
Abstract
The tumor microenvironment (TME) is a very complex niche that consists of multiple cell types, supportive matrix and soluble factors. Cells in the TME consist of both host cells that are present at tumor site at the onset of tumor growth and cells that are recruited in either response to tumor- or host-derived factors. PCa (PCa) thrives on crosstalk between tumor cells and the TME. Crosstalk results in an orchestrated evolution of both the tumor and microenvironment as the tumor progresses. The TME reacts to PCa-produced soluble factors as well as direct interaction with PCa cells. In return, the TME produces soluble factors, structural support and direct contact interactions that influence the establishment and progression of PCa. In this review, we focus on the host side of the equation to provide a foundation for understanding how different aspects of the TME contribute to PCa progression. We discuss immune effector cells, specialized niches, such as the vascular and bone marrow, and several key protein factors that mediate host effects on PCa. This discussion highlights the concept that the TME offers a potentially very fertile target for PCa therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The tumor microenvironment (TME) is a very complex niche that consists of multiple cell types, supportive matrix and soluble factors [1]. Cells in the TME consist of both host cells that are present at tumor site at the onset of tumor growth and cells that are recruited in either response to tumor- or host-derived factors. All host cells may play a role in the TME, dependent on tumor location, including stromal cells, stem cells, vascular cells, immune cells, etc. Traditionally, cancer therapies have targeted the tumor cells themselves; however, in the last few years, the concept of targeting the TME, in addition to the tumor cells, is being explored and exploited. Thus a comprehensive understanding of the TME is necessary in order to develop the most efficacious therapies.
Prostate cancer (PCa) thrives on crosstalk between tumor cells and the TME. Crosstalk results in an orchestrated evolution of both the tumor and microenvironment as the tumor progresses. The TME reacts to PCa-produced soluble factors as well as direct interaction with PCa cells. In return, the TME produces soluble factors, structural support and direct contact interactions that influence the establishment and progression of PCa. There are several excellent reviews on tumor:TME crosstalk [2, 3]. In this current review, we will focus on the host side of the equation to provide a foundation for understanding how different aspects of the TME contribute to PCa progression.
Immune Cells
Inflammation has been suggested to contribute to PCa initiation and progression for many years [4, 5]. Inflammation can potentially contribute to PCa pathophysiology through several mechanisms including generation of reactive oxygen species that lead to mutagenesis; production of cytokines by inflammatory cells; and enhancement of migration into the tumor of additional supportive host cells. Immune cells compromise the cellular arm of the inflammatory response and we highlight here some of these cells and how they have been thought to contribute to PCa.
T cells and PCa
Tumor infiltrating lymphocytes (TIL) are a well-recognized facet of tumor histology; however, how TILs contribute to the PCa progression is not well defined. One T cell that has received a lot of attention is the T-regulatory (T-reg) cell. In general terms Tregs are CD4(+) T cells defined as inhibitors of the T effector cell activity; however, specific definitions based on cell surface markers constantly change due to heterogeneity of the Treg population [6]. This constant evolution of defining the Treg phenotype can create a challenge in interpreting the literature and one must be aware of how the Treg is being defined in any particular publication. Tregs generally suppress anti-tumor responses. In support of the importance of Treg cells in PCa, Tregs, in this instance defined as CD4 + CD25high and in vitro immunosuppressive function, were found to be increased in PCa tissue compared to non-cancerous prostate [7]. Additionally, to determine the phenotype of TILs, Sfanos et al. performed multiple biopsies of PCa and then subjected them to flow cytometry [8]. They found that tumor infiltrating Tcells were skewed towards Treg (FoxP3+) and Th17 cells as opposed to CD4+ cells. This finding was extended in the peripheral blood, where Treg cells, defined as CD4 + CD25high, were increased in men with PCa compared to normal donors [7]. Further studies revealed that tumor infiltrating Treg cells, defined as CD8+ Foxp3+ cells, suppressed naive T-cell proliferation mainly through a cell contact-dependent mechanism [9] although in the murine TRAMP model of PCa CD4(+)CD25(+)Foxp3(+) Tregs were found to be dispensable for induction of tumor-specific tolerance [10]. In another evaluation of function by Yokokawa et al., levels of Tregs, defined as CD4 + CD25(high)FoxP3+, in the peripheral blood of healthy donors and patients with biochemically progressive, localized, and metastatic PCa were each measured by flow cytometry [11]. The functional activity of Tregs was determined by their ability to suppress the proliferation of CD4 + CD25- T cells. They found that Tregs in patients with PCa had significantly greater suppressive functionality than Tregs from healthy donors. These results indicate the presence of tumor or changes due to tumor therapies can modulate Treg function and suggest that larger clinical trials to validate these findings should be performed. Along these lines, the T cell response can be altered by androgen manipulation. It has been shown that androgen ablation in a mouse model induced a transient increased in CD4+ and CD8+ in residual tumor [12]. More than 2 months later, Treg cells, defined as FoxP3+ cells, were increasingly found within prostate epithelium, whereas cytotoxic T-lymphocytes, which were evenly distributed before androgen ablation, became sequestered within stroma. Thus, these manipulations could impact an immunotherapy regimen and should be considered upon treatment of PCa patients with immune-dependent regimens.
In addition to Treg cells another, CD4(+) T cell, Th17 cells, may impact PCa biology and therapeutics. Th17 cells are CD4(+) effector cells that produce extensive amounts of IL-17 which is a proinflammatory cytokine that attracts and activates granulocytes and monocytes. An initial clue to the importance of Th17 cells came from an early study that demonstrated that injection of a fusogenic glycoprotein in combination with an heat shock protein 70 (HSP70), as an immune adjuvant, induced an anti-tumor T cell immune response that was associated with increased IL-17 expression [13]. As indicated earlier, Sfanos et al. found that tumor infiltrating Tcells were skewed towards Treg (FoxP3+) and Th17 cells as opposed to CD4+ cells in PCa tissues. In order to determine the prognostic implications of the pretreatment level of Th17 cells compared with regulatory T-cell status in PCa patients receiving active whole cell immunotherapy, Derhovanessian et al. compared the proportion of Th17 cells and Treg cells in the peripheral blood of hormone-resistant non-bone metastatic PCa patients prior to immunotherapy [14]. They found that the frequency of Th17 cells inversely correlated with time to disease progression. Furthermore, those patients that responded to immunotherapy with significant reductions in PSA velocity showed a Th17 profile similar to healthy male controls and significantly different from patients that did not respond. In contrast, although the frequency of Tregs in peripheral blood was higher in men with PCa compared to healthy age-matched controls, there was no difference among responders and non-responders. These data indicated that Th17 cells may be prognostic for therapeutic response to active whole cell vaccine; whereas, Treg cells would have limited utility in this arena.
Macrophages and PCa
Macrophages found within the TME, termed tumor-associated macrophages (TAMs), influence diverse processes such as angiogenesis, tumor cell proliferation, and metastasis during tumor progression [15]. Studies using clinical cases have revealed TAMs may offer some prognostic benefit. The amount of TAMs present in PCa tumor biopsies has been shown to be a useful predictive factor for PSA failure or progression of PCa after hormonal therapy predictive for tumor response [16]. Similarly, the density of macrophages in the presence of metastasis in PCa tumors provided prognostic information [15]. These results suggest that macrophages contribute to PCa progression.
TAMs produce a variety of proinflammatory cytokines including monocyte chemoattractant protein-1 (MCP-1); macrophage inhibitory cytokine-1 (MIP-1); IL-6 and IL-8 that may impact the overall cancer cell’s growth and function, including invasive ability. For example, incubation of the LNCaP PCa cell line with media from activated macrophages induced an NF-KB-driven pro-inflammatory response in the LNCaP cell lines [17]. Additionally, therapies may impact the TAM’s ability to produce factors that favor PCa growth. For example, TAMs harvested from irradiated tumors induced more rapid tumor growth than TAMs from non-irradiated tumors in a mouse model of PCa [18]. This was associated with increased production of Arg-1, COX-2 and iNOS from irradiated TAMs. In another instance, in vitro studies were used to demonstrate that the bisphosphonate zoledronic acid suppressed MMP-9 expression by PCa TAMs and enhance the TAMs polarization to the anti-tumoral M1 phenotype [19]. To explore the role of macrophages in PCa, Halin et al. injected Dunning R-3327 AT-1 rat PCa cells into the prostates of syngenic and immunocompetent Copenhagen rats and analyzed vascular proliferation and macrophage density [20]. Endothelial proliferation increased with tumor size both in the tumor and importantly also in the extratumoral normal prostate tissue. Macrophages accumulated in the tumor and in the extratumoral normal prostate tissue and were most abundant in the invasive zone. Moreover, only extratumoral macrophages showed strong positive associations with tumor size and extratumoral vascular proliferation. Treatment with clodronate-encapsulated liposomes, which kills macrophages, reduced the monocyte/macrophage infiltration and resulted in a significant inhibition of tumor growth. This was accompanied by a suppressed proliferation in microvessels and in the extratumoral prostate tissue also in arterioles and venules. The tumor cells were found to produce multiple factors that promote monocyte recruitment, angiogenesis, and tissue remodeling. In another it was reported that co-culture of PC-3, DU145, and LNCaP PCa cells with isolated human monocytes significantly stimulated PCa cell invasion activity [21]. Taken together, these findings suggest that TAMs contribute to the progression of PCa in experimental models.
In some instances, PCa cells have been shown to co-opt normal cellular mechanisms to achieve progression. In the context of macrophage migration, CC chemokine ligand 2 (CCL2; also known as monocyte chemotactic protein-1 (MCP-1)) which promotes migration of monocytes as a chemotactic factor, has been shown to be secreted from human bone marrow endothelial cells [22] and its expression can be induced by PTHrP [23]. It was shown that CCL2 can promote PCa proliferation and invasion into the bone [24].
Due to their propensity to infiltrate tumors, some investigators have sought ways to use macrophages to target therapeutics to tumors. In one example, macrophages laden with hypoxia-regulated adenovirus expression construct were found to target hypoxic areas of PCa tumors in mice [25]. In the areas of hypoxia, the oncolytic adenovirus, which was restricted to prostate cells as it was driven by prostate-specific promoter elements, was activated and infected target tissues. In another study using macrophages as a delivery vehicle, Satoh et al. performed intratumoral injection of macrophages transduced with murine IL-12 recombinant adenoviral vector in the orthotopic 178–2 BMA mouse PCa model [26]. The adenoviral-IL-12-transduced macrophages induced significant suppression of primary tumor growth and spontaneous lung metastases compared. The tumors demonstrated significantly increased infiltration of CD4+ and CD8+ T cells compared with controls. These results indicate that the macrophage may be useful as a delivery vehicle for therapeutics and can upregulate immune function.
The Vascular Niche in PCa
Angiogenesis has been described as one of the “hallmarks of cancer” as the delivery of nutrients and oxygen, and the removal of waste products, are required for cellular function [27]. Therefore, the sprouting of new vessels (angiogenesis) is a critical component of the TME. Stimulators of angiogenesis include: vascular endothelial growth factor-A (VEGF-A), transforming growth factor beta (TGF-β), and interleukin-8 (IL-8); while inhibitors include: thrombospondin-1 (TSP-1) and angiostatin. In concert, these molecules normally regulate vessel growth in processes such as development, wound healing and the female reproductive cycle. In tumors, these vessels are co-opted to support the rapid division of tumor cells.
Angiogenesis specifically refers to the sprouting of new blood vessels from existing vasculature [27]. This process typically involves the proliferation of endothelial cells (EC) and migration towards a pro-angiogenic molecule, such as VEGF-A. Vasculogenesis concerns tube formation by EC, and is more typically associated with embryogenesis and development, but may still play a significant role in tumor vascularization. The expansion of existing vasculature depends on the proliferation of endothelial cells or the accumulation of circulating endothelial progenitor cells (CEP) [28]. CEP are immature endothelial cells, typically arising in the bone marrow, that can extravasate from existing vasculature and promote new vessel formation. Understanding the biology of CEP is currently an area of intense interest, as CEP can be released from the bone marrow in response to pro-angiogenic factors [29]. Likewise circulating endothelial cells (CEC), mature EC of existing vasculature, can be sloughed off, circulate, and promote neo-vascularization at distant sites. Ruzinova et al. suggested that differential expression if Id genes may regulate CEP recruitment in poorly differentiated PCa [30]. However, it appears that organ site may play a role in CEC/CEP recruitment, and thus the role of stromal cells in the microenvironment may have similar effects in tumor [31]. It is only recently that a standardized approach for identifying CEC/CEP in humans has been devised [32]. Even though angiogenesis is regulated at the microenvironment level, CEC may be indicative of systemic changes in angiogenesis. The use of CEC/CEP as a prognostic marker in PCa has been suggested, but the results have been conflicting [33]. Strijbos et al. and Blann et al. have shown no prognostic significance in the number or change of CEC during treatment, which is in conflict with earlier work by Benezra’s early findings that CEC are related to tumor grade [30, 34, 35]. It is plausible that different markers used for the classification of CEC/CEP are causing these discrepancies, relaying the need for further investigation with a consistent panel of markers in a large population [29, 32].
Division and invasion of ECs constitute the bulk of angiogenic relationships within a tumor. However, growth and division of tumor-supporting vessels is disorganized compared to the normal physiologic processes described above. This disorganized process yields vessels that are convoluted, leaky, and considerably more immature than normal vasculature. Vessel leakiness promoted by VEGF, previously known as vascular permeability factor (VPF), not only induces new vessel growth, but also allows for the migration of tumor cells into the vasculature, leading to systemic dissemination [36]. VEGF acts on ECs through a variety of VEGF receptors (VEGFR), but primarily VEGFR1 and VEGFR2. In relation to endothelial cells, VEGFR1 plays a role in cell motility and extravasation, whereas VEGFR2 primarily regulates proliferation and vascular permeability [37]. Numerous therapeutics targeting the ligand and receptor have been studies to mixed results in past clinical trials [38].
Recent evidence suggests that pericytes, cells in normal tissue that support ECs and mature vessels, are also present in a variety of tumor types, including PCa [39]. The presence of pericytes in the TME suggests that a mature vascular phenotype may be present within the tumor, even though these pericytes are still more susceptible than in normal tissues [40]. Interestingly, it has been suggested that circulating pericytes may come to reside within the tumor and may inhibit tumor cell intravasation; although further investigation into these roles is necessary [40]. Furthermore, targeting of pericytes may be a novel method of inhibiting angiogenesis in PCa [40, 41]. In a model of lung carcinoma, Cascone et al. found that resistance to bevacizumab, a humanized monoclonal antibody binding VEGF-A, was related to increased EGFR signaling in pericytes and an increase in normalized and mature vasculature [42]. Combination of VEGF and EGFR inhibitors reversed this phenotype, inhibiting angiogenesis and prolonging progression free survival, suggesting that pericytes may play a role in angiogenesis and drug resistance. In PCa, targeting of the NG2 proteoglycan on pericytes has been shown to inhibit neovascularization in mouse models [41].
Bone is the prominent metastatic site for PCa, and the unique vasculature within bone may be partially responsible for this. The presence of sinusoids in trabecular bone may lead to areas of low blood flow allowing for cellular aggregation [43]. Furthermore, the presence of fenestrated capillaries, normally required for the intravasation of developing myeloid cells into the blood, may facilitate extravasation of tumor for tumor cells due to the relatively large pores present (30–40 μm). The establishment of metastatic foci rapidly leads to the requirement of new blood vessels, a proverbial flipping of the “angiogenic switch.” Judah Folkman first suggested that tumors are not able to grow past 2 mm3 without the influx of new vasculature [44]. A reason for this is that tumor cells become hypoxic due to the inability of oxygen to diffuse more than this distance from blood vessels, leading to a decreased partial pressure of oxygen as distance for a vessel increases. Under normoxic conditions, HIF-1α is rapidly ubiquitinated by Von-Hippel Lindau (VHL) factor leading to HIF-1α’s degradation [45]. During hypoxia, VHL is hydroxylated, allowing HIF-1α to translocate to the nucleus and bind hypoxic response elements, thus stimulating the production of VEGF and other pro-angiogenic factors. It has been suggested that the “angiogenic switch” in PCa may be governed through CXCL12 and CXCR4 with the downstream regulator phosphoglycerate kinase 1 (PGK1) playing a significant role [46]. Recently, metastasis-associated protein 1 (MTA1) was found to stimulate VEGF production in PCa cells, especially within the bone TME and may be a marker of increase aggressiveness [47].
The bone microenvironment is unique, and offers specific conditions which may be conducive to PCa-induced angiogenesis. For example, it has recently been shown that MMP9 produced by osteoclasts induces vascular development in PCa bone metastases, but does not alter tumor growth or impact the lytic/blastic nature of the lesion [48]. Conversely, it has been shown that MMP9 produced by PCa metastases in the brain did not alter the number of vessels, yet the tumor cells retained their metastatic capabilities [49]. Therefore, the context of the TME plays a substantial role in the regulation of tumor function as the expression of a single molecule in different locations may lead to variable outcomes. Furthermore, differential gene expression based upon metastatic site has been observed, with a significant difference in microvessel density when bone and liver metastases were compared [50]. These effects underscore the importance of the TME in respect to angiogenesis and other tumor properties.
Despite the advance of numerous therapeutics targeting angiogenesis across a number of tumor types, clinical trials of these compounds have typically led to dissatisfying results [38]. One of the first anti-angiogenic therapies was metronomic therapy. Metronomic therapy gives small doses of chemotherapeutics (e.g. dexamethasone and cyclophosphamide) more often (e.g. daily), and has shown some success in treating hormone resistant PCa [51, 52]. Combination of thalidomide, a teratogen which inhibits endothelial cell division, with docetaxel has shown an increase in 18 month overall survival of 68.2% to 42.9% compared to single agent docetaxel [53]. Even though bevacizumab in combination with docetaxel did not lead to a significant prolongation of survival, it has recently been suggested that the addition of thalidomide to this combination may be more efficacious for the treatment of castrate resistant disease [54]. Current clinical trials are focusing on the use of small molecules which target multiple receptor tyrosine kinases and the constituents of their signaling pathways to inhibit angiogenesis and tumor cell proliferation; these studies have been recently summarized [55, 56]. These results suggest that inhibition of angiogenesis may play a role in the inhibition of metastatic PCa.
Prostate Fibroblastic Stroma and PCa
Fibroblasts compose a significant component of the underlying support structure of the prostate tissue. They produce matrix, primarily collagen, which forms the structural framework for epithelial tissue. At least four cellular sub-types have been identified within the fibroblastic component of the stromal tissue microenvironment, and these sub-types likely share common lineage. These include fibrocytes, replication competent fibroblasts, senescent fibroblasts, and myofibroblasts.
Fibrocytes are putative stem/progenitor cells which can generate proliferative/replicative fibroblasts. These, in turn, can continue to replicate, senesce, or differentiate into myofibroblasts [57–60]. Fibrocytes and/or replication competent stromal fibroblasts that endure sub-lethal levels of DNA damage may undergo senescence. Senescence is a physiological cellular state that is non-replicative but metabolically very active. Senescence may be triggered by conditions that produce cellular stress, e.g., exposure to reactive oxygen species, carcinogenic insult, irradiation, etc., or by cells reaching critical levels of telomere shortening and consequent replicative exhaustion. Many studies have shown that senescent cells accumulate in various tissues and organ sites with increasing age in vivo [61, 62]. It is reasonable to expect that this process occurs in the prostate, as well. Senescent fibroblasts are characterized by several changes in cell functions, including increased expression and secretion of inflammatory chemokines. For example, EGF-type growth cytokines and CXC-type chemokines were transcriptionally up-regulated in senescent compared to non-senescent prostate fibroblasts [63]. Moreover, either direct co-culture with, or exposure to, conditioned medium from senescent prostate fibroblasts stimulated the proliferation of cultured PCa cells. In another study, it was shown that senescent fibroblasts derived from lung, foreskin or breast tissues, or prostate stromal fibroblasts forced to undergo senescence, secreted significantly higher levels of many of these same cytokines and chemokines collectively termed ‘senescence associated secreted proteins’ (SASPs) [58]. Many of these same growth factors and SASPs, particularly CXC-type chemokines, are secreted by aging prostate stromal fibroblasts and stimulate the proliferation of both non-transformed and transformed prostate epithelial cells [64]. Thus, aging-associated senescence of fibroblastic cell types in the stroma may promote the proliferation of adjacent PCa cells.
In addition to undergoing senescence, fibrocytes and/or replication competent stromal fibroblasts can also differentiate into myofibroblasts. Myofibroblasts are contractile cells that express both alpha-smooth muscle actin and collagen [65]. They are transiently produced in many tissues as part of the normal wound response cascade and typically undergo apoptosis at the conclusion of that process [66]. However, myofibroblasts have been described as a persistent component of the stroma in association with malignant prostatic epithelium and, in this context, comprise part of an “activated” or “reactive stroma” [60, 67, 68] and are also called cancer associated fibroblasts (CAF) [69]. LNCaP PCa cells co-cultured with myofibroblasts exhibited a reduced death rate [70] and myofibroblast-rich “reactive stroma” characterizes tumor- and prostate intraepithelial neoplasia (PIN)-associated stroma [60, 67]. These latter studies also suggest that stromal cells immediately adjacent to PIN are induced to differentiate into myofibroblast-rich reactive stroma, and that reactive stroma progresses with the development of PCa. IL-8 (aka CXCL8) has been shown to be sufficient for induction of a replication competent fibroblast to myofibroblast transition [71], and that over-expression of keratinocyte-derived chemokine, the mouse homologue of IL-8, in mouse prostatic epithelium was sufficient to produce hyperplastic prostate epithelial acini associated with a periacinar reactive stroma [72]. Other studies have shown that, in addition to IL-8, TGF-beta and androgen can induce myofibroblast differentiation [73] and promote transformation of prostate cells in a murine model [74]. Taken together, these studies suggest that multiple stimuli provided by the prostate microenvironment can induce myofibroblast differentiation and accumulation. Perhaps not coincidentally, CXCL8 is a SASP, suggesting that fibroblastic senescence and myofibroblast differentiation may be mechanistically linked in the prostate and perhaps other organs. Further work may elucidate whether carcinoma-associated fibroblasts (CAFs), which have been shown to stimulate tumor progression both in vitro and In vivo [75], comprise a milieu of senescent fibroblasts and myofibroblasts as well as replicating fibroblasts and fibrocytic progenitor cells.
Bone Marrow Stroma and Osteoblast Niche in PCa
Bone marrow stromal cells (BMSC) and osteoblasts have been shown to modulate PCa activity. Growth of PCa cells with supernatant of BMSC cultures or direct contact with BMSC revealed induction of genes that were modified by either soluble factors, or direct contact or both indicating that there are multiple functions mediated by the BMSC [76]. In a study that looked at the impact of heterotypic direct contact between PC-3 cells and BMSC or an osteoblast cell line, it was found that urokinase plasminogen activator (uPA) was upregulated by both, but not prostate stromal cells indicating some specificity for the impact of BMSC and osteoblasts (both bone microenvironment-derived cells) on PCa [77]. These findings also implicated that BMSC and osteoblasts could enhance invasion through induction of uPA which has been shown to be induced by osteoblasts and enhance invasion [78]. In addition to uPA, TGF-β is produced by osteoblasts and been shown to promote both migration and invasion by PCa cells [79, 80]. In addition to impact on uPA expression, co-culture of human PCa cell lines with bone tissue induced endothelin-1 (ET-1) expression from PCa cells [81]. ET-1 stimulates osteoblast differentiation [82] and prostate-specific antigen (PSA) expression. In light of its protumorigenic effect, investigators have explored blocking ET-1 activity through targeting its receptor. This strategy has been shown to enhance taxane chemotherapy in a mouse model of PCa [83]. In contrast to its impact on osteoblastic activity, ET-1 may inhibit PCa growth through constriction of afferent arterioles [84]. Although preclinical studies suggest that targeting ET-1 activity may impact PCa progression, these findings have not translated clinically. Several phase II and III trials using an inhibitor of the ET-1 receptor (atresentan) that evaluated time to clinical and radiographic progression failed to demonstrate a significant benefit with atrasentan versus placebo [85]. Along these lines, a multi-site phase III, randomized, double-blind, placebo-controlled trial of atrasentan in patients with nonmetastatic hormone refractory PCa was recently completed [86]. The primary endpoint was the time to disease progression (TTP) that was defined as the onset of metastasis. However, no difference in TTP was noted between those receiving atresentan versus placebo. It was noted that there were large differences in TTP among different sites, suggesting that trial conduct may have influenced the results [86]. A new ET-1 receptor inhibitor (zibotentan) with greater specificity than atresentan is currently in phase III clinical trials for hormone refractory PCa [87]. Thus, the overall balance of ET-1’s effect on PCa is not clear at this time.
The bone is the most common site for PCa metastasis. To explore the role of the bone on the biology of metastasis Fu et al. determined how osteoblasts influence gene expression in PCa cells. They found that conditioned-media from osteoblasts induced a gene expression pattern in PCa cell lines similar that found in metastatic versus non-metastatic PCa cells [88]. These results suggest that osteoblasts produce soluble factors that contribute to the progression of PCa skeletal metastases, including their transition to an osteoblast-like phenotype. In addition to these pro-metastatic effects, osteoblasts have been shown to protect cells from chemotherapeutics such as adriamycin [89]. Thus, overall osteoblast can contribute to the PCa progression through induction of proliferation, enhancing a metastatic phenotype and protecting PCa cells from therapies.
In addition functioning to promote PCa progression, osteoblasts provide a critical role in initial seeding of PCa tumors into the bone. Osteoblasts compromise an important component of the hematopoietic stem cell (HSC) niche. One function they serve is to attract HSC hematopoietic stem cells to the niche through production of the chemotactic factor stromal-derived factor-1 (SDF-1). SDF-1 binds to its receptor, CXCR4, on the HSC and causing it to localize to the HSC niche. Sun et al. demonstrated that PCa cells use this same osteoblast-derived SDF-1 to localize to the HSC niche [90]. Shiozawa et al. reported that PCa cells compete with HSC for the HSC niche [91]. In a murine model using human PCa cells, they found that manipulating the size of the niche directly impacted the PCa occupancy of the niche and the associated establishment of metastases. Furthermore, the PCa cells induced terminal differentiation of HSC, resulting in their being less competition for the niche binding sites.
Cytokines and PCa
A variety of small soluble proteins are secreted by cells in the TME that can impact PCa growth. These cytokines can then act in a paracrine fashion on tumor cells to stimulate a variety of tumor phenotype activities including proliferation, chemoresistance, prevent apoptosis, enhance motility or invasion, etc. Here we focus on several cytokines implicated in the PCa TME. As there are too many protein factors to describe, we apologize in advance to those whose important work was not included but summarize key factors in Tables 1 and 2.
Hepatocyte Grow Factor (HGF)
HGF (also called Scatter Factor) mediates signaling through binding to c-Met, a transmembrane tyrosine kinase receptor. HGF is expressed by stromal cells including myofibroblasts and osteoblasts [92, 93] HGF has been shown to be elevated in men with PCa [94] and in combination with PSA provide an increased confidence of the presence of PCa [95]. Increased HGF pre-surgery was a predictor of lymph node involvement and post-surgical recurrence [96]. Inactivation of the androgen receptor induces c-Met expression [97]. This in turn allowed for c-HGF to induce oxidative stress-mediated DNA damage in PCa cells.
HGF has been shown to induce PCa proliferation [98] which appears to be mediated, in part, through Transient Receptor Potential Canonical-6 (TRPC6) calcium channels [99]. HGF also promotes scatter and invasion [100, 101]. It promotes scatter through downregulation of E-cadherin [102]. Invasion is mediated by HGF through HGF-induced anterograde lysosome trafficking that is dependent upon the PI3K pathway, microtubules and RhoA, resulting in increased cathepsin B secretion and invasion by the cells [103]. In addition to direct cancer effect, HGF may impact the bone microenvironment through induction of osteoblastic proteins in PCa as HGF upregulates bone morphogenetic protein receptor expression [104] and BMP-7 expression in PCa cells.
In a study to determine c-Met expression in PCa, both mRNA and protein levels of c-Met was determined for prostate tissues from 32 Japanese male subjects [105]. Although there was significantly higher c-Met protein expression in malignant (PCa treated with/without neoadjuvant endocrine therapy) than in non-malignant prostate tissues (normal prostate and benign prostate hyperplasia; BPH), unexpectedly, c-Met mRNA showed high expression in the non-malignant group. Thus, there was no parallelism between mRNA and protein expressions of c-Met. Immunohistochemical localization and expression of c-Met protein was found to be intense in cancer cells and weak in epithelia of normal and hyperplastic prostates.
Several methods to target the HGF:c-Met axis have been explored. Inhibiting HGF activity with NK4, a competitive antagonist composed of an internal fragment of HGF that binds c-Met, diminished tumor growth and invasion of PC-3 PCa cells co-injected with HGF-producing fibroblasts in a murine model [106]. Similarly, use of BMS-777607, a selective small molecule Met kinase inhibitor decreased HGF-induced scattering of PCa cell lines [107] although it had no impact on cell proliferation. It was recently found, using computer-assisted molecular design, that Oleocanthal, a secoiridoid isolated from extra-virgin olive oil, was a potential Met inhibitor [108]. This was confirmed in vitro in PCa cells in which Oleocanthal inhibited phosphorylation of c-Met kinase. Thus, a variety of methods to block HGF activity exist and may be translated into clinical utility.
Interleukin-6
Interleukin-6 (IL-6) is produced by inflammatory cells and osteoblasts and thus has ample opportunity to interact with PCa. There are multiple lines of clinical and experimental evidence that suggest IL-6 promotes PCa progression [109]. Several studies have demonstrated that IL-6 is elevated in the sera of patients with metastatic PCa [110]. For example, serum levels of IL-6 and its soluble receptor were assessed to determine if they could predict biochemical recurrence in radical prostatectomy patients [111]. Mulitivariate analysis reveated that preoperative serum levels of IL-6 higher than 1.2 pg/ml had a significantly increased probability of biochemical recurrence. Although a small study, these results provide the rationale to further evaluate the utility of IL-6 as a prognostic factor. Similar to this finding pre-diagnostic IL-6 was associated with time to prostate cancer progression/death among healthy weight PCa cases [112]. Overall, the preponderance of evidence suggests that IL-6 is elevated in men with PCa and is related to its clinical outcome.
In addition these clinical observations, in vitro cellular studies have provided evidence that IL-6 modulates PCa cell growth. Chung et al. showed that IL-6 promoted cell growth of hormone-refractory cells, but had no effect on the growth of hormone-dependent cell lines [113]. Addition of exogenous IL-6 to the culture media of LNCaP cells by several groups has resulted in a dose-dependent inhibition of cell growth with development of a neuroendocrine cell phenotype in some cases; whereas in some instances proliferation is increased [113–119]. The reasons for these differences have not been clarified to date but it appears that IL-6 is only inhibitory to LNCaP cells, but not other PCa cell lines [120].
In addition to increased cell proliferation contributing to increased tumor volume decreased apoptosis can also promote increased tumor volume. A previous report demonstrated that orchiectomy induced conversion of LuCaP 35 tumors to an AI phenotype [121]. It has been shown that targeting IL-6 with an anti-IL6 antibody promoted apoptosis in AI PC-3 PCa cells growing in mice [122]. IL-6 has been recognized in many different cell systems to have an anti-apoptotic effect including PCa cells [123, 124]. In PCa, IL-6 protects against apoptosis through activation of Stat 3 [123] and phosphatidylinositol (PI)-3 kinase [124]. Taken together, the observations that inhibition of IL-6 decreased cell proliferation and increased apoptosis suggests that both IL-6 stimulates both of these processes resulting in an additive effect to promote increased tumor volume in AI tumors.
IL-6 has been suggested to promote other aspects of PCa pathophysiology in addition to proliferation. For example, IL-6 may contribute to the induction of tumorigenesis in the prostate microenvironment. Rojas et al. demonstrated that IL-6, through activation of signal transducer and activator of transcription 3 (STAT3), stimulated autocrine activation of insulin-like type I growth factor receptor (IGF-IR) to confer tumorigenesis [125]. In addition to tumorigenesis, IL-6 has been suggested to induce neuroendocrine differentiation of PCa tumors [115]. Specifically, it was shown that PCa cell-produced BMP6 induced IL-6 expression from TAMs, which then promoted neuroendocrine differentiation of PCa cells. Interestingly, IL-6 itself, along with the cytokine CCL2, promotes survival of human CD11b + peripheral blood mononuclear cells and induces M2-type macrophage polarization [126]. In summary, IL-6 can contribute to PCa pathophysiology through multiple mechanisms.
Due to the preponderance of literature indicating that IL-6 contributes to PCa progression inhibition of IL-6 was evaluated in a clinical trial. Specifically, a phase II study evaluated the efficacy of a human-mouse chimeric monoclonal neutralizing IL-6 antibody (siltuximab, also known as CNTO 328) in men with androgen resistant PCa that had been treated with one prior chemotherapy with the primary endpoint being prostate specific antigen (PSA) response rate (defined by a 50% reduction of PSA) [127]. The response rate was small (3.8% of 53 men) and no men with measureable disease had a Response Evaluation Criteria in Solid Tumors (RECIST) response. A decline in C-reactive protein suggested that IL-6 was being effectively neutralized. Taken together, these results suggest that IL-6 neutralization in men with advanced androgen resistant disease may not have a significant therapeutic benefit. However, this does not eliminate the possibility that IL-6 neutralization may provide an adjunct benefit to a chemotherapy regimen, especially in light of IL-6’s ability to have an anti-apoptotic effect [128]. Based on all the clinical and preclinical evidence for IL-6’s role in PCa, further exploration for IL-6 inhibition is warranted; however, its efficacy may greatly depend on the stage of disease or other individualized factors.
Receptor Activator of NFkB Ligand
Osteoblasts promote osteoclastogenesis through expression of tumor necrosis factor receptor family member called receptor activator of NFκB ligand (RANKL). RANKL can be expressed either as a membrane bound molecule or in some instances is cleaved to form a function soluble RANKL peptide [129]. RANKL binds to the transmembrane receptor RANK on osteoclast precursors and initiates osteoclastogenesis [130, 131]. It was demonstrated that RANKL is required for generation of osteoclasts as transgenic rankl −/− and rank −/− mice had no osteoclasts and developed osteosclerotic bones [132, 133]. Osteoprotegrin (OPG) is a soluble glycoprotein that negatively regulates osteoclastogenesis through sequestering RANKL resulting in blocking its interaction with RANK [134]. Thus, the balance between RANKL and OPG determines the extent of bone resorption.
OPG, RANKL and/or RANK expression are dysregulated in a number of cancers in bone, including osteoclastoma [135] and PCa [136]. RANKL expression was identified in the epithelium and stroma of clinical PCa specimens [136] and increasing levels correlate with increased stage of disease [137] suggesting that it can serve as a prognostic factor [138]. However, large prospective clinical trials to investigate this have not yet been reported and it remains to be clearly demonstrated that RANKL does provide prognostic information.
In vitro and in vivo studies have demonstrated that PCa cells initiate osteoclastogenesis through RANKL [139, 140]. Inhibition of RANKL activity with OPG [139] or soluble RANK [141] decreased the numbers of mature osteoclasts and number of lesions in bone in mice carrying human PCa cells. Importantly, treatment with OPG has also been demonstrated to block pain-related behavior in mice carrying bone cancers [142, 143]. Overall, these studies suggest that in bone metastatic tumors, inhibition of the primary resorptive stage may be sufficient to inhibit tumor establishment and halt progression of disease, even in those tumors that have primarily an osteoblastic phenotype.
In addition to its ability to promote osteoclastogenesis, RANKL has been shown to interact with RANK on the PCa cells themselves and induce their migration and a pro-metastatic gene expression [144]. Furthermore, OPG has been shown to be a survival factor through its ability to inhibit TRAIL-mediated apoptosis in PCa cells [145]. Thus, the sum of evidence suggests that targeting RANKL may provide a dual effect of both direct inhibition of pro-metastatic tumor activity in addition to inhibiting tumor-induced bone resorption. These findings have provided the rationale to evaluate the clinical efficacy of targeting RANKL in cancer-related bone disease.
The lead anti-RANKL compound is a fully human monoclonal IgG(2) antibody that binds RANKL (denosumab) and has been evaluated in several clinical studies. A Phase II study in which as subset of patients had PCa bone metastases demonstrated that inhibition of RANKL activity with denosumab normalizes the typically elevated levels of urinary NTx (a marker of osteoclast activity) [146]. This provided key evidence that PCa patients experience increased bone resorption that was targetable with an osteoclast inhibitor. In a large (n = 1,468) multinational, phase III trial in men with PCa who were receiving androgen-deprivation therapy, bone mineral density (BMD) at the lumbar spine was significantly improved from baseline after 24 (primary endpoint) and 36 months of treatment with denosumab relative to that with placebo [147, 148]. A phase III randomized double-blind, double-dummy trial that compared denosumab with zoledronic acid (an anti-osteoclastic bisphosphonate) for prevention of skeletal-related events (SRE: defined as pathological fracture, radiation to bone, surgery to bone, or spinal cord compression) in men with bone metastases from castration-resistant PCa. The primary endpoint was time to first on-study SRE. Denosumab significantly delayed the time to first on-study SRE as well as the time to first and subsequent onstudy SRE. Also denosumab had greater suppression of the bone turnover markers uNTx and bone-specific alkaline phosphatase compared with zoledronic acid. These results demonstrated that targeting RANKL in men with advance PCa has a therapeutic effect and the superiority of denosumab over zoledronic acid in diminishing SREs [149]. Based on these and other preclinical and clinical data, densoumab was given United States Federal Drug Administration approval for treating men with PCa (and breast cancer) bone metastases. Ongoing studies to determine if there is an actual antitumor effect are ongoing, but this question yet remains to be answered.
Conclusions and Future Directions
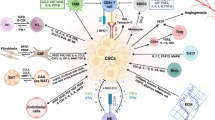
Multiple host factors contribute to PCa progression in the TME (Fig. 1). Immune cells, such as Tregs, Th17 and macrophages overall promote PCa progression through direct and indirect mechanisms. The vascular compartment contributes to PCa growth through provision of oxygen and nutrients. Furthermore, PCa cells enhance angiogenesis through attracting EC and promoting their growth in the TME. Similarly, the bone marrow stroma and osteoblasts provide growth factors, chemoattractants, such as SDF-1, and proteins that stimulate invasion, such as uPA. Additionally, the bone microenvironment provides a specialized niche for PCa seeding and growth, which also confers a protective effect against chemotherapy. Finally, cytokines mediate many of the host cells, effects on PCa cells. Proteins such as HGF and IL-6 have pleiotropic actions on PCa cells. These findings drive home the concept that the TME offers a potentially very fertile target for PCa therapy. Future studies directed on host factors, in addition to direct anti-tumor targeting, offer the greatest promise for achieve significant therapeutic gains.

Host cells and niches that compose the PCa tumor microenvironment (TME). Multiple immune cells such as T regulatory (Treg), T helper IL-17-expressing cells (Th17) and macrophages (Mϕ) contribute to the TME. The vascular microenvironment composed of endothelial cells and their circulating progenitors provides nutrition to PCa cells. The bone microenvironment houses osteoblasts (OB) that provide a site for PCa cells to seed to and produce factors that regulate PCa proliferation. Fibroblasts located in both the prostate itself and at multiple metastatic sites produce a variety of factors that promote PCa growth and invasion
References
Keller ET, Li LY (2011) The first Tianjin, China forum on tumor microenvironment. Cancer research 71:310–313
Niu YN, Xia SJ (2009) Stroma-epithelium crosstalk in prostate cancer. Asian J Androl 11:28–35
Yoneda T, Hiraga T (2005) Crosstalk between cancer cells and bone microenvironment in bone metastasis. Biochem Biophys Res Commun 328:679–687
Omabe M, Ezeani M (2011) Infection, inflammation and prostate carcinogenesis. Infect Genet Evol.
Vasto S, Carruba G, Candore G, Italiano E, Di Bona D, Caruso C (2008) Inflammation and prostate cancer. Future Oncol 4:637–645
Teng MW, Ritchie DS, Neeson P, Smyth MJ (2010) Biology and clinical observations of regulatory T cells in cancer immunology. Curr Top Microbiol Immunol
Miller AM, Lundberg K, Ozenci V, Banham AH, Hellstrom M, Egevad L, Pisa P (2006) CD4 + CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol 177:7398–7405
Sfanos KS, Bruno TC, Maris CH, Xu L, Thoburn CJ, DeMarzo AM, Meeker AK, Isaacs WB, Drake CG (2008) Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res 14:3254–3261
Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM, Thompson TC, Old LJ, Wang RF (2007) CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res 13:6947–6958
Degl’Innocenti E, Grioni M, Capuano G, Jachetti E, Freschi M, Bertilaccio MT, Hess-Michelini R, Doglioni C, Bellone M (2008) Peripheral T-cell tolerance associated with prostate cancer is independent from CD4 + CD25+ regulatory T cells. Cancer Res 68:292–300
Yokokawa J, Cereda V, Remondo C, Gulley JL, Arlen PM, Schlom J, Tsang KY (2008) Enhanced functionality of CD4 + CD25(high)FoxP3+ regulatory T cells in the peripheral blood of patients with prostate cancer. Clin Cancer Res 14:1032–1040
Akins EJ, Moore ML, Tang S, Willingham MC, Tooze JA, Dubey P (2010) In situ vaccination combined with androgen ablation and regulatory T-cell depletion reduces castration-resistant tumor burden in prostate-specific pten knockout mice. Cancer Res 70:3473–3482
Kottke T, Sanchez-Perez L, Diaz RM, Thompson J, Chong H, Harrington K, Calderwood SK, Pulido J, Georgopoulos N, Selby P, Melcher A, Vile R (2007) Induction of hsp70-mediated Th17 autoimmunity can be exploited as immunotherapy for metastatic prostate cancer. Cancer Res 67:11970–11979
Derhovanessian E, Adams V, Hahnel K, Groeger A, Pandha H, Ward S, Pawelec G (2009) Pretreatment frequency of circulating IL-17+ CD4+ T-cells, but not Tregs, correlates with clinical response to whole-cell vaccination in prostate cancer patients. Int J Cancer 125:1372–1379
Lissbrant IF, Stattin P, Wikstrom P, Damber JE, Egevad L, Bergh A (2000) Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int J Oncol 17:445–451
Nonomura N, Takayama H, Nakayama M, Nakai Y, Kawashima A, Mukai M, Nagahara A, Aozasa K, Tsujimura A (2010) Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int.
Wong CP, Bray TM, Ho E (2009) Induction of proinflammatory response in prostate cancer epithelial cells by activated macrophages. Cancer Lett 276:38–46
Tsai CS, Chen FH, Wang CC, Huang HL, Jung SM, Wu CJ, Lee CC, McBride WH, Chiang CS, Hong JH (2007) Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys 68:499–507
Tsagozis P, Eriksson F, Pisa P (2008) Zoledronic acid modulates antitumoral responses of prostate cancer-tumor associated macrophages. Cancer Immunol Immunother 57:1451–1459
Halin S, Rudolfsson SH, Van Rooijen N, Bergh A (2009) Extratumoral macrophages promote tumor and vascular growth in an orthotopic rat prostate tumor model. Neoplasia 11:177–186
Lindholm PF, Lu Y, Adley BP, Vladislav T, Jovanovic B, Sivapurapu N, Yang XJ, Kajdacsy-Balla A (2010) Role of monocyte-lineage cells in prostate cancer cell invasion and tissue factor expression. Prostate 70:1672–1682
Loberg RD, Day LL, Harwood J, Ying C, St John LN, Giles R, Neeley CK, Pienta KJ (2006) CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia 8:578–586
Lu Y, Xiao G, Galson DL, Nishio Y, Mizokami A, Keller ET, Yao Z, Zhang J (2007) PTHrP-induced MCP-1 production by human bone marrow endothelial cells and osteoblasts promotes osteoclast differentiation and prostate cancer cell proliferation and invasion in vitro. Int J Cancer 121:724–733
Zhang J, Lu Y, Pienta KJ (2010) Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst 102:522–528
Muthana M, Giannoudis A, Scott SD, Fang HY, Coffelt SB, Morrow FJ, Murdoch C, Burton J, Cross N, Burke B, Mistry R, Hamdy F, Brown NJ, Georgopoulos L, Hoskin P, Essand M, Lewis CE, Maitland NJ (2011) Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res 71:1805–1815
Satoh T, Saika T, Ebara S, Kusaka N, Timme TL, Yang G, Wang J, Mouraviev V, Cao G, el MA Fattah, Thompson TC (2003) Macrophages transduced with an adenoviral vector expressing interleukin 12 suppress tumor growth and metastasis in a preclinical metastatic prostate cancer model. Cancer Res 63:7853–7860
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science (New York NY) 275:964–967
Bertolini F, Shaked Y, Mancuso P, Kerbel RS (2006) The multifaceted circulating endothelial cell in cancer: towards marker and target identification. Nat Rev Cancer 6:835–845
Ruzinova MB, Schoer RA, Gerald W, Egan JE, Pandolfi PP, Rafii S, Manova K, Mittal V, Benezra R (2003) Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer cell 4:277–289
Duda DG, Cohen KS, Kozin SV, Perentes JY, Fukumura D, Scadden DT, Jain RK (2006) Evidence for incorporation of bone marrow-derived endothelial cells into perfused blood vessels in tumors. Blood 107:2774–2776
Mancuso P, Antoniotti P, Quarna J, Calleri A, Rabascio C, Tacchetti C, Braidotti P, Wu HK, Zurita AJ, Saronni L, Cheng JB, Shalinsky DR, Heymach JV, Bertolini F (2009) Validation of a standardized method for enumerating circulating endothelial cells and progenitors: flow cytometry and molecular and ultrastructural analyses. Clin Cancer Res 15:267–273
Georgiou HD, Namdarian B, Corcoran NM, Costello AJ, Hovens CM (2008) Circulating endothelial cells as biomarkers of prostate cancer. Nature clinical practice 5:445–454
Blann AD, Balakrishnan B, Shantsila E, Ryan P, Lip GY (2010) Endothelial progenitor cells and circulating endothelial cells in early prostate cancer: a comparison with plasma vascular markers. The Prostate
Strijbos MH, Gratama JW, Schmitz PI, Rao C, Onstenk W, Doyle GV, Miller MC, de Wit R, Terstappen LW, Sleijfer S (2010) Circulating endothelial cells, circulating tumour cells, tissue factor, endothelin-1 and overall survival in prostate cancer patients treated with docetaxel. Eur J Cancer 46:2027–2035
Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science (New York NY) 219:983–985
Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck TF, Pelletier N, Ferrara N (2001) Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem 276:3222–3230
Ebos JM, Kerbel RS (2011) Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nature reviews.
Bono AV, Pannellini T, Liberatore M, Montironi R, Cunico SC, Cheng L, Sasso F, Musiani P, Iezzi M (2010) Sorafenib’s inhibition of prostate cancer growth in transgenic adenocarcinoma mouse prostate mice and its differential effects on endothelial and pericyte growth during tumor angiogenesis. Analytical and quantitative cytology and histology/the International Academy of Cytology [and] American Society of Cytology 32:136–45
Welen K, Jennbacken K, Tesan T, Damber JE (2009) Pericyte coverage decreases invasion of tumour cells into blood vessels in prostate cancer xenografts. Prostate cancer and prostatic diseases 12:41–46
Ozerdem U (2006) Targeting of pericytes diminishes neovascularization and lymphangiogenesis in prostate cancer. Prostate 66:294–304
Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB, Oborn CJ, Park YY, Erez B, Jacoby JJ, Lee JS, Lin HY, Ciardiello F, Herbst RS, Langley RR, Heymach JV (2011) Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. The Journal of clinical investigation.
Edlund M, Sung SY, Chung LW (2004) Modulation of prostate cancer growth in bone microenvironments. J Cell Biochem 91:686–705
Folkman J (1971) Tumor angiogenesis: therapeutic implications. The New England journal of medicine 285:1182–1186
Czyzyk-Krzeska MF, Meller J (2004) von Hippel-Lindau tumor suppressor: not only HIF’s executioner. Trends in molecular medicine 10:146–149
Wang J, Wang J, Dai J, Jung Y, Wei CL, Wang Y, Havens AM, Hogg PJ, Keller ET, Pienta KJ, Nor JE, Wang CY, Taichman RS (2007) A glycolytic mechanism regulating an angiogenic switch in prostate cancer. Cancer research 67:149–159
Kai L, Wang J, Ivanovic M, Chung YT, Laskin WB, Schulze-Hoepfner F, Mirochnik Y, Satcher RL Jr, Levenson AS (2011) Targeting prostate cancer angiogenesis through metastasis-associated protein 1 (MTA1). Prostate 71:268–280
Bruni-Cardoso A, Johnson LC, Vessella RL, Peterson TE, Lynch CC (2010) Osteoclast-derived matrix metalloproteinase-9 directly affects angiogenesis in the prostate tumor-bone microenvironment. Mol Cancer Res 8:459–470
Littlepage LE, Sternlicht MD, Rougier N, Phillips J, Gallo E, Yu Y, Williams K, Brenot A, Gordon JI, Werb Z (2010) Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer research 70:2224–2234
Morrissey C, True LD, Roudier MP, Coleman IM, Hawley S, Nelson PS, Coleman R, Wang YC, Corey E, Lange PH, Higano CS, Vessella RL (2008) Differential expression of angiogenesis associated genes in prostate cancer bone, liver and lymph node metastases. Clinical & experimental metastasis 25:377–388
Emmenegger U, Francia G, Shaked Y, Kerbel RS (2010) Metronomic chemotherapy: principles and lessons learned from applications in the treatment of metastatic prostate cancer. Recent results in cancer research Fortschritte der Krebsforschung 180:165–183
Pasquier E, Kavallaris M, Andre N (2010) Metronomic chemotherapy: new rationale for new directions. Nature reviews 7:455–465
Dahut WL, Gulley JL, Arlen PM, Liu Y, Fedenko KM, Steinberg SM, Wright JJ, Parnes H, Chen CC, Jones E, Parker CE, Linehan WM, Figg WD (2004) Randomized phase II trial of docetaxel plus thalidomide in androgen-independent prostate cancer. J Clin Oncol 22:2532–2539
Ning YM, Gulley JL, Arlen PM, Woo S, Steinberg SM, Wright JJ, Parnes HL, Trepel JB, Lee MJ, Kim YS, Sun H, Madan RA, Latham L, Jones E, Chen CC, Figg WD, Dahut WL (2010) Phase II trial of bevacizumab, thalidomide, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer. J Clin Oncol 28:2070–2076
Bianchini D, Zivi A, Sandhu S, de Bono JS (2010) Horizon scanning for novel therapeutics for the treatment of prostate cancer. Ann Oncol 21(Suppl 7):vii43–vii55
Yu EM, Jain M, Aragon-Ching JB (2010) Angiogenesis inhibitors in prostate cancer therapy. Discovery medicine 10:521–530
Isaacs JT (2008) Prostate stem cells and benign prostatic hyperplasia. Prostate 68:1025–1034
Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6:2853–2868
Untergasser G, Gander R, Lilg C, Lepperdinger G, Plas E, Berger P (2005) Profiling molecular targets of TGF-beta1 in prostate fibroblast-to-myofibroblast transdifferentiation. Mech Ageing Dev 126:59–69
Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD, Rowley DR (2002) Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin Cancer Res 8:2912–2923
Campisi J (2011) Cellular senescence: putting the paradoxes in perspective. Curr Opin Genet Dev 21:107–112
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS (2010) The essence of senescence. Genes Dev 24:2463–2479
Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS (2006) The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res 66:794–802
Begley L, Monteleon C, Shah RB, Macdonald JW, Macoska JA (2005) CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell 4:291–298
Scotton CJ, Chambers RC (2007) Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest 132:1311–1321
Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G (2007) The myofibroblast: one function, multiple origins. Am J Pathol 170:1807–1816
Tuxhorn JA, McAlhany SJ, Dang TD, Ayala GE, Rowley DR (2002) Stromal cells promote angiogenesis and growth of human prostate tumors in a differential reactive stroma (DRS) xenograft model. Cancer Res 62:3298–3307
Schauer IG, Rowley DR (2011) The functional role of reactive stroma in benign prostatic hyperplasia. Differentiation.
Liao CP, Adisetiyo H, Liang M, Roy-Burman P (2010) Cancer-associated fibroblasts enhance the gland-forming capability of prostate cancer stem cells. Cancer Res 70:7294–7303
Singh H, Dang TD, Ayala GE, Rowley DR (2004) Transforming growth factor-beta1 induced myofibroblasts regulate LNCaP cell death. J Urol 172:2421–2425
Schauer IG, Ressler SJ, Tuxhorn JA, Dang TD, Rowley DR (2008) Elevated epithelial expression of interleukin-8 correlates with myofibroblast reactive stroma in benign prostatic hyperplasia. Urology 72:205–213
Schauer IG, Ressler SJ, Rowley DR (2009) Keratinocyte-derived chemokine induces prostate epithelial hyperplasia and reactive stroma in a novel transgenic mouse model. Prostate 69:373–384
Gerdes MJ, Larsen M, Dang TD, Ressler SJ, Tuxhorn JA, Rowley DR (2004) Regulation of rat prostate stromal cell myodifferentiation by androgen and TGF-beta1. Prostate 58:299–307
Franco OE, Jiang M, Strand DW, Peacock J, Fernandez S, Jackson RS 2nd, Revelo MP, Bhowmick NA, Hayward SW (2011) Altered TGF-beta signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res 71:1272–1281
Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR (1999) Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 59:5002–5011
Wang J, Levenson AS, Satcher RL Jr (2006) Identification of a unique set of genes altered during cell-cell contact in an in vitro model of prostate cancer bone metastasis. Int J Mol Med 17:849–856
Zhang S, Wang J, Bilen MA, Lin SH, Stupp SI, Satcher RL (2009) Modulation of prostate cancer cell gene expression by cell-to-cell contact with bone marrow stromal cells or osteoblasts. Clin Exp Metastasis 26:993–1004
Festuccia C, Giunciuglio D, Guerra F, Villanova I, Angelucci A, Manduca P, Teti A, Albini A, Bologna M (1999) Osteoblasts modulate secretion of urokinase-type plasminogen activator (uPA) and matrix metalloproteinase-9 (MMP-9) in human prostate cancer cells promoting migration and matrigel invasion. Oncol Res 11:17–31
Duivenvoorden WC, Hirte HW, Singh G (1999) Transforming growth factor beta1 acts as an inducer of matrix metalloproteinase expression and activity in human bone-metastasizing cancer cells. Clin Exp Metastasis 17:27–34
Festuccia C, Bologna M, Gravina GL, Guerra F, Angelucci A, Villanova I, Millimaggi D, Teti A (1999) Osteoblast conditioned media contain TGF-beta1 and modulate the migration of prostate tumor cells and their interactions with extracellular matrix components. Int J Cancer 81:395–403
Chiao JW, Moonga BS, Yang YM, Kancherla R, Mittelman A, Wu-Wong JR, Ahmed T (2000) Endothelin-1 from prostate cancer cells is enhanced by bone contact which blocks osteoclastic bone resorption. Br J Cancer 83:360–365
Guise TA, Yin JJ, Mohammad KS (2003) Role of endothelin-1 in osteoblastic bone metastases. Cancer 97:779–784
Akhavan A, McHugh KH, Guruli G, Bies RR, Zamboni WC, Strychor SA, Nelson JB, Pflug BR (2006) Endothelin receptor A blockade enhances taxane effects in prostate cancer. Neoplasia 8:725–732
Weydert CJ, Esser AK, Mejia RA, Drake JM, Barnes JM, Henry MD (2009) Endothelin-1 inhibits prostate cancer growth in vivo through vasoconstriction of tumor-feeding arterioles. Cancer Biol Ther 8:720–729
Murphy G (2005) Atrasentan for metastatic hormone refractory prostate cancer. Issues Emerg Health Technol:1–4
Nelson JB, Love W, Chin JL, Saad F, Schulman CC, Sleep DJ, Qian J, Steinberg J, Carducci M (2008) Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer 113:2478–2487
Shepard DR, Dreicer R (2010) Zibotentan for the treatment of castrate-resistant prostate cancer. Expert Opin Investig Drugs 19:899–908
Fu Z, Dozmorov IM, Keller ET (2002) Osteoblasts produce soluble factors that induce a gene expression pattern in non-metastatic prostate cancer cells, similar to that found in bone metastatic prostate cancer cells. Prostate 51:10–20
Reyes-Moreno C, Sourla A, Choki I, Doillon C, Koutsilieris M (1998) Osteoblast-derived survival factors protect PC-3 human prostate cancer cells from adriamycin apoptosis. Urology 52:341–347
Sun YX, Schneider A, Jung Y, Wang J, Dai J, Cook K, Osman NI, Koh-Paige AJ, Shim H, Pienta KJ, Keller ET, McCauley LK, Taichman RS (2005) Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res 20:318–329
Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, Kim JK, Patel LR, Ying C, Ziegler AM, Pienta MJ, Song J, Wang J, Loberg RD, Krebsbach PH, Pienta KJ, Taichman RS (2011) Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest.
Zhu X, Humphrey PA (2000) Overexpression and regulation of expression of scatter factor/hepatocyte growth factor in prostatic carcinoma. Urology 56:1071–1074
Humphrey PA, Zhu X, Zarnegar R, Swanson PE, Ratliff TL, Vollmer RT, Day ML (1995) Hepatocyte growth factor and its receptor (c-MET) in prostatic carcinoma. Am J Pathol 147:386–396
Naughton M, Picus J, Zhu X, Catalona WJ, Vollmer RT, Humphrey PA (2001) Scatter factor-hepatocyte growth factor elevation in the serum of patients with prostate cancer. J Urol 165:1325–1328
Hashem M, Essam T (2005) Hepatocyte growth factor as a tumor marker in the serum of patients with prostate cancer. J Egypt Natl Canc Inst 17:114–120
Gupta A, Karakiewicz PI, Roehrborn CG, Lotan Y, Zlotta AR, Shariat SF (2008) Predictive value of plasma hepatocyte growth factor/scatter factor levels in patients with clinically localized prostate cancer. Clin Cancer Res 14:7385–7390
Maeda A, Nakashiro K, Hara S, Sasaki T, Miwa Y, Tanji N, Yokoyama M, Hamakawa H, Oyasu R (2006) Inactivation of AR activates HGF/c-Met system in human prostatic carcinoma cells. Biochem Biophys Res Commun 347:1158–1165
Pisters LL, Troncoso P, Zhau HE, Li W, von Eschenbach AC, Chung LW (1995) c-met proto-oncogene expression in benign and malignant human prostate tissues. J Urol 154:293–298
Wang Y, Yue D, Li K, Liu YL, Ren CS, Wang P (2010) The role of TRPC6 in HGF-induced cell proliferation of human prostate cancer DU145 and PC3 cells. Asian J Androl 12:841–852
Nishimura K, Kitamura M, Takada S, Nonomura N, Tsujimura A, Matsumiya K, Miki T, Matsumoto K, Okuyama A (1998) Regulation of invasive potential of human prostate cancer cell lines by hepatocyte growth factor. Int J Urol 5:276–281
Nishimura K, Kitamura M, Miura H, Nonomura N, Takada S, Takahara S, Matsumoto K, Nakamura T, Matsumiya K (1999) Prostate stromal cell-derived hepatocyte growth factor induces invasion of prostate cancer cell line DU145 through tumor-stromal interaction. Prostate 41:145–153
Miura H, Nishimura K, Tsujimura A, Matsumiya K, Matsumoto K, Nakamura T, Okuyama A (2001) Effects of hepatocyte growth factor on E-cadherin-mediated cell-cell adhesion in DU145 prostate cancer cells. Urology 58:1064–1069
Steffan JJ, Williams BC, Welbourne T, Cardelli JA (2010) HGF-induced invasion by prostate tumor cells requires anterograde lysosome trafficking and activity of Na + −H + exchangers. J Cell Sci 123:1151–1159
Ye L, Lewis-Russell JM, Davies G, Sanders AJ, Kynaston H, Jiang WG (2007) Hepatocyte growth factor up-regulates the expression of the bone morphogenetic protein (BMP) receptors, BMPR-IB and BMPR-II, in human prostate cancer cells. Int J Oncol 30:521–529
Tsuka H, Mori H, Li B, Kanamaru H, Matsukawa S, Okada K (1998) Expression of c-MET/HGF receptor mRNA and protein in human non-malignant and malignant prostate tissues. Int J Oncol 13:927–934
Davies G, Mason MD, Martin TA, Parr C, Watkins G, Lane J, Matsumoto K, Nakamura T, Jiang WG (2003) The HGF/SF antagonist NK4 reverses fibroblast- and HGF-induced prostate tumor growth and angiogenesis in vivo. Int J Cancer 106:348–354
Dai Y, Siemann DW (2010) BMS-777607, a small-molecule met kinase inhibitor, suppresses hepatocyte growth factor-stimulated prostate cancer metastatic phenotype in vitro. Mol Cancer Ther 9:1554–1561
Elnagar AY, Sylvester PW, El Sayed KA (2011) (−)-Oleocanthal as a c-Met inhibitor for the control of metastatic breast and prostate cancers. Planta Med
Culig Z, Puhr M (2011) Interleukin-6: a multifunctional targetable cytokine in human prostate cancer. Mol Cell Endocrinol
George DJ, Halabi S, Shepard TF, Sanford B, Vogelzang NJ, Small EJ, Kantoff PW (2005) The prognostic significance of plasma interleukin-6 levels in patients with metastatic hormone-refractory prostate cancer: results from cancer and leukemia group B 9480. Clin Cancer Res 11:1815–1820
Alcover J, Filella X, Luque P, Molina R, Izquierdo L, Auge JM, Alcaraz A (2010) Prognostic value of IL-6 in localized prostatic cancer. Anticancer Res 30:4369–4372
Stark JR, Li H, Kraft P, Kurth T, Giovannucci EL, Stampfer MJ, Ma J, Mucci LA (2009) Circulating prediagnostic interleukin-6 and C-reactive protein and prostate cancer incidence and mortality. International journal of cancer Journal international du cancer 124:2683–2689
Chung TD, Yu JJ, Spiotto MT, Bartkowski M, Simons JW (1999) Characterization of the role of IL-6 in the progression of prostate cancer. Prostate 38:199–207
Alberti C (2010) Neuroendocrine differentiation in prostate carcinoma: focusing on its pathophysiologic mechanisms and pathological features. G Chir 31:568–574
Lee GT, Kwon SJ, Lee JH, Jeon SS, Jang KT, Choi HY, Lee HM, Kim WJ, Lee DH, Kim IY (2011) Macrophages induce neuroendocrine differentiation of prostate cancer cells via BMP6-IL6 Loop. The Prostate
Smith ND, Schulze-Hoepfner FT, Veliceasa D, Filleur S, Shareef S, Huang L, Huang XM, Volpert OV (2008) Pigment epithelium-derived factor and interleukin-6 control prostate neuroendocrine differentiation via feed-forward mechanism. J Urol 179:2427–2434
Okamoto M, Lee C, Oyasu R (1997) Autocrine effect of androgen on proliferation of an androgen-responsive prostatic carcinoma cell line, LNCaP: role of interleukin-6. Endocrinology 138:5071–5074
Qiu Y, Ravi L, Kung HJ (1998) Requirement of ErbB2 for signalling by interleukin-6 in prostate carcinoma cells. Nature 393:83–85
Wallner L, Dai J, Escara-Wilke J, Zhang J, Yao Z, Lu Y, Trikha M, Nemeth JA, Zaki MH, Keller ET (2006) Inhibition of interleukin-6 with CNTO328, an anti-interleukin-6 monoclonal antibody, inhibits conversion of androgen-dependent prostate cancer to an androgen-independent phenotype in orchiectomized mice. Cancer Res 66:3087–3095
Culig Z, Steiner H, Bartsch G, Hobisch A (2005) Interleukin-6 regulation of prostate cancer cell growth. J Cell Biochem 95:497–505
Corey E, Quinn JE, Buhler KR, Nelson PS, Macoska JA, True LD, Vessella RL (2003) LuCaP 35: a new model of prostate cancer progression to androgen independence. Prostate 55:239–246
Smith PC, Keller ET (2001) Anti-interleukin-6 monoclonal antibody induces regression of human prostate cancer xenografts in nude mice. Prostate 48:47–53
Chung TD, Yu JJ, Kong TA, Spiotto MT, Lin JM (2000) Interleukin-6 activates phosphatidylinositol-3 kinase, which inhibits apoptosis in human prostate cancer cell lines. Prostate 42:1–7
Lee SO, Lou W, Johnson CS, Trump DL, Gao AC (2004) Interleukin-6 protects LNCaP cells from apoptosis induced by androgen deprivation through the Stat3 pathway. Prostate 60:178–186
Rojas A, Liu G, Coleman I, Nelson PS, Zhang M, Dash R, Fisher PB, Plymate SR, Wu JD (2011) IL-6 promotes prostate tumorigenesis and progression through autocrine cross-activation of IGF-IR. Oncogene.
Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ (2009) CCL2 and interleukin-6 promote survival of human CD11b + peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem 284:34342–34354
Dorff TB, Goldman B, Pinski JK, Mack PC, Lara PN Jr, Van Veldhuizen PJ Jr, Quinn DI, Vogelzang NJ, Thompson IM Jr, Hussain MH (2010) Clinical and correlative results of SWOG S0354: a phase II trial of CNTO328 (siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin Cancer Res 16:3028–3034
Cavarretta IT, Neuwirt H, Zaki MH, Steiner H, Hobisch A, Nemeth JA, Culig Z (2008) Mcl-1 is regulated by IL-6 and mediates the survival activity of the cytokine in a model of late stage prostate carcinoma. Adv Exp Med Biol 617:547–555
Sabbota AL, Kim HR, Zhe X, Fridman R, Bonfil RD, Cher ML (2010) Shedding of RANKL by tumor-associated MT1-MMP activates Src-dependent prostate cancer cell migration. Cancer research 70:5558–5566
Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, Kanno T, Murakami A, Tsuda E, Morinaga T, Higashio K (1998) Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 139:1329–1337
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93:165–176
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J (1999) RANK is essential for osteoclast and lymph node development. Genes Dev 13:2412–2424
Kong YY, Boyle WJ, Penninger JM (1999) Osteoprotegerin ligand: a common link between osteoclastogenesis, lymph node formation and lymphocyte development. Immunol Cell Biol 77:188–193
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Boyle WJ et al (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319
Atkins GJ, Haynes DR, Graves SE, Evdokiou A, Hay S, Bouralexis S, Findlay DM (2000) Expression of osteoclast differentiation signals by stromal elements of giant cell tumors. J Bone Miner Res 15:640–649
Brown JM, Corey E, Lee ZD, True LD, Yun TJ, Tondravi M, Vessella RL (2001) Osteoprotegerin and rank ligand expression in prostate cancer. Urology 57:611–616
Chen G, Sircar K, Aprikian A, Potti A, Goltzman D, Rabbani SA (2006) Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer 107:289–298
Perez-Martinez FC, Alonso V, Sarasa JL, Manzarbeitia F, Vela-Navarrete R, Calahorra FJ, Esbrit P (2008) Receptor activator of nuclear factor-kappaB ligand (RANKL) as a novel prognostic marker in prostate carcinoma. Histol Histopathol 23:709–715
Zhang J, Dai J, Qi Y, Lin DL, Smith P, Strayhorn C, Mizokami A, Fu Z, Westman J, Keller ET (2001) Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest 107:1235–1244
Morrissey C, Kostenuik PL, Brown LG, Vessella RL, Corey E (2007) Host-derived RANKL is responsible for osteolysis in a C4-2 human prostate cancer xenograft model of experimental bone metastases. BMC Cancer 7:148
Zhang J, Dai J, Yao Z, Lu Y, Dougall W, Keller ET (2003) Soluble receptor activator of nuclear factor kappaB Fc diminishes prostate cancer progression in bone. Cancer Res 63:7883–7890
Luger NM, Honore P, Sabino MA, Schwei MJ, Rogers SD, Mach DB, Clohisy DR, Mantyh PW (2001) Osteoprotegerin diminishes advanced bone cancer pain. Cancer Res 61:4038–4047
Honore P, Luger NM, Sabino MA, Schwei MJ, Rogers SD, Mach DB, O’Keefe PF, Ramnaraine ML, Clohisy DR, Mantyh PW (2000) Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nat Med 6:521–528
Armstrong AP, Miller RE, Jones JC, Zhang J, Keller ET, Dougall WC (2008) RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate 68:92–104
Holen I, Croucher PI, Hamdy FC, Eaton CL (2002) Osteoprotegerin (OPG) is a survival factor for human prostate cancer cells. Cancer Res 62:1619–1623
Fizazi K, Lipton A, Mariette X, Body JJ, Rahim Y, Gralow JR, Gao G, Wu L, Sohn W, Jun S (2009) Randomized phase II trial of denosumab in patients with bone metastases from prostate cancer, breast cancer, or other neoplasms after intravenous bisphosphonates. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 27:1564–1571
Muir VJ, Scott LJ (2010) Denosumab: in cancer treatment-induced bone loss. BioDrugs 24:379–386
Smith MR, Egerdie B, Hernandez Toriz N, Feldman R, Tammela TL, Saad F, Heracek J, Szwedowski M, Ke C, Kupic A, Leder BZ, Goessl C (2009) Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 361:745–755
Fizazi K, Carducci M, Smith M, Damiao R, Brown J, Karsh L, Milecki P, Shore N, Rader M, Wang H, Jiang Q, Tadros S, Dansey R, Goessl C (2011) Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet 377:813–822
Kim J, Lahl K, Hori S, Loddenkemper C, Chaudhry A, deRoos P, Rudensky A, Sparwasser T (2009) Cutting edge: depletion of Foxp3+ cells leads to induction of autoimmunity by specific ablation of regulatory T cells in genetically targeted mice. J Immunol 183:7631–7634
Roato I, D’Amelio P, Gorassini E, Grimaldi A, Bonello L, Fiori C, Delsedime L, Tizzani A, De Libero A, Isaia G, Ferracini R (2008) Osteoclasts are active in bone forming metastases of prostate cancer patients. PloS one 3:e3627
Bruni-Cardoso A, Johnson LC, Vessella RL, Peterson TE, Lynch CC (2010) Osteoclast-derived matrix metalloproteinase-9 directly affects angiogenesis in the prostate tumor-bone microenvironment. Molecular cancer research: MCR 8:459–470
Juarez P, Guise TA (2010) Tgf-Beta pathway as a therapeutic target in bone metastases. Curr Pharm Des 16:1301–1312
Tomas D, Spajic B, Milosevic M, Demirovic A, Marusic Z, Kruslin B (2010) Intensity of stromal changes predicts biochemical recurrence-free survival in prostatic carcinoma. Scand J Urol Nephrol 44:284–290
Wang J, Ying G, Jung Y, Lu J, Zhu J, Pienta KJ, Taichman RS (2010) Characterization of phosphoglycerate kinase-1 expression of stromal cells derived from tumor microenvironment in prostate cancer progression. Cancer Res 70:471–480
Dai J, Hall CL, Escara-Wilke J, Mizokami A, Keller JM, Keller ET (2008) Prostate cancer induces bone metastasis through Wnt-induced bone morphogenetic protein-dependent and independent mechanisms. Cancer Res 68:5785–5794
Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR (2005) Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res 65:8887–8895
Pinzone JJ, Hall BM, Thudi NK, Vonau M, Qiang YW, Rosol TJ, Shaughnessy JD Jr (2009) The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 113:517–525
Bagnato A, Loizidou M, Pflug BR, Curwen J, Growcott J (2011) Role of the endothelin axis and its antagonists in the treatment of cancer. Br J Pharmacol 163:220–233
Lin Y, Wang F (2010) FGF signalling in prostate development, tissue homoeostasis and tumorigenesis. Biosci Rep 30:285–291
Hurle RA, Davies G, Parr C, Mason MD, Jenkins SA, Kynaston HG, Jiang WG (2005) Hepatocyte growth factor/scatter factor and prostate cancer: a review. Histol Histopathol 20:1339–1349
Tawara K, Oxford JT, Jorcyk CL (2011) Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag Res 3:177–189
Katiyar SK (2006) Matrix metalloproteinases in cancer metastasis: molecular targets for prostate cancer prevention by green tea polyphenols and grape seed proanthocyanidins. Endocr Metab Immune Disord Drug Targets 6:17–24
Fili S, Karalaki M, Schaller B (2009) Mechanism of bone metastasis: the role of osteoprotegerin and of the host-tissue microenvironment-related survival factors. Cancer Lett 283:10–19
Zhang X, Wang W, True LD, Vessella RL, Takayama TK (2009) Protease-activated receptor-1 is upregulated in reactive stroma of primary prostate cancer and bone metastasis. Prostate 69:727–736
Liao J, McCauley LK (2006) Skeletal metastasis: established and emerging roles of parathyroid hormone related protein (PTHrP). Cancer Metastasis Rev 25:559–571
Castellano D, Sepulveda JM, Garcia-Escobar I, Rodriguez-Antolin A, Sundlov A, Cortes-Funes H (2011) The role of RANK-ligand inhibition in cancer: the story of denosumab. Oncologist 16:136–145
Hirbe AC, Morgan EA, Weilbaecher KN (2010) The CXCR4/SDF-1 chemokine axis: a potential therapeutic target for bone metastases? Curr Pharm Des 16:1284–1290
Jones E, Pu H, Kyprianou N (2009) Targeting TGF-beta in prostate cancer: therapeutic possibilities during tumor progression. Expert Opin Ther Targets 13:227–234
Li Y, Cozzi PJ (2007) Targeting uPA/uPAR in prostate cancer. Cancer Treat Rev 33:521–527
Botelho F, Pina F, Lunet N (2010) VEGF and prostatic cancer: a systematic review. Eur J Cancer Prev 19:385–392
Robinson DR, Zylstra CR, Williams BO (2008) Wnt signaling and prostate cancer. Curr Drug Targets 9:571–580
Acknowledgements
This work was supported by National Cancer Institute Grant P01 CA093900.
Conflict of Interest
The authors declare that they have no conflict of interest with the organization that sponsored this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sottnik, J.L., Zhang, J., Macoska, J.A. et al. The PCa Tumor Microenvironment. Cancer Microenvironment 4, 283–297 (2011). https://doi.org/10.1007/s12307-011-0073-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-011-0073-8