
Abstract
The anatomy and physiology of adults with congenital heart disease place them at increased risk for thrombosis. This chapter discusses the most common thromboses, namely, deep venous thrombosis, pulmonary embolism, arrhythmias and thromboembolic stroke, thrombosis associated with prosthetic valves, and special concerns of the adult with Fontan circulation. The chapter closes with a discussion of therapeutics for thrombosis prevention and management.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Adult with congenital heart disease
- Anticoagulation
- Thrombosis
- Deep venous thrombosis
- Pulmonary embolism
- Stroke
- Arrhythmias
- Prosthetic valves
- Fontan
1 Propensity to Thrombosis in Adults with Congenital Heart Disease
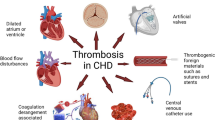
It has long been recognized that adults with congenital heart disease (CHD), in particular cyanotic congenital heart disease (CCHD), are prone to thrombosis whether or not they have Eisenmenger physiology [1,2,3]. Although cerebrovascular events and pulmonary embolism (PE) are the most widely discussed, thrombosis of additional sites in the single-ventricle population, namely, the Fontan pathway [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19] and deep veins of the lower extremities [20], may be equally devastating.
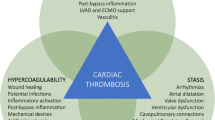
It is not surprising that adult congenital heart disease (ACHD) patients are prone to thrombosis. Throughout their lifetime, they have been exposed to the triad of risk factors for thrombosis initially described by Virchow in 1856 [21], namely, (a) stasis of blood, (b) hypercoagulability, and (c) endothelial injury. As in pediatric heart disease [14, 22, 23], an expansion of Virchow’s triad [24] appears warranted:
-
1.
Altered blood flow (an expansion of “stasis of blood”)
In addition to stasis of blood, ACHD patients have the potential to be exposed to “altered blood flow”:
-
(a)
Stasis of blood may occur in dilated heart chambers as well as in dilated native or prosthetic outflow tracts. These areas of stasis may serve as a nidus for thrombus formation.
-
(b)
Turbulent flow may occur across stenotic native or prosthetic heart valves, intracardiac devices/leads, stents, and/or obstructed native or prosthetic outflow tracts activating platelets either directly [25, 26] or by increasing sheer stress that can result in platelet activation independent of endothelial damage [27]. Activated platelets adhere to these “abnormal,” artificial vascular surfaces and may result in thrombus formation. The same mechanisms are involved in the turbulent flow of mechanical support (cardiopulmonary bypass (CPB), extracorporeal membrane oxygenation (ECMO), ventricular assist devices (VAD)) where thrombosis can occur despite systemic anticoagulation. Platelets are activated. Thrombin generation is increased via the extrinsic system in response to cytokines, ischemia, sheer stress, and activated platelets and via the contact system in response to contact of blood with the circuit. A profound derangement in the coagulation/fibrinolytic balance occurs with an ineffective counterbalance by anticoagulant factors such as antithrombin III. The procoagulant state may persist well past decannulation [28,29,30,31]. Although children, especially infants with their immature coagulation system, may be more prone to such effects, it seems most likely that at least some of these factors are at play in ACDH patients as well.
-
(a)
-
2.
Blood component abnormalities (expansion of hypercoagulability)
Blood component abnormalities have been described in children with acyanotic, acquired, as well as single-ventricle heart disease and include altered anticoagulation protein levels, increased thrombin generation potential, decreased endogenous inhibitors of coagulation, and decreased fibrinolytic proteins, among others. Such coagulopathies were first identified in children and adolescents with a Fontan circulation but more recently have been described through all stages of single-ventricle palliation, i.e., even before the Fontan completion [32,33,34,35,36,37,38,39,40,41]. Coagulopathies in Fontan patients are further described in the section on Fontans below.
As is well known to physicians caring for adults with CHD, erythrocytosis and hyperviscosity associated with chronic cyanosis are risk factors for thrombosis as well. This may be exacerbated by iron deficiency anemia which makes red cells more rigid and less deformable as well as by dehydration [3, 14, 34, 42].
-
3.
Endothelial injury and dysfunction
Turbulent flow along normal vascular endothelial surfaces results in endothelial injury and dysfunction which is a precursor to thrombosis. Endothelial injury exposes tissue factor and subendothelial collagen-stimulating platelet aggregation and coagulation at the site of injury. Altered levels of endothelial markers in plasma have been found to precede intravascular coagulation and thrombosis in one study of patients with functionally univentricular hearts [43]. In addition, adults with CHD most likely have had numerous cardiac catheterizations, exposure to CPB, and central lines throughout their medical history that have the potential of further damaging an already compromised endothelium.
-
4.
Inflammation and bloodstream infection
Adults with CHD are at risk for exposure to inflammation and blood stream infections which pose further risk for thrombosis. In addition to creating turbulent flow and endothelial disruption, mechanical support (CPB, ECMO, VAD) results in an inflammatory state [29,30,31], activating platelets and exposing tissue factor by cytokine release [26]. Recent pediatric studies have implicated sepsis with increased thrombus formation especially in the presence of an indwelling central venous line [44,45,46].
Patients with CHD often have one or more of these thrombosis risk factors at play, while during mechanical circulatory support (CPB, ECMO, VAD), all four of these conditions coexist.
1.1 Eisenmenger Physiology
A special note should be made about patients with Eisenmenger physiology who have a well- known risk of bleeding as well as clotting, thereby making anticoagulation controversial and often contradicted [Giannakoulas 2015]. In addition to the four factors described above, pulmonary hypertension in and of itself may result in endothelial dysfunction in the pulmonary arteries increasing the risk of pulmonary emboli. Cell aggregation secondary to decreased pulmonary blood flow may also be a thrombotic risk [10, 47]. On the other side of the spectrum, pulmonary hemorrhage from rupture of one of the pulmonary vessels is often the cause of death in this patient population [48,49,50].
This chapter will discuss common thrombotic concerns in the adult with CHD, namely, deep venous thrombosis (DVT), pulmonary embolism (PE), arrhythmias as a risk factor for stroke, and special concerns in the Fontan patient. We will conclude with a discussion of both currently available and “under-investigation” antithrombotic therapies.
2 Deep Venous Thrombosis and Pulmonary Embolism in Adults with Congenital Heart Disease
A recent editorial by Giannakoulas and Boutsikou [51] reviewed the prevalence of thrombosis in adults with CCHD from ten studies over the past 25 years. The prevalence of arterial cerebral thrombosis ranged from 0 to 47%, whereas the prevalence of pulmonary thrombosis ranged from 13.2 to 35%. Only two studies looked at both cerebral and pulmonary thromboses, and as expected, the studies differed in whether they were prospective/retrospective and most importantly in whether the thrombosis was clinically evident or clinically silent and documented on screening studies only. The most recent of the included studies by Jensen [52] documented cerebral thrombosis at 47% and pulmonary thrombosis at 31% in a prospective study and commented that the large discrepancy between clinical history and imaging findings suggests a high prevalence of silent thrombotic events.
Although single-ventricle anatomy and cyanosis are significant risk factors for early postoperative thrombosis in infants and children (especially DVT associated with central venous lines) [53, 54], there is limited data on the significance of these risk factors in the adult with CHD.
It is estimated that just over half of all medical patients in US acute care hospitals are at risk for venous thromboembolism (VTE) including DVT and PE [55] with those in the critical care setting at particularly increased risk [56]. Pulmonary embolism is considered the most common preventable cause of hospital death [57], and the Institute of Medicine defines the failure to provide adequate thromboprophylaxis to hospitalized, at-risk patients a medical error [58]. Although the prevalence of VTE in critically ill adults with CHD in the ICU is not known, based on the propensity for thrombosis in adults with CHD and the increased risk of VTE with age, it can be inferred that adults with CHD incur as much or more risk for VTE than those without CHD.
2.1 Risk Assessment and VTE Prophylaxis in Hospitalized Patients
Hospitalized patient characteristics considered high risk for VTE are malignancy, stroke, major general surgery and orthopedic surgery, trauma and fractures, spinal cord injury, obesity, pregnancy, heart failure, myocardial infarction, sepsis, respiratory failure, renal disease, older age, prolonged immobility >3 days, hypercoagulable states, and history of previous VTE [59, 60]. Although there are a number of schemes devised to assess thrombosis risk in the non-congenital heart disease population [61,62,63], they are not validated in those with CHD, and in general, we recommend that all adults with CHD in the ICU receive prophylactic anticoagulation unless they are at increased risk for bleeding (i.e., acute gastrointestinal or intracerebral bleed, profound thrombocytopenia).
Pharmacologic VTE prophylaxis is recommended over mechanical means (i.e., sequential compression devices) with the agent of choice being low molecular weight heparin (LMWH), though unfractionated heparin (UFH) can be used for those with renal failure. If the patient is at high risk for bleeding, sequential compression devices and elastic graduated compression stockings are recommended over no prophylaxis. Thromboprophylaxis should continue until the patient is ambulatory or discharged.
In the general population, prophylaxis has been found to decrease the risk of VTE in acutely ill medical patients, but there is no proven mortality benefit in medical patients [64]. In contrast, mortality benefit has been seen in surgical patients [65], although the reasons for this difference are not known. Outcomes for VTE prophylaxis in the adult with CHD are yet to be defined.
2.2 Management of Deep Venous Thrombosis and Pulmonary Embolism in ACHD
The mainstay of deep vein thrombosis treatment is anticoagulation in the absence of contraindications (i.e., active hemorrhage, recent intracerebral bleed, trauma, thrombocytopenia). The purpose of anticoagulation is to prevent complications such as clot extension, acute pulmonary embolism, and recurrent DVT. Anticoagulation is specifically indicated for proximal DVT which has a higher risk of embolization and death compared to distal (e.g., below the knee) DVT [66] as well as for symptomatic distal DVTs. Other factors should be considered when deciding between surveillance and anticoagulation of asymptomatic distal DVTs such as high D-dimer levels, clot burden and extent, prior DVT, prolonged immobility, as well as bleeding risk. Inferior vena cava filter can be considered for those with a lower extremity proximal DVT and an absolute contraindication to anticoagulation [67]. Thrombolytic therapy or catheter-based extraction is reserved for rare cases such as those with massive iliofemoral DVT (phlegmasia cerulea dolens).
The diagnosis of PE in the single-ventricle patient by conventional means such as computed tomography with contrast or ventilation-perfusion scans is limited by the alteration in pulmonary arterial blood flow (lack of a systemic venous mixing chamber), and thus the diagnosis may rely on pulmonary angiography. The treatment of acute PE is guided by severity, hemodynamic stability, and bleeding risk. Therapeutic anticoagulation is indicated for patients with confirmed PE and no contraindications to anticoagulation. In general, those with hemodynamic instability with concern for massive PE should be considered for more aggressive therapies such as thrombolysis and/or mechanical embolectomy [68, 69].
The choice of anticoagulation for the treatment of VTE must take into account a number of factors including liver disease, renal disease, route of administration and dosing, compliance, and reversibility, keeping in mind that the critically ill patient in the ICU may have a predisposition toward hemorrhage. Direct oral anticoagulants (DOACs) are safe and effective alternatives to standard anticoagulation (heparin and warfarin). Table 28.1 summarizes factors to consider in selecting anticoagulation for patients with acute VTE [68]. Extensive discussion regarding diagnosis and treatment of VTE is beyond the scope of this chapter, and management should follow recommendations from clinical guidelines [68, 69].
3 Arrhythmia and Thromboembolic Stroke in Adults with Congenital Heart Disease
Atrial arrhythmias such as intra-atrial reentry tachycardia, typical atrial flutter, and atrial fibrillation occur in 15% of adult patients with congenital heart disease [70]. The incidence of atrial arrhythmias increases with age and underlying disease complexity, representing a major source of morbidity for adults with CHD including thromboembolic stroke [70, 71]. In the critical care setting, there is an increased risk of atrial tachyarrhythmias due to inflammation, surges in catecholamines, ischemia, volume shifts, and atrial stretch [72]. Additional superimposed risk factors include immobility, presence of prosthetic material, indwelling leads, intracardiac shunts or septal patch margin defects, sluggish flow and venous stasis, cyanosis, and hypercoagulability [3, 12], making the risk of stroke considerable.
The presence of atrial fibrillation increases the risk of thromboembolic stroke by fivefold, but stroke risk also depends on the presence of other risk factors [73]. The anticoagulation management of atrial fibrillation or atrial flutter in the critically ill adult with CHD must balance the risk of bleeding against the risk of stroke. The CHA2DS2-VASc score and the HAS-BLED score are means of assessing stroke and bleeding risk, respectively. The CHA2DS2-VASc score is a risk stratification scheme for adults with non-valvular atrial fibrillation to assess stroke risk which considers the following: age, gender, heart failure, hypertension, history of stroke or transient ischemic attack, vascular disease, and diabetes [74] (Table 28.2). Those with CHA2DS2-VASc score ≥ 2 are considered at high risk for stroke and in whom benefit from anticoagulation exceeds bleeding risk. Though not necessarily meant to be used in the ICU setting, it can potentially help determine overall stroke risk. HAS-BLED is a risk score intended to assess bleeding risk which must always be considered when placing a patient on anticoagulation therapy [74]. The HAS-BLED score assigns 1 point for the presence of each of the following bleeding risk factors: hypertension, abnormal renal and/or liver function, previous stroke, bleeding history, labile INR, elderly and concomitant drugs, and/or alcohol abuse with a score of ≥3 indicating a high risk of bleeding (Table 28.3).
The applicability of both schemes in ACHD has been examined [75,76,77]. The Anticoagulation Therapy in Congenital Heart Disease (TACTIC) study was a retrospective multicenter trial examining the incidence of thromboembolic events in adults with CHD and atrial arrhythmias and the predictive capacity of the CHA2DS2-VASc and HAS-BLED scores [75]. There were 482 patients managed on a variety of antiplatelet/anticoagulation schemes, including no therapy. The thromboembolic event rate was 8.7% over an average follow-up time of 11.3 ± 9.4 years after the qualifying arrhythmia. The CHA2DS2-VASc score was not predictive of thromboembolism, but higher congenital heart lesion complexity correlated with higher thromboembolic risk. Bleeding events occurred in 8.3% of patients, and HAS-BLED was an independent predictor of major bleed on multivariable analysis.
That the CHA2DS2-VASc score did not adequately predict thromboembolic risk in adults with CHD may be a reflection of a distinct, heterogeneous patient population with wide variation of underlying lesions and younger age. The average age of ACHD patients who experience atrial arrhythmias and the age at which they present with stroke are significantly younger than that of the general population [70, 78, 79]. The authors suggest the CHA2DS2-VASc score could be modified to include disease complexity as defined by the Bethesda criteria [80] which is consistent with recent guidelines on arrhythmia management in ACHD that recommend disease complexity be considered in guiding anticoagulation strategy. In this document, long-term anticoagulation is a Class I recommendation for intra-atrial reentry tachycardia or atrial fibrillation in patients with highly complex CHD and Class IIA recommendation for those with moderate forms of CHD. For patients with simple non-valvular forms of CHD with intra-atrial reentry tachycardia or atrial fibrillation, anticoagulation, aspirin, or no thromboprophylaxis is a Class IIB recommendation [81].
In practice, there is no risk stratification scheme that completely captures stroke risk for adults with CHD and atrial arrhythmias, especially in the ICU setting, but underlying disease complexity and risk factors as described by the CHA2DS2-VASc score as well as HAS-BLED score can be used in guiding therapy especially as the patient transitions out of the ICU setting and into ambulatory care. In general, if a new-onset atrial fibrillation or flutter continues for more than 48 h in the ICU, anticoagulation is recommended, and any attempt at rhythm control should be preceded by transesophageal echocardiography to rule out intracardiac thrombosis. Anticoagulation should be instituted and continued for at least 4–6 weeks afterward. However, bleeding risk in the ICU patient may be prohibitive due to platelet dysfunction from renal failure, thrombocytopenia, liver disease/cirrhosis, coagulopathy, etc., and benefit must be balanced against risk.
Patients who are admitted to the ICU on anticoagulation for stroke prophylaxis in the setting of atrial arrhythmias should have anticoagulation continued. Consideration should be made to switching to a reversible short-acting agent such as intravenous heparin if invasive procedures are anticipated. Gaps in anticoagulation can be managed for short periods of time as day-to-day stroke risk is relatively low.
4 Prosthetic Valves in Adults with Congenital Heart Disease
Valve replacement surgery is one of the most common types of cardiac surgery in ACHD [82]. Thromboembolic complications and anticoagulation-related bleeding are major contributors to morbidity and mortality after valve replacement surgery [83].
Management of patients with prosthetic valves in the ICU may involve the postoperative management of those who undergo valve replacement or long-term complications such as valve thrombosis, embolic phenomenon, or infective endocarditis. Furthermore, the anticoagulation management of patients with mechanical valve prostheses can post special challenges for patients in the ICU who are admitted for noncardiac reasons, and interruption of anticoagulation may be necessary.
4.1 Thromboembolism Prophylaxis
The highest risk of thromboembolic complications occurs within the first 3 months of surgery for both bioprosthetic and mechanical valve prostheses [84]. Atrial arrhythmias, a common comorbidity found in conjunction with valvular heart disease, can further increase the risk of thromboembolic stroke. After implantation of a mechanical prosthetic valve, anticoagulation with warfarin, a vitamin K antagonist, is recommended which has been proven to decrease risk of thrombosis and thromboembolism [85]. The addition of antiplatelet therapy can decrease the risk of systemic embolism or death in patients with mechanical heart valves [86]. The American College of Cardiology (ACC)/American Heart Association (AHA) recommends anticoagulation in patients with mechanical valve prostheses with warfarin at a target INR of 2.5 in the aortic and 3.0 in the mitral positions in the absence of other risk factors (Class I Ib) as well as a low-dose antiplatelet agent (Class IA) [87]. The On-X mechanical prosthesis (On-X Life Technologies Inc., Austin, Texas) in the aortic position can be maintained at a lower target INR 1.5–2.0 (Class IIb) after 3 months of standard therapy [88]. Direct oral anticoagulants (DOACs) should not be used in patients with mechanical valves. A recent trial investigating dabigatran vs. warfarin after aortic or mitral mechanical valve replacement was terminated early after demonstrating excess thromboembolic events and bleeding in the dabigatran arm [89].
The risk of thromboembolism after bioprosthetic valve surgery is not as high as mechanical valve replacement surgery, but there is an increased risk of events in the first 3–6 months after surgery [84, 90]. Early thrombogenicity of bioprosthetic valves can be attributed to incomplete endothelialization [91], so therefore some form of antiplatelet and/or antithrombotic therapy is recommended after bioprosthetic valve surgery. Anticoagulation with a warfarin for 3 months after bioprosthetic mitral or aortic valve replacement surgery with a target INR 2.5 is considered a reasonable strategy (IIa recommendation) to address risk of early postoperative thrombotic risk [88]. The role of DOACs in the anticoagulation management of patients with bioprosthetic valves is not yet defined.
4.2 Bridging Anticoagulation Therapy
Anticoagulation management including cessation and bridging therapy when there is interruption should take into account the type of procedure for which interruption is being considered, bleeding risk, as well as type, location, and number of prosthetic heart valves [88]. Minor procedures such as cataract removal, cutaneous biopsy, or dental procedures generally do not require cessation of anticoagulation. Critically ill patients in the ICU may have predisposition toward hemorrhage as discussed above including sepsis, thrombocytopenia, and renal and/or liver failure. The ACC/AHA guidelines do not recommend bridging when anticoagulation is interrupted in patients with mechanical aortic valves and no other risk factors for thrombosis [87].
When oral anticoagulation with warfarin is temporarily interrupted, bridging therapy with intravenous UFH or subcutaneous LMWH can be considered for higher-risk patients including those with mechanical mitral valve prostheses or patients with aortic mechanical valve prosthesis and an additional risk factor (e.g., atrial fibrillation, previous thromboembolism, hypercoagulable state, older-generation mechanical valve, left ventricular systolic dysfunction, or >1 mechanical valve) [92], but decisions should be made on an individual basis. Data supports the use of LMWH for bridging as safe and effective in mechanical heart valves [93, 94] although intravenous unfractionated heparin UFH may be preferable in the ICU setting given its short half-life. Figure 28.1 summarizes the ACC/AHA recommendations for antithrombotic therapy in prosthetic valves [87].

ACC/AHA recommendations for anticoagulation of prosthetic heart valves [87, 88]. Note: Updates in 2017 include [88]: • IIa recommendation from IIb for VKA after bioprosthetic AVR, • On-X AVR without risk factors target INR 1.5–2.0 (IIb, LOE B-R), and • IIb recommendation for VKA INR target 2.5 for 3 months after TAVR
4.3 Prosthetic Valve Thrombosis and Stenosis
Mechanical prosthetic valve obstruction can be caused by thrombosis, pannus, or a combination thereof and can present as progressive dyspnea or in extremis with severe pulmonary edema [95]. Transthoracic echocardiogram is the first-line imaging modality (including Doppler gradient across the valve), but transesophageal echocardiography is often necessary to assess leaflet motion. Computed tomography and fluoroscopy are useful to evaluate for thrombosis, but differentiation between thrombosis and pannus is challenging and can be difficult to distinguish [96]. Clinical history is important in the evaluation including time from surgery, history of inadequate anticoagulation, and acuity of symptoms as thrombosis more often presents acutely compared to pannus. Infective endocarditis must be excluded. Once thrombosis is diagnosed, anticoagulation should be instituted. Mechanical left-sided prosthetic valve obstruction has a high mortality rate and requires urgent therapy including fibrinolysis or surgery (Class Ib) [88].
Bioprosthetic valve thrombosis usually presents in a more subacute manner with progressive stenosis and can occur years after surgery [97]. Evaluation is similar as above with the use of transesophageal echocardiography to better evaluate for thrombosis. Again, infective endocarditis must be ruled out. Bioprosthetic valve thrombosis has been shown to respond to warfarin [98] which can be considered as an initial treatment in patients who are hemodynamically stable (Class IIa) [88].
Patients with a prosthetic valve and stroke should undergo work-up for cardioembolic stroke including evaluation of the prosthetic valve by echocardiogram. Specifically, transesophageal echocardiography is recommended to better evaluate valve function, evidence of thrombosis or vegetation, or any other source of embolism including a careful interrogation of the left atrium, left atrial appendage, and aorta for atherosclerotic plaque. If thromboembolism occurred in the setting of inadequate antithrombotic therapy, institution of adequate therapy is the first step [87]. If thromboembolism occurred despite adequate antithrombotic therapy, consideration should be given to augmenting therapy which includes the addition of low-dose aspirin if maintained only on therapeutic warfarin, increasing the goal INR, or adding warfarin for those who were maintained on aspirin alone (i.e., bioprosthetic valves) [87].
5 Special Consideration of Thrombosis in Adult Patients with Fontan Circulation
It has long been recognized that thrombosis in the patient with Fontan circulation is associated with significant mortality and morbidity. A recent systemic review by Alsaied in 2017 of 28 studies, 6707 patients, and a mean follow-up time from Fontan of 8.23 ± 5.42 years found thrombosis and bleeding to be the fifth most common cause of death accounting for 10% of the 1000 deaths reported [19].
The overall incidence of thrombosis in the patient with Fontan circulation is about 12% in recent reports [15, 18] and 5.2% in a meta-analysis looking at extracardiac Fontans only [13]. Earlier reports describe a 3–33% incidence with stroke in 1.4–3.6% [14]. The discrepancy in reported incidence is secondary to the variability in the population studied, the time from the Fontan procedure, the method of thrombosis detection, the location of the thrombosis investigated (i.e., all thrombosis vs. pulmonary emboli vs. cerebral vascular event), and whether the study investigated clinically evident or silent thrombosis. Although these reviews are an important source of observational data, their heterogeneous designs limit the ability to draw conclusions from which clinical practice guidelines can be generated. They have, however, generated the following important observations:
-
There appears to be two peaks in the incidence of thrombosis post Fontan: early (0–6 months) [99] and late (5–15 years post Fontan) [4,5,6, 9, 11, 16].
-
Thrombosis post Fontan is most commonly found in the Fontan itself, intracardiac (systemic venous atrium, pulmonary venous atrium, hypoplastic ventricle) and pulmonary arteries, although it has been reported as well in the ligated pulmonary artery stump, hypoplastic aortic root, and coronary arteries and associated with heparin-induced thrombocytopenia (HIT) [7, 15, 17]. Silent pulmonary emboli have also been described [8].
-
Risk factors supported by focused retrospective observational studies include atriopulmonary type of Fontan connection, bilateral bidirectional cavopulmonary anastomoses, hypoplastic cardiac chambers with flow stasis, presence of a blind-ended pulmonary artery stump, and a history of previous thrombosis. Additional potential factors supported by general retrospective observational studies or expert opinion include protein-losing enteropathy, prolonged pleural effusions, prolonged immobilization, ventricular dysfunction, arrhythmia, presence of thrombogenic foreign material, atrial-level fenestration, Kawashima connection, and an abnormal thrombophilia profile [14]. Additional risk factors for stroke identified in adults with CHD that include the Fontan include systemic arterial hypertension, atrial fibrillation, iron deficiency anemia, microcytosis, and prior phlebotomy [3]. Additional risk factors for pulmonary embolus include age, female gender, low oxygen saturation, ventricular dysfunction, and low systolic flow velocities in the pulmonary arteries [10, 47].
It is not surprising that the adult with Fontan circulation has a propensity to thrombosis, especially in the ICU setting. The factors described above of altered blood flow, blood component abnormalities, and endothelial dysfunction are intrinsic to the Fontan. Several studies have evaluated coagulation proteins in single-ventricle patients and have shown an infantile pattern, namely, lower levels of both pro- and anticoagulant proteins, namely, low protein C and suppression of the thrombomodulin-protein C-protein S pathway [32, 35, 36, 40]. In addition several studies have documented elevated factor VIII after the Fontan [34, 37, 41]. Odegard in 2009 showed lower than control levels of both pro- and anticoagulation proteins from Norwood stage I up to Fontan [39]. After the Fontan protein C stayed low, and factor VIII was found to be significantly higher than controls; 42% of Fontan patients had F VIII >160%. There was no correlation with hemodynamics, hepatic synthetic function, or hepatocellular dysfunction. The speculation is that high factor VIII, low protein C plus Fontan physiology puts patients at risk of thrombosis over a lifetime.
Knowing that the Fontan patient is prone to thrombosis, what data is available regarding thromboprophylaxis? Two recent meta-analyses warrant mention. In 2011 Marrone reviewed 20 studies, 1075 patients who had extracardiac Fontans [13]. The average time from Fontan in the studies was 2 months to 12 years. The overall thrombotic rate was 5.2%, 4.5% in those who received aspirin alone and 5% in those receiving warfarin with or without aspirin. Alsaied in 2015 reviewed 10 studies, 1200 patients with all types of Fontans [15]. The average time from Fontan in the combined studies was 7.1 years. The overall thrombotic rate was 11.3%: 18.6% in those receiving no thromboprophylaxis, 8.6% in those receiving aspirin only, and 9% in those receiving warfarin. From these meta-analyses, it appears that some form of thromboprophylaxis is protective but that warfarin is not superior to aspirin. Whether or not thrombosis is less frequent in the extracardiac Fontan warrants further investigation.
When considering the adult Fontan patient, several limitations in these meta-analyses must be acknowledged. The average follow-up from the Fontan in the two meta-analyses was less than 15 years, giving little information about the risk of thrombosis and effect of thromboprophylaxis in the third and fourth decade of life. Second, the studies address neither time in therapeutic range for warfarin patients nor aspirin resistance in the patients receiving aspirin. Third, additional risk factors for thrombosis were not address in either meta-analysis.
Carefully considering the currently available, albeit limited, data, the American Heart Association’s 2013 Scientific Statement on Prevention and Treatment of Thrombosis in Pediatric and Congenital Heart Disease makes the following recommendations regarding prevention and treatment of thrombosis in the patient with a palliated single ventricle at all stages [14]:
-
1.
Patients with a palliated single ventricle should undergo clinical assessment for the anatomic and hemodynamic risk factors for thrombus. Risk factors for thrombus (as listed earlier in this chapter) should be ameliorated (i.e., arrhythmias, ventricular dysfunction, prolonged immobilization) and minimized (blind-ended pulmonary artery stump, prolonged immobilization) when possible (Class I; Level of Evidence B).
-
2.
For patients with a palliated single ventricle, serial clinical assessment and monitoring for changes in anatomic and hemodynamic thrombotic risk factor are indicated because risk factors may change over time (Class I; Level of Evidence C). New risk factor for thrombus should be ameliorated (i.e., arrhythmias, ventricular dysfunction, prolonged immobilization) and minimized (prolonged immobilization) when possible (Class I; Level of Evidence C).
-
3.
Patients with a palliated single ventricle should be monitored for thrombosis with periodic transthoracic echocardiography (with focused attention to the identification of thrombi) as part of routine follow-up assessments (Class I; Level of Evidence C).
-
4.
For patients with a palliated single ventricle, if thrombosis is suspected on clinical grounds or from transthoracic echocardiography, diagnostic confirmation with transesophageal echocardiography, MRI, computed tomography, computed tomographic angiography, nuclear medicine lung perfusion scan, or venography/angiography can be useful (Class IIa; Level of Evidence C).
-
5.
For patients with a palliated single ventricle, other imaging modalities (in addition to echocardiography) used to detect thrombosis such as transesophageal echocardiography or MRI may be considered for surveillance for patients with anatomic or hemodynamic risk factors (Class IIb; Level of Evidence C).
-
6.
Initiation of antithrombotic therapy or an increase in the magnitude of antithrombotic therapy for prophylaxis (change in agent, i.e., from antiplatelet to anticoagulant or higher target levels) is probably reasonable if anatomic or hemodynamic risk factors become present at any stage in the single-ventricle pathway (Class IIa; Level of Evidence C).
And in addition specifically for Fontan patients:
-
7.
Long-term antiplatelet therapy for prevention of thrombosis is reasonable after the Fontan procedure (Class IIa; Level of Evidence C).
-
8.
Prophylaxis with warfarin or LMWH may be reasonable in infants and children for 3–12 months after the Fontan procedure (Class IIb; Level of Evidence C).
-
9.
Long-term therapy with warfarin may be reasonable after the Fontan procedure for patients with anatomic or hemodynamic risk factors (Class IIb; Level of Evidence C).
Several additional comments are warranted regarding thromboprophylaxis in the Fontan patient. No method of thromboprophylaxis is 100% effective with a thrombosis rate of currently about 12% [15, 18]. Thrombosis prevention in the Fontan patient is more than pharmacotherapy. Understanding and mitigating risk factors as well as diagnosing and treating thrombosis as early as possible are essential. Thrombosis risk in the Fontan patient is not constant over time. As risk factors increase, antithrombotic therapy should be reassessed and readjusted. Continued assessment of risk over time [9] with adjustment of antithrombotic therapy based on current risk assessment is essential. Finally, as is well known to ACHD physicians, warfarin is a medication with a narrow therapeutic window and many drug-drug interactions. The DOACs are discussed below. They may have promise in the adult with Fontan circulation [100], but safety and efficacy have not yet been established especially in the Fontan with liver disease, thrombocytopenia, and/or protein-losing enteropathy where concerns of bleeding are real.
6 Anticoagulant Therapy
The following section will provide specific information regarding mechanism of action, pharmacokinetics, dosing, monitoring, and associated harms for commonly used anticoagulants. It is important to note that the predominant risk associated with anticoagulation therapy is hemorrhage. To minimize this bleeding risk, concurrent antiplatelet therapy should be avoided, if possible.
6.1 Unfractionated Heparin
Unfractionated heparin (UFH) is extracted from bovine lung or porcine intestine and is composed of a heterogeneous mixture of highly sulfated glycosaminoglycans [101]. UFH itself has no intrinsic anticoagulant effect but instead acts through the binding of antithrombin (AT) potentiating the AT anticoagulant activity over 1000-fold inactivating coagulant factors IIa (thrombin), Xa, XIa, and XIIa [102,103,104,105]. UFH is parentally available as either a continuous intravenous infusion or a subcutaneous injection. The half-life is short and estimated at 1.5 ± 0.5 h [106]. Unfortunately, UFH interacts with other plasma proteins, endothelial cells, and macrophages which can alter the pharmacokinetics with a resultant high inter- and intra-patient dose response.
Intravenous UFH therapy is typically started as a bolus (80 units/kg, maximum bolus 4000–5000 units) followed by the initiation of a continuous infusion (18 units/kg/h, maximum initial infusion 1000–2000 U/h). If a patient has a low AT level (i.e., congenital deficiency, acquired deficiency secondary to losses from nephrotic syndrome, draining chylous effusion, consumption, or asparaginase therapy), higher UFH doses may be required. The two most common assays used to monitor UFH include the aPTT and the UFH anti-Xa level. Both tests have pitfalls when used to monitor UFH as well as poor correlation. The therapeutic goal for the aPTT is 1.5–2.5 times control (60–85 seconds), and the UFH anti-Xa goal is 0.3–0.7 units/mL. See Table 28.4 for the dose titration of UFH using these laboratory markers.
The aPTT is not a direct measure of the UFH effect and can be impacted by many other parameters that could result in either an over or underestimation of the true UFH effect. For example, an elevated factor VIII or fibrinogen (acute phase reactants) will shorten the aPTT making it appear that the patient is heparin resistant. Alternatively a deficiency in a coagulation factor (i.e., from liver failure or consumption) or the presence of an antiphospholipid antibody will prolong the aPTT making it appear that the patient is therapeutic on UFH. The UFH anti-Xa level provides a more direct measure that is not impacted by the above factors, although the UFH anti-Xa level does not reflect additional anticoagulant targets of UFH. The activated clotting time (ACT) is used to monitor higher heparin doses given to patients undergoing cardiopulmonary bypass or cardiac catheterization. The ACT is a whole-blood clotting time that is simple to perform with a rapid turnaround time. It is more sensitive to a wider range of heparin doses than the PTT and is impacted by the same factors as the PTT, and additionally thrombocytopenia prolongs the ACT.
Full reversal of heparin can be obtained with the use of protamine sulfate, a basic protein that binds heparin and forms a salt [105]. Approximately 1 mg of protamine will neutralize 100 units of UFH [105]. Calculations should be made based on the total amount of heparin received in the prior 2–2.5 h [105]. Adverse events such as hypotension and bradycardia can be minimized with slow administration of protamine.
The benefits of UFH include the short half-life and the ability to completely reverse the anticoagulation effect. Additionally UFH can be used in the setting of renal failure where LMWHs cannot. In clinically unstable patients at risk for hemorrhage who require anticoagulation, UFH should be strongly considered. Transition to an alternative anticoagulant should occur once the patient is clinically stable. Nonhemorrhagic complications associated with UFH include HIT/HITT (see separate section) and osteopenia with long-term use [107, 108].
6.2 Low Molecular Weight Heparin
Low molecular weight heparins (LMWH) are derived from UFH by chemical or enzymatic depolymerization and contain shorter-length polysaccharide chains. Similar to UFH, LMWH exert an anticoagulant effect through binding AT and potentiating the AT anticoagulant activity, but as compared to UFH, there is a reduced inhibitory activity against factor IIa (thrombin) relative to factor Xa. LMWH are administered as a subcutaneous injection. The PK properties are more stable as compared to UFH. The half-life is 3–6 h and the LMWH anti-Xa levels peak 3–5 h after dosing. LMWH is predominantly cleared by the kidneys.
Dosing will vary based on the exact LMWH that is used. Common preparations include enoxaparin, 1 mg/kg/dose every 12 h, or 1.5 mg/kg once daily; in obesity, use ideal body weight to calculate the dose and tinzaparin 175 anti-Xa IU/kg daily . Additionally, similar to UFH if a patient has a low AT level, higher doses of the LMWH may be required.
Routine monitoring is not recommended except for extreme obesity or in the setting of renal insufficiency. When monitoring the LMWH, anti-Xa peak level is used with a goal of 0.5–1 units/mL (drawn 4–6 h postinjection). LMWH can be partially reversed (approximately 70%) with protamine. LMWH should not be used in the setting of renal failure. Nonhemorrhagic complications associated with LMWH are similar to UFH. HIT has been reported with the use of LMWH, but it is thought to be less frequent than UFH [109]. Long-term LMWH use is associated with less bone loss than UFH [105].
6.3 Heparin-Induced Thrombocytopenia
Heparin-induced thrombocytopenia (HIT) is an immunologic complication of heparin therapy. Antibodies are formed against complexes of heparin and an endogenous platelet protein, platelet factor 4 (PF4) [110]. It is postulated that the binding of antibodies to the heparin-PF4 complex results in increased platelet reactivity with a resultant prothrombotic state [110]. This disorder is characterized by thrombocytopenia (HIT) and sometimes thrombosis (HITT) that can occur in any vascular bed and can be catastrophic. Interestingly, while the immune reaction is relatively common post-heparin exposure (8–50%), with the highest in those undergoing cardiac surgery (25–50%), only a small portion of patients will actually develop the clinical consequences of thrombocytopenia and thrombosis [111, 112]. Overall the incidence of HIT is estimated to be approximately 0.02–3% of patients [111,112,113,114]. The incidence is influenced by the clinical setting, type of heparin used, and dose [115]. Prophylactic dose UFH has a higher risk of HIT versus prophylactic LMWH [114]. Untreated HIT has a mortality of 20–30% [116, 117].
The diagnosis of HIT is based on clinical criteria. Multiple scoring systems have been developed [118,119,120]. The most commonly used is the 4T’s clinical probability score (Table 28.5) [120]. Classically, HIT is characterized by symptoms appearing 5–10 days post-heparin exposure with a 50% fall in the platelet count (rarely is the platelet count <50,000/μL), and there can be venous or arterial thrombosis. Laboratory testing can be supportive in making the diagnosis and includes ELISA for the heparin-P4 antibodies and the serotonin-releasing assay. The ELISA is readily available with a fast turnaround time. This test is highly sensitive but has a significant false-positive rate. The ELISA is most helpful in ruling out the diagnosis with a negative assay. The serotonin-releasing assay is highly specific and sensitive; it measures platelet reactivity in the presence of the patient’s plasma. Unfortunately, it is only performed in a few highly specialized laboratories so is not readily available for most clinicians.
The treatment of HIT includes the removal of all heparin from the patient including from central lines and avoidance of LMWH. Anticoagulation should be initiated (even without thrombosis) with a non-heparin anticoagulant such as a direct thrombin inhibitor (i.e., bivalirudin or argatroban) [121]. In this setting, warfarin should never be initiated by itself due to an increased risk of skin necrosis and further thrombotic events. Warfarin can be initiated once the platelet count has normalized and overlapped with the non-heparin anticoagulant until the INR is therapeutic. The median time for platelet recovery is approximately 4 days. Fondaparinux, a synthetic pentasaccharide LMWH, is also commonly used off-label for the treatment of HIT. Although rarely HIT has been reported with the use of fondaparinux [122]. The direct oral anticoagulants (DOACs) have not been studied in HIT, so their use in this clinical setting would be considered off-label and without sufficient clinical supporting data.
The duration of anticoagulation in HIT is determined by the presence of thrombosis. For those patients with only thrombocytopenia, there remains a high risk of thrombosis for 2–4 weeks after developing HIT. Anticoagulation should be continued during this high-risk time [121]. For patients with a thrombosis, anticoagulation should be continued for 3 months [121]. In general, HIT antibodies are transient with a median time of disappearance of 50–85 days without clear evidence of an amnestic immune response with reexposure [123]. In general, patients with a history of HIT should not be reexposed to UFH or LMWH. The one exception is for procedures where the use of UFH is favored over other anticoagulants like CPB if repeat HIT antibody testing is negative. In those patients with a history of HIT who have demonstrated resolution of the HIT antibodies, a short exposure to UFH during CPB is considered acceptable [121]. This strategy does not apply to those patients with persistent antibodies, however.
6.4 Direct Thrombin Inhibitors: Argatroban or Bivalirudin
Argatroban and bivalirudin are parenteral direct thrombin inhibitors and are typically used in the setting of HIT/HITT when heparin needs to be avoided. Bivalirudin reversibly binds the active catalytic and substrate binding site of thrombin. It has a half-life of 25 min and has approximately 20% renal clearance. The initial dose is 0.15–0.2 mg/kg/h. It is typically monitored by the aPTT titrated to 1.5–3 times control, but the ACT has also been used. A limited number of laboratories are running direct thrombin inhibitor assays, although this has not yet become standard of care.
Argatroban reversibly binds the active catalytic site of thrombin. It also has a very short half-life of 45 min. It is predominantly metabolized in the liver so should not be used in the setting of hepatic failure. The initial dose is 2 mcg/kg/min. It is titrated via the aPTT to 1.5–3 times control; the ACT has also been used. No antidote exists for the reversal of bivalirudin or argatroban, but both have very short half-lives, so discontinuation of the infusion should be sufficient for most forms of bleeding.
6.5 Fondaparinux
Fondaparinux is a synthetic analog of the AT-binding pentasaccharide found in heparin and LMWH. The structure was modified to enhance its affinity for AT increasing the specific activity and half-life. Unlike UFH or LMWH, fondaparinux has no inhibitory activity against factor IIa (thrombin) and only inactivates factor Xa.
Fondaparinux is administered via a subcutaneous injection. The half-life is longer than LMWH at 17 h. There is minimal binding to other proteins. The clearance is exclusively renal. A steady state is reached after the third or fourth daily dose. Dosing is guided by weight: <50 kg 5 mg daily, 50–100 kg 7.5 mg daily, and >100 kg 10 mg daily. Routine coagulation monitoring is not recommended. If a level is needed, specific assays calibrated for fondaparinux should be used [124]. Similar to LMWH, monitoring is achieved by measuring fondaparinux anti-Xa peak level with a goal of 0.5–1 mg/L (drawn 4 h post).
There are no reversal agents available for fondaparinux. Unlike other forms of heparin, protamine does not bind fondaparinux. There are reports of using various hemostatic therapies (prothrombin complex concentrates, activated prothrombin complex concentrates, or recombinant factor VIIa) to treat life-threatening bleeding secondary to fondaparinux [125]. Due to the long half-life and lack of a reversal agent, this medication should only be utilized in patients who are clinically stable with a low risk of hemorrhage. It should not be used in patients with renal insufficiency.
6.6 Warfarin
Warfarin is a vitamin K antagonist and interferes with the cyclic conversion of vitamin K through the inhibition of vitamin K epoxide reductase. This results in a decrease in the posttranslational γ-carboxylation of vitamin K-dependent clotting factors which is imperative for biologic function. This impacts coagulation factors II, VII, IX, and X and anticoagulant proteins C and S [126]. Warfarin absorption is primarily gastric and is complete within 4 h of administration. It has a very long half-life of 36–42 h. In general, warfarin is initiated with an anticoagulation “bridge” because patients are initially prothrombotic due to the variable half-lives of anti- and procoagulant proteins lowered by warfarin.
Initial dosing is individualized considering the patient’s cardiac function, nutritional status, prior warfarin dosing, current medications, and hepatic function. Typical maintenance dosing is 2–5 mg/day. Patients with Fontan circulation may require less warfarin. Patients >60 years of age may require less warfarin due to changes in warfarin metabolism. The warfarin dose is titrated via the international normalized ratio (INR). The INR goal is dictated by the clinical indication for warfarin but is commonly 2–3.
There are multiple choices available for warfarin reversal including vitamin K (oral, intravenous, or subcutaneous), non-activated prothrombin complex concentrate (PCC, Kcentra), or fresh frozen plasma. How warfarin reversal is completed is determined by the severity of bleeding and the patient’s thrombotic risk associated with reversal [127]. In the setting of severe, life-threatening bleeding, vitamin K replacement should be augmented with the use of either Kcentra or fresh frozen plasma. Kcentra contains concentrated non-activated coagulation factors II, VII, IX, and X and anticoagulant proteins C and S and is FDA approved for the urgent reversal of warfarin. Kcentra dosing is determined by the INR and the patient’s body weight: INR 2–<4, 25 units FIX/kg (maximum 2500 units); INR 4–6, 35 units FIX/kg (maximum 3500 units); and INR 6, 50 units FIX/kg (maximum 5000 units). The benefit to using a Kcentra as opposed to fresh frozen plasma is that a much smaller volume is given to the patient and replaces only those coagulation and anticoagulation proteins impacted by warfarin.
6.7 Direct Oral Anticoagulants
The first direct oral anticoagulant (DOAC) received FDA approval in 2010. All of the DOACs bind directly to a key coagulant protein to inhibit fibrin formation. There are two broad categories for mechanism of action including direct anti-Xa inhibitors (rivaroxaban, apixaban, and edoxaban) and direct thrombin (IIa) inhibitors (dabigatran). The advantage to all of the DOACs is more predictable pharmacokinetics with a wide therapeutic window and limited drug-drug interactions as compared to warfarin. They also have a relative rapid onset of action and shorter half-lives as compared to warfarin. Dosing of DOACs varies by the specific drug.
Table 28.6 provides general dosing schema for each DOAC. For all of the DOACs, dosing needs to be adjusted in the setting of impaired renal clearance, and in significant renal impairment, DOAC use is not recommended.
The key advantage to predictable pharmacokinetics and a wide therapeutic window for DOACs is that routine therapeutic monitoring is not needed [128]. The disadvantage is that methods to monitor these medications are not readily available [128]. There are times when drug monitoring might be needed including in the setting of emergency surgery, breakthrough thrombosis, renal insufficiency, or bleeding [129]. Specialized laboratory assays for DOACs have been developed including a dilute thrombin time for dabigatran and anti-Xa assays that are drug calibrated for the anti-Xa inhibitors. The PT or PTT cannot reliably be used to monitor these medications.
In the setting of bleeding, all of the DOACs have relatively short half-lives of 10–12 h, so supportive care can be given, while the medication is wearing off in most cases. In the setting of life-threatening hemorrhage, reversal for dabigatran or consideration for other hemostatic therapies for oral Xa inhibitors needs to be considered. Currently, idarucizumab is the only FDA-approved DOAC reversal agent, and it is specific for dabigatran. Idarucizumab is a monoclonal antibody fragment that binds dabigatran with an affinity that is 350 times greater than thrombin [130]. It can bind both free and thrombin-bound dabigatran and neutralizes its activity [130]. Clinically it is given as a one-time intravenous infusion with a complete reversal of the anticoagulant effect in minutes [131]. A rebound anticoagulant effect after the initial idarucizumab transfusion has been reported and needs to be considered in the setting of renal failure [132, 133]. Dabigatran can also be partially removed with hemodialysis [134]. For the Xa inhibitors, there is a reversal agent in development, andexanet alfa. It is a human factor Xa decoy protein but is not currently FDA approved [135]. There are reports of using various hemostatic therapies (prothrombin complex concentrates, activated prothrombin complex concentrates, or recombinant factor VIIa) to treat life-threatening bleeding secondary to DOACs [136, 137].
DOACs have been demonstrated to be efficacious and in the USA are approved for the use in stroke prevention from non-valvular atrial fibrillation, the treatment of DVT and PE, secondary prevention of DVT, and DVT prevention post-elective knee and hip surgery [138]. As mentioned above, at this time DOACs should not be used in the setting of mechanical heart valves. A clinical trial comparing dabigatran to warfarin in patients with either an aortic or mitral mechanical heart valve was halted early secondary to increased thrombotic and bleeding events in those patients receiving dabigatran as compared to warfarin [89]. A phase 2 clinical trial is currently underway using rivaroxaban in the setting of mechanical aortic valve replacement (ClincalTrials.gov Identifier: NCT02128841). There is no data to support the routine use of DOACs for thrombosis prevention in Fontan patients though case reports have been published and a multicenter trial is underway.
7 Thrombolytic Therapy
The strongest indication for thrombolytic therapy includes either a life- or limb-threatening thrombotic event including stroke, prosthetic valve thrombosis, or a massive pulmonary embolism [139,140,141,142,143,144]. Significant bleeding (including intracranial hemorrhage) and thromboembolism are known complications of thrombolysis. Thrombolysis can be performed locally or systemically. Contraindications to thrombolytic therapy generally include active bleeding, an inability to maintain the platelet count >75,000/μL or fibrinogen >100 mg/dL, a major operation or site of hemorrhage within 7–10 days, seizures within 48 h, central nervous system surgery/ischemia/trauma/hemorrhage within 30 days, or uncontrolled hypertension. These contraindications are not absolute, and the relative risks of thrombolytic therapy should be weighed against the potential benefits in each clinical situation.
When instituting thrombolytic therapy, baseline laboratory values should be obtained (CBC, PT, PTT, fibrinogen, and D-dimer) to assess for additional bleeding risks. For systemic sustained thrombolysis, these labs should be obtained prior to therapy and followed every 4–6 h during therapy. An increase in the D-dimer and a drop in the fibrinogen level are indicative of a “lytic” state. To minimize the risk of bleeding, if the fibrinogen level drops below 100 mg/dL, consider either holding thrombolytic therapy or infusing cryoprecipitate as an external source of fibrinogen. The platelet count should be kept >75,000/μL. Tissue plasminogen activator (tPA) is currently the only thrombolytic agent available in the USA. Concomitant use of heparin has been used at either low or therapeutic dosing with therapeutic heparin usually continued after the thrombolytic is stopped. Due to the very high risk of hemorrhage during and immediately after thrombolysis, invasive procedures should be minimized.
References
Ihenacho HN, Fletcher DJ, Breeze GR, Stuart J. Consumption coagulopathy in congenital heart-disease. Lancet. 1973;1:231–4.
Perloff JK, Marelli AJ, Miner PD. Risk of stroke in adults with cyanotic congenital heart disease. Circulation. 1993;87:1954–9.
Ammash N, Warnes CA. Cerebrovascular events in adult patients with cyanotic congenital heart disease. J Am Coll Cardiol. 1996;28:768–72.
Rosenthal DN, Friedman AH, Kleinman CS, Kopf GS, Rosenfeld LE, Hellenbrand WE. Thromboembolic complications after Fontan operations. Circulation. 1995;92:II287–93.
Coon PD, Rychik J, Novello RT, Ro PS, Gaynor JW, Spray TL. Thrombus formation after the Fontan operation. Ann Thorac Surg. 2001;71:1990–4.
Seipelt RG, Franke A, Vazquez-Jimenez JF, et al. Thromboembolic complications after Fontan procedures: comparison of different therapeutic approaches. Ann Thorac Surg. 2002;74:556–62.
Porcelli R, Moskowitz BC, Cetta F, et al. Heparin-induced thrombocytopenia with associated thrombosis in children after the Fontan operation: report of two cases. Tex Heart Inst J. 2003;30:58–61.
Varma C, Warr MR, Hendler AL, Paul NS, Webb GD, Therrien J. Prevalence of “silent” pulmonary emboli in adults after the Fontan operation. J Am Coll Cardiol. 2003;41:2252–8.
Kaulitz R, Ziemer G, Rauch R, et al. Prophylaxis of thromboembolic complications after the Fontan operation (total cavopulmonary anastomosis). J Thorac Cardiovasc Surg. 2005;129:569–75.
Broberg CS, Ujita M, Prasad S, et al. Pulmonary arterial thrombosis in eisenmenger syndrome is associated with biventricular dysfunction and decreased pulmonary flow velocity. J Am Coll Cardiol. 2007;50:634–42.
Khairy P, Fernandes SM, Mayer JE Jr, et al. Long-term survival, modes of death, and predictors of mortality in patients with Fontan surgery. Circulation. 2008;117:85–92.
Khairy P. Thrombosis in congenital heart disease. Expert Rev Cardiovasc Ther. 2013;11:1579–82.
Marrone C, Galasso G, Piccolo R, et al. Antiplatelet versus anticoagulation therapy after extracardiac conduit Fontan: a systematic review and meta-analysis. Pediatr Cardiol. 2011;32:32–9.
Giglia TM, Massicotte MP, Tweddell JS, et al. Prevention and treatment of thrombosis in pediatric and congenital heart disease: a scientific statement from the American Heart Association. Circulation. 2013;128:2622–703.
Alsaied T, Alsidawi S, Allen CC, Faircloth J, Palumbo JS, Veldtman GR. Strategies for thromboprophylaxis in Fontan circulation: a meta-analysis. Heart. 2015;101:1731–7.
Ohuchi H, Yasuda K, Miyazaki A, et al. Prevalence and predictors of haemostatic complications in 412 Fontan patients: their relation to anticoagulation and haemodynamics. Eur J Cardiothorac Surg. 2015;47:511–9.
Keraliya AR, Murphy DJ, Steigner ML, Blankstein R. Thrombus in hypoplastic aorta: an uncommon cause of acute myocardial infarction. J Cardiovasc Comput Tomogr. 2016;10:263–4.
Atz AM, Zak V, Mahony L, et al. Longitudinal outcomes of patients with single ventricle after the Fontan procedure. J Am Coll Cardiol. 2017;69:2735–44.
Alsaied T, Bokma JP, Engel ME, et al. Factors associated with long-term mortality after Fontan procedures: a systematic review. Heart. 2017;103:104–10.
Valente AM, Bhatt AB, Cook S, et al. The CALF (congenital heart disease in adults lower extremity systemic venous health in Fontan patients) study. J Am Coll Cardiol. 2010;56:144–50.
Virchow R. Thrombose und Embolie. Gefässentzündung und septische Infektion. Gesammelte Abhandlungen zur wissenschaftlichen Medicin. 1856. pp. 219–752.
Giglia TM, Witmer C. Bleeding and thrombosis in pediatric cardiac intensive care. Pediatr Crit Care Med. 2016;17:S287–95.
Giglia TM, Witmer C. Hematologic aspects of pediatric and adolescent heart disease: bleeding, clotting and blood component abnormalities in Moss & Adams Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adult, 9th ed. In: Allen HDS, Robert E, Penny DJ, Feltes TF, Cetta F, editors. Moss & Adams’ heart disease in infants, children, and adolescents, including the fetus and young adult. Philadelphia: Lippincott Williams & Wilkins; 2016. p. 1900.
Wolberg AS, Aleman MM, Leiderman K, Machlus KR. Procoagulant activity in hemostasis and thrombosis: Virchow’s triad revisited. Anesth Analg. 2012;114:275–85.
Waniewski J, Kurowska W, Mizerski JK, et al. The effects of graft geometry on the patency of a systemic-to-pulmonary shunt: a computational fluid dynamics study. Artif Organs. 2005;29:642–50.
Ravn HB, Hjortdal VE, Stenbog EV, et al. Increased platelet reactivity and significant changes in coagulation markers after cavopulmonary connection. Heart. 2001;85:61–5.
Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289–97.
Jaggers JJ, Neal MC, Smith PK, Ungerleider RM, Lawson JH. Infant cardiopulmonary bypass: a procoagulant state. Ann Thorac Surg. 1999;68:513–20.
Guay J, Ruest P, Lortie L. Cardiopulmonary bypass induces significant platelet activation in children undergoing open-heart surgery. Eur J Anaesthesiol. 2004;21:953–6.
Heying R, van Oeveren W, Wilhelm S, et al. Children undergoing cardiac surgery for complex cardiac defects show imbalance between pro- and anti-thrombotic activity. Crit Care (London England). 2006;10:R165.
Jaggers J, Lawson JH. Coagulopathy and inflammation in neonatal heart surgery: mechanisms and strategies. Ann Thorac Surg. 2006;81(6):S2360.
Tomita H, Yamada O, Ohuchi H, et al. Coagulation profile, hepatic function, and hemodynamics following Fontan-type operations. Cardiol Young. 2001;11:62–6.
Jahangiri M, Shore D, Kakkar V, Lincoln C, Shinebourne E. Coagulation factor abnormalities after the Fontan procedure and its modifications. J Thorac Cardiovasc Surg. 1997;113:989–92; discussion 92–3
Jahangiri M, Kreutzer J, Zurakowski D, Bacha E, Jonas RA. Evaluation of hemostatic and coagulation factor abnormalities in patients undergoing the Fontan operation. J Thorac Cardiovasc Surg. 2000;120:778–82.
Odegard KC, McGowan FX Jr, DiNardo JA, et al. Coagulation abnormalities in patients with single-ventricle physiology precede the Fontan procedure. J Thorac Cardiovasc Surg. 2002;123:459–65.
Odegard KC, McGowan FX Jr, Zurakowski D, et al. Coagulation factor abnormalities in patients with single-ventricle physiology immediately prior to the Fontan procedure. Ann Thorac Surg. 2002;73:1770–7.
Odegard KC, McGowan FX Jr, Zurakowski D, et al. Procoagulant and anticoagulant factor abnormalities following the Fontan procedure: increased factor VIII may predispose to thrombosis. J Thorac Cardiovasc Surg. 2003;125:1260–7.
Odegard KC, Zurakowski D, Hornykewycz S, et al. Evaluation of the coagulation system in children with two-ventricle congenital heart disease. Ann Thorac Surg. 2007;83:1797–803.
Odegard KC, Zurakowski D, DiNardo JA, et al. Prospective longitudinal study of coagulation profiles in children with hypoplastic left heart syndrome from stage I through Fontan completion. J Thorac Cardiovasc Surg. 2009;137:934–41.
Horigome H, Murakami T, Isobe T, Nagasawa T, Matsui A. Soluble P-selectin and thrombomodulin-protein C-Protein S pathway in cyanotic congenital heart disease with secondary erythrocytosis. Thromb Res. 2003;112:223–7.
Rask O, Hanseus K, Ljung R, Strandberg K, Berntorp E. Lower incidence of procoagulant abnormalities during follow-up after creation of the Fontan circulation in children. Cardiol Young. 2009;19:152–8.
Levin E, Wu J, Devine DV, et al. Hemostatic parameters and platelet activation marker expression in cyanotic and acyanotic pediatric patients undergoing cardiac surgery in the presence of tranexamic acid. Thromb Haemost. 2000;83:54–9.
Binotto MA, Maeda NY, Lopes AA. Evidence of endothelial dysfunction in patients with functionally univentricular physiology before completion of the Fontan operation. Cardiol Young. 2005;15:26–30.
Thornburg CD, Smith PB, Smithwick ML, Cotten CM, Benjamin DK Jr. Association between thrombosis and bloodstream infection in neonates with peripherally inserted catheters. Thromb Res. 2008;122:782–5.
Randolph AG, Cook DJ, Gonzales CA, Andrew M. Benefit of heparin in peripheral venous and arterial catheters: systematic review and meta-analysis of randomised controlled trials. BMJ. 1998;316:969–75.
O'Connor MJ, Ravishankar C, Ballweg JA, et al. Early systemic-to-pulmonary artery shunt intervention in neonates with congenital heart disease. J Thorac Cardiovasc Surg. 2011;142:106–12.
Silversides CK, Granton JT, Konen E, Hart MA, Webb GD, Therrien J. Pulmonary thrombosis in adults with Eisenmenger syndrome. J Am Coll Cardiol. 2003;42:1982–7.
Saha A, Balakrishnan KG, Jaiswal PK, et al. Prognosis for patients with Eisenmenger syndrome of various aetiology. Int J Cardiol. 1994;45:199–207.
Daliento L, Somerville J, Presbitero P, et al. Eisenmenger syndrome. Factors relating to deterioration and death. Eur Heart J. 1998;19:1845–55.
Niwa K, Perloff JK, Kaplan S, Child JS, Miner PD. Eisenmenger syndrome in adults: ventricular septal defect, truncus arteriosus, univentricular heart. J Am Coll Cardiol. 1999;34:223–32.
Giannakoulas G, Boutsikou M. The Gordian knot of thromboembolism in congenital heart disease. Heart. 2015;101:1523–4.
Jensen AS, Idorn L, Thomsen C, et al. Prevalence of cerebral and pulmonary thrombosis in patients with cyanotic congenital heart disease. Heart. 2015;101:1540–6.
Manlhiot C, Menjak IB, Brandao LR, et al. Risk, clinical features, and outcomes of thrombosis associated with pediatric cardiac surgery. Circulation. 2011;124:1511–9.
Giglia TM, Petrosa WL, Veneziale K, et al. Use of PC4 database in a prospective cohort study of hospital-acquired thrombosis in pediatric cardiac in-patients. J Am Coll Cardiol. 2016;67:976.
Anderson FA Jr, Zayaruzny M, Heit JA, Fidan D, Cohen AT. Estimated annual numbers of US acute-care hospital patients at risk for venous thromboembolism. Am J Hematol. 2007;82:777–82.
Lim W, Meade M, Lauzier F, et al. Failure of anticoagulant thromboprophylaxis: risk factors in medical-surgical critically ill patients*. Crit Care Med. 2015;43:401–10.
Horlander KT, Mannino DM, Leeper KV. Pulmonary embolism mortality in the United States, 1979–1998: an analysis using multiple-cause mortality data. Arch Intern Med. 2003;163:1711–7.
The Surgeon General’s Call to Action to Prevention Deep Vein Thrombosis and Pulmonary Embolism. In: Services UDoHaH, editor. http://www.surgeongeneral.gov/topics/deepvein/calltoaction/call-to-action-ondvt-2008.pdf.2008.
Anderson FA Jr, Spencer FA. Risk factors for venous thromboembolism. Circulation. 2003;107:I9–16.
Di Nisio M, van Es N, Buller HR. Deep vein thrombosis and pulmonary embolism. Lancet. 2016;388:3060–73.
Barbar S, Noventa F, Rossetto V, et al. A risk assessment model for the identification of hospitalized medical patients at risk for venous thromboembolism: the Padua Prediction Score. J Thromb Haemost. 2010;8:2450–7.
Spyropoulos AC, Anderson FA Jr, FitzGerald G, et al. Predictive and associative models to identify hospitalized medical patients at risk for VTE. Chest. 2011;140:706–14.
Nendaz M, Spirk D, Kucher N, et al. Multicentre validation of the Geneva Risk Score for hospitalised medical patients at risk of venous thromboembolism. Explicit ASsessment of Thromboembolic RIsk and Prophylaxis for Medical PATients in SwitzErland (ESTIMATE). Thromb Haemost. 2014;111:531–8.
Kakkar AK, Cimminiello C, Goldhaber SZ, Parakh R, Wang C, Bergmann JF. Low-molecular-weight heparin and mortality in acutely ill medical patients. N Engl J Med. 2011;365:2463–72.
Collins R, Scrimgeour A, Yusuf S, Peto R. Reduction in fatal pulmonary embolism and venous thrombosis by perioperative administration of subcutaneous heparin. Overview of results of randomized trials in general, orthopedic, and urologic surgery. N Engl J Med. 1988;318:1162–73.
Galanaud JP, Sevestre-Pietri MA, Bosson JL, et al. Comparative study on risk factors and early outcome of symptomatic distal versus proximal deep vein thrombosis: results from the OPTIMEV study. Thromb Haemost. 2009;102:493–500.
White RH, Brunson A, Romano PS, Li Z, Wun T. Outcomes after vena cava filter use in noncancer patients with acute venous thromboembolism: a population-based study. Circulation. 2016;133:2018–29.
Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest. 2016;149:315–52.
Jaff MR, McMurtry MS, Archer SL, et al. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation. 2011;123:1788–830.
Bouchardy J, Therrien J, Pilote L, et al. Atrial arrhythmias in adults with congenital heart disease. Circulation. 2009;120:1679–86.
Engelfriet P, Boersma E, Oechslin E, et al. The spectrum of adult congenital heart disease in Europe: morbidity and mortality in a 5 year follow-up period. The Euro Heart Survey on adult congenital heart disease. Eur Heart J. 2005;26:2325–33.
Walkey AJ, Hogarth DK, Lip GYH. Optimizing atrial fibrillation management: from ICU and beyond. Chest. 2015;148:859–64.
Pisters R, Lane DA, Marin F, Camm AJ, Lip GY. Stroke and thromboembolism in atrial fibrillation. Circulation. 2012;76:2289–304.
Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the euro heart survey on atrial fibrillation. Chest. 2010;137:263–72.
Khairy P, Aboulhosn J, Broberg CS, et al. Thromboprophylaxis for atrial arrhythmias in congenital heart disease: a multicenter study. Int J Cardiol. 2016;223:729–35.
Masuda K, Ishizu T, Niwa K, et al. Increased risk of thromboembolic events in adult congenital heart disease patients with atrial tachyarrhythmias. Int J Cardiol. 2017;234:69–75.
Heidendael JF, Bokma JP, de Groot JR, Koolbergen DR, Mulder BJ, Bouma BJ. Weighing the risks: thrombotic and bleeding events in adults with atrial arrhythmias and congenital heart disease. Int J Cardiol. 2015;186:315–20.
Lanz J, Brophy JM, Therrien J, Kaouache M, Guo L, Marelli AJ. Stroke in adults with congenital heart disease: incidence, cumulative risk, and predictors. Circulation. 2015;132:2385–94.
Mandalenakis Z, Rosengren A, Lappas G, Eriksson P, Hansson PO, Dellborg M. Ischemic stroke in children and young adults with congenital heart disease. J Am Heart Assoc. 2016;5:e003071.
Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Develop Guidelines on the Management of Adults With Congenital Heart Disease). Developed in Collaboration With the American Society of Echocardiography, Heart Rhythm Society, International Society for Adult Congenital Heart Disease, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;52:e143–263.
Khairy P, Van Hare GF, Balaji S, et al. PACES/HRS Expert Consensus Statement on the Recognition and Management of Arrhythmias in Adult Congenital Heart Disease: developed in partnership between the Pediatric and Congenital Electrophysiology Society (PACES) and the Heart Rhythm Society (HRS). Endorsed by the governing bodies of PACES, HRS, the American College of Cardiology (ACC), the American Heart Association (AHA), the European Heart Rhythm Association (EHRA), the Canadian Heart Rhythm Society (CHRS), and the International Society for Adult Congenital Heart Disease (ISACHD). Heart Rhythm. 2014;11:e102–65.
Holst KA, Dearani JA, Burkhart HM, et al. Reoperative multivalve surgery in adult congenital heart disease. Ann Thorac Surg. 2013;95:1383–9.
Brennan JM, Edwards FH, Zhao Y, et al. Long-term safety and effectiveness of mechanical versus biologic aortic valve prostheses in older patients: results from the Society of Thoracic Surgeons Adult Cardiac Surgery National Database. Circulation. 2013;127:1647–55.
Heras M, Chesebro JH, Fuster V, et al. High risk of thromboemboli early after bioprosthetic cardiac valve replacement. J Am Coll Cardiol. 1995;25:1111–9.
Cannegieter SC, Rosendaal FR, Briet E. Thromboembolic and bleeding complications in patients with mechanical heart valve prostheses. Circulation. 1994;89:635–41.
Massel DR, Little SH. Antiplatelet and anticoagulation for patients with prosthetic heart valves. Cochrane Database Syst Rev. 2013;(7):CD003464.
Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:e521–643.
Nishimura RA, Otto CM, Bonow RO, et al. AHA/ACC focused update of the 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Circulation. 2017;135:e1159–e95.
Eikelboom JW, Connolly SJ, Brueckmann M, et al. Dabigatran versus warfarin in patients with mechanical heart valves. N Engl J Med. 2013;369:1206–14.
Russo A, Grigioni F, Avierinos JF, et al. Thromboembolic complications after surgical correction of mitral regurgitation incidence, predictors, and clinical implications. J Am Coll Cardiol. 2008;51:1203–11.
Roudaut R, Serri K, Lafitte S. Thrombosis of prosthetic heart valves: diagnosis and therapeutic considerations. Heart. 2007;93:137–42.
Douketis JD, Spyropoulos AC, Spencer FA, et al. Perioperative management of antithrombotic therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e326S–50S.
Seshadri N, Goldhaber SZ, Elkayam U, et al. The clinical challenge of bridging anticoagulation with low-molecular-weight heparin in patients with mechanical prosthetic heart valves: an evidence-based comparative review focusing on anticoagulation options in pregnant and nonpregnant patients. Am Heart J. 2005;150:27–34.
Spyropoulos AC, Turpie AG, Dunn AS, et al. Perioperative bridging therapy with unfractionated heparin or low-molecular-weight heparin in patients with mechanical prosthetic heart valves on long-term oral anticoagulants (from the REGIMEN registry). Am J Cardiol. 2008;102:883–9.
Barbetseas J, Nagueh SF, Pitsavos C, Toutouzas PK, Quinones MA, Zoghbi WA. Differentiating thrombus from pannus formation in obstructed mechanical prosthetic valves: an evaluation of clinical, transthoracic and transesophageal echocardiographic parameters. J Am Coll Cardiol. 1998;32:1410–7.
Tanis W, Habets J, van den Brink RB, Symersky P, Budde RP, Chamuleau SA. Differentiation of thrombus from pannus as the cause of acquired mechanical prosthetic heart valve obstruction by non-invasive imaging: a review of the literature. Eur Heart J Cardiovasc Imaging. 2014;15:119–29.
Egbe AC, Pislaru SV, Pellikka PA, et al. Bioprosthetic valve thrombosis versus structural failure: clinical and echocardiographic predictors. J Am Coll Cardiol. 2015;66:2285–94.
Pislaru SV, Hussain I, Pellikka PA, et al. Misconceptions, diagnostic challenges and treatment opportunities in bioprosthetic valve thrombosis: lessons from a case series. Eur J Cardiothorac Surg. 2015;47:725–32.
Manlhiot C, Brandao LR, Kwok J, et al. Thrombotic complications and thromboprophylaxis across all three stages of single ventricle heart palliation. J Pediatr. 2012;161:513–9.e3.
Pujol C, Niesert AC, Engelhardt A, et al. Usefulness of direct oral anticoagulants in adult congenital heart disease. Am J Cardiol. 2016;117:450–5.
Linhardt RJ, Gunay NS. Production and chemical processing of low molecular weight heparins. Semin Thromb Hemost. 1999;25(Suppl 3):5–16.
Damus PS, Hicks M, Rosenberg RD. Anticoagulant action of heparin. Nature. 1973;246:355–7.
Rosenberg RD, Lam L. Correlation between structure and function of heparin. Proc Natl Acad Sci U S A. 1979;76:1218–22.
Lindahl U, Backstrom G, Hook M, Thunberg L, Fransson LA, Linker A. Structure of the antithrombin-binding site in heparin. Proc Natl Acad Sci U S A. 1979;76:3198–202.
Garcia DA, Baglin TP, Weitz JI, Samama MM. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e24S–43S.
Estes JW. The kinetics of heparin. Ann N Y Acad Sci. 1971;179:187–204.
Murphy MS, John PR, Mayer AD, Buckels JA, Kelly DA. Heparin therapy and bone fractures. Lancet. 1992;340:1098.
Sackler JP, Liu L. Heparin-induced osteoporosis. Br J Radiol. 1973;46:548–50.
Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med. 1995;332:1330–5.
Warkentin TE, Hayward CP, Boshkov LK, et al. Sera from patients with heparin-induced thrombocytopenia generate platelet-derived microparticles with procoagulant activity: an explanation for the thrombotic complications of heparin-induced thrombocytopenia. Blood. 1994;84:3691–9.
Warkentin TE, Sheppard JA, Horsewood P, Simpson PJ, Moore JC, Kelton JG. Impact of the patient population on the risk for heparin-induced thrombocytopenia. Blood. 2000;96:1703–8.
Pouplard C, May MA, Iochmann S, et al. Antibodies to platelet factor 4-heparin after cardiopulmonary bypass in patients anticoagulated with unfractionated heparin or a low-molecular-weight heparin : clinical implications for heparin-induced thrombocytopenia. Circulation. 1999;99:2530–6.
Smythe MA, Koerber JM, Mattson JC. The incidence of recognized heparin-induced thrombocytopenia in a large, tertiary care teaching hospital. Chest. 2007;131:1644–9.
Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood. 2005;106:2710–5.
Arepally GM. Heparin-induced thrombocytopenia. Blood. 2017;129:2864–72.
Ahmed I, Majeed A, Powell R. Heparin induced thrombocytopenia: diagnosis and management update. Postgrad Med J. 2007;83:575–82.
Warkentin TE, Greinacher A. Heparin-induced thrombocytopenia and cardiac surgery. Ann Thorac Surg. 2003;76:2121–31.
Cuker A, Arepally G, Crowther MA, et al. The HIT Expert Probability (HEP) Score: a novel pre-test probability model for heparin-induced thrombocytopenia based on broad expert opinion. J Thromb Haemost. 2010;8:2642–50.
Lillo-Le Louet A, Boutouyrie P, Alhenc-Gelas M, et al. Diagnostic score for heparin-induced thrombocytopenia after cardiopulmonary bypass. J Thromb Haemost. 2004;2:1882–8.
Lo GK, Juhl D, Warkentin TE, Sigouin CS, Eichler P, Greinacher A. Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006;4:759–65.
Linkins LA, Dans AL, Moores LK, et al. Treatment and prevention of heparin-induced thrombocytopenia: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e495S–530S.
Bhatt VR, Aryal MR, Shrestha R, Armitage JO. Fondaparinux-associated heparin-induced thrombocytopenia. Eur J Haematol. 2013;91:437–41.
Warkentin TE, Kelton JG. Temporal aspects of heparin-induced thrombocytopenia. N Engl J Med. 2001;344:1286–92.
Paolucci F, Frasa H, Van Aarle F, et al. Two sensitive and rapid chromogenic assays of fondaparinux sodium (Arixtra) in human plasma and other biological matrices. Clin Lab. 2003;49:451–60.
Elmer J, Wittels KA. Emergency reversal of pentasaccharide anticoagulants: a systematic review of the literature. Transfus Med (Oxford, England). 2012;22:108–15.
Furie B, Bouchard BA, Furie BC. Vitamin K-dependent biosynthesis of gamma-carboxyglutamic acid. Blood. 1999;93:1798–808.
Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th). Chest. 2008;133:160S–98S.
Baruch L. Laboratory monitoring of anticoagulant medications: focus on novel oral anticoagulants. Postgrad Med. 2013;125:135–45.
Wright C, Brown R, Cuker A. Laboratory measurement of the direct oral anticoagulants: indications and impact on management in clinical practice. Int J Lab Hematol. 2017;39 Suppl 1:31–6.
Schiele F, van Ryn J, Litzenburger T, Ritter M, Seeliger D, Nar H. Structure-guided residence time optimization of a dabigatran reversal agent. MAbs. 2015;7:871–80.
Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. New Engl J Med. 2015;373:511–20.
Stecher A, Vene N, Mavri A, Mijovski MB, Krevel B, Gradisek P. Late rebound of dabigatran levels after idarucizumab reversal in two patients with severe renal failure. Eur J Anaesthesiol. 2017;34:400–2.
Simon A, Domanovits H, Ay C, Sengoelge G, Levy JH, Spiel AO. The recommended dose of idarucizumab may not always be sufficient for sustained reversal of dabigatran. J Thromb Haemost. 2017;15:1317–21.
Khadzhynov D, Wagner F, Formella S, et al. Effective elimination of dabigatran by haemodialysis. A phase I single-centre study in patients with end-stage renal disease. Thromb Haemost. 2013;109:596–605.
Connolly SJ, Milling TJ Jr, Eikelboom JW, et al. Andexanet alfa for acute major bleeding associated with factor Xa inhibitors. N Engl J Med. 2016;375:1131–41.
Yates S, Sarode R. Novel thrombin and factor Xa inhibitors: challenges to reversal of their anticoagulation effects. Curr Opin Hematol. 2013;20:552–7.
Siegal DM, Garcia DA, Crowther MA. How I treat target-specific oral anticoagulant-associated bleeding. Blood. 2014;123:1152–8.
Cowell RP. Direct oral anticoagulants: integration into clinical practice. Postgrad Med J. 2014;90:529–39.
Sun JC, Davidson MJ, Lamy A, Eikelboom JW. Antithrombotic management of patients with prosthetic heart valves: current evidence and future trends. Lancet. 2009;374:565–76.
Whitlock RP, Sun JC, Fremes SE, Rubens FD, Teoh KH. Antithrombotic and thrombolytic therapy for valvular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e576S–600S.
Desai S, Kavinsky C. Localized left atrial administration of tPA for the treatment of mechanical mitral valve thrombosis. Catheter Cardiovasc Interv. 2008;72:151–5.
Johnson JA, Rauh RA, Phillips SD, Cetta F. Thrombolytic therapy in the treatment of right-sided prosthetic valve thrombosis in adults with congenital heart disease. Congenit Heart Dis. 2011;6:266–8.
Taherkhani M, Hashemi SR, Hekmat M, Safi M, Taherkhani A, Movahed MR. Thrombolytic therapy for right-sided mechanical pulmonic and tricuspid valves: the largest survival analysis to date. Tex Heart Inst J. 2015;42:543–7.
Ozturk S, Erdem F, Ozturk S, Ayhan S. Successful thrombolytic therapy in a patient with congenital corrected transposition of the great arteries. Acta Med Acad. 2016;45:158–62.
Friberg L, Rosenqvist M, Lip GY. Evaluation of risk stratification schemes for ischaemic stroke and bleeding in 182 678 patients with atrial fibrillation: the Swedish Atrial Fibrillation cohort study. Eur Heart J. 2012;33:1500–10.
Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138:1093–100.
Monagle P, AKC C, Goldenberg NA, et al. Antithrombotic therapy in neonates and children: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e737S–801S.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Giglia, T.M., Witmer, C.M., Kim, Y.Y. (2019). Thrombosis and Modern Anticoagulation Options for the Adult with Congenital Heart Disease. In: da Cruz, E., Macrae, D., Webb, G. (eds) Intensive Care of the Adult with Congenital Heart Disease. Congenital Heart Disease in Adolescents and Adults. Springer, Cham. https://doi.org/10.1007/978-3-319-94171-4_28
Download citation
DOI: https://doi.org/10.1007/978-3-319-94171-4_28
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-94170-7
Online ISBN: 978-3-319-94171-4
eBook Packages: MedicineMedicine (R0)