
Abstract
Hepatocellular carcinoma (HCC) typically arises within a background of underlying chronic liver disease. Persistent inflammation is the key element underpinning its development, regardless of the underlying aetiology of chronic liver disease. Several immunological mechanisms can impact the HCC development and progression, as well as response to treatments. Furthermore, there is increasing hope that targeting the immune response itself can be exploited as a therapeutic strategy. Many patients develop antigen-specific adaptive immune responses; however, the background of the liver as a tolerogenic organ and the tumour cells foster an immunosuppressive niche that prevents antigen-mediated clearance of tumour cells. Inhibitory immune mechanisms, such as the presence of regulatory T cells, macrophages and neutrophils with a pro-tumour phenotype, release of anti-inflammatory factors and expression of inhibitory receptors (e.g., cytotoxic T-lymphocyte-associated protein 4 or programmed cell death 1 receptor), contribute for the maintenance of that immunosuppressive microenvironment. This review summarises the knowledge on the contribution of the different immune system elements towards tumour development and progression, as well as current immunotherapeutic approaches being explored in the field.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara Key Learning Points-
1.
The liver is constantly exposed to antigens and pathogen-derived molecules from the gut and has intrinsic tolerogenic mechanisms to ensure that chronic and systemic immune responses do not occur.
-
2.
Hepatocellular carcinoma (HCC) is known to develop on a background of chronic liver disease and inflammation.
-
3.
Despite evidence of immune responses against tumour-associated antigens (TAA), the HCC microenvironment fosters an immunosuppressive niche that escapes immune surveillance.
-
4.
The inflammatory niche created in HCC is a critical target for immunotherapy, including vaccines, oncolytic immunotherapy, cell-based therapy, cytokines/cytokine inhibitors and immune checkpoint inhibitors.
-
1.
There has been a steady increase in patients presenting with HCC arising in the absence of significant liver disease or underlying inflammation—the cause of which is uncertain.
-
2.
Cancers arising in the absence of chronic liver disease tend to be associated with the metabolic syndrome. Obesity and type 2 diabetes may also affect immune responses, although these are less well studied.
-
3.
Understanding how to activate a suppressed antitumour immune response safely and effectively is challenging.
-
4.
The cancer immunotherapy era is an exciting one, but likely to be hindered by the present lack of biomarkers to guide selection of mono or combination therapies and monitor response.
-
5.
The use of therapies activating aspects of the antitumour immune response in immunosuppressed patients will require careful consideration.
Introduction
Hepatocellular carcinoma (HCC) accounts for 70–85% of the total primary liver cancer burden. It usually arises in a background of chronic liver disease consequent to hepatitis B virus (HBV)/hepatitis C virus (HCV) infection, alcoholic-related liver disease (ARLD) or non-alcoholic fatty liver disease (NAFLD), which is tightly linked to the metabolic syndrome and obesity [1]. While HCC is the fifth most common cancer in men and the ninth most common one in women, it is the second most common cause of cancer-related death worldwide [2], reflecting late-stage presentation and limited therapeutic options. Surgery, liver transplantation and local ablative therapies can be curative in early disease, but most patients are offered palliative treatments or supportive care. Currently, the only first-line FDA-approved treatment for advanced-stage HCC is sorafenib, a multikinase inhibitor, which offers a median overall survival (OS) benefit of just 10 weeks. Hence, there is a clear and urgent need for new therapies—with a recent focus in the oncology field on immune checkpoint inhibitors.
The liver is continually exposed to a multitude of antigens, gut-derived pathogens, toxins and environmental and bacterial products—entering the liver from the gastrointestinal tract via the portal vein. The liver has therefore developed constitutive tolerogenic mechanisms to prevent persistent gut-associated immune stimulation and systemic and chronic inflammation. A common feature underpinning HCC development, however, is chronic inflammation—consequent to the persistent hepatocyte injury associated with the aetiologies described above, which occurs in approximately 90% of the cases. The combination of chronic inflammation and the intrinsic tolerogenic properties of the liver creates an environment that facilitates cancer development, with progression promoted by additional immunosuppressive manipulation by the tumour itself.
In this chapter we will discuss some of the knowledge we have to date on how immune tolerance is evaded during liver disease and what we know about its contribution to HCC development and progression. We will also highlight some of the current therapeutic approaches designed to harness the immune system as a therapy for HCC.
Pathobiology of HCC-Related Aetiologies
ARLD and NAFLD
ARLD is the most common aetiology of HCC in industrialised countries, being responsible for 32–45% of cases [3]. However, in the last decades, the incidence of NAFLD-related HCC has been increasing worldwide, possibly because of the obesity and type 2 diabetes epidemic [4]. The mechanisms leading to HCC in either ARLD or NAFLD are similar, and several reviews have focused on these [1, 3,4,5]. A key aspect is the chronic damage and hence chronic stimulation of the immune system that overrides liver tolerance. Increased exposure to gut pathogens and persistent hepatotoxicity result in the production of regulatory miRNAs, pro-inflammatory mediators and damage-associated molecular patterns (DAMPs) that activate the immune response. Alcohol increases levels of miR-212 in gut epithelial cells, leading to decreased expression of ZO-1, a tight junction protein, disrupting gut integrity and allowing for translocation of bacterial endotoxins to the liver. There, the endotoxins impact Kupffer cells (liver-resident macrophages), hepatocytes and endothelial cells; Kupffer cells are activated, upregulate miR-155 and release pro-inflammatory mediators including tumour necrosis factor (TNF), contributing to hepatic inflammation. Additionally, alcohol induces oxidative stress: in hepatocytes, levels of miR-34a and miR-217 increase, resulting in hepatic steatosis via SIRT1 and, in endothelial cells, levels of miR-199a decrease, leading to endothelin-1 and hypoxia-inducible factor α (HIF-1 α) release, all contributing to the amplification of inflammation [3, 5].
In NAFLD, high-fat and carbohydrate (mainly fructose) intake can exacerbate cytokine production and increase hepatic de novo lipogenesis (via SREBP and ChREBP transcription factors), thus promoting lipid peroxidation and DNA damage. The underlying dysfunctional adipose tissue releases additional factors: TNFα and interleukin (IL)-6 enhance c-Jun N-terminal kinase (JNK)/nuclear factor kappa B (NF-κB) and Janus kinase (JAK)/signal transducer and activator of transcription (STAT)3 pathways, while leptin activates Akt/mTOR, leading to expression of genes involved in cell proliferation, migration and survival. Low levels of adiponectin hinder its anti-inflammatory activity and antagonising effect on leptin. In obesity, fatty liver may also be susceptible to carcinogens as a result of impaired ATP production, defective autophagy mechanisms, deregulation of energy and hormonal balance, hypoxia and systemic inflammation. Increased susceptibility of the steatotic liver to carcinogenic insults can be due to several local and systemic pathological changes that occur including metabolic imbalances and the “metabolic syndrome”, hyperinsulinemia and the presence of insulin-like growth factor receptors in HCC, the systemic effects of dysregulated cytokines and adipokines, immune dysregulation and alteration in gut microbiota [1, 4].
HBV and HCV
Viral hepatitis plays a significant role in up to 80% of all HCC globally, with HBV being responsible for two-thirds of all cases; HCV is responsible for 25% of HCC-related deaths. HCV is the primary cause of end-stage liver disease worldwide, and, unlike HBV-related acute hepatitis, it only resolves in about 10–40% of cases [6].
Histological changes are similar between both HBV and HCV infections, namely, hepatocyte death, inflammation, steatosis and progressive fibrosis, leading to cirrhosis and HCC. Specific mechanisms causing disease progression include expression of viral hepatitis B surface antigen (HBsAg) on the surface of hepatocytes, resulting in stimulation of the host’s immune system, chronic inflammation, increased production of reactive oxygen species (ROS) and oxidative DNA damage. Integration of the viral DNA into the host genome can also result in genomic instability, chromosomal loss and abnormal gene activation. These effects are compounded by the ability of viral proteins to interfere with the regulation of cell cycle proteins and promote apoptotic escape. Moreover, persistent chemokines, cytokines, proteases and ROS produced by the inflammatory cell infiltrate promote the carcinogenic process further by inducing cell survival and proliferation [6].
Chronic Inflammation, Immune Suppression and HCC Progression
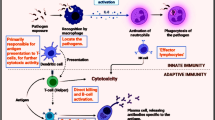
In tumour-bearing hosts, cancer progression is driven by mechanisms promoting immune tolerance to tumour-associated antigens (TAA), including a failure to recognise malignant cells and suppression of the immune cells responsible for the death and clearance of the tumour. Despite a lack of knowledge concerning these pathways, available data thus far highlights the multiple immune responses implicated in HCC progression and allows for identification of promising new targets for future therapy [7, 8]. These cancer-related changes in the immune response comprise of changes in the number and/or function of immune cells, changes in cytokine levels and immune receptor/ligand expression, some of which will be reviewed here (Fig. 3.1).

Crosstalk between multiple immune mechanisms determines the outcome of tumour cell death or growth. The different immunotherapy approaches are summarised and aim to promote tumour elimination or suppress tumour progression. Key: TAA tumour-associated antigen, TIL tumour-infiltrating lymphocytes, NK natural killer cell, CD80/86 B7 costimulatory molecules expressed on the surface of antigen-presenting cells, TRAIL TNF-related apoptosis-inducing ligand, IL-1 interleukin-1, TNF tumour necrosis factor, IFNγ interferon γ, Treg T regulatory cells, MDSC myeloid-derived suppressor cells, TAM tumour-associated macrophage, CTLA-4 cytotoxic T-lymphocyte-associated protein 4, PD-1 programmed death 1, PD-L1 programmed death ligand 1, TIM3 T-cell immunoglobulin and mucin domain-containing protein 3, IL-4 interleukin-4, IL-10 interleukin-10
Senescence and the Senescence-Associated Secretory Phenotype (SASP)
Cellular senescence is a stress-response mechanism aimed at inducing proliferative arrest in a cell at risk of malignant transformation. In the liver, such process can be triggered by chronic inflammation, leading to recurring events of hepatocyte death, compensatory regeneration of hepatocytes and either replicative senescence or oncogene-driven senescence. A recent study by Eggert et al. [9] was designed to further address the implications of hepatocyte senescence in HCC development and progression. The SASP, which comprises of cytokines and chemokines secreted by senescent cells, is designed to recruit and activate myeloid cells and clear senescent hepatocytes, thus preventing tumorigenesis. However, this is dependent upon the context—SASP of senescent hepatocytes may also promote the growth of established HCC via recruitment of immunosuppressive immature myeloid cells which in turn inhibit NK-cell antitumour function, can dampen antitumour T cell responses and even promote tumour growth by production of growth factors, proteases and cytokines [9]. Hence, peritumoral tissue senescence contributes to accelerated tumour growth in mice and to decreased overall and recurrence-free survival in humans.
Immune Cells Involved in the Immunosuppressive HCC Niche
T Cells
The role of T cells in HCC is complex in that the outcome and prognosis differ depending on the type of T cell present and its ability to contribute to antitumour immunity. CD8+ T cells recognise antigens presented on major histocompatibility (MHC) complex I and kill tumour cells by secretion of cytolytic granules. CD4+ Th1 cells are able to kill tumour cells via the TNF-related apoptosis-inducing ligand (TRAIL) pathway. They secrete the cytokine interferon gamma (IFN-γ) which activates antigen-presenting cells (APC) and promotes CD8+ and NK-cell activation. CD4+ Th2 cells on the other hand are thought to be more immunosuppressive, producing the cytokines IL-4, IL-5, IL-10 and IL-13 involved in eosinophil recruitment and B-cell proliferation. CD4+ regulatory T cells (Treg) promote self-tolerance and prevention of autoimmunity, and these cells are often increased in patients’ tumours and blood. Induced by transforming growth factor (TGF)-β, Treg cells can supress CD8+ and NK cytotoxic killing.
Significant changes in gene expression occur in the liver microenvironment, which influence HCC progression. A unique gene signature comprising 17 immune-related genes was shown to strongly predict the development of venous metastases and relapse in HCC patients [10]. Here, a global shift from a Th1 to a Th2 cytokine setting was observed, most likely compounded by the elevated expression of macrophage colony-stimulating factor (CSF1). In this immunosuppressive environment of the metastatic HCC, pro-inflammatory cytokines such as IL-1, TNF and IFN-γ were significantly downregulated, whereas the anti-inflammatory cytokines such as IL-4, IL-5, IL-8, and IL-10 were strongly upregulated. These results centred on HBV-positive metastatic HCC. However, changes in the proportions of T-cell subtypes and function associated with HCC are well established in many mouse models of HCC and in human samples.
A recent study by Ma et al. [11] elucidated a role for CD4+ T cells in NAFLD-associated HCC. Here authors showed, in both mouse models and human samples, that dysregulation of lipid metabolism typical of NAFLD originates a selective loss of intrahepatic CD4+ but not CD8+ T cells, leading to accelerated hepatocarcinogenesis. CD4+ T cells had a greater mitochondrial mass than CD8+ T cells and produced higher levels of mitochondrial-derived ROS, which ultimately caused their death. Linoleic acid, a fatty acid accumulated in NAFLD, was found to be largely responsible for this mitochondrial dysfunction. The in vivo use of antioxidants reversed NAFLD-induced HCC. This novel link between obesity-associated lipid accumulation and selective CD4+ T-cell loss suggests a crucial role for CD4+ T cells in the disease progression from NAFLD to HCC.
In the chronically inflamed liver (particularly due to chronic viral infection) and HCC, it is common to find lymphocytic immune cell aggregates consisting predominantly of T and B cells, which form distinct structures known as ectopic lymphoid structures (ELS). The pro-tumorigenic role of ELS in HCC was recently reported, demonstrating that these lymphocyte structures—driven by NF-κB activation—provide a cellular and cytokine microniche that supports the growth and egress of malignant hepatocyte progenitor cells [12]. The authors identified HCC with similar chromosomal alterations, pointing towards a common source of malignant progenitor cells originating in ELS [12].
In HBV or HCV hepatitis-associated HCC, T- and B-cell production of the pro-inflammatory cytokines lymphotoxin (LT) α and β is markedly upregulated (alongside their receptor, LTβR) [13]. LTαβ acts mainly on hepatocytes expressing the LTβR, leading to elevated LT signalling, increased NF-κB activation and the release of chemokine C-C motif ligand (CCL)2, CCL7, chemokine C-X-C motif ligand (CXCL)1 and CXCL10 chemokines. The resulting increase in inflammatory cell recruitment leads to hepatocyte secretion of cytotoxic cytokines (IL-6, IL-1β, LTαβ), tissue damage, hepatocyte proliferation and death. In this scenario, hepatocytes are increasingly more predisposed to genomic instability leading to HCC. Furthermore, the authors also showed that LTβR inhibition in LTαβ-transgenic mice with hepatitis suppresses HCC formation [13].
Neutrophils
Neutrophils are often thought to be innocent bystanders in cancer development and progression. However, controversial roles have emerged in recent years [14,15,16]. Friedlander and colleagues (2009) identified N1 antitumour neutrophils as being those that “fight infection and cancer”, while N2 pro-tumour neutrophils—which display increased arginase and a loss of oxidative burst and phagocytic capacity—are present in the cancer microenvironment and promote tumour progression [17]. Subsequently, a study led by Wilson et al. [18] further highlighted the pro-tumour role of neutrophils using a diethylnitrosamine (DEN)-induced model of HCC. A tumour suppressor function for hepatocellular nfkb1 that controls hepatocyte production of neutrophil chemokines was also described. The chemokine network comprising of S100A9, CXCL1 and CXCL2 was responsible for neutrophil recruitment to the liver, where neutrophils induced ROS-mediated telomere damage in hepatocytes and increased the development of HCC. In nfkb1 knockout mice, several features were exacerbated—namely, steatosis, neutrophil recruitment, fibrosis, hepatocyte telomere damage and ultimately HCC. By antibody-mediated depletion of neutrophils or disruption of the chemokine network, these effects were abrogated and HCC development attenuated.
In another recent neutrophil study, researchers aimed at evaluating the role of tumour-associated neutrophils (TAN) in the progression of HCC and sorafenib resistance [19]. Here, they showed that CCL2 and CCL17 were highly expressed by TAN and peripheral blood neutrophils (PBN) when exposed to conditioned media from HCC cell lines. The number of CCL2+ or CCL17+ TANs correlated with tumour size, microvascular invasion, tumour encapsulation, tumour differentiation and stage. Also, patients whose tumours presented lower levels of CCL2+ or CCL17+ TAN had longer survival times than those with higher numbers of these cells. CCL2 enhanced the recruitment of macrophages, whereas CCL17 induced the recruitment of Treg cells (but not CD4+ CD25– or CD8+ lymphocytes). Mechanistically, the authors identified the PI3K/Akt and p38/MAPK signalling pathways as crucial mediators of the transformation of PBN into TAN in HCC. Regarding the neutrophil impact in sorafenib treatment, it was demonstrated that sorafenib-induced hypoxia activated NF-κB signalling, thus enhancing CXCL5 secretion by HCC cells, which initiated TAN recruitment. Depletion of TAN resulted in a reduction in tumour volume and enhancement of the effects of sorafenib [19].
Myeloid-Derived Suppressor Cells (MDSC)
MDSC are a heterogeneous group of immature myeloid cells known for their immunosuppressive and pro-tumoural functions—they can induce tumour angiogenesis by vascular endothelial growth factor (VEGF) secretion, and they are able to disrupt both innate and adaptive antitumour activity [8, 20]. For example, Li et al. have shown that MDSC abrogate natural killer (NK)-cell cytotoxicity, NKG2D expression and IFN-γ production via membrane-bound TGF-β. Moreover, the authors demonstrated that depletion of MDSC restored NK function [21].
Macrophages
Macrophages are known to exist in a continuous spectrum of phenotypes, although they are usually referred to by the simplified nomenclature of M1 (classically activated, antitumour) and M2 (alternatively activated, pro-tumour) macrophages [22]. The transition between pro- and antitumour phenotypes is fluid and dependent upon signals from the local microenvironment but can have a profound effect on tumoural immunity via production of pro/anti-inflammatory mediators and expression of inhibitory molecules against T cells and NK cells such as PD-L1. In a mouse model of HCC, TGF-β has been shown to skew macrophages towards an M2 pro-tumour phenotype, inducing the expression of IL-6, and T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), which is an inhibitory receptor for T cells. TIM-3 expression by tumour-associated macrophages (TAM) also correlated with tumour grade and poor survival in HCC patients [23]. It appears that HCC cells can halt the maturation of infiltrating monocytes into macrophages by secreting cytokines that promote immunosuppressive TAM function, promoting evasion of antitumour immunity.
Immunotherapy Approaches: Harness the Immune System to Modulate HCC Progression
Natural occurring adaptive immune responses towards HCC have been previously described and recently reviewed by Makarova-Rusher et al. [7]. Most patients develop adaptive immune responses against TAA, such as α-fetoprotein (AFP), telomerase reverse transcriptase (TERT), melanoma antigen gene-A (MAGE-A) and foetal oncoprotein glypican-3 (GPC3). However, the interactions between the HCC cells and the immune system mainly foster an immunosuppressive microenvironment that prevents antigen-mediated clearance of tumour cells via some of the mechanisms discussed above. A number of clinical trials have evaluated approaches aimed at enhancing the immune response against TAA or dampening the suppressive signals, as summarised in Table 3.1.
Vaccines
AFP was the first TAA to be targeted in the clinic for HCC treatment, in 2003. The clinical trial reported measurable (albeit transient) CD8+ T-cell responses following patient immunisation with a vaccine to four HLA-restricted AFP peptides [24]. Improvement in clinical outcomes in vaccine trials can be achieved by co-administering dendritic cells (DC)—which are professional antigen-presenting cells—pulsed with autologous tumour lysates [25].
Adoptive Cell Transfer
Adoptive cell transfer (ACT) is an autologous infusion of ex vivo-selected, ex vivo-activated and ex vivo-expanded tumour-infiltrating lymphocytes (TIL), which are obtained from a patient’s tumour or peripheral blood. Cytokine-induced killer cells (CIK) and genetically modified T cells can also be used, including TAA-specific T cells, e.g., GPC3 (Table 3.1). In a clinical trial developed by Shimizu K et al. [27], patients were treated with an autologous tumour lysate-pulsed DC vaccine and activated T-cell transfer, after curative resection. Preliminary data support remarkable differences in OS between patients submitted to surgery alone (41.0 months) and to combination treatment (97.7 months).
Oncolytic Viruses
The use of oncolytic viruses as vectors for the delivery of transgenes is a relatively recent approach in the treatment of different types of cancer, including HCC. The JX-594 (Pexa-Vec) is an oncolytic poxvirus engineered to carry the human gene for granulocyte-macrophage colony-stimulating factor (GM-CSF) and has been used to stimulate antitumour responses. This particular virus selectively replicates in cancer cells due to a disruption of the viral thymidine kinase gene. Infected cells lyse and release TAA, which can be taken up by antigen-presenting cells, with the additional expression of GM-CSF heightening the antitumour immune responses. In liver cancer, a study in which patients were randomised to one of two doses of vaccinia demonstrated encouraging results, particularly for the higher dose. Notably, both doses produced equivalent response rates between injected and distant non-injected tumours, supporting the establishment of a systemic immune response [26]. An ongoing trial (NCT03071094) is set to evaluate the safety and efficacy of combining this oncolytic vaccinia with an immune checkpoint inhibitor (anti-PD-1) as a first-line treatment for advanced HCC.
Cytokines
Inflammatory changes associated with liver disease and HCC often display a clear dysregulation in the balance between immunosuppressive (e.g., IL-10, IL-4, IL-5) and immune-activating (e.g., TNF, IFN-γ, IL-1) cytokines, promoting Treg expansion and a reduction in DC function. Trials with the immune modulator IFNα showed early promise which was not realised in a larger trial [28]. Treatments with cytokine inhibitors for the treatment of HCC are ongoing, with mixed results reported thus far. TGF-β is known for regulating cell differentiation, proliferation and death, as well as for its immunosuppressive functions towards T cells, NK cells and neutrophils [8, 17]. In an ongoing phase II non-randomised clinical trial with a novel small molecule inhibitor of TGF-β receptor I, LY2157299, preliminary results suggest AFP expression may influence response [29]. This molecule is currently being studied in a phase II, non-randomised trial as a single agent and in combination with sorafenib or ramucirumab, an anti-vascular endothelial growth factor receptor (VEGFR) 2 monoclonal antibody (NCT01246986).
Immune Checkpoint Inhibitors
T cells and NK cells require organised activation and recognition signals before they are able to mediate tumour cell killing. However, essential inhibitory signalling also exists to prevent unwanted T- and NK-cell responses and “self-harm”. Unfortunately, tumour cells can hijack this inhibitory pathway in order to evade immune cell destruction.
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is an inhibitory checkpoint receptor expressed on T cells, upregulated in patients with viral hepatitis. Upon contact with the activation molecules B7-1 and B7-2 expressed on antigen-presenting cells, CTLA-4 transmits co-inhibitory signals to the T cell, impairing its activity and preventing T-cell immunity [7, 8]. A CTLA-4-targeted antibody therapy has been clinically evaluated in a phase II, noncontrolled, multicentre clinical trial for patients with advanced HCC and chronic HCV infection. As a single agent, the trial reported a partial response rate of 17.6%, stable disease rate of 76.4% and median OS of 8.2 months, with evidence also of antiviral activity [30].
Another immune checkpoint pathway is that regulated by programmed cell death 1 receptor (PD-1). PD-1 is upregulated in T cells in HCC and its ligands—programmed death ligand 1 or 2 (PD-L1/PD-L2)—are involved in immune suppression of T cells by inducing their apoptosis or dysfunction [7, 8]. Monoclonal antibodies that target this pathway have been approved by the FDA as treatments for other cancer types, with encouraging results in patients with HCC [31, 32]. In September 2017, the Food and Drug Administration (FDA) granted accelerated second-line approval for nivolumab, for the treatment of HCC in patients who have progressed on sorafenib. Approval was based on a 154-patient subgroup of the CHECKMATE-040 (NCT01658878) trial. As a condition of accelerated approval, further trials will be required to verify the clinical benefit of the antibody for this indication. Ongoing clinical trials with immune checkpoint inhibitors in combination with other therapies are summarised in Table 3.1, e.g., phase I trial for ramucirumab and durvalumab (anti-PD-L1) in the setting of locally advanced and unresectable or metastatic gastrointestinal or thoracic malignancies, including HCC (NCT02572687) [8].
In addition to CTLA-4 and PD-1, there are other immune checkpoint inhibitors expressed on activated T cells and NK cells including KIR, TIM-3 and LAG-3 [8] and further highlight the promise of dual or triple therapy in patients with high expression of these receptors.
Conclusions and Future Perspectives
As we begin to understand how the chronic inflammatory responses associated with chronic liver disease, associated with HCC-induced immune tolerance, we are entering an exciting era of immunotherapy—with perhaps tangible hope for the first time that effective anti-HCC therapies delivering long-term survival are on the horizon. How we will select and monitor these therapies and use them safely in different groups of patients is not yet clear, as the field is hampered by the lack of either tissue are circulating biomarkers to guide clinical decision making. Progress in these fields is also set to have a substantial impact on the future for patients with HCC.
References
Marengo A, Rosso C, Bugianesi E. Liver cancer: connections with obesity, fatty liver, and cirrhosis. Annu Rev Med. 2016;67:103–17.
Knudsen ES, Gopal P, Singal AG. The changing landscape of hepatocellular carcinoma: etiology, genetics, and therapy. Am J Pathol. 2014;184(3):574–83.
Stickel F. Alcoholic cirrhosis and hepatocellular carcinoma. Adv Exp Med Biol. 2015;815:113–30.
Reeves HL, Zaki MY, Day CP. Hepatocellular carcinoma in obesity, type 2 diabetes, and NAFLD. Dig Dis Sci. 2016;61(5):1234–45.
Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol. 2013;10(9):542–52.
Alison MR, Nicholson LJ, Lin WR. Chronic inflammation and hepatocellular carcinoma. Recent Results Cancer Res. 2011;185:135–48.
Makarova-Rusher OV, Medina-Echeverz J, Duffy AG, Greten TF. The yin and yang of evasion and immune activation in HCC. J Hepatol. 2015;62(6):1420–9.
Obeid JM, Kunk PR, Zaydfudim VM, Bullock TN, Slingluff CL Jr, Rahma OE. Immunotherapy for hepatocellular carcinoma patients: is it ready for prime time? Cancer Immunol Immunother. 2017;67(2):161–74.
Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30(4):533–47.
Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10(2):99–111.
Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531(7593):253–7.
Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol. 2015;16(12):1235–44.
Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009;16(4):295–308.
Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015;10(4):562–73.
Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016;16(7):431–46.
Powell DR, Huttenlocher A. Neutrophils in the tumor microenvironment. Trends Immunol. 2016;37(1):41–52.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell. 2009;16(3):183–94.
Wilson CL, Jurk D, Fullard N, Banks P, Page A, Luli S, et al. NFkappaB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat Commun. 2015;6:6818.
Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. 2016;150(7):1646–58 e17.
Medina-Echeverz J, Eggert T, Han M, Greten TF. Hepatic myeloid-derived suppressor cells in cancer. Cancer Immunol Immunother. 2015;64(8):931–40.
Li HQ, Han YM, Guo QL, Zhang MG, Cao XT. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. 2009;182(1):240–9.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61.
Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut. 2015;64(10):1593–604.
Butterfield LH, Ribas A, Meng WS, Dissette VB, Amarnani S, Vu HT, et al. T-cell responses to HLA-A*0201 immunodominant peptides derived from alpha-fetoprotein in patients with hepatocellular cancer. Clin Cancer Res. 2003;9(16 Pt 1):5902–8.
Lee WC, Wang HC, Hung CF, Huang PF, Lia CR, Chen MF. Vaccination of advanced hepatocellular carcinoma patients with tumor lysate-pulsed dendritic cells: a clinical trial. J Immunother. 2005;28(5):496–504.
Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19(3):329–36.
Shimizu K, Kotera Y, Aruga A, Takeshita N, Katagiri S, Ariizumi SI, et al. Postoperative dendritic cell vaccine plus activated T-cell transfer improves the survival of patients with invasive hepatocellular carcinoma. Hum Vaccin Immunother. 2014;10(4):970–6.
Chen LT, Chen MF, Li LA, Lee PH, Jeng LB, Lin DY, et al. Long-term results of a randomized, observation-controlled, phase III trial of adjuvant interferon Alfa-2b in hepatocellular carcinoma after curative resection. Ann Surg. 2012;255(1):8–17.
Faivre SJ, Santoro A, Kelley RK, Merle P, Gane E, Douillard J-Y, et al. A phase 2 study of a novel transforming growth factor-beta (TGF-β1) receptor I kinase inhibitor, LY2157299 monohydrate (LY), in patients with advanced hepatocellular carcinoma (HCC). J Clin Oncol. 2014;32(suppl 3):abstract LBA173.
Sangro B, Gomez-Martin C, de la Mata M, Inarrairaegui M, Garralda E, Barrera P, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81–8.
El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502.
Sangro B, Crocenzi TS, Welling TH, Iñarrairaegui M, Prieto J, Fuertes C, et al. Phase I dose escalation study of nivolumab (Anti-PD-1; BMS-936558; ONO- 4538) in patients (pts) with advanced hepatocellular carcinoma (HCC) with or without chronic viral hepatitis. J Clin Oncol. 2013;31(suppl):abstr TPS3111.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Maurício, J., Reeves, H., Wilson, C.L. (2019). Roles of the Immune System in the Development and Progression of Hepatocellular Carcinoma. In: Cross, T., Palmer, D. (eds) Liver Cancers. Springer, Cham. https://doi.org/10.1007/978-3-319-92216-4_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-92216-4_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-92215-7
Online ISBN: 978-3-319-92216-4
eBook Packages: MedicineMedicine (R0)