
Abstract.
Ascochyta blight is an economically important disease of chickpea caused by the fungus Ascochyta rabiei. The fungus shows considerable variation for pathogenicity in nature. However, studies on the genetics of pathotype-specific resistance are not available for this plant-pathosystem. The chickpea landrace ILC 3279 has resistance to pathotype I and II of the pathogen. In order to understand the inheritance of pathotype-specific resistance in this crop, both Mendelian and quantitative trait loci analyses were performed using a set of intraspecific, recombinant inbred lines derived from a cross between the susceptible accession ILC 1272 and the resistant ILC 3279, and microsatellite markers. We identified and mapped a major locus (ar1, mapped on linkage group 2), which confers resistance to pathotype I, and two independent recessive major loci (ar2a, mapped on linkage group 2 and ar2b, mapped on linkage group 4), with complementary gene action conferring resistance to pathotype II. Out of two pathotype II-specific resistance loci, one (ar2a) linked very closely with the pathotype I-specific resistance locus, indicating a clustering of resistance genes in that region of the chickpea genome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ascochyta blight disease, caused by Ascochyta rabiei (Pass) Lab., is a destructive foliar fungal disease of chickpea (Cicer arietinum L.) in West and Central Asia, North Africa, North America and Australia. Incidence and epidemics of this disease are weather dependent. In some years, cool and wet weather conditions favor the disease's development and often result in up to 100% yield loss (Reddy et al. 1990; Singh et al. 1992; Singh and Reddy 1993). Fungicide treatments for control of the disease exist, but are often impractical and uneconomical (Reddy et al. 1990). One of the most effective, economical and environmentally safe ways to manage the disease is to cultivate resistant varieties (Singh et al. 1992).
Many resistant sources have been identified for this blight disease from the chickpea germplasm bank (Singh et al. 1981; Singh and Reddy 1993). Some have been used for studying inheritance of resistance to the disease. These studies showed that in some desi chickpeas, and in several kabuli chickpeas (ILC 72, ILC 183, ILC 200 and ILC 935), a single dominant gene controlled resistance (Vir et al. 1975; Singh and Reddy 1983). In some other cultivars, it has been reported that resistance was governed by two dominant genes (Tewari and Pandey 1986), one recessive gene (Tewari and Pandey 1986), two recessive genes (Kusmenoglu 1990) or by two to three quantitative loci (Santra et al. 2000), depending on the screening techniques and the cultivar used.
Several reports suggest that a high level of variability exists for the pathogenicity trait in A. rabiei populations (Vir and Grewal 1974; Nene and Reddy 1987; Malik and Rahman 1992; Porta-Puglia 1992). For instance, Vir and Grewal (1974) reported finding more than ten pathotypes among field isolates from India. Nene and Reddy (1987) reported five pathogenic groups and several strains among isolates from, respectively, Pakistan and Turkey. A recent survey of pathogenicity in Syria (Udupa et al. 1998) revealed the occurrence of three pathotypes: pathotype I (less aggressive); pathotype II (aggressive) and pathotype III (most aggressive). Although several studies on inheritance of ascochyta blight resistance in chickpea are available, and A. rabiei's pathogenic variability is well documented, studies on the genetics of resistance to individual pathotypes, and of the localization of their resistance genes on the genome, have not been made.
Two approaches are commonly used for studying the genetics of pathotype-specific resistance in crop plants. The first of these involves the development and use of multiple sets of near-isogenic lines (NILs) to separate and, thereby, identify several resistance genes (Mackill and Bonman 1992). The second method involves the use of molecular markers applied to permanent populations, such as recombinant inbred lines (RILs), and may be used to allow the location of major resistance genes and quantitative trait loci to specific chromosomal regions (Wang et al. 1994). The development of NILs involves a number of generations of selection and crossing; developing lines with several major genes in a given background is a very difficult task. Therefore, molecular marker application on RILs is the more practical approach for inheritance studies and mapping of the pathotype-specific resistance of chickpea.
It has been shown that a chickpea landrace ILC 3279, originating in the former USSR, has resistance to both pathotype I and II of A. rabiei (Udupa et al. 1998). A RIL population derived from this landrace and a susceptible cultivar would provide a unique opportunity to study the genetics of resistance to different pathotypes of the pathogen. Here, we report the inheritance and mapping of pathotype-specific resistance in such a RIL population of chickpea, and show that resistance to pathotype I is conferred by a major locus and resistance to pathotype II is conferred by two major loci.
Materials and methods
Population development and phenotypic analysis
An intraspecific cross of chickpea, between pure lines ILC 1272 (susceptible to A. rabiei and originating in Turkey) and ILC 3279 (resistant to A. rabiei and originating in the former USSR), was used to develop a RIL population. The cross was advanced by single-seed descent from F2 to F6–F7 in a greenhouse and/or in a field, and the F6 or F7 lines were used for genetic analysis of blight resistance. Isolates of A. rabiei pathotype I (isolate no. 5) and pathotype II (isolate no. 6) were used for inoculation (Udupa et al. 1998). The inoculum preparation, temperature, relative humidity, light conditions, screening method and disease scoring scale used in this study were the same as that described by Udupa et al. (1998). The individual RILs, along with the parents, were tested for resistance to each of the pathotypes respectively, under growth-chamber conditions. Seedlings of 10 days old were inoculated. Readings for disease severity on a single plant basis were taken on the 14th day after inoculation, using a 1 to 9 scale, where: 1 = no symptoms; 2 = a small round tissue depression or spot; 3 = elongated spots; 4 = a coalescent spot; 5 = stem girdling; 6 = stem breaking; 7 = lesion growths downward from breaking points; 8 = a whole plant nearly dead and 9 = plant dead.
Genotypic analysis
Total DNA was extracted from the leaf tissue (Udupa et al. 1999) of each RIL and its parents, and was used to evaluate microsatellite polymorphism. 140 microsatellite marker primer pairs (Hüttel et al. 1999; Udupa et al. 1999; Winter et al. 1999) were used to amplify the simple sequence-length polymorphic DNA (Udupa et al. 1999; Winter et al. 1999) between the parents. The amplified DNA fragments were analyzed using ALFexpress DNA Sequencer (Amersham Pharmacia Biotech; Udupa et al. 1999) or DNA fragments were visualized via silver staining, using a silver staining kit (Promega, USA) as described by the supplier. The polymorphic primer pairs were further tested on RILs.
Statistical analysis
Mapmaker/Exp version 3.0 (Whitehead Institute, Cambridge, Mass., USA; Lincoln et al. 1993) was used to create a linkage map. Markers were included on the map only if the LOD value obtained was >3. The Kosambi mapping function (Kosambi 1944) was used to convert recombination frequencies to map distances in centiMorgans (cMs). The QTL Cartographer version 1.15a program (developed by J. Basten, B.S. Weir and Z.-B. Zeng, NC State University, Raleigh, N.C., USA) was used to identify putative disease resistance QTLs using simple linear regression models, where the genetic markers served as independent variables and phenotypes served as dependent variables. This resulted in estimation of F- and Likelihood ratio (LR)-statistics for each marker. In order to minimize the number of type-I errors leading to QTL false positives, and to compensate for non-random selection of plants used for molecular mapping, we chose a strict probability level of P < 0.001 as the threshold used to indicate a significant association of a QTL with a particular marker locus based on the F-statistic. Likelihood ratio (LR) test values were used as the χ2 value with 1 df (Maddala 1992). Interval mapping analysis (with a permutation test, 1,000 permutations) was carried out using QTL Cartographer, to confirm the presence of putative disease resistance QTL on the framework map. A LOD score of >3.0 was chosen to indicate significant results in the interval analyses. In order to determine the effect of each of the pathotypes on RILs and their parents, an ANOVA was performed using the program Agrobase 21 (Agronomix Software Inc., Winnipeg, Canada).
Results
Parental polymorphism and genetic linkage map
Out of 140 microsatellite markers tested, 67 markers revealed polymorphism between the parents ILC 1272 and ILC 3279, and 52 of them were mapped on the genome. For most of the mapped markers, marker segregation ratios in this intraspecific population were closer to Mendelian expectations than in the previous inter-specific chickpea population of Winter et al. (2000), which showed a 39.8% distorted segregation. In this intraspecific population, χ2 values for 46 markers were less than 3.84 at P = 0.05; for four markers they were less than 6.63 at P = 0.01, and were more than 3.84 at P = 0.05. Only two markers, namely CaSTMS 10 and TAA169, showed segregation distortion (χ2 values were more than 6.63 at P = 0.01), and these two distorted markers were not used for QTL analysis. The present genetic map, based on microsatellite markers with eight linkage groups (corresponding to the chromosome number) and constructed using the Kosambi function, covered 419 cM in length with an average distance of 7.9 cM between adjacent markers (Fig. 1). Although the overall distribution of markers on individual linkage groups was largely similar to the interspecific map of Winter et al. (2000), marker orientation and linkage distance between markers differed.

Linkage map and the positions of loci for resistance to A. rabiei. The locus ar1 for resistance to pathotype I and the vertical bars to right of the linkage groups indicate QTLs (ar2a and ar2b) defined by LOD score >3.0 supporting the confidence interval for resistance to pathotype II
Inheritance and mapping of resistance to pathotype I
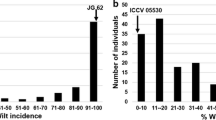
In relation to pathotype I, the cultivar ILC 1272 was the susceptible parent (mean disease rating of 8.00) and ILC 3279 the resistant parent (mean disease rating of 3.67). The average disease score of the individual 97 recombinant inbred lines (RILs) derived from the cross ILC 1272 × ILC 3279 followed a bimodal distribution (Fig. 2a). Differences in disease scores among RILs and the parents were highly significant (ANOVA, F-value = 14.77, df = 98, 122; P < 0.001). Based on symptoms, the RILs were classified into susceptible and resistant types. The RILs with the "stem breaking" symptom or symptoms more severe than this (disease scores of six or above) were considered to be "susceptible". Up to the symptom "stem girdling" they were considered "resistant" (disease score of five and below), using average scores. Of the 97 RILs, 53 were resistant and 44 were susceptible. A similar result was observed if LSD values (LSD = 1.62 at P = 0.05) were considered for the classification of resistant RILs, as suggested by Tekeoglu et al. (2000). Segregation of susceptible/resistant phenotypes of the RILs fitted a 1:1 ratio (χ2 = 0.51, df = 1, P < 0.05). This segregation ratio suggests that resistance to pathotype I is controlled by a single locus (suggested name: ar1). Our previous study, with limited F2:3 families, showed that the majority of the segregants are susceptible (unpublished results), indicating that the resistance gene is recessive. Further genetic mapping of this trait with the microsatellite markers localized this gene to one end of linkage group 2 (LG 2), with a linkage distance of 24 cM from GA16 and a LOD score of 3.2. When the disease scores were analyzed as quantitative traits, using a simple linear regression model, a highly significant association was also observed with the same marker (likelihood ratio test = 19.8, df = 1, P < 0.0001; F = 21.5, df =1, 95; P < 0.0001), further confirming that a single locus controls this trait.

Frequency distribution of RIL mean scores for resistance to pathotype I (A) and pathotype II (B) in the RIL population of the cross between ILC 1272 (susceptible) and ILC 3279 (resistant). Arrows show means of the parents
Inheritance and mapping of resistance to pathotype II
The cultivar ILC 3279 is resistant (having a mean disease score of 3.83), and the cultivar ILC 1272 susceptible (disease score of 8–9 with a mean of 8.00), to pathotype II. The distribution of average disease scores of 96 RILs are presented in Fig. 2b. Differences in disease scores among RILs and the parents were highly significant (ANOVA, F-value = 12.27, df = 97, 164, P < 0.001). The RILs were classified as "susceptible" (a total of 70 RILs) and "resistant" (a total of 26 RILs), employing the same criteria used for pathotype I. The segregation data of the RILs fits the expected segregation ratio of 3 susceptible: 1 resistant (χ2 = 0.22, df =1, P < 0.05) for two recessive complementary genes (suggested names: ar2a and ar2b) conferring resistance.
A significant relationship was observed between individual disease scores of pathotype I and pathotype II (correlation coefficient = 0.58). Furthermore, out of 52 pathotype I resistant RILs, 27 and 25 RILs were, respectively, susceptible and resistant to pathotype II (segregation ratio fits the expected ratio of 1:1; χ2 = 0.08, df =1, P < 0.05). Almost all RILs susceptible to pathotype I (44 RILs) are also susceptible to pathotype II (43 RILs) with only one exception (a RIL susceptible to pathotype I which is resistant to pathotype II). These observations indicate that, in chickpea, resistance to pathotypes I and II is genetically linked.
Analysis of trait-marker association was performed for each microsatellite marker independently, using the χ2 test for the independence of segregation for the resistance genes and individual microsatellite markers. Analysis revealed significant linkage with the ar1 gene of LG 2, and with the microsatellite markers of LG 4 (data not shown). This observation was further confirmed by simple linear regression analysis of the markers with the disease score of the RILs (Table 1). The LR ratio was highest with the TR20 microsatellite locus. However, precise localization of the genes to a particular interval between the markers in LG 2 and LG 4 could not be accomplished with these analyses, because of the recessive and complementary nature of the gene action of the resistance genes.
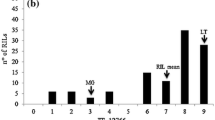
Using the strategy of interval mapping and the permutation test, we localized (mapped) pathotype II resistance loci ar2a and ar2b with a peak of significance close to the pathotype I resistance locus ar1 (on LG 2) and the TA72 microsatellite marker (2 cM; on LG 4) respectively (Fig. 3). Even though ar1 and ar2a mapped in the same region, the segregation data showed that the two loci are not the same. This conclusion is drawn from the observation that, out of 44 pathotype I-susceptible RILs, one showed resistance reaction to pathotype II, indicating recombination between the ar1 and ar2a loci. Since susceptibility is dominant over resistance, other recombinant classes could not be visualized. Based on the observed recombinant class, other classes could be estimated (by multiplying by 3). Linkage distance was estimated using the Kosombi function between the two loci (recombination: 4.12%; map units: 4.1 cM). The QTL ar2b has two peaks, one on either side of marker TA72 to the interval TA130–TA72–TR20, 2 cM apart (Fig. 3) with a LOD score of >3.0. This indicates the possibility that two closely linked loci contribute to the pathotype II-specific resistance response [as observed by Banerjee et al. (2001) for the RPS2-resistance locus of Arabidopsis thaliana].

Plots of LOD scores for resistance to pathotype II in the RIL population derived from a cross between ILC 1272 and ILC 3279. The plots show the location of putative quantitative loci identified for the resistance on linkage groups 2 (upper graph) and 4 (lower graph)
Discussion
Molecular mapping in chickpeas is mainly limited by the availability of polymorphic markers. With the development of sequence-tagged microsatellite site markers a new marker system became available for chickpea genome mapping, which show a much higher level of polymorphism than other marker systems (Udupa et al. 1999). Using 140 such markers, polymorphism was studied between parental lines which differ for ascochyta blight resistance.
Although, on average, only about 50% of polymorphic microsatellites were detected, we were able to study the inheritance and mapping of pathotype-specific resistance (pathotype I and II) to ascochyta blight for the first time in cultivated chickpea. Our study demonstrated that resistance to pathotype I is controlled by a major locus; and resistance to pathotype II is controlled by two recessive loci with complementary gene action.
On the subjects of inheritance and molecular mapping of ascochyta blight resistance, the present study differs from others reported previously. We used a different population type (intraspecific cross derived RILs), and a different screening method (under controlled conditions). We tested for pathotype-specific resistance (pathotypes I and II) and used different marker types (microsatellites). Previously, interspecific cross derived RILs have been used in this type of study (Santra et al. 2000). Moreover, previous studies have undertaken field screening under natural conditions (Kusmenoglu 1990; Santra et al. 2000; Tekeoglu et al. 2000) and used different kinds of markers (Santra et al. 2000). Interspecific crosses generate wider variability for many morphological traits, which could interfere in scoring for the disease. For mapping the disease resistance loci, accurate screening methods (which provide a suitable temperature, relative humidity and light conditions for the pathogen to develop the disease) and a control on the pathogen (pathotype) infecting the RILs are required. Our screening method, under controlled environmental conditions (growth chambers), differed from tests in the field and in plastic houses, in that co-infection of seedlings with other pathotypes of the pathogen and other pathogens was avoided. In addition, our method controlled temperature, relative humidity and light conditions better than other methods, which, if not appropriately controlled, can often interfere with assays and may increase the number of false scorings.
In linkage analysis, each false-scored qualitative phenotype would be interpreted as two recombination events flanking the locus (Wright et al. 1998). The "maximum-likelihood" location of a discrete phenotype that included a low frequency of errors would either be in an interval that was large enough to include several "double recombinants", or at a sufficient "recombinational distance" from the end of a linkage group that false-scored resistance or might be attributed to recombination (Wright et al. 1998). Therefore, analysis of resistance as a quantitative phenotype may further improve the reliability of genetic mapping (Wright et al. 1998).
The importance of analyzing quantitative phenotypes is especially well illustrated by molecular dissection of resistance to various pathogens in several crops (Young 1996; Ghislain et al. 2001; Iqbal et al. 2001; Wang et al. 2001; Wilson et al. 2001). In many cases, in comparison with QTL analysis, classical Mendelian genetics detected a lower number of regions involved in imparting resistance. (Wright et al. 1998). However, in our present study, both classical Mendelian genetics and QTL analysis have shown similar results; that is: (1) resistance to pathotype I is controlled by a major locus and that of pathotype II by two major independent recessive loci with complementary gene action; (2) both methods mapped these respective major genes to the same regions in the linkage groups. The similarity between the results of the two types of genetic analysis indicates the strong reliability of our experimental data.
Resistance to ascochyta blight in chickpea accession ILC 72 was reported to be governed by a single dominant gene in a previous study by Singh and Reddy (1983). They also suggested that the presence of some additional minor genes played a part in imparting the resistance. Observations of Kusmenoglu (1990) suggested that resistance to ascochyta blight was conferred by two recessive genes. Using the same source of resistance, Tekeoglu et al. (2000) reported three major recessive genes controlling resistance, possibly in conjunction with many minor modifying genes. In the same RIL populations, Santra et al. (2000) demonstrated the presence of two or three QTLs conferring resistance to ascochyta blight, depending upon on the year of planting. Since these studies were conducted under field conditions, there was very little control on the inoculum (pathotypes of A. rabiei and other species of pathogens), inoculum density, temperature, humidity and plant age. Therefore, the general incongruity which exists between these studies and ours is mainly the result of the use of a different source of resistant and susceptible parents, and of a different screening method and pathotypes.
In plants, the majority of resistance genes appear to be linked and organized as complex clusters (Dickinson et al. 1993; Anderson et al. 1996; Dixon et al. 1996, 1998; Parniske et al. 1997; Song et al. 1997; Meyers et al. 1998; Shen et al. 1998). Our results in chickpea also supported the view that disease resistance genes are linked, and showed that ar1 (pathotype I-specific resistance gene) and ar2a (one of the pathotype II-specific resistance genes) are located on LG 2. Furthermore, the comparison of our results with the mapping results of fusarium wilt resistance genes in chickpea suggests that fusarium wilt resistance genes are also located on LG 2 (Winter et al. 2000).
Genetic dissection of ascochyta blight resistance in this study provides insight into pathotype-specific resistance in chickpea. Further fine mapping of the chickpea genome will help in the isolation of these resistance genes/clusters and in marker-assisted selection to improve ascochyta blight resistance in chickpea.
References
Anderson PA, Okubura PA, Arroyogarcia R, Meyers BC, Michelmore RW (1996) Molecular analysis of irradiation-induced and spontaneous deletion mutants at a disease resistance locus in Lactuca sativa. Mol Gen Genet 251:316–325
Banerjee D, Zhang X, Bent AF (2001) The leucine-rich repeat domain can determine effective interaction between RPS2 and other host factors in Arabidopsis RPS2-mediated disease resistance. Genetics 158:439–450
Dickinson MJ, Jones DA, Jones JDG (1993) Close linkage between the Cf-2/Cf-5 and Mi resistance loci in tomato. Mol Plant-Microbe Interact 6:341–347
Dixon MS, Jones DA, Keddie JS, Thomas CM, Harrison K, Jones JD (1996) The tomato Cf-2 disease resistance locus comprises two functional genes encoding leucine-rich repeat proteins. Cell 84:451–459
Dixon MS, Hatzixanthis K, Jones DA, Harrison K, Jones JDG (1998) The tomato Cf-5 disease resistance gene and six homologs show pronounced allelic variation in leucine-rich repeat copy number. Plant Cell 10:1915–1925
Ghislain M, Trognitz B, Herrera MR, Solis J, Casallo G, Vásquez C, Hurtado O, Castillo R, Portal L, Orrillo M (2001) Genetic loci associated with field resistance to late blight in offspring of Solanum phureja and S. tuberosum grown under short-day conditions. Theor Appl Genet 103:33–442
Hüttel B, Winter P, Weising K, Choumane W, Weigand F, Kahl G (1999) Sequence-tagged microsatellite-site markers for chickpea (Cicer arietinum L.). Genome 42:210–217
Iqbal MJ, Meksem K, Njiti VN, Kassem MA, Lightfoot DA (2001) Microsatellite markers identify three additional quantitative trait loci for resistance to soybean sudden-death syndrome (SDS) in Essex × Forrest RILs. Theor Appl Genet 102:187–192
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Kusmenoglu I (1990) Ascochyta blight of chickpea: inheritance and relationship to seed size, morphological traits and isozyme variation. MS thesis, Washington State University, Pullman, Washington
Lincoln SE, Daly MJ, Lander ES (1993) MAPMAKER/EXP version 3.0: A Tutorial and Reference Manual, 3rd edn. Whitehead Institute for Biomedical Research, Cambridge, Massachusetts, USA
Mackill DJ, Bonman JM (1992) Inheritance of blast resistance in near-isogenic lines of rice. Phytopathology 82:746–749
Maddala GS (1992) Introduction to Econometrics, 2nd edn. MacMillan Publishing Company, New York, pp 119–120
Malik BA, Rahman MS (1992) Breeding for ascochyta blight-resistance desi chickpea in Pakistan. In: Singh KB, Saxena MC (eds) Disease resistance breeding in chickpea. ICARDA, Aleppo, Syria, pp 77–85
Meyers BC, Chin DB, Shen KA, Sivaramakrishnan S, Lavelle DO, Zhang Z, Michelmore RW (1998) The major resistance gene cluster in lettuce is highly duplicated and spans several megabases. Plant Cell 10:1817–1832
Nene YL, Reddy MV (1987) Chickpea diseases and their control. In: Singh KB, Saxena MC (eds) Disease resistance breeding in chickpea. ICARDA, Aleppo, Syria, pp 233–270
Parniske M, Hammond-Kosack KE, Golstein C, Thomas CM, Jones DA, Harrison K, Wulff BB, Jones JD (1997) Novel disease resistance specificities result from sequence exchange between tandemly repeated genes at the Cf-4/9 locus of tomato. Cell 91:821–832
Porta-Puglia A (1992) Variability in Ascochyta rabiei. In: Singh KB, Saxena MC (eds) Disease resistance breeding in chickpea. ICARDA, Aleppo, Syria, pp 135–143
Reddy MV, Nene YL, Singh G, Bashir M (1990) Strategies for management of foliar diseases of chickpea. In:Chickpea in the nineties. Proc 2nd Int Workshop on Chickpea Improvement, 4–8 Dec. 1989, ICRISAT Center, India, pp 117–127
Santra DK, Tekeoglu M, Ratnaparkhe M, Kaiser WJ, Muehlbauer FJ (2000) Identification and mapping of QTLs conferring resistance to ascochyta blight in chickpea. Crop Sci 40:1606–1612
Shen KA, Meyers BC, Islam-Faridi MN, Chin DB, Stelly DM, Michelmore RW (1998) Resistance gene candidates identified by PCR with degenerate oligonucleotide primers map to clusters of resistance genes in lettuce. Mol Plant-Microbe Interact 11:815–823
Singh KB, Reddy MV (1983) Inheritance of resistance to ascochyta blight in chickpea. Crop Sci 23:9–10
Singh KB, Reddy MV (1993) Resistance to six races of Ascochyta rabiei in the world germplasm collection of chickpea. Crop Sci 33:186–189
Singh KB, Hawtin GC, Nene YL, Reddy MV (1981) Resistance in chickpeas to Ascochyta rabiei. Plant Dis 65:586–587
Singh KB, Reddy MV, Haware MP (1992) Breeding for resistance to ascochyta blight in chickpea. In: Singh KB, Saxena MC (eds) Disease resistance breeding in chickpea. ICARDA, Aleppo, Syria, pp 23–54
Song WY, Pi LY, Wang GL, Gardner J, HolstenT, Ronald PC (1997) Evolution of the rice Xa21 disease resistance gene family. Plant Cell 9:1279–1287
Tekeoglu M, Santra DK, Kaiser WJ, Muehlbauer FJ (2000) Ascochyta blight resistance in three chickpea recombinant inbred line populations. Crop Sci 40:1251–1256
Tewari SK, Pandey MP (1986) Genetics of resistance to ascochyta blight of chickpea (Cicer arietinum L.). Euphytica 35:211–215
Udupa SM, Weigand F, Saxena MC, Kahl G (1998) Genotyping with RAPD and microsatellite markers resolves pathotype diversity in the ascochyta blight pathogen of chickpea. Theor Appl Genet 97:299–307
Udupa SM, Robertson LD, Weigand F, Baum M, Kahl G (1999) Allelic variation at (TAA) n microsatellite loci in a world collection of chickpea (Cicer arietinum L.) germplasm. Mol Gen Genet 261:354–363
Vir S, Grewal JS (1974) Physiological specialization in Ascochyta rabiei, the causal organism of gram blight. Indian Phytopathol 27:265–266
Vir S, Grewal JS, Gupta VP (1975) Inheritance of resistance to ascochyta blight in chickpea. Euphytica 24:209–211
Wang D, Arelli PR, Shoemaker RC, Diers BW (2001) Loci underlying resistance to Race 3 of soybean cyst nematode in Glycine soja plant introduction 468916. Theor Appl Genet 103:561–566
Wang GL, Mackill DJ, Bonman JM, McCouch SR, Champoux MC, Nelson RJ (1994) RFLP mapping of genes conferring complete and partial resistance to blast from durably resistant rice cultivar. Genetics 136:1421–1434
Wilson IW, Schiff CL, Hughes DE, Somerville SC (2001) Quantitative trait loci analysis of powdery mildew disease resistance in the Arabidopsis thaliana accession Kashmir-1. Genetics 158:1301–1309
Winter P, Pfaff T, Udupa SM, Hüttel B, Sharma PC, Sahi S, Arreguin-Espinoza R, Weigand F, Muehlbauer FJ, Kahl G (1999) Characterization and mapping of sequence-tagged microsatellite sites in the chickpea (Cicer arietinum L.) genome. Mol Gen Genet 262:90–101
Winter P, Beko-Iseppon A.-M, Hüttel B, Ratnaparkhe M, Tullu A, Sonnante G, Pfaff T, Tekeoglu M, Santra D, Sant VJ, Rajesh PN, Kahl G, Muehlbauer FJ (2000) A linkage map of the chickpea (Cicer arietinum L.) genome based on recombinant inbred lines from a C. arietinum × C. reticulatum cross: localization of resistance genes for Fusarium wilt races 4 and 5. Theor Appl Genet 101:1155–1163
Wright RJ, Thaxton PM, El-Zik, KM, Paterson AH (1998) D-subgenome bias of Xcm resistance genes in tetraploid gossypium (cotton) suggests that polyploid formation has created novel avenues for evolution. Genetics 149:1987–1996
Young ND (1996) QTL mapping and quantitative disease resistance in plants. Annu Rev Phytopathol 34:479–501
Acknowledgements.
The authors' research was supported by grants to ICARDA from the German Federal Ministry of Economic Cooperation and Development (BMZ, Bonn, Germany) and the Arab Fund for Economic and Social Development (AFESD, Kuwait).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Hagemann
Rights and permissions
About this article
Cite this article
Udupa, S.M., Baum, M. Genetic dissection of pathotype-specific resistance to ascochyta blight disease in chickpea (Cicer arietinum L.) using microsatellite markers. Theor Appl Genet 106, 1196–1202 (2003). https://doi.org/10.1007/s00122-002-1168-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-002-1168-x