blower PE et al (2008) MicroRNAs modulate the chemosensitivity of tumor cells. Mol Cancer Ther 7(1):1–9
Zheng T et al (2010) Role of microRNA in anticancer drug resistance. Int J Cancer 126(1):2–10
Dranoff G (2004) Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer 4(1):11–22
Vasto S et al (2012) Inflammation and cancer of the prostate, in prostate cancer: a comprehensive perspective. Springer Nature, pp 115–122
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and Cancer. Cell 140(6):883–899
Giri D, Ittmann M (2000) Interleukin-1α is a paracrine inducer of FGF7, a key epithelial growth factor in benign prostatic hyperplasia. Am J Pathol 157(1):249–255
Furbert-Harris P et al (2003) Inhibition of prostate cancer cell growth by activated eosinophils. Prostate 57(2):165–175
Nguyen DP, Li J, Tewari AK (2014) Inflammation and prostate cancer: the role of interleukin 6 (IL-6). BJU Int 113(6):986–992
Culig Z, Bartsch G, Hobisch A (2002) Interleukin-6 regulates androgen receptor activity and prostate cancer cell growth. Mol Cell Endocrinol 197(1–2):231–238
Deeble PD et al (2001) Interleukin-6- and cyclic AMP-mediated signaling potentiates neuroendocrine differentiation of LNCaP prostate tumor cells. Mol Cell Biol 21(24):8471–8482
Sottnik JL et al (2011) The PCa tumor microenvironment. Cancer Microenviron 4(3):283–297
Ancrile B, Lim KH, Counter CM (2007) Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev 21(14):1714–1719
Yonish-Rouach E et al (1991) Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 352(6333):345–347
Hodge DR, Hurt EM, Farrar WL (2005) The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 41(16):2502–2512
Culig Z et al (2005) Interleukin-6 regulation of prostate cancer cell growth. J Cell Biochem 95(3):497–505
van Zaanen HC et al (1998) Chimaeric anti-interleukin 6 monoclonal antibodies in the treatment of advanced multiple myeloma: a phase I dose-escalating study. Br J Haematol 102(3):783–790
Borsellino N, Belldegrun A, Bonavida B (1995) Endogenous interleukin 6 is a resistance factor for cis-diamminedichloroplatinum and etoposide-mediated cytotoxicity of human prostate carcinoma cell lines. Cancer Res 55(20):4633–4639
Hudes G et al (2013) A phase 1 study of a chimeric monoclonal antibody against interleukin-6, siltuximab, combined with docetaxel in patients with metastatic castration-resistant prostate cancer. Investig New Drugs 31(3):669–676
Balkwill F (2002) Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev 13(2):135–141
Karin M, Lin A (2002) NF-κB at the crossroads of life and death. Nat Immunol 3(3):221–227
Nguyen DP et al (2014) Recent insights into NF-kappaB signalling pathways and the link between inflammation and prostate cancer. BJU Int 114(2):168–176
Suh J et al (2002) Mechanisms of constitutive NF-κB activation in human prostate cancer cells. Prostate 52(3):183–200
Shukla S et al (2004) Nuclear factor-κB/p65 (Rel a) is constitutively activated in human prostate adenocarcinoma and correlates with disease progression. Neoplasia 6(4):390–400
Fuxe, J. and M.C.I. Karlsson, Epithelial–Mesenchymal transition: a link between cancer and inflammation, in cancer and inflammation mechanisms : chemical, biological, and clinical aspects, Y. U. Hiraku, S. O. Kawanishi, and H. Oshima, Editors. p. 1 online resource (409 pages)
Heldin CH, Landstrom M, Moustakas A (2009) Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol 21(2):166–176
Xu J, Lamouille S, Derynck R (2009) TGF-beta-induced epithelial to mesenchymal transition. Cell Res 19(2):156–172
Wu D, Pan W (2010) GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci 35(3):161–168
Buijs JT et al (2007) TGF-beta and BMP7 interactions in tumour progression and bone metastasis. Clin Exp Metastasis 24(8):609–617
Fuxe J, Vincent T, Garcia A (2010) De Herreros, Transcriptional crosstalk between TGF-beta and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle 9(12):2363–2374
Maitah MY et al (2011) Up-regulation of sonic hedgehog contributes to TGF-beta1-induced epithelial to mesenchymal transition in NSCLC cells. PLoS One 6(1):e16068
Oft M et al (1996) TGF-beta1 and ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev 10(19):2462–2477
Zavadil J et al (2004) Integration of TGF-beta/Smad and Jagged1/notch signalling in epithelial-to-mesenchymal transition. EMBO J 23(5):1155–1165
Massague J (2008) TGFbeta in Cancer. Cell 134(2):215–230
Dalal BI, Keown PA, Greenberg AH (1993) Immunocytochemical localization of secreted transforming growth factor-beta 1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am J Pathol 143(2):381–389
Kawada M et al (2008) Transforming growth factor-beta1 modulates tumor-stromal cell interactions of prostate cancer through insulin-like growth factor-I. Anticancer Res 28(2A):721–730
Bates RC, Mercurio AM (2003) Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell 14(5):1790–1800
Wu Y et al (2009) Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 15(5):416–428
Wyckoff J et al (2004) A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 64(19):7022–7029
Jordan CT, Guzman ML, Noble M (2006) Cancer stem cells. N Engl J Med 355(12):1253–1261
Clarke MF et al (2006) Cancer stem cells--perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res 66(19):9339–9344
Tanno T, Matsui W (2014) In: Hiraku YU, Kawanishi SO, Oshima H (eds) Stem cell theory and inflammation-related cancer, in cancer and inflammation mechanisms : Chemical, biological, and clinical aspects, p 1 online resource (409 pages)
Widera D et al (2006) Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci 7:64
Audet J et al (2001) Distinct role of gp130 activation in promoting self-renewal divisions by mitogenically stimulated murine hematopoietic stem cells. Proc Natl Acad Sci U S A 98(4):1757–1762
Tothova Z et al (2007) FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128(2):325–339
Ito K et al (2007) Regulation of reactive oxygen species by Atm is essential for proper response to DNA double-strand breaks in lymphocytes. J Immunol 178(1):103–110
Ito K et al (2006) Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 12(4):446–451
Liu Y et al (2009) p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4(1):37–48
Adelman DM, Maltepe E, Simon MC (1999) Multilineage embryonic hematopoiesis requires hypoxic ARNT activity. Genes Dev 13(19):2478–2483
Gilbertson RJ, Rich JN (2007) Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7(10):733–736
Guzman ML et al (2005) The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 105(11):4163–4169
Sullivan R, Graham CH (2008) Chemosensitization of cancer by nitric oxide. Curr Pharm Des 14(11):1113–1123
Tiligada E (2006) Chemotherapy: induction of stress responses. Endocr Relat Cancer 13(Suppl 1):S115–S124
Pervaiz S, Clement MV (2004) Tumor intracellular redox status and drug resistance--serendipity or a causal relationship? Curr Pharm Des 10(16):1969–1977
Zavadil J, Bottinger EP (2005) TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24(37):5764–5774
Vesuna F et al (2009) Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 11(12):1318–1328
Gupta PB et al (2009) Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138(4):645–659
Mani SA et al (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133(4):704–715
Nguyen QD et al (2005) Commutators of PAR-1 signaling in cancer cell invasion reveal an essential role of the rho-rho kinase axis and tumor microenvironment. Oncogene 24(56):8240–8251
Yang MH et al (2008) Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol 10(3):295–305
Merchant AA, Matsui W (2010) Targeting hedgehog--a cancer stem cell pathway. Clin Cancer Res 16(12):3130–3140
Jamieson CH et al (2004) Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351(7):657–667
Acknowledgements
We would like to acknowledge Victoria Hackert for her help in collecting relevant materials. Both KKY and SSY are Prostate Cancer Foundation Young Investigators. AKT is supported by grants from PCF and Deane Prostate Health.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Tewari, A.K., Stockert, J.A., Yadav, S.S., Yadav, K.K., Khan, I. (2018). Inflammation and Prostate Cancer. In: Schatten, H. (eds) Cell & Molecular Biology of Prostate Cancer. Advances in Experimental Medicine and Biology, vol 1095. Springer, Cham. https://doi.org/10.1007/978-3-319-95693-0_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-95693-0_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95692-3
Online ISBN: 978-3-319-95693-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)