
Abstract
Along with the rapid development of high-throughput sequencing technology, a large amount of multi-omics data sets are generated, which provide more opportunities to understand the mechanism of complex diseases. In this study, an improved particle swarm optimization with dynamic scale-free network, named DSFPSO, is proposed for detecting multi-omics features. The highlights of DSFPSO are the introduced scale-free network and velocity updating strategies. The scale-free network is employed to DSFPSO as its population structure, which can dynamically adjust the iteration processes. Three types of velocity updating strategies are used in DSFPSO for fully considering the heterogeneity of particles and their neighbors. Both gene function analysis and pathway analysis on colorectal cancer (CRC) data show that DSFPSO can detect CRC-associated features effectively.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
With the development of high-throughput sequencing technology, a vast amount of biological data of different categories have been generated by The Cancer Genome Atlas (TCGA). They provide us more opportunities to learn the biological mechanism of complex diseases [1].
Detecting features from biological data is an effective way to illuminate the underlying mechanism of diseases. A variety of feature extraction methods have been widely used to analyze the gene expression data. For instance, least absolute shrinkage and selection operator (LASSO), penalized matrix decomposition (PMD) and sparse principal component analysis (SPCA) are commonly used methods of feature extraction. Roth V. used the generalized LASSO method to feature selection problems for microarray data [2]. Liu carried differential expression analysis on RNA-seq count data based on PMD [3]. Lass et al. applied SPCA to clustering and feature selection problems [4]. Although LASSO, PMD and SPCA have achieved satisfactory performance on explaining the gene expression, they still have some defects in multi-omics feature extraction. These conventional feature extraction methods which can only identify genomic feature from single type of genomic feature cannot handle the integrated TCGA datasets.
Recently, many particle swarm optimization (PSO) based methods have been proposed for determining SNP-SNP interactions [5], gene features selection [6], and cancer classifications [7]. PSO is a population-based search algorithm of adaptive evolution, which proposed by Kennedy and Eberhart in 1995 [8]. Owing to its simple structure and fast convergence, PSO has become an important evolutionary algorithm. In recent years, numerous studies have been carried out to improve the performance of PSO. Kennedy and Mendes have conducted a deep research on population structure and particle behavior, founding that topology has a profound impact on particle behavior [9]. Liu et al. proposed SFPSO (Scale-Free PSO) [10]. Gao proposed SIPSO (Selectively-informed Particle Swarm Optimization), which employed scale-free network to simulate the population structure and greatly improved the optimization process [11]. The DMSPSO proposed by Zhao, used random dynamic changed population structure which greatly improved the ability of local search [12].
However, conventional improvement on PSO algorithm suffers from the limited particle population structure. For example, SFPSO and SIPSO generate the population structure before experiments which cannot embody the dynamic changes in the process of iteration. DMSPSO achieves the dynamic changes in population structure to a certain extent, but the population structure building becomes a completely random process which is unable to fit in with the actual optimization problems.
In this paper, we propose an improved PSO-based algorithm with dynamic scale-free network, named DSFPSO, to detect multi-omics features. The innovations of DSFPSO are the introduction of scale-free network and velocity updating strategies. We employ scale-free network as its population structure which can be dynamically adjusted in the process of iteration. Three types of velocity updating strategies are used in DSFPSO for fully considering the heterogeneity of particles and the connecting between neighbors. Specifically, to utilize the difference of gene expression based on different levels of multi-omics data, we employ the ranking function to extract the most effective gene features. To evaluate the validity of DSFPSO, experiments applied on CRC are handled by DSFPSO and other compared methods. The identified genes are appraised by gene function analysis and pathway analysis. Results show that the novel method can identify CRC-associated features effectively.
2 Methods
2.1 Standard PSO Algorithm
PSO is similar to other evolutionary algorithms which use the concepts of “groups” and “evolution” [13]. The speed of each particle can be dynamically adjusted according to the particle itself and its peers’ experience based on the fitness value. Based on the fitness of the position, each particle will move to a better place and obtain the optimal solution of optimization problems.
Standard PSO algorithm can be illustrated as follows.
-
Step1:
Initialize the particle velocity and position;
-
Step2:
Evaluate the fitness of each particle;
-
Step3:
Decide whether to update personal and group best positions by comparing the fitness;
-
Step4:
Update the position and speed of the particles;
-
Step5:
If not meet the ending condition, then return to Step2.
2.2 DSFPSO on Multi-omics Data
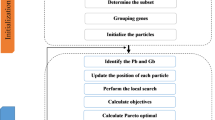
The flowchart of the proposed method is shown in Fig. 1. We will describe DSFPSO in details on six aspects.

The flowchart of DSFPSO.
2.2.1 Initializing Particles with Multi-omics Data
According to the characteristics of the omics data, we integrate the data as genomics and clinical information matrices. The whole genome matrix is the search space of particles while the clinical information matrix is used for the test of particle fitness.
Based on the above mapping of multi-omics data, the position of particle \( i \) at iteration \( t \) can be illustrated as
where \( I,K,T,M \) represents the number of particles, combination dimension of genomic features, iteration, and gene features in the genome datasets, respectively. \( x_{ik}^{t} \) is the selected genomic feature of particle \( i \) at iteration \( t \) in \( k \) dimensional space.
The speed of particle \( i \) at iteration \( t \) can be defined as
where \( v_{ik}^{t} \) is the speed of \( x_{ik}^{t} \).
Similarly, before the first iteration, \( Position_{t} (i) \), \( Velocity_{t} (i) \), \( Pbest_{t} (i) \), \( Neibest_{t} (i) \), \( Gbest_{t} (i) \) are assigned a random value in their domain respectively.
2.2.2 Analysis of the Fitness Function
Since mutual information does not need to assume the distribution of genomics data and can effectively measure the nonlinear relationship between genetic characteristics [14], we employ it as fitness function, which can be formulated as
Therefore, higher mutual information value denotes strong association between the genetic characteristic combination and the clinical information.
2.2.3 Updating the Dynamic Scale-Free Network
In order to fully utilize the properties of particles and experimental data, we have adopted a new strategy of link growth and selecting.
In one iteration, we make the out of network particles in fitness descending order and select new particles with higher fitness from these particles to join the network. Then these new particles will choose excellent neighbors from the network particles with the same sort processing.
In the dynamic process of scale-free network building, the particles position and population structure will be dynamically updated with the join of new particles in the solution space. Furthermore, we select the excellent new particles according to fitness value instead of the basic scale-free network adding new points without selection, which greatly improve the reliability of particles information exchange.
2.2.4 Updating the Particle Speed
In DSFPSO, the scale-free network building is synchronized with the solving iteration. Accordingly, particles have the difference of “in” and “out” of the network in the process of scale-free network building, so the two kinds of particles should be treated differently using different velocity updating strategies. The velocity updating equations can be formulated as
where \( \eta \) is learning rate, \( c_{1} \) and \( c_{2} \) are acceleration coefficients. \( w_{ik}^{t} \) is dynamic inertia weight balancing the capability between global and local search, \( rand(a,b) \) is random number between \( a \) and \( b \), \( N(i) \) denotes the neighbors of the particle \( i \), \( K_{i} \) is the number of neighbors for particle \( i \).
Based on the speed updating of particles, the position updating equation can be formulated as
2.2.5 Updating Personal Best Position, Neighbor Best Position and Group Best Position
In DSFPSO, particle’s personal best position will be updated by the position with the maximum mutual information. The specific equations can be formulated as
Similarly, the group best position updating equations can be written as
And the neighbor best position updating equations can be written as
2.2.6 Finding Final Results
In genomics data, each gene may have several genetic characteristics due to the differences of gene expression. In the results of DSFPSO, a gene may have a variety of genomic characteristics or may not. In this paper, we resort scoring strategies to extract gene features based on the score of gene expression [15]. The scoring function can be described as
where \( rank(i) \) represents the rank value of genomic features \( i \), \( n \) is the total order value of all the gene characteristics,\( G \) is the expression set of each gene.
3 Results
3.1 TCGA CRC Data
TCGA CRC data can be obtained from its web portal (https://tcga-data.nci.nih.gov/docs/publications/tcga/). Data used in this paper is the integrated data which has been preprocessed by Lee [16] (http://genomeportal.stanford.edu/tcga-crc/pages/datainformation). Considering the experiment needs, we carry discretization on somatic mutations and methylation data,which greatly improved the stability of the experiment.
The CRC data of TCGA used in this paper from 197 samples contains 5,188 genomic features of 1325 genes, including copy number variation, somatic mutations, methylation data and gene expression data (Fig. 2).

The CRC data of TCGA.
3.2 Gene Enrichment Analysis
ToppGene is a one-stop portal for gene list enrichment analysis and candidate gene prioritization based on functional annotations and protein interactions network.
To show the effectiveness of DSFPSO, we carry out GO enrichment analysis using ToppGene (https://toppgene.cchmc.org/enrichment.jsp) and compare the results on the same data set, including PSO,SIPSO, LASSO, PMD and SPCA. We input the top 500 genes identified by these methods into the ToppGene Suite, respectively, whose threshold value of the p-value is set to 0.001 and other parameters are set as default. Table 1 lists the top 10 closely related GO terms found by ToppGene. From this table, we can see that the term of “positive regulation of gene expression” has the lowest P-Value (9.38E-19), so it is considered as the most probable enrichment item. Furthermore, we notice that in the term of “regulation of multicellular organismal development” PSO outperforms DSFPSO and in the term of “regulation of transcription by RNA polymerase II” PMD outperforms DSFPSO. In general, DSFPSO shows better performance than SIPSO, PSO, LASSO, PMD and SPCA in majority results.
3.3 KEGG Pathway Analysis
KEGG (Kyoto Encyclopedia of Genes and Genomes) is a database which systematacially analyzes the function of gene to reveal the genetic and chemical blueprint of life [17].
In this study, we use DAVID (https://David-d.ncifcrf.gov/) on KEGG pathway to analyze the results. The top 10 CRC-associated pathways are shown in Table 2. Among them, Pathways in cancer and Colorectal cancer are obviously correlated with cancers. [18] indicates that PI3 K-Akt signaling pathway play an important role in inflammation-induced colorectal carcinogenesis. PI3 K-Akt signaling pathway links intimately with cellular metabolism and has great influence on cancer biological behavior [19]. The FoxO signaling pathway plays a central role in diverse physiological processes including cellular energy storage, growth and survival, among others [20]. [21] suggests that FOXO3a is a relevant mediator of the cytotoxic effects of cisplatin in colon cancer cells. Adherens junction pathway plays a critical role in cellular adhesion, glandular differentiation, and cellular proliferation. The function of this pathway correlated proteins is compromised in a number of intestinal diseases, including ulcerative colitis that has an increased incidence for colorectal cancer [22].
3.4 Analysis of Gene Function
In order to evaluate the algorithm’s performance and explore the correlation between genes and the pathogenesis of colorectal cancer, we carry out detailed analysis on 10 CRC-related genes among top identified 50 genes. The gene function descriptions are shown in Table 3.
CSMD1 alterations can correlate with earlier clinical presentation in colorectal tumors, thus further implicating CSMD1 as a tumor suppressor gene [23]. Loss of CSMD1 may contribute to the poor prognosis of colorectal cancer patients [24]. [25] indicates that KBTBD11 influences colorectal cancer risk, especially in interaction with an MYC-regulated SNP rs6983267. WRN promoter methylation connects mucinous differentiation, microsatellite instability and CpG island methylator phenotype in colorectal cancer [26]. SUZ12 mRNA expression in the CRC tissues is significantly increased than in the non-cancerous tissue. Increased SUZ12 mRNA expression is directly correlated with primary tumor size, regional lymph node metastases, distant metastasis and AJCC stage. Furthermore, CRC patients with higher level of SUZ12 showed a worse disease-free survival (DFS) [27]. CDX2 is mutated in a colorectal cancer with normal APC/β-catenin signaling [28, 29] shows that CDX2 specifies intestinal development and homeostasis and is considered a tumor suppressor in colorectal carcinogenesis.
4 Conclusions
Considering traditional PSO algorithms usually take equal treatment of all particles and ignore the disadvantages related to the heterogeneity of population structure, we propose an improved PSO algorithm named as DSFPSO to identify gene features of complex diseases. This algorithm dynamically adjusts population structure according to the particles status in the process of iteration.
With fitness of particles as a standard for preferred link selection, DSFPSO realizes the true meaning of PSO for dynamic scale-free network. Moreover,this is the first time for PSO algorithm introduced into multi-omics data analysis with CRC data provided by TCGA as the experiment data and filtering results through scoring strategies. Experimental results show that DSFPSO can be convergent to global optimization quickly and find CRC-associated genes, which will provide valid references for early diagnosis, effective treatment and prognostic guidance of colorectal cancer. To explore correlations among differentially expressed genes is left as our future work.
References
Bersanelli, M., Mosca, E., Remondini, D., et al.: Methods for the integration of multi-omics data: mathematical aspects. BMC Bioinformatics 17(2), S15 (2016)
Roth, V.: The generalized LASSO: a wrapper approach to gene selection for microarray data. Sekretariat für Forschungsberichte, Inst. für Informatik III (2002)
Liu, J.X., Gao, Y.L., Xu, Y., et al.: Differential expression analysis on RNA-Seq count data based on penalized matrix decomposition. IEEE Trans. Nanobiosci. 13(1), 12–18 (2014)
Luss, R., d’Aspremont, A.: Clustering and feature selection using sparse principal component analysis. Optim. Eng. 11(1), 145–157 (2010)
Zhang, W., Shang, J., Li, H., Sun, Y., Liu, J.X.: SIPSO: Selectively Informed Particle Swarm Optimization Based on Mutual Information to Determine SNP-SNP Interactions. In: Huang, D.-S., Bevilacqua, V., Premaratne, P. (eds.) ICIC 2016. LNCS, vol. 9771, pp. 112–121. Springer, Cham (2016). https://doi.org/10.1007/978-3-319-42291-6_11
Chuang, L.Y., Chang, H.W., Tu, C.J., et al.: Improved binary PSO for feature selection using gene expression data. Comput. Biol. Chem. 32(1), 29–38 (2008)
Kar, S., Sharma, K.D., Maitra, M.: Gene selection from microarray gene expression data for classification of cancer subgroups employing PSO and adaptive K-nearest neighborhood technique. Expert Syst. Appl. 42(1), 612–627 (2015)
He, R.: An improved particle swarm optimization based on self-adaptive escape velocity. J. Softw. 16(12), 2036–2044 (2005)
Kennedy, J., Mendes, R.: Population structure and particle swarm performance. In: Proceedings of the 2002 Congress on evolutionary computation 2002 CEC 2002, vol. 2, pp. 1671–1676. IEEE (2002)
Liu, C., Du, W.B., Wang, W.X.: Particle swarm optimization with scale-free interactions. PLoS ONE 9(5), e97822 (2014)
Gao, Y., Du, W., Yan, G.: Selectively-informed particle swarm optimization. Sci. Rep. 5, 9295 (2015)
Zhao, S.Z., Liang, J.J., Suganthan, P.N., et al.: Dynamic multi-swarm particle swarm optimizer with local search for large scale global optimization. In: IEEE Congress on 2008 Evolutionary Computation (IEEE World Congress on Computational Intelligence), pp. 3845–3852. IEEE (2008)
Shi, Y.: Particle swarm optimization: developments, applications and resources evolutionary computation. In: Proceedings of the 2001 Congress on 2001, vol. 1, pp. 81–86. IEEE (2001)
Shang, J., Sun, Y., Li, S., et al.: An improved opposition-based learning particle swarm optimization for the detection of SNP-SNP interactions. Biomed Res. Int. 2015, 524821 (2015)
Liu, J.X., Gao, Y.L., Zheng, C.H., et al.: Block-constraint robust principal component analysis and its application to integrated analysis of TCGA data. IEEE Trans. Nanobiosci. 15(6), 510–516 (2016)
Lee, H.J., Flaherty, P., Ji, H.P.: Systematic genomic identification of colorectal cancer genes delineating advanced from early clinical stage and metastasis. BMC Med. Genomics 6(1), 1 (2013)
Kanehisa, M., Araki, M., Goto, S., et al.: KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36(suppl_1), D480–D484 (2007)
Li, N., Bu, X., Tian, X., et al.: Fatty acid synthase regulates proliferation and migration of colorectal cancer cells via HER2-PI3 K/Akt signaling pathway. Nutr. Cancer 64(6), 864–870 (2012)
Josse, C., Bouznad, N., Geurts, P., et al.: Identification of a microRNA landscape targeting the PI3 K/Akt signaling pathway in inflammation-induced colorectal carcinogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 306(3), G229–G243 (2014)
Zhang, Y., Gan, B., Liu, D., et al.: FoxO family members in cancer. Cancer Biol. Ther. 12(4), 253–259 (2011)
De Mattos, S.F., Villalonga, P., Clardy, J., et al.: FOXO3a mediates the cytotoxic effects of cisplatin in colon cancer cells. Mol. Cancer Ther. 7(10), 3237–3246 (2008)
Mees, S.T., Mennigen, R., Spieker, T., et al.: Expression of tight and adherens junction proteins in ulcerative colitis associated colorectal carcinoma: upregulation of claudin-1, claudin-3, claudin-4, and β-catenin. Int. J. Colorectal Dis. 24(4), 361–368 (2009)
Webb, E.L., Rudd, M.F., Sellick, G.S., et al.: Search for low penetrance alleles for colorectal cancer through a scan of 1467 non-synonymous SNPs in 2575 cases and 2707 controls with validation by kin-cohort analysis of 14 704 first-degree relatives. Hum. Mol. Genet. 15(21), 3263–3271 (2006)
Zhang, R., Song, C.: Loss of CSMD1 or 2 may contribute to the poor prognosis of colorectal cancer patients. Tumor Biol. 35(5), 4419–4423 (2014)
Gong, J., Tian, J., Lou, J., et al.: A polymorphic MYC response element in KBTBD11 influences colorectal cancer risk, especially in interaction with a MYC regulated SNP rs6983267. Ann. Oncol. 29(3), 632–639 (2018)
Kawasaki, T., Ohnishi, M., Suemoto, Y., et al.: WRN promoter methylation possibly connects mucinous differentiation, microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod. Pathol. 21(2), 150 (2008)
Liu, Y.L., Gao, X., Jiang, Y., et al.: Expression and clinicopathological significance of EED, SUZ12 and EZH2 mRNA in colorectal cancer. J. Cancer Res. Clin. Oncol. 141(4), 661–669 (2015)
Da Costa, L.T., He, T.C., Yu, J., et al.: CDX2 is mutated in a colorectal cancer with normal APC/β-catenin signaling. Oncogene 18(35), 5010 (1999)
Brabletz, T., Spaderna, S., Kolb, J., et al.: Down-regulation of the homeodomain factor Cdx2 in colorectal cancer by collagen type I: an active role for the tumor environment in malignant tumor progression. Cancer Res. 64(19), 6973–6977 (2004)
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 61502272, 61572284); Project of Shandong Province Higher Educational Science and Technology Program (J18KA373); the Scientific Research Foundation of Qufu Normal University (BSQD20130119); the Science and Technology Planning Project of Qufu Normal University (xkj201524).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Ethics declarations
The authors declare that there is no conflict of interests regarding the publication of this paper.
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this paper

Cite this paper
Li, H., Li, SJ., Shang, J., Liu, JX., Zheng, CH. (2018). An Improved Particle Swarm Optimization with Dynamic Scale-Free Network for Detecting Multi-omics Features. In: Zhang, F., Cai, Z., Skums, P., Zhang, S. (eds) Bioinformatics Research and Applications. ISBRA 2018. Lecture Notes in Computer Science(), vol 10847. Springer, Cham. https://doi.org/10.1007/978-3-319-94968-0_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-94968-0_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-94967-3
Online ISBN: 978-3-319-94968-0
eBook Packages: Computer ScienceComputer Science (R0)