
Abstract
As precise phenotyping is essential and the cost of generating phenotyping data at every generation is very expensive, recent advances in genomics technologies and the availability of a wide range of genotyping platforms have made the cost of genotyping much less expensive compared with phenotyping. The recent developments in sequencing technologies have manifold increased the repertoire of various types of markers that are available in chickpea including SSRs, SNPs, DArTs, hundreds of thousands transcript reads and BAC-end sequences saturated genetic maps, QTL maps as well as physical maps, and the sequencing of both kabuli and desi type has greatly helped in using marker-assisted technologies to be applied in plant breeding. Germplasm resequencing for identification of genome-wide SNPs and their subsequent utilization in genomic selection has the potential to break the yield barrier being experienced in chickpea and many other crops. Genomic-assisted breeding for marker-assisted backcrossing (MABC) for introgressing QTL region, marker-assisted recurrent selection, gene pyramiding, marker-assisted selection (MAS), and genomic selection can now be taken up in chickpea. The conventional plant breeding should take these tools to make greater genetic gains, increase selection potential, and have faster breeding cycles so that the genetic improvement gains are increased in chickpea.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Chickpea
- Genomic-assisted breeding (GAB)
- Marker-assisted backcrossing (MABC)
- QTL
- Marker-assisted recurrent selection (MARS)
- Gene pyramiding
- Marker-assisted selection (MAS)
- Genomic selection
13.1 Introduction
One of the most pressing problems of the world today is adequate nutrition for exploding human population as 870 million people go hungry worldwide (http://www.fao.org). The growing world population together with the lack of expansion or even reduction of available arable lands needed to maintain agricultural sustainability implies that the relative importance of plant breeding to raise crop yield potential and adaptiveness is now greater than in the past (Araus et al. 2002). Plant adaptation is a key factor that will determine the future severity of the effects of climatic change on food grain production. Relatively inexpensive changes, such as shifting planting dates or switching to an existing crop variety, may moderate the negative impact of climatic change. However, improvements in crop productivity to meet the requirement of growing demand mentioned above will not be easy without further technological breakthroughs that allow yield ceilings to be shifted through the development of new crop varieties (Rosenzweig and Parry 1994).
In India, from time immemorial, many legumes and pulses have been consumed as part of a primarily cereal-based diet. In the last 50 years, per capita availability of pulses has been steadily coming down from 70 grams/capita/day to 34 grams/capita/day (http://www.faostat3.fao.org). This is solely on account of stagnant crop yields. Annual pulse production has barely crawled in the last 23 years from 13.3 million tonnes in 1985–1986 to 17.8 million tonnes at present. On the other hand, for the majority vegetarian population of India, the sole protein supplement is getting out of reach with a nutritional calamity looming large. The major challenges impeding the pulse production and productivity are limited genetic diversity in the primary gene pool, genotype and environment interaction, multiple biotic and abiotic stresses, and limited screening methods for the precise phenotyping of the target traits (Kumar et al. 2014).
Chickpea, an oldest and widely cultivated pulse crop of the world, commonly known as Bengal gram or Garbanzo (Cicer arietinum L.). It is a highly self-pollinated crop, with 2n = 2x = 16 (Arumuganathan and Earle 1991) and genome size of 738 MB with an estimated 28,269 genes (Varshney et al. 2013b). It belongs to genus Cicer, tribe Cicereae, family Fabaceae, and subfamily Papilionaceae. It originated in southeastern Turkey (Ladizinsky and Adler 1976). The name Cicer is of Latin origin, derived from the Greek word “kikus” meaning force or strength. Nutritionally, chickpeas contain 23% protein, 64% total carbohydrates (with 47% starch and 6% soluble sugar), 5% fat, 6% crude fiber, and 3% ash. It is also a rich source of vitamins, minerals like phosphorus, calcium, magnesium, iron, zinc and dietary fiber or Non-Starch Polysaccharides (NSP). Chickpea is grown widely for diverse uses throughout the Indian subcontinent, Mediterranean basin, the Middle East, and Africa and is becoming an important legume crop in new regions like Australia and North America, because of its nutritional value, diversified uses, and ability to grow better with low inputs under harsh edaphic and arid environments than many other crops. It is an important component of the cropping system of subsistence farmers in the Indian subcontinent, West Asia and North Africa. Some of these attributes together with its ability to derive more than 70% of its nitrogen from symbiotic nitrogen fixation (Saxena et al. 1988) make chickpea a promising crop for sustainable agriculture.
Chickpea is a major rabi pulse with significant contribution toward pulse economy of the world. It is cultivated on 13.2 million hectares of land with 11.62 million tonnes of production across the world. India is the largest producer of chickpea with an area of 7.58 million hectares which produces 8.32 million tonnes of chickpea (FAOSTAT 2012; Agriculture statistics at a glance 2013). India accounts nearly 70% at a global level and 75% at Asian level in terms of world’s chickpea area and production. In spite of being major producer, Indian productivity (912 kg ha−1) is very low as compared to other major chickpea-growing countries like Myanmar (1407 kg/ha), Ethiopia (1549 kg/ha), USA (1825 kg/ha), Canada (1825 kg/ha), and Mexico (1500 kg/ha). Productivity enhancement in chickpea can help to release this negative trade balance as it contributes more than 45% to Indian pulse economy. Many approaches have been advocated for much needed chickpea productivity enhancement which can create additional genetic variation especially for traits of economic importance and enable effective utilization of available germplasm in chickpea improvement programs for enhanced and sustained chickpea production across the continents. Various biotic and abiotic stresses hamper chickpea production. Among the biotic stresses, Ascochyta blight, Fusarium wilt, Helicoverpa pod borer, and Botrytis gray mold are very important yield reducers. Drought, heat, cold, and salinity stress are the major abiotic factors that significantly affect chickpea yields.
As almost all the traits with agronomic values are genetically complex, which are affected by many genes, environments, and their interactions (Cramer et al. 2011; Grishkevich and Yanai 2013), identification of involved genetic factors such as quantitative trait loci (QTL) has been playing a vital role in manipulating the traits of interest and understanding of genetic architecture (Holland 2007; Xu 2010). However, conventional breeding requires assaying all the individuals for the target traits collected from a sample population making it expensive and time-consuming and needs to be supplemented with genomic-assisted breeding (GAB) (Varshney et al. 2005, 2007). Due to lack of chickpea genomic information until recently, it was considered an orphan legume for implementing GAB. On the other hand, recent advancements in the comparative genomics and genomic approaches have generated the genome sequence and genomic resources transforming chickpea to a resource-rich crop similar to other major food crops (Thudi et al. 2014).
13.2 Genomic-Assisted Breeding for Abiotic Stress Tolerance in Chickpea
13.2.1 MAS for Drought Tolerance
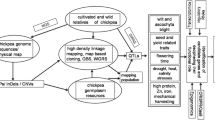
Being stable, unaffected by environment, and easily assessable with no disparity of growth and developmental stages, molecular markers are now considered ideal for diversity studies, QTL identification, fingerprinting, gene tagging, constructing linkage maps, positional cloning, evolutionary studies, and marker-assisted selection (Bharadwaj et al. 2010; Shubha et al. 2011; Pooja et al. 2014; Chaudhary et al. 2014; Maqbool et al. 2016). The very first chickpea genetic map based on SSR markers was developed by Winter et al. (1999), and they reported 174 paired primers. NIPGR (The National Institute for Plant Genome Research, India) developed 280 SSR markers with the help of microsatellite enrichment (Sethy et al. 2006). SSR markers developed for chickpea have been used for genetic map construction (Radhika et al. 2007; Bhardwaj et al. 2002; Shefali et al. 2015), assessment of interspecific genetic diversity (Udupa et al. 1999; Bharadwaj et al. 2010; Yadav et al. 2011), QTL mapping for agronomic parameters (Udupa and Baum 2003; Subodh et al. 2015), and assessment of drought tolerance (Maqbool et al. 2016).
Chickpea accession ICC 4958 was used for development of novel 311 SSR primers (Nayak et al. 2010). Expressed sequence tags (ESTs) have also been mined for SSR primers (Varshney et al. 2009). Tentative unique sequences (TUSs) were used for designing 3172 SSR paired primers out of which 728 were nonredundant SSR paired primers, after identification of 26,252 SSR sequences (Hiremath et al. 2011). BAC libraries were used by Lichtenzveig et al. (2005) for development of 233 SSR markers. Thudi et al. (2011) designed 1344 SSR primers after identification of 6845 SSRs by mining of 46,270 BAC-end sequences. Heuttel et al. (1999) developed 28 SSR primers, Winter et al. (1999) developed 174 SSR primers, Sethy et al. (2003) developed 10, Sethy et al. (2006) developed 85, Qadir et al. (2007) developed 63, and Nayak et al. (2010) developed 311 SSR primers for chickpea. The recent genome sequencing project of chickpea enabled the discovery of 81,845 SSRs, of which 48,298 SSRs were found to be suitable for development of SSR primers for PCR amplification (Varshney et al. 2013b). These SSR markers are exclusively being used for improvement of drought tolerance in chickpea. “QTL hotspot” has been successfully introgressed into the genetic background of the elite varieties JG11, KAK2, and Chefe. Three SSR markers (TAA170, ICCM0249, and STMS11) were used for foreground selection, and 10 amplified fragment length polymorphism (AFLP) primer combinations were used for background selection after each generation of backcrossing while introgressing “QTL hotspot” into JG 11 genetic background. A total of 29 introgression lines were developed with ~93% recurrent parent genome recovery after three backcross cycles followed by two generations of selfing (Varshney et al. 2013c). The introgression lines developed from JG11 x ICC 4958 were found to possess higher root length density, root dry weight, and rooting depth compared to both the donor and recipient parents; these are the most important target traits for enhancing drought tolerance in chickpea (Varshney et al. 2013a, b). Furthermore, preliminary analysis of phenotypic evaluation of these lines in India (Patancheru, Dharwad, Nandyal, Durgapura, and Gulbarga), Kenya, and Ethiopia indicated that several lines with >10% increase in yield under rain-fed conditions and ~20% increase in yield under irrigated conditions were available. Based on the preliminary results, other national partners like IIPR, IARI in India, and Egerton University (Kenya) and the Ethiopian Institute of Agricultural Research (Ethiopia) in sub-Saharan Africa initiated introgressing this region into genetic backgrounds of elite cultivars in their regions. The RILs of ICC 4958 X Annigeri have been extensively studied for root traits. An SSR marker (TAA 170) was identified for a major QTL that accounted for 33% of the variation for root weight and 33% of the variation for root length (Chandra 2006).
For efficient and effective germplasm management and conservation, the concept of core and minicore collections have been advocated (Upadhyaya and Ortiz 2001), and trait-specific germplasm has been identified to aid breeding and genomic-assisted selection (Upadhyaya et al. 2012; Meena et al. 2010). Further attempts were also made to characterize the chickpea germplasm at the molecular level in several studies (Iruela et al. 2002; Croser et al. 2003; Nguyen et al. 2004; Rao et al. 2007; Upadhyaya et al. 2008; Sefera et al. 2011; Choudhary et al. 2012a; Sarika et al. 2014) separately from phenotypic characterization (Krishnamurthy et al. 2013a, 2013b; Tapan et al. 2015; Supriya et al. 2017; Neeraj et al. 2016). Characterization of chickpea germplasm for targeted trait-specific germplasm and genomic-assisted selection (Upadhyaya et al. 2012; Roorkiwal et al. 2013) has been reported by numerous researchers. Trait mapping and TILLING approach based on next-generation sequencing (Thudi et al. 2014) have been undertaken to identify genes involved in drought tolerance.
Screening of the chickpea minicore collection for root traits was recently conducted in two different seasons with the double objective of characterizing the genetic variability of drought-avoidance root traits and selecting suitable mapping population parents for molecular mapping of these traits. The complete minicore germplasm collection of C. arietinum (211 accessions) along with five popular cultivars as references (216 total entries) were evaluated in PVC cylinders in the first season, along with an additional seven popular cultivars and ten accessions of wild annual species (233 total entries) in the second season. The statistical differences of entries were significant (P < 0.001) for both root and shoot traits (Serraj et al. 2004). The root and shoot growth of the wild species was relatively poor compared to C. arietinum lines. Some of the germplasm accessions with deep root systems were ICC1356, ICC 3512, ICC4872, ICC13523, and ICC15697. Germplasm accession ICC8261 had the highest root length density and an extremely high root/shoot ratio and rooting depth in both seasons. ICC4958 which is previously the only source used as a deep and large root system parent or control in most drought avoidance studies was confirmed to be an extremely prolific rooting genotype. The new genotypes identified can be used as valuable alternative sources for diversification of mapping populations with varying growth duration and to obtain the required polymorphism for successfully mapping root traits in chickpea.
Accumulation of more superior alleles through marker-assisted recurrent selection (MARS) has also been adopted for enhancing the level of drought tolerance (Varshney et al. 2012) that increases the frequency of numerous desirable alleles having additive effects in recurrent crosses (Bernardo and Charcosset 2006). MARS has proven to be successful in private breeding programs in enhancing genetic gains and is effective at improving quantitative traits in maize (Zea mays L.), soybean and sunflower (Helianthus annuus L.) (Johnson 2003; Eathington et al. 2007). In brief, MARS is a modern breeding approach that enables us to increase the frequency of several beneficial alleles with an additive effect and small individual effects in recurrent crosses (Bernardo and Charcosset 2006). Although several multinational companies are using MARS in crops like maize and soybean, only a few public sector institutes have started to use MARS in crops like wheat (Charmet et al. 2001), sorghum (Sorghum bicolor (L.) Moench), barley (Abdallah Oukarroum et al. 2009), and rice (Grenier et al. 2012). The use of MARS In chickpea breeding has been reported only at ICRISAT. Four elite “desi” chickpea genotypes were used in pairwise crossing for pyramiding desirable alleles which subsequently led to elite genotypes with enhanced drought tolerance (Thudi et al. 2014). The four superior desi genotypes were selected based on their performance: ICCV 04112, ICCV 05107, ICCV 93954 (released as JG 11 in India), and ICCV 94954 (released as JG 130 in India). Two crosses were made by using elite by elite lines (JG 11 ICCV 04112 and JG 130 ICCV 05107). To pyramid the superior alleles of the favorable QTLs identified based on F3 genotyping data and F5 phenotyping data (from Ethiopia, Kenya and India), a set of eight lines were selected for each cross using OptiMAS ver. 1.0 (Valente et al. 2013). It is anticipated that at the end of the project, RC3F4 progenies will be available for evaluation at multiple locations. These efforts will lead to the development of superior lines with more enhanced drought tolerance. Some efforts have been initiated to use MARS in the case of chickpea for assembling favorable alleles for drought tolerance using ICCV 04112 ICCV 93954 and ICCV 05107 ICCV 94954 crosses. Nevertheless, IARI and IIPR also have initiated MARS in chickpea by using Pusa 372 JG 130 and DCP 92–3 ICCV 10 crosses. These efforts are expected to develop superior lines with enhanced drought tolerance for other ecological regions.
Nevertheless, for understanding the genetics of complex traits like drought tolerance, trait mapping is essential for identifying the genes underlying drought tolerance. Based on the evaluation of the minicore collection for terminal drought tolerance, germplasm lines with prolific root systems were identified, and three recombinant inbred line mapping populations (Annigeri, ICC 4958, ICC 4958 ICC 1882, and ICC 283 ICC 8261) were developed at ICRISAT (Gaur et al. 2008). Comprehensive QTL analysis has provided several stable, consistent, and robust main-effect QTLs for 13 out of 20 drought tolerance traits explaining 10–58.20% of phenotypic variation (Varshney et al. 2014b). Markers flanking these QTLs can be deployed for enhancing drought tolerance as well as individual trait improvement through MABC breeding. A genomic region referred to as “QTL hotspot,” spanning ~29 cM on Cicer arietinum Linkage Group 04 (CaLG04) of an intraspecific genetic map (ICC 4958 ICC 1882), was found to harbor 12 out of 25 main-effect QTLs for 12 traits explaining ~58.20% of phenotypic variation (Varshney et al. 2014b). Seven SSR markers (ICCM0249, NCPGR127, TAA170, NCPGR21, TR11, GA24, and STMS11) present in QTL hotspot are the most important markers for marker-assisted introgression of this genomic region into elite genetic backgrounds for enhancing drought tolerance through MABC. The data were analyzed for the estimation of genetic components of variance for root traits. These mapping populations are expected to facilitate identification of markers for additional QTLs for root traits.
Similarly, several other mapping populations were also developed for gaining insights into most prevalent biotic and abiotic stresses (Gaur et al. 2014). Next-generation multiparent advanced generation intercross (MAGIC) population is one of a next-generation multiple mapping population comprising of 4–20 parents in cross-combination and a good source of increasing genetic variability. A group of eight chickpea genotypes from different origins was used as parents for the development of a MAGIC population at ICRISAT. Using MAGIC population is beneficial because inclusion of several parents ensures the segregation of QTLs for multiple traits, deployment for understanding complex traits, and the discovery and characterization of novel genes (Glaszmann et al. 2010).
Sequence information and identification of novel genes for agronomically important traits can be done using a number of methods including EST databases (Sreenivasulu et al. 2002). Sanger sequencing and next-generation sequencing techniques have been used for transcriptomic studies of chickpea. Initially EST abundance was assessed for tissue-specific expression, stress-responsive expression, and development-related expression. Chickpea genotypes were grown under drought and salt stresses, and complementary DNA libraries were generated which comprised 20,162 ESTs (Varshney et al. 2009). Another transcriptomic library comprising of 103,215 transcripts (Hiremath et al. 2011) and 53,409 contigs (Garg et al. 2011) have been generated for drought responsiveness. Gene discovery is very limited in chickpea, and few efforts have been made to identify the ESTs associated with stress responses through transcriptomic studies (Varshney et al. 2009). Jain and Chattopadhyay (2010) studied the transcript profiling differences between two chickpea genotypes under different drought treatments and concluded that highly expressing ESTs in tolerant genotypes were encoding proteins involved in transcription, signal transduction, protein metabolism, and cellular organization. Differential downregulation and upregulation of transcriptome has been reported by Deokar et al. (2011) in tolerant and susceptible chickpea genotypes subjected to drought stress. In silico expression studies were also done to know the differential expression of tolerant and susceptible chickpea genotypes under drought stress (Varshney et al. 2009).
Microarray, suppression subtractive hybridization, EST sequencing, and super serial analysis of gene expression (SAGE) have been used for functional genomic analysis of chickpea genotypes in stress-responsive conditions (Buhariwalla et al. 2005; Matsumura et al. 2005, and Molina et al. 2008). The drought- and salinity-responsive transcriptome of chickpea was evaluated using the SuperSAGE technique, reporting that 3000 transcripts were responsive to drought and salinity stresses (Kahl et al. 2007). Transcriptome analysis of chickpea roots was carried out using deep SuperSAGE (combination of next-generation sequencing techniques with SAGE) under normal and water stress conditions, and 17,493 unique transcripts were identified which were drought responsive (Molina et al. 2008). Comprehensive transcriptome analyses demonstrated that osmolyte accumulation, transcription regulation, signal transduction, and ROS scavenging were remodeled under drought stress and were therefore potential target phenomena for improvement of drought tolerance (Molina et al. 2008).
Furthermore, for creating novel alleles and for functional validation of candidate drought-responsive genes, a “target-induced local lesions in genome” (TILLING) population, comprising 10,000 M2 chickpea lines, was also developed by ICRISAT and IARI. A next-generation sequence-based TILLING approach is being adopted to mine novel and potential alleles for some genes associated with terminal drought tolerance (ICRISAT, unpubl. data). Application of marker-assisted selection (MAS) for drought tolerance is still low with little success reported (Oyier 2012; Varshney 2016). The selection based on markers flanking the identified genomic regions in chickpea is expected to accelerate efforts in breeding drought-tolerant varieties. Twenty introgression lines (IL4s) of chickpea harboring the root QTL hotspot from the donor parent ICC 4958 were phenotyped for root and morphological traits under rain-fed conditions. Absence of differences among the ILs for morphological traits indicates similar genetic background of ILs being derived through marker-assisted backcrossing. Marker analysis of the 20 ILs showed presence of the recurrent parent allele in most of the ILs with root QTL.
13.2.1.1 Genomic Selection Approaches for Drought Tolerance
As precise phenotyping is essential and the cost of generating phenotyping data at every generation is very expensive, recent advances in genomics technologies and the availability of a wide range of genotyping platforms have made the cost of genotyping much less expensive compared with phenotyping. Genomic selection is a modern breeding approach that is unlike MABC and MARS; it predicts the breeding values (i.e., the genomic as estimated breeding values) of lines based on historical phenotyping data and the genotyping data. Genomic selection has proven to be successful in several animal breeding programs (Schefers and Weigel 2012; Eggen 2012) as well as in crop plants like maize (Zhao et al. 2012). Efforts to deploy genomic selection in chickpea are underway at ICRISAT. In this regard, a collection of 320 elite breeding lines was selected as the “training population.” In addition to compiling historical phenotyping data for ~10 years at >10 locations, research has extensively phenotyped the training population for several traits of agronomic importance at ICRISAT (Patancheru) and IARI (New Delhi) during the cropping season of 2011–2012 and 2012–2013 under rain-fed and irrigated conditions. In parallel, the training population was genotyped using KBioscience Competitive Allele-Specific Polymerase chain reaction (KASPar) assays (651) and diversity array technology (15,360 features). Collected phenotypic data and generated genome-wide marker profiling data (>3000 markers) were used with a range of statistical methods including ridge regression-best linear unbiased prediction, kinship-based ridge regression, BayesCp, BayesB, Bayesian least absolute shrinkage and selection operator (LASSO), and random forest prediction to predict genomic-estimated breeding values (Roorkiwal et al. 2013). Resequencing of the germplasm lines and parents of different mapping populations will enable the identification of genome-wide single-nucleotide polymorphism (SNP) markers that can be effectively utilized in genomic selection.
13.2.1.2 Future Perspectives
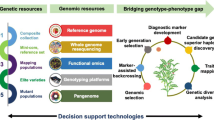
As drought is a complex phenomenon, no single approach for all locations may be applicable for enhancing drought tolerance. In this context, an integrated effort deploying need-based approaches is essential. Furthermore, for accelerating the adoption of the molecular breeding for enhanced drought tolerance in chickpea, the development of markers that are easily assayable and technically less demanding, and that do not require high capital equipment for genotyping, termed “breeder-friendly markers,” is essential. For instance, conversion of SNPs to Illumina Veracode, cleaved amplified polymorphic sequences or KASPar assays will enable their wider application in breeding programs. In addition, the development of decision support tools is essential for enhancing the precision of selection and to accelerate GAB in crop plants in general. In this area, ICRISAT has developed several important user-friendly decision support tools like the integrated SNP mining and utilization pipeline, the molecular breeding design tool, and the genotyping data management system. Several other tools that aid in genomic-assisted selection have been integrated and made available on an integrated breeding platform (https://www.integratedbreeding.net/molecular-breeding, accessed 6 June 2014). Further well-structured molecular breeding programs are essential for the effective deployment of GAB approaches for crop improvement (Varshney et al. 2013d). To achieve this, training in modern plant breeding skills and fostering integrated breeding strategies and sharing of knowledge and expertise among collaborative partners, especially in developing countries with limited infrastructure and human resources, are the needs of the hour.
13.2.1.3 Candidate Genes for Abiotic Stress Tolerance
Research has revealed several genes are known to be involved in salinity tolerance; the association analysis based on candidate gene sequencing approach is meagerly reported. The salinity-tolerant candidate genes which are supposed to play an important role include ASR (abscisic acid stress and ripening gene), DREB (drought-responsive element binding proteins), ERECTA, SuSy (sucrose synthase), DHN, AKIN, CAD, EREBP, LEA, and Myb transcription factor.
Abscisic acid Stress and Ripening (ASR) gene-ASR is a stress-inducible gene that plays also plays major role in fruit ripening and maturation, has been reported exclusively in plants. Iusem et al. (1993) reported the first Asr gene from cultivated tomato, and since then Asr genes have been found in various species of dicotyledonous and monocotyledonous plants. They also play a vital role in abiotic stress mechanisms like drought, salt, cold, and limited light (Schneider et al. 1997; Huang et al. 2000; Maskin et al. 2001; Jeanneau et al. 2002; Kalifa et al. 2004). Stress endurance through induction of stress-related genes was reported for the dehydration-responsive element binding proteins (DREB) transcription factor. Both the forms, DREB1 and DREB2, are reported to be involved in two separate signal transduction pathways under low temperature and dehydration, respectively. They belong to the ERF (ethylene-responsive element binding factors) family of transcription factors. The roles of DREB proteins in biotic and abiotic stress tolerance were reviewed in detail by Agarwal et al. (2006). DREB (Dehydration response element binding) homologue in chickpea was also amplified using primer pairs designed using unigene showing match against DREB gene (Srinivasan et al. 1999; Amit et al. 2011). About 1200 bp amplicon for DREB was reported by Roorkiwal et al. (2012).
The ERECTA gene codes for a protein kinase receptor, one of a very large and complex family of signaling molecules called protein kinases, and their receptors, which mediates plants responses to disease, predation, and stress. ERECTA regulates leaf organogenesis and reduces the density of stomata on the leaf under-surface, hence reduces the evapotranspiration. In Arabidopsis, the ERECTA gene has been shown to control organ growth and flower development by promoting cell proliferation (Shpak et al. 2004). In Arabidopsis ERECTA gene is known to be involved in inflorescence development and organ growth by promoting cell proliferation. Transgenic Arabidopsis plants that ectopically overexpress the ERECTA gene improve plant transpiration efficiency and drought tolerance by affecting stomatal density, epidermal cell expansion, mesophyll cell proliferation, and cell contact. In addition, Masle et al. (2005) isolated Arabidopsis ERECTA gene, a putative leucine-rich repeat receptor-like kinase that regulated transpiration efficiency located on Arabidopsis chromosome 2. The ERECTA gene can change both leaf stomatal number and leaf structure, and regulate the flowering time, and is proved to regulate plant transpiration efficiency and consequently to have a bright prospect in improving crop drought resistance and using water at high efficiency. The role of the ERECTA gene was identified by screening Arabidopsis inbred lines and mutant plants, thereby identifying the ERECTA homologues in both dicot and monocot crop species. The contribution of ERECTA gene toward water use efficiency was confirmed using complementation assays on wilting mutant Arabidopsis plants (Masle et al. 2005). The transformation of ERECTA gene in the crop species would be major breakthrough in the area of agriculture, with respect to drought tolerance and agronomic performance.
Sucrose synthase (SuSy) and sucrose phosphate synthase (SPS)—sucrose synthase and the sucrose phosphate synthase are the key enzymes involved in the sugar metabolism pathway. SuSy enzyme belongs to the family of glycosyltransferases, especially, hexosyltransferases. It is also commonly known as UDP glucose-fructose glucosyltransferase, sucrose synthase, and uridine diphosphoglucose-fructose glucosyltransferase. The enzyme sucrose synthase (UDP-D- glucose: D-fructose 2a-glucosyltransferase) catalyzes the reversible conversion of sucrose uridine diphosphate into fructose and UDP-glucose. S-adenosylmethionine synthetase1 gene homologue in chickpea, primers were designed using contig sequence showing similarity against S-adenosylmethioninesynthetase 1 (SAM1) gene of Arabidopsis thaliana. PCR amplification yielded about 300 bp amplicons across eight chickpea genotypes. Serine/threonine protein kinase (STPK) gene homologue was amplified using the STPK specific primer pair designed considering unigene sequence having similarity with Arabidopsis thaliana putative serine/threonine protein kinase. Amplicon size of STPK gene approximately 450 bp STPK has been shown to play an important role in response to abiotic stress response and seed development in peanut (Rudrabhatla and Rajasekharan 2004).
Although the reaction is reversible, it is thought that the enzyme is mainly involved in the breakdown of sucrose (Huber and Huber 1996; Geigenberger and Stitt 1993; Geigenberger et al. 1995). Hence the activity of sucrose synthase can be important in controlling either starch or cellulose biosynthesis by supplying UDP-glucose as a precursor or as an immediate substrate (Chourey et al. 1991; Delmer and Amor 1995). Ingram et al. (1997) reported the isolation and characterization of cDNA clones encoding SPS from Craterostigma plantagineum, a resurrection plant in which the accumulation of sucrose is considered to play an important role in tolerance to severe protoplasmic dehydration. It is suggested that the overall regulation of SPS is strongly influenced by the changing composition of the cytoplasm in C. plantagineum leaves during the dehydration-rehydration cycle.
Researchers have isolated the AKIN homologues in various plant species including Arabidopsis, rice, potato, and tobacco and established their role in abiotic stress response (Purcell et al. 1998). AKIN homologue was amplified using AKIN specific primer pair designed considering unigene sequence showing match with Arabidopsis AKIN (SNF-1-related protein kinase) with approximate amplicon size of 1100 bp. Amplification of AMADH (aminoaldehyde dehydrogenase) homologue yielded a product of ~1200 bp (Roorkiwal and Sharma 2012). Protective/curative role of AMADH gene in response to stress events caused by mechanical injury has been reported earlier in pea seedlings (Petrivalsky et al. 2007).
DHNs are one of several proteins that have been specifically associated with qualitative and quantitative changes in cold hardiness (Close 1996). Dehydrin homologue was amplified using primer pair designed for known dehydrin gene using chickpea unigene with approximate amplicon size of 380 bp (Roorkiwal and Sharma 2012).
The role of plant Myb proteins has been well characterized by using different genetic approaches. In most of the cases, the Myb domain binds to a specific DNA sequence (C/TAACG/TG) to facilitate transcriptional activation (Biedenkapp et al. 1988). Myb genes were amplified using unigene sequence showing match against Glycine max Myb transcription factor (Roorkiwal and Sharma 2012).
Cinnamyl-alcohol dehydrogenase (CAD) gene homologue was isolated from eight chickpea genotypes using primers designed for contig showing match with cinnamyl-alcohol dehydrogenase (CAD) gene of Arabidopsis thaliana (Roorkiwal and Sharma 2012). CAD is expected to play a key role in plant defense against various abiotic and biotic stresses (Raes et al. 2003). For isolation of ethylene-responsive element binding protein (EREBP) gene homologue in chickpea, primers were designed using contig sequence showing similarity against ethylene-responsive transcription factor from Arabidopsis thaliana. Amplification carried out across eight chickpea genotypes produced about 400 bp amplicons (Roorkiwal and Sharma 2012). The AP2/EREBP genes play various roles in developmental processes and in stress-related responses in plants.
Late embryogenesis abundant (LEA) genes represent a gene family that plays important role in vegetative tissues in response to drought, salinity, cold stress, and exogenous application of abscisic acid (Dure et al. 1989). Primers designed using contig showing sequence similarity with LEA domain-containing protein Arabidopsis thaliana were used to isolate late embryogenesis abundant (LEA) gene in chickpea with approximate amplicon size of 600 bp (Roorkiwal and Sharma 2012).
13.2.2 Salinity
Large land tracts worldwide are being degraded due to salinity, particularly in irrigated areas estimating to about 20 percent (Neeraj et al. 2016). Every day for more than 20 years, an average of 2000 hectares of irrigated land in arid and semiarid areas across 75 countries have been degraded by salt, according to a study by UN University’s Canadian-based Institute for Water, Environment and Health (UNU-INWEH 2014). Higher salt concentrations severely affected germination, root and shoot length, and water uptake in chickpea (Neeraj et al. 2016). There is degradation and lysis of germinated root in such salt soils in chickpea. NaCl has more impact than Na2SO4. The seeds of chickpea for both landrace have a maximum tolerant level of salinity with 10dS/m. At this salt concentration, significant effect is seen in the seeds compared to the control. But at concentration of 15ds/m of Na2 SO4, the germination and growth of seeds are severely affected, and only few seeds start to germinate or raise shoot and root, which dried later (Haileselasie and Teferii 2012).
The tolerance to salinity by chickpea clearly indicated that the sodium to potassium ion concentrations was lower in tolerant lines as compared to sensitive lines. Pod abortion was higher in the salt-sensitive genotypes. However, no effect was seen on pollen viability, in vitro pollen germination, and in vivo pollen tube growth (Turner et al. 2013). The tolerant lines exhibited greater pod number and biomass compared to saline sensitive lines.
Vadez et al. (2007) reported a strong relationship (r 2 = 0.50) between the seed yield and salinity. In a study on the seedling parameters in a diverse set of chickpea genotypes under saline stress vs normal conditions, Neeraj et al. 2016 reported maximum reduction in seedling roots weight when germinated in saline conditions. The roots play a major role in establishment of seedling and stem growth, and the highly susceptible lines failed to germinate in saline soils. There was an overall decrease in seedling characters like seedling shoot weight and root biomass. The resistant checks CSG 8962 and JG 62 along with the lines ICCV 00104 and ICCV 06101 showed minimum reduction in seedling characters under salt stress conditions. The yield under saline stress showed a positive correlation with all physiological parameters like RWC and MSI but negative correlation with Na:K ratio under salt stress condition. The traits like higher mean seed yield per plant under saline stress, higher pods per plant, higher RWC, higher MSI, and a low stem Na:K ratio are associated with tolerance to salinity in chickpea. Greater genetic gains can be obtained by using these parameters in selection for salinity tolerance.
Only few studies have reported the presence of QTLs for salinity tolerance with sufficiently large marker interval (Samineni 2016 and Vadez et al. 2012). The narrow genetic base in chickpea further limits the efforts to develop stress-tolerant cultivars. The identification of genes associated with salinity stress responses can greatly facilitate the development of improved chickpea cultivars with enhanced salinity tolerance using molecular breeding approaches. The availability of large-scale genomic resources is essential for understanding the biology of complex abiotic stress-like salinity. The availability of next-generation sequencing technologies provides a high-throughput means to study gene expression profiles at the whole-genome level (Garg et al. 2016; Roorkiwal et al. 2014). It has been realized that comparative differential gene expression analysis between genotypes with contrasting response to the stresses can provide a better understanding of the molecular mechanisms underlying tolerance and provide better candidate gene information involved in imparting tolerance to salt stress (Cotsaftis et al. 2011; Lenka et al. 2011). A total 46 QTLs for salinity tolerance was identified using mapping population from ICCV 2 x JG 11. Out of 49 QTLs, 19 QTLs were for phonological traits (7 QTL for days to flowering and 12 QTLs for days to maturity) and 27 QTLs for yield and yield-related traits. Minor QTLs were detected for harvest index (HI) on CaLG04d in salinity treatment, while finding of controlled experiment revealed CaLG07 harbors QTLs for yield, pod number, filled pod number, and seed number (Pushpavalli et al. 2015). QTLs for salinity tolerance are located in the genomic region of CaLG05 flanked by two makers, i.e., CaM0463 and ICCM 272, which contained 17 main QTLs for seven traits (DF, DM, ADM, stem and leaf weight, 100-seed weight, HI, and yield). Genomic region on CaLG07 contains seven QTLs for five different traits, viz., DF, DM, seed number, pod number, and yield. Genomic region on CaLG08 contained eight QTLs for three traits DF, DM, and HI. Out of the abovementioned genomic regions, CaLG05 and CaLG07 genomic regions were most important as they contained QTLs for traits that were remarkably related to yield under salt stress conditions (Puspavalli et al. 2015).
13.2.3 Cold/Chilling
Chilling and freezing are the two categories under the cold stress. The genetic response of genotypes to different stresses appears to be mostly common (Seki et al. 2002). Pollen sterility and flower abortion are the most common effects of cold stress in chickpea when it occurs in the reproductive phase. Tolerance to cold stress becomes all the more important in West Asia and North Africa, Australia, Europe, Canada, and Northern India. Freezing (mean daily temperature < −1.5 °C) and chilling temperatures (mean daily temperature between −1.5 and 15 °C) are known to affect chickpea at various stages of development from germination to maturation (Croser et al. 2003). In these climates and late sown crop of Northern India, chilling temperature in the vegetative stage and cold stress at flowering due to sudden frost greatly reduce the yield of the crop. The breeding procedures focusing on development of cultivars for these regions need to target cold tolerance both at seedling and flowering stages. Screening of germplasm at ICARDA has identified several cold-tolerant lines from the cultivated (Singh et al. 1995) and wild species (Robertson et al. 1995).
Flower abortion due to cold stress at temperatures of 15 °C and below are reported in Australia (Siddique and Sedgeley 1986), the Mediterrranean (Singh 1993), and India (Savithri et al. 1980; Srinivasan et al. 1998). Flower abortion due to cold stress in chickpea is associated with lower levels of sucrose, glucose, and fructose in anthers and pollen (Nayyar et al. 2005). Total sugars and starch were found to be higher in cold-tolerant genotypes compared to the susceptible ones whereas oxidative stress was low (Kumar et al. 2014). Sharma and Nayyar (2014) analyzing a total of 9205 EST bands in cold-tolerant chickpea genotype ICC16349 found that the cold stress altered expression of 127 ESTs (90 upregulated, 37 downregulated) in anthers. Ninety-two of these (two third proportion) were novel with unknown protein identity and function. The remaining about one third (35) belonged to several functional categories such as pollen development, signal transduction, ion transport, transcription, carbohydrate metabolism, translation, energy, and cell division. Limited genes were involved in regulating cold tolerance in chickpea anthers. Moreover, the cold tolerance was manifested by upregulation of majority of the differentially expressed transcripts. The anthers appeared to employ dual cold tolerance mechanism based on their protection from cold by enhancing triacylglycerol and carbohydrate metabolism and maintenance of normal pollen development by regulating pollen development genes. Functional characterization of about two third of the novel genes is needed to have precise understanding of the cold tolerance mechanisms in chickpea anthers (Sharma and Nayyar 2014). Chilling temperatures during early reproductive growth cause yield losses in chickpea in parts of the Indian sub-continent and Australia. The plants continue to produce flowers but fail to set pods when mean daily temperature falls below 150 C. ICRISAT scientists have developed several breeding lines (e.g., ICCV 88502, ICCV 88503, ICCV 88506, ICCV 88510, ICCV 88516) that are able to set pods at lower temperature (mean daily temperature between 12and 15 °C). A pollen selection method was developed in Australia and applied to transfer chilling tolerance from ICCV 88516 to chilling sensitive cultivars, leading to development and release of chilling-tolerant cultivars Sonali and Rupali (Clarke and Siddique 2004). RFLP markers for chilling tolerance were identified and subsequently converted to SCAR markers. These were used successfully to select chilling-tolerant progeny from a cross between Amethyst and ICCV 88516 but were ineffective in other crosses (Millan et al. 2006).
13.3 Genomic-Assisted Breeding for Biotic Stress Tolerance in Chickpea
13.3.1 Fusarium Wilt
Among the biotic stresses that caused major damage to chickpea production, Fusarium wilt caused by Fusarium oxysporum f.sp. ciceris is the major yield reducer. Losses to the tune of up to 90% have been reported by Singh (1993). Havare and Neme (1982) have given the race distribution of this pathogen in India. Of the eight races, they identified only IA, 2, 3, and 4 to be prevalent in India. Lines with resistance to this pathogen have been identified and have been used in crop improvement programs. The line WR315 among all the resistant donors is most widely used and has been reported to be resistant to all the races except race 3 (Haware et al. 1997). Molecular markers have been identified for most of the Foc genes (Table 13.1). It has been observed that most of these resistant genes are present in two different linkage groups on different chromosomes, while Teresa Millan et al. (2006) mostly reported it to be present on linkage group 2. Tekeoglu et al. (2000) also reported some of these genes to be present on linkage group 3 too. Improvement of Pusa 256 through marker-assisted backcrossing for introgression of Foc 2 gene using TA37 and TA110 was done by Pratap et al. (2017). Similarly, C214 was improved for resistance against race 1 by Varshney et al. (2014a). Bharadwaj et al. (2011) also reported identification of markers linked to resistance for different races of Fusarium wilt and development of introgression lines in a Generation Challenge Project (GCP) along with ICRISAT
13.3.2 Ascochyta Blight
The chickpea-growing regions of higher latitudes, i.e., colder regions with cooler, cloudy, and humid weather during reproductive stages, get affected by Ascochyta blight (AB). Different workers have reported different pathotypes and subtypes for this disease. Udupa et al. (1998) have reported three pathotypes while Nene and Reddy (1987) reported five pathotypes. Resistant lines have been identified, and some of these like ILC 3279, ILC 195, ILC 482, and ILC 72 developed by ICARDA have been released as varieties. Molecular markers for AB-resistant QTLs and molecular markers linked to them by large number of workers (Table 13.1). The marker-assisted breeding program using the identified QTLs has just been initiated at ICAR-Indian Institute of Plant Research (IIPR), Kanpur; Punjab Agricultural University (PAU), Ludhiana; and ICAR-Indian Agricultural Research Institute (IARI), New Delhi.
13.3.3 Botrytis Gray Mold
In the recent past, Botrytis gray mold (BGM) is emerging as an important disease of chickpea in the eastern part of Indian subcontinent including Nepal and Bangladesh and North India including Pakistan and Australia (Haware and Mc Donald 1992). This fungus has a very wide host range, and the source of absolute resistance has not been found in Cicer arietinum L. germplasm (Pande et al. 2001). Three QTLs were mapped on two linkage groups LG3 and LG6 by Anuradha et al. (2011). Resistance to this pathogen has been identified in wild Cicer sp. Cicer bijugum (Haware et al. 1992). Punjab Agricultural University (PAU), Ludhiana, has developed some lines through pre-breeding having tolerance/resistance to Botrytis gray mold. Comprehensive evaluation of these lines, their derivatives, and previously reported sources in field and in controlled conditions for their level of tolerance/resistance is being done by ICAR-Indian Institute of Plant Research (IIPR), Kanpur; Punjab Agricultural University (PAU), Ludhiana; ICAR-Indian Agricultural Research Institute (IARI), New Delhi; and G.B. Pant University of Agriculture and Technology, GBPUA&T (Pantnagar).
13.4 Molecular Markers
The most recent quindecennial (2002–2017) has seen the advancement of substantial level of genomic assets in chickpea. Simple sequence repeats (SSR) markers, most favored markers for molecular breeding, were accessible in exceptionally predetermined number in this crop until 2005. Paucity of polymorphic molecular markers in chickpea (Cicer arietinum L.) has been a major limitation in the improvement of this important legume. However it is not so anymore. The concerted efforts by chickpea workers and generous funding and efforts by Indian Council of Agricultural Research (ICAR), Generation Challenge Programme, The Bill and Melinda Gates Foundation, Department of Biotechnology (DBT), etc. have led to the development of large-scale molecular markers, construction of comprehensive linkage map, and draft genome sequencing. ICRISAT, NIPGR, and NRCPB have been in forefront in the development of marker repertoire (Sethy et al. 2006, Varshney et al. BMC Genomics 2009). 2000 genomic SSR markers chickpea have been developed (Varshney et al. 2007; Nayak et al. 2010; Thudi 2011), ESTs (Varshney et al. 2009), 454/FLX transcript reads (Hiremath et al. 2011; Garg et al. 2011,) and BAC-end sequences (Thudi et al. 2011). 26,082 potential SNPs have been identified (Hiremath et al. 2011) based on alignment of ~37 million Illumina/Solexa tags. Similarly, at National Institute of Plant Genome Research (NIPGR), a set of 487 novel functional markers including 125 EST-SSRs, 151 intron-targeted primers (ITPs), 109 expressed sequence tag polymorphisms (ESTPs), and 102 SNP markers has been developed (Choudhary et al. 2012b). Though DArT markers were developed in pigeon pea, their use was mostly restricted to introgression studies as these were very less polymorphic in the cultivated pigeon pea (Thudi et al. 2011). KASPar assays for 2005 SNPs in chickpea (Hiremath et al. 2012) were developed. High throughput SNP genotyping platform utilizing DArT and next generation sequencing (NGS) technology like pyrosequencing (Alderborn et al. 2000; Ching and Rafalski 2002; Varshney et al. 2009), mass spectrum analysis (Rodi et al. 2002), Affymetrix chip (Borevitz et al. 2003), Golden Gate assay (Fan et al. 2003; Rostoks et al. 2006), Roche 454/FLX, AB Biosystem, and Illumina/Solexa are used for whole-genome transcription identification techniques to spot genomic regions and genes underlying plant stress responses (Varshney et al. 2009; Varshney et al. 2010) to develop massive scale SNPs and using for genotyping to develop highly saturated genetic and transcript maps (Gujaria et al. 2011). Approximately 15300 (by DArT Pvt. Ltd, Australia And ICRISAT)DArT are available in chickpea featuring 21500 array, 300 panel resulted in 5400 polymorphic features and ~200 maker loci on genetic map (Varshney et al. 2010) (Table 13.2).
13.5 Conclusion
Advances in sequencing and genotyping technologies helped in generation of several thousand markers including SSRs, SNPs, and DArTs and hundreds of thousands transcript reads and BAC-end sequences in chickpea. Comprehensive transcriptome assemblies and genome sequences have either been developed or underway for other important traits including quality, herbicide tolerance, salinity, etc. Based on these resources, dense genetic maps, QTL maps as well as physical maps for chickpea have also been developed. As a result, chickpea graduated from “orphan” or “less-studied” crops to “genomic resource-rich” crops. Genomic-assisted breeding approaches in the form of marker-assisted selection (MAS) and marker-assisted backcrossing (MABC) for introgressing QTL region for drought tolerance-related traits; Fusarium wilt resistance and Ascochyta blight resistance in chickpea have also been initiated. However, it is critical to use other modern breeding approaches like marker-assisted recurrent selection (MARS), advanced-backcross (AB-backcross) breeding, and genomic selection (GS) to utilize the full potential of genomic-assisted breeding for crop improvement.
References
Agarwal PK, Agarwal P, Reddy MK, Sopory SK (2006) Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Rep 25(12):1263–1274
Alderborn A, Kristofferson A, Hammerling U (2000) Determination of single-nucleotide polymorphisms by real-time pyrophosphate DNA sequencing. Genome Res 10(8):1249–1258
Allen DJ, Lenné JM (1998) The pathology of food and pasture legumes. CAB International, New York
Anuradha C, Gaur PM, Pande S, Gali KK, Ganesh M, Kumar J, Varshney RK (2011) Mapping QTL for resistance to botrytis grey mould in chickpea. Euphytica 182(1):1–9
Araus JL, Slafer GA, Reynolds MP, Royo C (2002) Plant breeding and drought in C3 cereals: what should we breed for? Ann Bot 89(7):925–940
Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Report 9(3):208–218
Bernardo R, Charcosset A (2006) Usefulness of gene information in marker-assisted recurrent selection: a simulation appraisal. Crop Sci 46(2):614–621
Bharadwaj C, Chauhan SK, Rajguru G, Srivastava R, Satyavathi T, C Yadav S, Rizvi AH, Kumar J, Solanki RK (2010) Diversity analysis of chickpea (Cicer arietinum) cultivars using STMS markers. J Agri Sci 9:947
Bharadwaj C, Tripathi S, Varshney R, Kumar J (2011) Deployment of molecular markers for developing high yielding wilt resistance chickpea genotypes. In: Theme 2: 2.5: Integrated crop breeding. Poster Abstracts. 2011. General Research Meeting, Generation Challenge Programme. 21–25 September, 2011, Hyderabad, India. P13
Bharadwaj C, Srivastava R, Chauhan SK, Satyavathi CT, Kumar J, Faruqui A, Yadav S, Rizvi AH, Kumar T (2013) Molecular diversity and phylogeny in geographical collection of chickpea (Cicer sp.) accessions. J Genet 92(2):94–100
Bhardwaj C, Chauhan SK, Yadav S, Satyavathi TC, Singh R, Kumar J, Srivastava R, Rajguru G (2002) Molecular marker based linkage map of chickpea (Cicer arietinum) developed from desi× kabuli cross 81(2):116–118
Biedenkapp H, Borgmeyer U, Sippel AE, Klempnauer KH (1988) Viral myb oncogene encodes a sequence-specific DNA-binding activity. Nature 335(6193):835–837
Borevitz JO, Liang D, Plouffe D, Chang HS, Zhu T, Weigel D, Berry CC, Winzeler E, Chory J (2003) Large-scale identification of single-feature polymorphisms in complex genomes. Genome Res 13(3):513–523
Buhariwalla HK, Jayashree B, Eshwar K, Crouch JH (2005) Development of ESTs from chickpea roots and their use in diversity analysis of the Cicer genus. BMC Plant Biol 5(1):16
Chandra, S., Buhariwalla, H. K., Kashiwagi, J., Harikrishna, S., Sridevi, K. R., Krishnamurthy, L., Serraj, R., Crouch, J. H. Identifying QTL-linked markers in marker-deficient crops T. Fisher (Ed.), Proceedings of the 4th international crop science congress, Brisbane, Australia, 26 September–1 October 2004 2006
Charmet G, Robert N, Perretant MR, Gay G, Sourdille P, Groos C, Bernard S, Bernard M (2001) Marker assisted recurrent selection for cumulating QTLs for bread-making related traits. Euphytica 119:89–93. https://doi.org/10.1023/A:1017577918541
Ching A, Rafalski A (2002) Rapid genetic mapping of ESTs using SNP pyrosequencing and indel analysis. Cell Mol Biol Lett 7(2B):803–810
Cho S, Chen W, Muehlbauer FJ (2004) Pathotype-specific genetic factors in chickpea (Cicer arietinum L.) for quantitative resistance to ascochyta blight. Theor Appl Genet 109(4):733–739
Choudhary P, Khanna SM, Jain PK (2012a) Genetic structure and diversity analysis of the primary gene pool of chickpea using SSR markers. Genet Molecul Res 11(2):891–905
Choudhary S, Gaur R, Gupta S, Bhatia S (2012b) EST-derived genic molecular markers: development and utilization for generating an advanced transcript map of chickpea. Theor Appl Genet 124(8):1449–1462
Choudhary P, Khanna SM, Jain PK, Bharadwaj C, Kumar J, Lakhera PC, Srinivasan R (2013) Molecular characterization of primary gene pool of chickpea based on ISSR markers. Biochem Genet 51(3–4):306–322
Chourey PS, Taliercio EW, Kane EJ (1991) Tissue-specific expression and anaerobically induced posttranscriptional modulation of sucrose synthase genes in Sorghum bicolor M. Plant Physiol 96(2):485–490
Clarke HJ, Siddique KHM (2004) Response of chickpea genotypes to low temperature stress during reproductive development. Field Crop Res 90(2–3):323–334
Close TJ (1996) Dehydrins: emergence of a biochemical role of a family of plant dehydration proteins. Physiol Plant 97(4):795–803
Collard BCY, Pang ECK, Ades PK, Taylor PWJ (2003) Preliminary investigation of QTLs associated with seedling resistance to ascochyta blight from Cicer echinospermum, a wild relative of chickpea. Theor Appl Genet 107(4):719–729
Cotsaftis O, Plett D, Johnson AA, Walia H, Wilson C, Ismail AM, Close TJ, Tester M, Baumann U (2011) Root-specific transcript profiling of contrasting rice genotypes in response to salinity stress. Mol Plant 4(1):25–41
Cramer GR, Urano K, Delrot S, Pezzotti M, Shinozaki K (2011) Effects of abiotic stress on plants: a systems biology perspective. BMC Plant Biol 11(1):163
Croser JS, Clarke HJ, Siddique KHM, Khan TN (2003) Low-temperature stress: implications for chickpea (Cicer arietinum L.) improvement. Crit Rev Plant Sci 22(2):185–219
Delmer DP, Amor Y (1995) Cellulose biosynthesis. Plant Cell 7(7):987
Deokar AA, Kondawar V, Jain PK, Karuppayil SM, Raju NL, Vadez V, Varshney RK, Srinivasan R (2011) Comparative analysis of expressed sequence tags (ESTs) between drought-tolerant and-susceptible genotypes of chickpea under terminal drought stress. BMC Plant Biol 11(1):70
Dure L, Crouch M, Harada J, Ho THD, Mundy J, Quatrano R, Thomas T, Sung ZR (1989) Common amino acid sequence domains among the LEA proteins of higher plants. Plant Mol Biol 12(5):475–486
Eggen A (2012) The development and application of genomic selection as a new breeding paradigm. Anim Front 2(1):10–15
Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F, Butler SL, Deloukas P, Galver L (2003) Highly parallel SNP genotyping. In: January (ed) Cold Spring Harbor symposia on quantitative biology, vol 68. Cold Spring Harbor Laboratory Press, New York, pp 69–78
Flandez-Galvez H, Ades PK, Ford R, Pang ECK, Taylor PWJ (2003a) QTL analysis for ascochyta blight resistance in an intraspecific population of chickpea (Cicer arietinum L.). Theor Appl Genet 107(7):1257–1265
Flandez-Galvez H, Ford R, Pang ECK, Taylor PWJ (2003b) An intraspecific linkage map of the chickpea (Cicer arietinum L.) genome based on sequence tagged microsatellite site and resistance gene analog markers. Theor Appl Genet 106(8):1447–1456
Garg R, Patel RK, Tyagi AK, Jain M (2011) De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res 18(1):53–63
Garg R, Shankar R, Thakkar B, Kudapa H, Krishnamurthy L, Mantri N, Varshney RK, Bhatia S, Jain M (2016) Transcriptome analyses reveal genotype-and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci Rep 6:19228
Gaur PM, Krishnamurthy L, Kashiwagi J (2008) Improving drought-avoidance root traits in chickpea (Cicer arietinum L.)-current status of research at ICRISAT. Plant Product Sci 11(1):3–11
Gaur R, Sethy NK, Choudhary S, Shokeen B, Gupta V, Bhatia S (2011) Advancing the STMS genomic resources for defining new locations on the intraspecific genetic linkage map of chickpea (Cicer arietinum L.). BMC Genomics 12(1):117
Gaur PM, Thudi M, Samineni S, Varshney RK (2014) Advances in chickpea genomics. In: Legumes in the Omic Era. Springer, New York, pp 73–94
Geigenberger P, Stitt M (1993) Sucrose synthase catalyses a readily reversible reaction in vivo in developing potato tubers and other plant tissues. Planta 189(3):329–339
Geigenberger P, Krause KP, Hill LM, Reimholz R, MacRae E, Quick WP, Sonnewald U, Stitt M (1995) The regulation of sucrose synthesis in leaver and tubers of potato plants. In: International Symposium on sucrose metabolism. American Society Plant Physiologists
Glaszmann JC, Kilian B, Upadhyaya HD, Varshney RK (2010) Accessing genetic diversity for crop improvement. Curr Opin Plant Biol 13(2):167–173
Grenier, C., Châtel, M., Ospina, Y., Cao, T.V., Guimaraes, E.P., Martinez, C., Tohmé, J., Courtois, B. and Ahmadi, N., (2012). Population Improvement Through Recurrent Selection in Rice Prospect for Maker Assisted Recurrent Selection and Genome-Wide Selection W011
Grishkevich V, Yanai I (2013) The genomic determinants of genotype× environment interactions in gene expression. Trends Genet 29(8):479–487
Gujaria N, Kumar A, Dauthal P, Dubey A, Hiremath P, Prakash AB, Farmer A, Bhide M, Shah T, Gaur PM, Upadhyaya HD (2011) Development and use of genic molecular markers (GMMs) for construction of a transcript map of chickpea (Cicer arietinum L.). Theor Appl Genet 122(8):1577–1589
Gupta S, Kumar T, Verma S, Bharadwaj C, Bhatia S (2015) Development of gene-based markers for use in construction of the chickpea (Cicer arietinum L.) genetic linkage map and identification of QTLs associated with seed weight and plant height. Mol Biol Rep 42(11):1571–1580
Haileselasie TH, Teferii G (2012) The effect of salinity stress on germination of chickpea (Cicer arietinum L.) land race of Tigray. Curr Res J Biol Sci 4(5):578–583
Haware, M. P., Tripathi, H. S., Rathi, Y. P. S., Lenne, J. M., & Jayanthi, S. (1997). Integrated management of Botrytis gray mold of chickpea: cultural, chemical, biological, and resistance options. In Recent advances in research on botrytis gray mold of chickpea: summary proceedings of the Third Working Group Meeting to Discuss Collaborative Research on Botrytis Gray Mold of Chickpea, 15–17 Apr 1996, Pantnagar, Uttar Pradesh, India. Patancheru 502 324, Andhra Pradesh: International Crops Research Institute for the Semi-Arid Tropics. 68 pp (p. 9). ISBN 92–9066–373-1. Order code CPE 112
Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, Tuteja R, Kumar A, BhanuPrakash A, Mulaosmanovic B, Gujaria N, Krishnamurthy L (2011) Large-scale transcriptome analysis in chickpea (Cicer arietinum L.), an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnol J 9(8):922–931
Hiremath PJ, Kumar A, Penmetsa RV, Farmer A, Schlueter JA, Chamarthi SK, Whaley AM, Carrasquilla-Garcia N, Gaur PM, Upadhyaya HD, Kishor K (2012) Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol J 10(6):716–732
Holland JB (2007) Genetic architecture of complex traits in plants. Curr Opin Plant Biol 10(2):156–161
Huang JC, Lin SM, Wang CS (2000) A pollen-specific and desiccation-associated transcript in Lilium longiflorum during development and stress. Plant Cell Physiol 41(4):477–485
Huber SC, Huber JL (1996) Role and regulation of sucrose-phosphate synthase in higher plants. Annu Rev Plant Biol 47(1):431–444
Hüttel B, Winter P, Weising K, Choumane W, Weigand F, Kahl G (1999) Sequence-tagged microsatellite site markers for chickpea (Cicer arietinum L.). Genome 42(2):210–217
Ingram J, Chandler JW, Gallagher L, Salamini F, Bartels D (1997) Analysis of cDNA clones encoding sucrose-phosphate synthase in relation to sugar interconversions associated with dehydration in the resurrection plant Craterostigma plantagineum Hochst. Plant Physiol 115(1):113–121
Iruela M, Rubio J, Cubero JI, Gil J, Millan T (2002) Phylogenetic analysis in the genus Cicer and cultivated chickpea using RAPD and ISSR markers. Theor Appl Genet 104(4):643–651
Iusem ND, Bartholomew DM, Hitz WD, Scolnik PA (1993) Tomato (Lycopersicon esculentum) transcript induced by water deficit and ripening. Plant Physiol 102(4):1353
Jain D, Chattopadhyay D (2010) Analysis of gene expression in response to water deficit of chickpea (Cicer arietinum L.) varieties differing in drought tolerance. BMC Plant Biol 10(1):24
Jeanneau M, Gerentes D, Foueillassar X, Zivy M, Vidal J, Toppan A, Perez P (2002) Improvement of drought tolerance in maize: towards the functional validation of the Zm-Asr1 gene and increase of water use efficiency by over-expressing C4–PEPC. Biochimie 84(11):1127–1135
Johnson R (2003) Marker-assisted selection. In: Plant breeding reviews: part 1: long-term selection: maize, 24, pp 293–309
Kahl G, Molina C, Udupa SM, Rotter B, Horres R, Jungmann R, Belarmino LC, L'Taief B, Drevon J, Baum M, Winter P (2007) Super SAGE: exploring the stress transcriptome in chickpea. In: Plant and animal genome XV conference, pp 13–17
Kalifa Y, Gilad A, Konrad Z, Zaccai M, Scolnik PA, Dudy BZ (2004) The water-and salt-stress-regulated Asr1 (abscisic acid stress ripening) gene encodes a zinc-dependent DNA-binding protein. Biochem J 381(2):373–378
Konsam S, Chellapilla B, Ram G, Chellapilla TS, Jain PK (2014) Molecular diversity of chickpea ('Cicer arietinum'L.) genotypes differing in their Raffinose family oligosaccharides viz., raffinose and stachyose content as revealed through SSR markers. Aust J Crop Sci 8(8):1175
Krishnamurthy L, Kashiwagi J, Tobita S, Ito O, Upadhyaya HD, Gowda CL, Gaur PM, Sheshshayee MS, Singh S, Vadez V, Varshney RK (2013a) Variation in carbon isotope discrimination and its relationship with harvest index in the reference collection of chickpea germplasm. Funct Plant Biol 40(12):1350–1361
Krishnamurthy L, Kashiwagi J, Upadhyaya HD, Gowda CLL, Gaur PM, Singh S, Purushothaman R, Varshney RK (2013b) Partitioning coefficient—a trait that contributes to drought tolerance in chickpea. Field Crop Res 149:354–365
Kumar S, Hamwieh A, Manickavelu A, Kumar J, Sharma TR, Baum M (2014) Advances in lentil genomics. In: Legumes in the omic era. Springer, New York, pp 111–130
Ladizinsky G, Adler A (1976) Genetic relationships among the annual species of Cicer L. Theor Appl Genet 48(4):197–203
Lenka SK, Katiyar A, Chinnusamy V, Bansal KC (2011) Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol J 9(3):315–327
Lichtenzveig J, Scheuring C, Dodge J, Abbo S, Zhang HB (2005) Construction of BAC and BIBAC libraries and their applications for generation of SSR markers for genome analysis of chickpea, Cicer arietinum L. Theor Appl Genet 110(3):492–510
Maqbool MA, Aslam M, Ali H, Shah TM (2016) Evaluation of advanced chickpea (Cicer arietinum L.) accessions based on drought tolerance indices and SSR markers against different water treatments. Pak. J Bot 48(4):1421–1429
Maskin L, Gudesblat GE, Moreno JE, Carrari FO, Frankel N, Sambade A, Rossi M, Iusem ND (2001) Differential expression of the members of the Asr gene family in tomato (Lycopersicon esculentum). Plant Sci 161(4):739–746
Masle J, Gilmore SR, Farquhar GD (2005) The ERECTA gene regulates plant transpiration efficiency in Arabidopsis. Nature 436(7052):866
Matsumura H, Ito A, Saitoh H, Winter P, Kahl G, Reuter M, Krüger DH, Terauchi R (2005) SuperSAGE. Cell Microbiol 7(1):1–18
Meena HP, Kumar J, Upadhyaya HD, Bharadwaj C, Chauhan SK, Verma AK, Rizvi AH (2010) Chickpea mini core germplasm collection as rich sources of diversity for crop improvement. J SAT Agric Res 8:1–5
Millan T, Rubio J, Iruela M, Daly K, Cubero JI, Gil J (2003) Markers associated with Ascochyta blight resistance in chickpea and their potential in marker-assisted selection. Field Crop Res 84(3):373–384
Millan T, Clarke HJ, Siddique KH, Buhariwalla HK, Gaur PM, Kumar J, Gil J, Kahl G, Winter P (2006) Chickpea molecular breeding: new tools and concepts. Euphytica 147(1–2):81–103
Molina C, Rotter B, Horres R, Udupa SM, Besser B, Bellarmino L, Baum M, Matsumura H, Terauchi R, Kahl G, Winter P (2008) SuperSAGE: the drought stress-responsive transcriptome of chickpea roots. BMC Genomics 9(1):553
Nayak SN, Zhu H, Varghese N, Datta S, Choi HK, Horres R, JŘngling R, Singh J, Kishor PK, Sivaramakrishnan S, Hoisington DA (2010) Integration of novel SSR and gene-based SNP marker loci in the chickpea genetic map and establishment of new anchor points with Medicago truncatula genome. Theor Appl Genet 120(7):1415–1441
Nayyar H, Bains T, Kumar S (2005) Low temperature induced floral abortion in chickpea: relationship to abscisic acid and cryoprotectants in reproductive organs. Environ Exp Bot 53(1):39–47
Neeraj K, Bharadwaj C, Satyavathi CT, Madan P, Tapan K, Tripti S, Jain PK, Patil BS, Soren KR (2016) Yield correlation of chickpea (Cicer arietinum L.) genotypes based on physiological and morphological traits for salt tolerance. Int J Trop Agric 34(3):693–699
Nene YL, Reddy MV (1987) Chickpea diseases and their control. In: Chickpea diseases and their control, pp 233–270
Nguyen TT, Taylor PWJ, Redden RJ, Ford R (2004) Genetic diversity estimates in Cicer using AFLP analysis. Plant Breed 123(2):173–179
Oukarroum A, Schansker G, Strasser RJ (2009) Drought stress effects on photosystem I content and photosystem II thermotolerance analyzed using Chl a fluorescence kinetics in barley varieties differing in their drought tolerance. Physiol Plant 137(2):188–199
Pande S, Singh G, Rao JN, Bakr MA, Chaurasia PCP, Joshi S, Johansen C, Singh SD, Kumar J, Rahman MM, Gowda CL (2001) Integrated management of botrytis gray mold of chickpea. International Crops Research Institute for the Semi-Arid Tropics, India
Petřivalský M, Brauner F, Luhová L, Gagneul D, Šebela M (2007) Aminoaldehyde dehydrogenase activity during wound healing of mechanically injured pea seedlings. J Plant Physiol 164(11):1410–1418
Pratap A, Chaturvedi SK, Tomar R, Rajan N, Malviya N, Thudi M, Saabale PR, Prajapati U, Varshney RK, Singh NP (2017) Marker-assisted introgression of resistance to fusarium wilt race 2 in Pusa 256, an elite cultivar of desi chickpea. Mol Gen Genomics 292(6):1237–1245
Pushpavalli R, Krishnamurthy L, Thudi M, Gaur PM, Rao MV, Siddique KH, Colmer TD, Turner NC, Varshney RK, Vadez V (2015) Two key genomic regions harbour QTLs for salinity tolerance in ICCV 2× JG 11 derived chickpea (Cicer arietinum L.) recombinant inbred lines. BMC Plant Biol 15(1):124
Qadir SA, Datta S, Singh NP, Kumar S (2007) Development of highly polymorphic SSR markers for chickpea (Cicer arietinum L.) and their use in parental polymorphism. Indian J Genet Plant Breed 67(4):329–333
Radhika P, Gowda SJM, Kadoo NY, Mhase LB, Jamadagni BM, Sainani MN, Chandra S, Gupta VS (2007) Development of an integrated intraspecific map of chickpea (Cicer arietinum L.) using two recombinant inbred line populations. Theor Appl Genet 115(2):209–216
Rajesh PN, Muehlbauer FJ (2008) Discovery and detection of single nucleotide polymorphism (SNP) in coding and genomic sequences in chickpea (Cicer arietinum L.). Euphytica 162(2):291–300
Rakshit S, Winter P, Tekeoglu M, Muñoz JJ, Pfaff T, Benko-Iseppon AM, Muehlbauer FJ, Kahl G (2003) DAF marker tightly linked to a major locus for Ascochyta blight resistance in chickpea (Cicer arietinum L.). Euphytica 132(1):23–30
Rao LS, Rani PU, Deshmukh PS, Kumar PA, Panguluri SK (2007) RAPD and ISSR fingerprinting in cultivated chickpea (Cicer arietinum L.) and its wild progenitor Cicer reticulatum Ladizinsky. Genet Resour Crop Evol 54(6):1235–1244
Robertson, L. D., Singh, K. B., & Ocampo, B. (1995). A catalog of annual wild
Rodi CP, Darnhofer-Patel B, Stanssens P, Zabeau M, van den Boom D (2002) A strategy for the rapid discovery of disease markers using the MassARRAY system. BioTechniques 32:S62–S69
Roorkiwal M, Sharma PC (2012) Sequence similarity based identification of abiotic stress responsive genes in chickpea. Bioinformation 8(2):92
Rosenzweig C, Parry ML (1994) Potential impact of climate change on world food supply. Nature 367(6459):133–138
Rostoks N, Ramsay L, MacKenzie K, Cardle L, Bhat PR, Roose ML, Svensson JT, Stein N, Varshney RK, Marshall DF, Graner A (2006) Recent history of artificial outcrossing facilitates whole-genome association mapping in elite inbred crop varieties. Proc Natl Acad Sci 103(49):18656–18661
Rudrabhatla P, Rajasekharan R (2004) Functional characterization of peanut serine/threonine/tyrosine protein kinase: molecular docking and inhibition kinetics with tyrosine kinase inhibitors. Biochemistry 43(38):12123–12132
Sam RE, Theodore MC, Marlin DE, Robert SR, Jason K (2009).(2007) Molecular markers in a commercial breeding program. Crop Sci 47(S3):154–163
Savithri KS, Ganapathy PS, Sinha SK (1980) Sensitivity to low temperature in pollen germination and fruit-set in Cicer arietinum L. J Exp Bot 31(2):475–481
Saxena NP, Johansen C, Sethi SC, Talwar HS, Krishnamurthy L (1988) Improving harvest index in chickpea through incorporation of cold tolerance. Int Chickpea Newsletter 19:17–19
Schefers JM, Weigel KA (2012) Genomic selection in dairy cattle: integration of DNA testing into breeding programs. Anim Front 2(1):4–9
Schneider A, Salamini F, Gebhardt C (1997) Expression patterns and promoter activity of the cold-regulated gene ci21A of potato. Plant Physiol 113(2):335–345
Sefera T, Abebie B, Gaur PM, Assefa K, Varshney RK (2011) Characterisation and genetic diversity analysis of selected chickpea cultivars of nine countries using simple sequence repeat (SSR) markers. Crop Pasture Sci 62(2):177–187
Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, Oono Y, Kamiya A, Nakajima M, Enju A, Sakurai T, Satou M (2002) Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J 31(3):79–292
Serraj R, Krishnamurthy L, Kashiwagi J, Kumar J, Chandra S, Crouch JH (2004) Variation in root traits of chickpea (Cicer arietinum L.) grown under terminal drought. Field Crop Res 88(2–3):115–127
Sethy NK, Shokeen B, Bhatia S (2003) Isolation and characterization of sequence-tagged microsatellite sites markers in chickpea (Cicer arietinum L.). Mol Ecol Resour 3(3):428–430
Sethy NK, Shokeen B, Edwards KJ, Bhatia S (2006) Development of microsatellite markers and analysis of intraspecific genetic variability in chickpea (Cicer arietinum L.). Theor Appl Genet 112(8):1416–1428
Sharma KD, Muehlbauer FJ (2007) Fusarium wilt of chickpea: physiological specialization, genetics of resistance and resistance gene tagging. Euphytica 157(1–2):1–14
Sharma KD, Nayyar H (2014) Cold stress alters transcription in meiotic anthers of cold tolerant chickpea (Cicer arietinum L.). BMC Res Notes 7(1):717
Shpak ED, Berthiaume CT, Hill EJ, Torii KU (2004) Synergistic interaction of three ERECTA-family receptor-like kinases controls Arabidopsis organ growth and flower development by promoting cell proliferation. Development 131(7):1491–1501
Singh KB (1993) Problems and prospects of stress resistance breeding in chickpea
Singh S (2014) PM Gaur, SK Chaturvedi, NP Singh, and JS Sandhu. Broadening the genetic base of grain legumes, 51
Singh KB, Malhotra RS, Saxena MC (1995) Additional sources of tolerance to cold in cultivated and wild Cicer species. Crop Sci 35(5):1491–1497
Soren KR, Patil PG, Das A, Bohra A, Datta S, Chaturvedi SK, Nadarajan N (2012) Advances in pulses genomic research. Indian Institute of Pulses Research, Kanpur, p 25
Sreenivasulu N, Kishor PK, Varshney RK, Altschmied L (2002) Mining functional information from cereal genomes–the utility of expressed sequence tags. Curr Sci:965–973
Srinivasan A, Johansen C, Saxena NP (1998) Cold tolerance during early reproductive growth of chickpea (Cicer arietinum L.): characterization of stress and genetic variation in pod set1. Field Crop Res 57(2):181–193
Supriya S, Bharadwaj C, Vinay S, Neeraj K, Kv B, Patil BS, Soren KR, Chaturvedi SK, Manish R, Chauhan SK, Rajeev V (2017) Morpho-physiological grouping of chickpea (Cicerarietinum L.) genotypes on the basis of their response to drought stress. Int J Trop Agric 35(1):5–13
Tapan K, Bharadwaj C, Rizvi AH, Ashutosh S, Shailesh T, Afroz A, Chauhan SK (2015) Chickpea landraces: a valuable and divergent source for drought tolerance. Int J Tropical Agri 33(2 (part II)):633–638
Tekeoglu M, Santra DK, Kaiser WJ, Muehlbauer FJ (2000) Ascochyta blight resistance inheritance in three chickpea recombinant inbred line populations. Crop Sci 40(5):1251–1256
Thudi M, Bohra A, Nayak SN, Varghese N, Shah TM, Penmetsa RV, Thirunavukkarasu N, Gudipati S, Gaur PM, Kulwal PL, Upadhyaya HD (2011) Novel SSR markers from BAC-end sequences, DArT arrays and a comprehensive genetic map with 1,291 marker loci for chickpea (Cicer arietinum L.). PLoS One 6(11):e27275
Thudi M, Gaur PM, Krishnamurthy L, Mir RR, Kudapa H, Fikre A, Kimurto P, Tripathi S, Soren KR, Mulwa R, Bharadwaj C (2014) Genomics-assisted breeding for drought tolerance in chickpea. Funct Plant Biol 41(11):1178–1190
Turner NC, Colmer TD, Quealy J, Pushpavalli R, Krishnamurthy L, Kaur J, Singh G, Siddique KH, Vadez V (2013) Salinity tolerance and ion accumulation in chickpea (Cicer arietinum L.) subjected to salt stress. Plant Soil 365(1–2):347–361
Udupa SM, Baum M (2003) Genetic dissection of pathotype-specific resistance to ascochyta blight disease in chickpea (Cicer arietinum L.) using microsatellite markers. Theor Appl Genet 106(7):1196–1202
Udupa SM, Weigand F, Saxena MC, Kahl G (1998) Genotyping with RAPD and microsatellite markers resolves pathotype diversity in the ascochyta blight pathogen of chickpea. Theor Appl Genet 97(1–2):299–307
Upadhyaya HD, Ortiz R (2001) A mini core subset for capturing diversity and promoting utilization of chickpea genetic resources in crop improvement. Theor Appl Genet 102(8):1292–1298
Upadhyaya HD, Dwivedi SL, Baum M, Varshney RK, Udupa SM, Gowda CL, Hoisington D, Singh S (2008) Genetic structure, diversity, and allelic richness in composite collection and reference set in chickpea (Cicer arietinum L.). BMC Plant Biol 8(1):106
Upadhyaya HD, Kashiwagi J, Varshney RK, Gaur PM, Saxena KB, Krishnamurthy L, Gowda CLL, Pundir RPS, Chaturvedi SK, Basu PS, Singh IP (2012) Phenotyping chickpeas and pigeonpeas for adaptation to drought. Front Physiol 3:179
Vadez V, Krishnamurthy L, Serraj R, Gaur PM, Upadhyaya HD, Hoisington DA, Varshney RK, Turner NC, Siddique KHM (2007) Large variation in salinity tolerance in chickpea is explained by differences in sensitivity at the reproductive stage. Field Crop Res 104(1–3):123–129
Vadez V, Krishnamurthy L, Thudi M, Anuradha C, Colmer TD, Turner NC et al (2012) Assessment of ICCV 2× JG 62 chickpea progenies shows sensitivity of reproduction to salt stress and reveals QTL for seed yield and yield components. Mol Breed 30(1):9–21
Valente F, Gauthier F, Bardol N, Blanc G, Joets J, Charcosset A, Moreau L (2013) OptiMAS: a decision support tool for marker-assisted assembly of diverse alleles. J Hered 104(4):586–590
Varshney RK (2016) Exciting journey of 10 years from genomes to fields and markets: some success stories of genomics-assisted breeding in chickpea, pigeonpea and groundnut. Plant Sci 242:98–107
Varshney RK, Graner A, Sorrells ME (2005) Genomics-assisted breeding for crop improvement. Trends Plant Sci 10(12):621–630
Varshney RK, Hoisington DA, Upadhyaya HD, Gaur PM, Nigam SN, Saxena K, Vadez V, Sethy NK, Bhatia S, Aruna R, Gowda MC (2007) Molecular genetics and breeding of grain legume crops for the semi-arid tropics. In: Genomics-assisted crop improvement. Springer, Dordrecht, pp 207–241
Varshney RK, Close TJ, Singh NK, Hoisington DA, Cook DR (2009a) Orphan legume crops enter the genomics era! Curr Opin Plant Biol 12(2):202–210
Varshney RK, Hiremath PJ, Lekha P, Kashiwagi J, Balaji J, Deokar AA, Vadez V, Xiao Y, Srinivasan R, Gaur PM, Siddique KH (2009b) A comprehensive resource of drought-and salinity-responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L.). BMC Genomics 10(1):523
Varshney RK, Glaszmann JC, Leung H, Ribaut JM (2010) More genomic resources for less-studied crops. Trends Biotechnol 28(9):452–460
Varshney RK, Ribaut JM, Buckler ES, Tuberosa R, Rafalski JA, Langridge P (2012) Can genomics boost productivity of orphan crops? Nat Biotechnol 30(12):1172
Varshney RK, Mohan SM, Gaur PM, Gangarao NVPR, Pandey MK, Bohra A, Sawargaonkar SL, Chitikineni A, Kimurto PK, Janila P, Saxena KB (2013a) Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotechnol Adv 31(8):1120–1134
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar'an B, Millan T (2013b) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31(3):240
Varshney RK, Gaur PM, Chamarthi SK, Krishnamurthy L, Tripathi S, Kashiwagi J, Samineni S, Singh VK, Thudi M, Jaganathan D (2013c) Fast-track introgression of “QTL-hotspot” for root traits and other drought tolerance traits in JG 11, an elite and leading variety of chickpea. Plant Genome 6(3)
Varshney RK, Mohan SM, Gaur PM, Chamarthi SK, Singh VK, Srinivasan S, Swapna N, Sharma M, Pande S, Singh S, Kaur L (2014a) Marker-assisted backcrossing to introgress resistance to Fusarium wilt race 1 and Ascochyta blight in C 214, an elite cultivar of chickpea. The plant genome 7(1)
Varshney RK, Thudi M, Nayak SN, Gaur PM, Kashiwagi J, Krishnamurthy L, Jaganathan D, Koppolu J, Bohra A, Tripathi S, Rathore A (2014b) Genetic dissection of drought tolerance in chickpea (Cicer arietinum L.). Theor Appl Genet 127(2):445–462
Verma S, Gupta S, Bandhiwal N, Kumar T, Bharadwaj C, Bhatia S (2015) High-density linkage map construction and mapping of seed trait QTLs in chickpea (Cicer arietinum L.) using genotyping-by-sequencing (GBS). Sci Rep 5:17512
Winter P, Pfaff T, Udupa SM, Hüttel B, Sharma PC, Sahi S, Arreguin-Espinoza R, Weigand F, Muehlbauer FJ, Kahl G (1999) Characterization and mapping of sequence-tagged microsatellite sites in the chickpea (Cicer arietinum L.) genome. Mol Gen Genet MGG 262(1):90–101
Xu Y (2010) Molecular dissection of complex traits: practice. In: Molecular plant breeding, pp 249–285
Yadav S, Bharadwaj C, Chauhan SK, Rizvi AH, Kumar J, Satyavathi CT (2011) Analysis of genetic diversity in Cicerspecies using molecular markers. Indian J Genet Plant Breed 71(3):272–275
Zhao Y, Gowda M, Liu W, Würschum T, Maurer HP, Longin FH, Ranc N, Reif JC (2012) Accuracy of genomic selection in European maize elite breeding populations. Theor Appl Genet 124(4):769–776
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Bharadwaj, C. et al. (2018). Chickpea Genomics. In: Gosal, S., Wani, S. (eds) Biotechnologies of Crop Improvement, Volume 3. Springer, Cham. https://doi.org/10.1007/978-3-319-94746-4_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-94746-4_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-94745-7
Online ISBN: 978-3-319-94746-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)