
Abstract
Myeloid-derived suppressor cells are key components of tumor-induced immune suppression. They are composed of a heterogeneous population of immature myeloid cells that abrogates innate and adaptive immune responses. Myeloid-derived suppressor cells accumulate not only in peripheral blood, secondary lymphoid organs and tumors, but also in the liver in preclinical tumor models and in hepatocellular carcinoma patients. The liver, continuously exposed to food and microbial antigens from the intestine, avoids autoimmune damage through the use of specialized mechanisms of immune tolerance. In the context of cancer, myeloid-derived suppressor cells profit the intrinsic tolerogenic properties of the liver to accumulate and exert various immune-suppressive and tumor-promoting mechanisms which go from inducing immune cell dysfunction to supporting the generation of liver metastases. In this review, we seek to describe the phenotype, function, accumulation and therapeutic targeting of hepatic myeloid-derived suppressor cells both in preclinical settings and in the context of human hepatocellular carcinoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tumors are heterogeneous tissues composed of multiple distinct cell types that participate in tangled interactions with each other. The tumor microenvironment manipulates the host’s immune system to remain unnoticeable. Tumors recruit immunosuppressive cell subsets to control and bypass the host’s anti-tumor immune responses and to promote cancer invasiveness and metastasis. Among other cell subsets, myeloid-derived suppressor cells (MDSCs) have been long recognized as key players in tumor-induced immune suppression.
MDSCs represent a diverse population of immature cells of myeloid origin at different states of differentiation. In the context of cancer, immune-suppressive MDSCs accumulate in peripheral blood, bone marrow, tumor and secondary lymphoid organs. In healthy patients, these immature myeloid cells give rise to mature dendritic cells, macrophages or granulocytes. However, cancer-derived MDSCs remain immature and promote tolerance to tumor growth by suppressing innate and adaptive immune responses [1].
The liver performs multiple tasks: uptake of blood borne pathogens, metabolite detoxification and filtration of bacterial agents, thus exposing the liver to a high antigenic load. The local hepatic immune environment is committed to the induction of immune tolerance in order to avoid the generation of autoimmune disorders [2]. The liver serves as an ideal site for tumor-induced immune suppression. Tumors secrete diverse factors that circulate in the bloodstream resulting in the migration and accumulation of immature myeloid progenitors from the bone marrow into the hepatic tissue. This process has been described in mice bearing extrahepatic tumors of diverse origin such as mammary, lung, colon, skin and thymus [3–7]. Mouse models of liver metastases show that immature myeloid-suppressive cells from the bone marrow cluster into the liver [6, 8–10]. MDSCs also accumulate when tumors are injected orthotopically into the liver [11–13]. Intrahepatic liver tumor incidence correlates with MDSC infiltration both in carcinogen-induced [5] and in genetic-engineered mouse models [5, 14].
This review will cover the phenotype, mechanisms of immune suppression and therapeutic interventions involving liver-infiltrating MDSCs in preclinical models of extrahepatic and intrahepatic tumors. It will also address MDSCs in patients suffering hepatocellular carcinoma (HCC).
Phenotype of tumor-induced hepatic MDSCs
Tumor-induced MDSCs express the myeloid surface marker CD11b and the granulocyte/monocyte marker Gr1 [15]. Several studies describe an accumulation of CD11b+Gr1+ cells in the liver of mice bearing either extrahepatic or intrahepatic tumors [3, 16]. Hepatic CD11b+Gr1+ absolute cell number is estimated in 2x106 cells per gram liver, more than 20 % of non-parenchymal cells in mice bearing established extrahepatic tumors [4]. Similarly, MDSCs infiltrating transplantable intrahepatic tumors constitute 20 % of liver non-parenchymal cells [5, 12]. Fluorescence-activating cell sorting of CD11b+Gr1low and Gr1high cells from mice bearing intraperitoneal colorectal tumors reveals the different monocytic and granulocytic MDSC populations. MDSC subsets were further characterized by Giemsa staining and by differential expression of the glycoprotein Ly6C and myeloid differentiation antigen Ly6G [6]. As described for splenic and bone marrow-derived MDSCs, extrahepatic tumor-induced hepatic MDSCs express low levels of major histocompatibility complex-II (MHC-II), co-stimulatory molecules CD40, CD80, CD86 and low levels of glycoprotein CD1d [7]. Several studies used the surface marker CD244 [17] to distinguish liver granulocytic MDSCs from granulocytes [4, 7]. Tumor-induced hepatic MDSCs are phenotypically similar to MDSCs accumulating in tumors or in peripheral organs.
Suppression of T cell function as hallmark of tumor-induced hepatic MDSCs
A major limitation in the study of MDSC biology is the lack of specific markers to distinguish MDSCs from neutrophils [18], which represent a major intrahepatic population in acute or chronic inflammation. Unlike naïve hepatic CD11b+Gr1+ cells or neutrophils, tumor-induced hepatic MDSCs suppress antigen-specific and antigen-independent T cell proliferation in vitro [3, 5–7, 14, 19]. Liver MDSCs induced by extrahepatic tumors or isolated from either intrahepatic lesions or liver metastases suppress T cell proliferation in vitro to a similar extent. MDSC-mediated suppression of T cell responses is considered as the ultimate marker of their functional activity.
The effects of tumor-induced hepatic MDSCs on preventing T cell function in vivo are a matter of discussion in the field. Connolly et al. transfer ovalbumin (OVA)-T cell receptor (TCR)-specific splenocytes into naïve mice vaccinated 24 h later either with dendritic cells pulsed with OVA or with a mixture of OVA-pulsed dendritic cells with hepatic MDSCs. Addition of hepatic MDSCs decreases OVA Tetramer+ CD8+ T cell expansion caused by OVA-pulsed dendritic cell immunization [6]. Using oncogene-driven liver tumor models of distinct immunogenicity where SV40 oncogene/antigen retention in the liver causes local T cell tolerance, hepatic MDSC accumulation in the liver does not result in hepatic cytotoxic T lymphocyte unresponsiveness to tumor antigens in an in vivo cytotoxicity assay [14]. These results have to be interpreted carefully, since both experiments do not recapitulate the in vivo situation, where few antigen-specific intrahepatic CD8+ T cells and low antigen loads are present.
Immunomodulatory roles of tumor-induced hepatic MDSCs
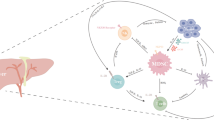
Hepatic MDSCs influence both innate and adaptive immune responses by depleting nutrients utilized by lymphocytes, promoting oxidative stress and reducing lymphocyte trafficking to the liver (Fig. 1). These effects mediated by hepatic MDSCs are linked to metabolic consequences in the liver.

Immune modulatory functions exerted by tumor-induced hepatic MDSCs. a In mice bearing extrahepatic tumors, liver-infiltrating CD11b+Gr1+ MDSCs suppress T cell responses either by depletion of l-arginine via ARG1 or by production of reactive oxygen species through NOX2. Tumor-induced MDSCs in the liver exert impaired antigen-presenting capacity due to low expression levels of MHC-II, glycoprotein CD1d and various co-stimulatory molecules. Hepatic MDSC-derived TGF-β1 induces NK cell anergy by decreased IFN-γ production and impaired ADCC. b In mouse models of orthotopic liver tumors or hepatic metastases, hepatic MDSCs suppress T cell responses by depletion of l-arginine and induce NK cell anergy through diverse mechanisms including NKG2D downregulation. Liver MDSCs cause B cell dysfunction by inducing downregulation of CD80 on B cells in a contact-dependent manner, resulting in poor CD4+ T cell activation. In a mouse model of orthotopic liver tumors, hepatic MDSCs express co-inhibitory receptor PD-L1
Liver metabolism is tightly controlled to regulate glucose, lipid and amino acid processing. l-arginine contributes to liver lipid metabolism and is catabolized in the liver by arginase I into ornithine and urea. Interestingly, arginase I has been shown to participate in tumor-induced immune dysfunction and immune escape [20]. Increased activity of arginase I by MDSCs leads to enhanced l-arginine catabolism and abolishes T cell function. It has been recently demonstrated that tumor-induced hepatic MDSCs catabolize l-arginine through arginase I, thereby depleting l-arginine locally and suppressing T cell proliferation [5, 7, 21]. Hepatic MDSC-mediated catabolism of l-arginine contributes to T cell dysfunction.
Reactive oxygen species (ROS) and nitrogen species (RNS) are chemically reactive by-products of oxygen and nitrogen metabolism in cell signaling and homeostasis. ROS and RNS metabolism is shared by all myeloid cells. In the context of cancer, MDSCs release superoxide (O2 −), hydrogen peroxide (H2O2), peroxynytrite (PNT) and nitric oxide (NO) catabolized by NADPH oxidases, arginase I and inducible nitric oxide synthase (iNOS) in different MDSC subsets. Release of PNT induces the nitration of the T cell TCR. Then, T cells are not able to bind antigen-specific peptides, leading to T cell dysfunction and antigen-specific immune tolerance [22].
Tumor-induced hepatic MDSCs release high amounts of ROS compared to their naïve hepatic counterparts [7]. The production of ROS and RNS is tightly regulated in the liver microenvironment since their release leads to hepatocyte killing and liver inflammation. A recent study shows that accumulation of MDSCs in the liver induced by extrahepatic tumors results in moderate liver damage [4]. This effect is more prevalent in mice with growing tumor burden and increasing serum transaminases that correlate with more intense hepatic MDSC infiltration. Tumor-induced hepatic accumulation of MDSCs influences the degree of liver damage in the steady state.
Tumor-induced infiltration of MDSCs into the liver affects the intrahepatic lymphocyte pool. Natural killer (NK) cells comprise more than 15 % of intrahepatic lymphocytes in mice and human [23]. Li et al. [16] show in orthotopic liver tumor models that intrahepatic NK cell infiltration diminishes while MDSCs increase. Hepatic NK cell numbers do not vary in mice bearing subcutaneous tumors [4]. Tumor-induced hepatic MDSCs (regardless of the site of tumor implantation) inhibit NK cell function and cytotoxicity, reflected by low levels of interferon gamma (IFN-γ). Hepatic MDSCs impair antibody-dependent cellular cytotoxicity (ADCC), and liver tumor-infiltrating NK cells decrease surface NKG2D (NK activating receptor) expression in contact with MDSCs. This mechanism of MDSC-induced immune suppression is dependent on membrane-bound transforming growth factor beta 1(TGF-β1), a multifunctional cytokine involved in HCC progression [24]. This body of work support that tumor-induced hepatic MDSCs influence the intrahepatic NK cell pool by promoting NK cell anergy.
B cells constitute (along with NK cells) the main intrahepatic lymphoid population. Although extrahepatic tumors do not modulate the liver B cell pool [4], B lymphocytes are essential to control local tumor growth in the liver in different preclinical models [25, 26]. Hepatic B cells downregulate surface co-stimulatory CD80 expression by interaction with intrahepatic MDSCs in a colorectal liver metastasis model, leading to decreased T cell activation [25]. CD80 downregulation in hepatic B cells is reversible in the absence of MDSCs and dependent on Stat3 activity, a crucial transcription factor regulating MDSC biology [27]. Therefore, hepatic B cell activity depends on the interaction with the hepatic MDSC pool in the context of liver cancer.
Accumulation of MDSCs in the liver: a chemokine romance
The mechanisms underlying the accumulation of MDSCs in tumors and in peripheral compartments are governed by the cytokine/chemokine system [28, 29]. Chemokines induce migration of tumor-induced immature myeloid-suppressive cells generated in the bone marrow to the liver (Fig. 2). Hepatic MDSCs express chemokine receptors such as CXCR2, CCR2, CXCR4 and CXCR5 [13, 30]. Extrahepatic and liver tumors secrete large amounts of CXCL1, CCL2, CCL5, CXCL5, S100A8 and S100A9 [4, 5, 13, 26, 31]. These data point out the importance of tumor-derived factors in modulating the immune myeloid microenvironment.

Schematic representation of tumor-induced MDSC generation and migration into the liver. Upon growth, tumor masses secrete various factors into systemic circulation, reaching various organs such as the bone marrow and the liver. Once in the bone marrow, tumor-derived factors promote the generation of immature MDSCs, which express various chemokine receptors such as CXCR4, CCR2, CXCR2 and CXCR5. The liver releases cytokines and chemokines such as CXCL1, CCL2 and CCL5 in response to tumor-derived factors, resulting in MDSC recruitment from the bone marrow and accumulation in the liver
Several studies provide a link between chemokines and MDSC accumulation into the liver. Antibody blockade of CXCL1 reduces the influx of MDSCs to the liver in mice bearing subcutaneous HCC [5]. Mice deficient in the CXCL1 receptor CXCR2 show reduced accumulation of MDSCs in mice bearing intraperitoneal tumors [6]. γδT cells in the liver tumor microenvironment secrete interleukin-17A (a pleiotropic cytokine associated with the defense against microbial pathogens but also expressed in several tumors [32]). Interleukin-17A induces CXCL5 secretion by the liver tumor mass, thus attracting CXCR2-expressing MDSCs [13]. CCR2/CCL2 chemokine axis plays a pivotal role in the migration of innate immune cells to the liver in acute and chronic inflammation [33–35]. In a recent study, antibody blockade or genetic ablation of CCR2 reduces MDSC numbers and significantly decreases the occurrence of colorectal metastasis into the liver [8]. In this model, myeloid cell ablation using CD11b-DTR mice reveals that CD11b+ Gr1low monocytic MDSCs promote metastasis to the liver. In this setting, CD11b+ Gr1low cells decrease the expression of angiopoietin-like 7 in cancer cells, enhancing their metastatic potential by inducing tumor blood vessel formation [36]. Therefore, tumor-induced CD11b+ Gr1low monocytic MDSCs accumulate in the liver in a CCR2-dependent manner and promote metastasis formation by facilitating tumor angiogenesis. In summary, these data highlight the chemokine system as an attractive target with therapeutic potential to decrease tumor-induced MDSC recruitment to the liver.
Presence and functions of MDSCs in HCC patients
Numerous studies have reported the presence of MDSCs in peripheral blood of patients with cancer [37]. Regarding HCC, MDSCs accumulate in peripheral blood and in the tumor of HCC patients [38–40]. Human MDSCs are defined as CD11b+CD33+HLA-DRlow/neg. Monocytic and granulocytic MDSC subsets are distinguished by the expression of the monocyte marker CD14. Likewise, monocytic MDSCs are characterized in HCC tumor tissue and PBMCs as CD14+HLA-DRlow cells. Further phenotypic analysis did not find differences between CD14+ HLA-DRlow monocytic MDSCs and CD14+HLA-DR+ monocytes. Functional studies showing suppression of T cell proliferation and impaired T cell-derived IFN-γ production due to CD14+HLA-DRlow cells highlight human monocytic MDSCs as immunosuppressive [38]. Recent studies show that activated hepatic stellate cells (a cell subset involved in the development of liver fibrosis in pathological conditions) promote monocytic CD14+HLA-DRlow cell conversion from mature CD14+ monocytes in a contact-dependent manner via CD44 expression [41] and catalase production [42].
Granulocytic CD14−CD15+CD11b+CD33+ MDSCs from patient PBMCs are sensitive to sample cryopreservation, a limiting factor for performing functional assays [43]. Mesenchymal stem cells (multipotent stromal cells very abundant in adipose tissue that can get mobilized and derive into a variety of cell types, including hepatocytes) induce granulocytic CD14−CD11b+CD33+ MDSCs in vitro through the production of hepatocyte growth factor, associated with angiogenesis and tumorigenesis [44]. Recently, granulocytic MDSCs have been reported in PBMCs from HCC patients [39], although no functional studies were performed using this cell subset.
Human MDSCs exert immune suppressor mechanisms, highlighting the translational relevance of MDSCs in human liver disease. CD14+ HLA-DRlow monocytic MDSCs from HCC patient PBMCs catabolize l-arginine [38] and therefore abrogate IFN-γ production by T cells. Addition of exogenous l-arginine to the co-culture restores IFN-γ secretion. Human monocytic MDSCs isolated from gastrointestinal cancer patient PBMCs express higher Arginase I mRNA levels than CD14+ HLA-DRhigh monocytes [21]. l-arginine catabolism by HCC-induced human MDSCs influences T cell responses in vitro.
MDSCs from HCC patients influence the function of diverse immune cell subsets. NK cells isolated from peripheral blood and tumor of HCC patients display reduced cytotoxicity compared to naïve controls [40]. CD14+ HLA-DRlow monocytic MDSCs from HCC patient PBMCs reduce NK cell cytotoxicity and IFN-γ production in a contact-dependent fashion by blocking the expression of the activating NK cell receptor NKp30 [40]. Human monocytic MDSCs control NK cell activation in HCC.
Tregs are key components involved in the maintenance of peripheral immune tolerance. In the context of cancer, Tregs establish tumor-induced tolerance and thus promote immune escape [45]. Treg abundance among PBMCs and in the liver has been linked to decreased CD8+ T cell numbers, lacks T cell activation and is associated with poor prognosis in HCC patients [46–48]. A recent study shows highly suppressive Tregs expressing glycoprotein A repetition predominant (GARP) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) in the peripheral blood of HCC patients [39]. Tumor-induced MDSCs promote the expansion of natural (nTregs) and influence the conversion of CD4+ T cells into inducible FoxP3+ Tregs and IL-10—expressing CD4+ T cells (Tr1 cells). We described the interplay between MDSCs and Tregs in the context of HCC [38]. We show that CD14+ HLA-DRlow monocytic MDSCs from HCC patient PBMCs convert CD4+ T cells into FoxP3+ iTregs. Human monocytic MDSCs also induce IL-10 secretion by CD4+ T cells, promoting the generation of Tr1 cells. The human monocytic MDSC compartment promotes Treg expansion and function in HCC.
Therapeutic interventions affecting tumor-induced hepatic MDSCs
In the context of cancer, several therapeutic regimes have proven to modulate the tumor-induced hepatic MDSC pool. These strategies either reduce MDSC infiltration in the liver or exploit MDSC properties such as enhanced migration and plasticity in the liver using diverse therapeutic agents to induce pro-inflammatory responses.
The Gr1 (clone RB6-8C5) antibody binds with high affinity to Ly6G molecules and with less affinity to Ly6C and is widely used to deplete CD11b+Gr1+ cells in tumor-bearing models. Although Gr1 depletion is effective in the tumor and in the periphery, tumor-induced MDSCs repopulate the liver in 24 h making Gr1 depletion less effective in the liver [49]. Tumor-induced hepatic MDSCs die upon Gr1 depletion, but there is an enhanced repopulation that comes from the bone marrow. Although the reason underlying this lack of depletion remains unknown, there is confidence in the efficacy of MDSC-specific peptibodies in depleting tumor-induced hepatic MDSCs [50].
Chimeric antigen receptor-modified T (CAR-T) cells constitute a successful approach in the treatment of lymphoid cancers. The therapeutic efficacy of CAR-T cells engineered against the carcinoembryonic antigen in hepatic metastases is compromised due to the accumulation of MDSCs [10]. In this model, hepatic MDSC accumulation is dependent on GM-CSF, a growth factor that supports MDSC recruitment and blocks MDSC maturation [1]. Hepatic MDSCs express programmed death ligand 1 (PD-L1), a co-inhibitory receptor that engages PD-1 in T cells and inhibits TCR-mediated T cell activation and proliferation. Therefore, PD-L1 on liver-infiltrating MDSCs suppresses CAR-T cell proliferation and function. Decreasing MDSC accumulation in the liver using anti-Gr1, anti-GM-CSF or blocking MDSC suppressive function using anti-PD-L1 synergizes with anti-carcinoembryonic antigen CAR-T cell therapy and results in anti-tumor responses. Blockade of molecules involved in MDSC biology and function enhances adoptive T cell therapy against colorectal cancer metastases.
Multitarget receptor tyrosine kinase (RTK) inhibitor sorafenib is the only approved systemic drug for the treatment of unresectable HCC, but provides limited survival benefits. Sorafenib slows tumor growth in subcutaneous HCC by decreasing Treg and MDSC accumulation in the spleen [51]. Interestingly, sorafenib has been recently shown to decrease tumor burden. However, sorafenib induces a compensatory accumulation of immune-suppressive MDSCs, tumor-associated macrophages and Tregs related to intratumoral hypoxia and CXCL12 in orthotopic HCC models [52]. Combining sorafenib with CXCR4 chemokine receptor blocker AMD3100 achieved better responses and decreased immune-suppressive cell recruitment to the liver. Combination of sorafenib, AMD3100 and checkpoint blockade inhibitor anti-programmed death-1 (PD-1) showed synergistic anti-tumor responses in treatment of orthotopic HCC by promoting CD8+ T cell infiltration from the tumor rim to the center.
Sunitinib is a RTK inhibitor with inferior overall survival in HCC patients when compared to sorafenib in a phase III clinical trial [53]. Sunitinib modulates the tumor microenvironment by decreasing tumor-infiltrating Treg and MDSC numbers in an intrahepatic colorectal metastases model [54]. This decrease was impaired in tumor-bearing mice deficient for the tyrosine kinase receptor c-kit, thus demonstrating a direct effect of sunitinib in MDSC reduction. Moreover, tumor-infiltrating MDSCs lose the expression of PD-L1 upon sunitinib treatment. Treatment of intrahepatic colorectal metastases using sunitinib combined with anti-CD137 and an adenovirus coding for the immune stimulatory cytokine interleukin-12 resulted in prolonged survival compared to anti-CD137 and adenovirus expressing interleukin-12 alone.
Oncolytic vectors are viruses that preferentially infect and lyse cancer cells. Oncolytic therapy against cancer is an attractive therapeutic modality. However, oncolytic vectors lack tumor tropism. Taking advantage of the migration capacity of MDSCs, a study used MDSCs as carriers for oncolytic vesicular stomatitis virus therapy of intrahepatic tumors [55]. Eisenstein et al. showed that MDSCs are the best cell subset in migrating to intrahepatic tumors. When MDSCs are infected with vesicular stomatitis virus, the combination significantly prolonged survival of tumor-bearing mice compared to free virus [55]. Interestingly, vesicular stomatitis virus infection enhances iNOS rather than arginase I catabolism in MDSCs and promotes tumor cell killing, therefore polarizing MDSCs toward a pro-inflammatory phenotype. MDSCs infected with oncolytic viruses serve as a trojan horse in the liver tumor microenvironment.
The accumulation of tumor-induced MDSCs in the liver can lead to unwanted side effects in therapeutic settings. Tumor-bearing mice suffer severe liver damage compared to naïve counterparts in two models of immune-mediated hepatitis. Hypothetically, tumor-induced hepatic MDSCs would contribute to suppress hepatitis in the liver, but the opposite was observed [7]. Tumor-induced hepatic MDSC pool increased upon acute hepatitis and MDSCs acquired not only functional but also phenotypic characteristics of pro-inflammatory myeloid cells. The severity of hepatic damage was caused by enhanced production of ROS by tumor-induced hepatic MDSCs [7]. Therefore, hepatic MDSCs are polarized toward inflammatory myeloid cells upon local inflammation.
Antibody-based immunotherapies are of great interest in the oncology field. As the use of immunotherapies increases, so do the concerns about their adverse effects in clinical settings. Agonistic antibodies to tumor necrosis factor receptor molecules such as activating CD137 and CD40 antibodies have shown promising results along with liver-related toxicities in both preclinical and early clinical settings. CD137 agonistic antibody induces acute liver damage by increasing the infiltration of intrahepatic CD8+ T cells. Systemic administration of interleukin-6 after CD137 agonistic antibody injection promotes recruitment of MDSCs to the liver. MDSC accumulation in the liver decreases the proliferation of CD8+ T cells and alleviates liver damage [56]. Our group and others reported that systemic administration of CD40 agonistic antibody increased liver damage transiently, highlighting the importance of finding administration routes without liver toxicity. CD40 agonistic antibody-induced liver damage is mediated by tumor-induced hepatic MDSCs in subcutaneous and liver metastases models and is dependent on ROS production [21]. CD40 agonistic antibody-induced expression of co-stimulatory molecules CD40 and CD80, enhanced loss of arginase I activity and stimulated ROS production by hepatic MDSCs resulting in increased hepatocyte cell death in vitro. Importantly, decreased arginase I mRNA levels and enhanced ROS production were assessed in CD14+ HLA-DRlow monocytic MDSCs from peripheral blood mononuclear cells (PBMCs) as well as impaired T cell suppression in vitro. CD40 agonistic antibody treatment promotes hepatic MDSC polarization into pro-inflammatory myeloid cells.
In conclusion, therapeutic modulation of tumor-induced hepatic MDSCs in preclinical models has important consequences in local tumor growth in terms of decreasing their suppressive function. Strategies inducing hepatic MDSC polarization would elicit potent local immune responses with the adverse effect of increasing liver toxicity. Additional data on CD40 agonists in intrahepatic tumors would determine whether the therapeutic activity overlaps liver toxicity, offering a new immune modulatory compound in the fight against HCC.
There are few clinical studies addressing potential strategies to target MDSCs in human cancer [37]. A recent study shows how 5-phosphodiesterase inhibitor tadalafil induces the reduction in MDSC and Treg frequencies in peripheral blood and a decrease in Arginase I mRNA expression by MDSCs in head and neck cancer patients [57]. There are no data available in the context of HCC. Similar to the preclinical field, efforts must be directed to thoroughly characterize MDSCs in human HCC. This would turn into developing novel therapies aimed to target MDSC plasticity toward mature, pro-inflammatory cells that would potentiate anti-tumor immune responses.
Outlook and future directions
During the last decade, our basic understanding of tumor-induced hepatic MDSCs has broadened extensively. Liver MDSCs comprise a singular, immune-suppressive myeloid cell subset. MDSCs display unique dynamics in hepatic repopulation after depletion compared to other peripheral organs and lose their immune-suppressive capacity upon inflammatory insults to the liver.
There is a lack of studies focused on analyzing the differences in the immunobiology of tumor-induced MDSCs that accumulate in organs such as the pancreas and the liver and their peripheral counterparts in spleen and blood. A study shows that pancreatic CD11b+ Gr1+ cells isolated from pancreatic tumors in a model of spontaneous pancreatic ductal adenocarcinoma suppress antigen-specific T cell proliferation in vitro to a larger extent than their splenic counterparts [58]. In line with this, preliminary data from our laboratory show that tumor-induced hepatic MDSCs from two unrelated extrahepatic tumor models are more suppressive than splenic MDSCs in vitro (Fig. 3). Although the underlying mechanism remains unknown, progress in our understanding of the organ-specific accumulation of tumor-derived factors might help to unravel differences between liver accumulating and peripheral MDSCs in terms of immune regulation.

Differences between tumor-induced splenic and hepatic MDSCs in suppressing antigen-specific CD8+ T cell proliferation. a Hepatic and splenic CD11b+ Gr1+ cells from EL-4 tumor-bearing mice (n = 4) were FACS sorted. b Hepatic and splenic CD11b+ cells from B16 GM-CSF tumor-bearing mice (n = 2) were MACS sorted. In both conditions, sorted cells were cultured with 105 CFSE-labeled OT-I splenocytes at different ratios in the presence of OVA257–264 peptide (0.1 μg/ml). Proliferation of CFSE+ CD8+ T cells was evaluated after 48 h by flow cytometry. Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.005; Student’s t test
Progress must be made in deciphering the expression and impact of regulatory molecules discovered in liver pathologies on tumor-induced hepatic MDSCs. Molecules such as the alarmin interleukin-33 and the leptin galectin-3 have been associated with liver fibrosis and cancer [59, 60], but their role on hepatic MDSCs remains unexplored and could unravel novel mechanisms of MDSC-mediated immune suppression in the liver.
Albeit notable, the findings highlighted in this review have been obtained mainly using preclinical models with extrahepatic or intrahepatic implanted tumors. The majority of these models do not reflect the clinical situation, where most of the HCC patients have underlying liver cirrhosis as a consequence of lifelong chronic liver inflammation. Obesity is recognized as a major risk of HCC [61]. Although there is a study linking obesity to MDSC accumulation in the liver [62], the lack of models mimicking obesity-induced HCC complicates the study of hepatic MDSCs. Therefore, developing new preclinical tumor models that recapitulate human pathogenesis in liver cancer not only would shed light on the regulatory mechanisms exerted by hepatic MDSCs, but would also point out potential targets to approach in therapeutic settings.
Concentrating resources in addressing these important topics would have direct implications in the study of MDSCs in patients suffering HCC. It has to be considered the gap between preclinical and clinical MDSC research in HCC. Unfortunately, few studies use human MDSCs isolated from HCC biopsies and human peripheral blood is the standardized source for MDSCs. More progress toward understanding the mechanisms underlying MDSC accumulation in the peripheral blood of cancer patients and a thorough definition of human MDSC phenotype must be achieved. Clinical studies aiming at specifically targeting MDSCs from HCC patients will help broaden the palette of immune-targeted therapies against HCC.
Abbreviations
- ADCC:
-
Antibody-dependent cell cytotoxicity
- CAR:
-
Chimeric antigen receptor
- DC:
-
Dendritic cell
- HCC:
-
Hepatocellular carcinoma
- H2O2 :
-
Hydrogen peroxide
- IFN-γ:
-
Interferon gamma
- iNOS:
-
Inducible nitric oxide synthase
- MDSC:
-
Myeloid-derived suppressor cell
- MHC-II:
-
Major histocompatibility complex-II
- mTOR:
-
Mammalian target of rapamycin
- NOX2:
-
NADPH oxidase 2
- NK:
-
Natural killer
- NO:
-
Nitric oxide
- OVA:
-
Ovalbumin
- PBMC:
-
Peripheral blood mononuclear cell
- PD-L1:
-
Programmed death ligand 1
- PNT:
-
Peroxynitrite
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- Treg :
-
Regulatory T cell
- TGF-β1:
-
Transforming growth factor beta 1
References
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12:253–268. doi:10.1038/nri3175
Crispe IN (2009) The liver as a lymphoid organ. Annu Rev Immunol 27:147–163. doi:10.1146/annurev.immunol.021908.132629
Ilkovitch D, Lopez DM (2009) The liver is a site for tumor-induced myeloid-derived suppressor cell accumulation and immunosuppression. Cancer Res 69:5514–5521. doi:10.1158/0008-5472.CAN-08-4625
Eggert T, Medina-Echeverz J, Kapanadze T, Kruhlak MJ, Korangy F, Greten TF (2014) Tumor induced hepatic myeloid derived suppressor cells can cause moderate liver damage. PLoS One 9:e112717. doi:10.1371/journal.pone.0112717
Kapanadze T, Gamrekelashvili J, Ma C et al (2013) Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J Hepatol 59:1007–1013. doi:10.1016/j.jhep.2013.06.010
Connolly MK, Mallen-St Clair J, Bedrosian AS et al (2010) Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. J Leukoc Biol 87:713–725. doi:10.1189/jlb.0909607
Kapanadze T, Medina-Echeverz J, Gamrekelashvili J et al (2015) Tumor-induced CD11b Gr-1 myeloid-derived suppressor cells exacerbate immune mediated hepatitis in mice in a CD40-dependent manner. Eur J Immunol 45(4):1148–1158. doi:10.1002/eji.201445093
Zhao L, Lim SY, Gordon-Weeks AN et al (2013) Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology 57:829–839. doi:10.1002/hep.26094
Kitamura T, Fujishita T, Loetscher P, Revesz L, Hashida H, Kizaka-Kondoh S, Aoki M, Taketo MM (2010) Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc Natl Acad Sci USA 107:13063–13068. doi:10.1073/pnas.1002372107
Burga RA, Thorn M, Point GR et al (2015) Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. doi:10.1007/s00262-015-1692-6
Gonzalez-Aparicio M, Alzuguren P, Mauleon I et al (2011) Oxaliplatin in combination with liver-specific expression of interleukin 12 reduces the immunosuppressive microenvironment of tumours and eradicates metastatic colorectal cancer in mice. Gut 60:341–349. doi:10.1136/gut.2010.211722
Gauttier V, Judor JP, Le Guen V, Cany J, Ferry N, Conchon S (2014) Agonistic anti-CD137 antibody treatment leads to antitumor response in mice with liver cancer. Int J Cancer 135:2857–2867. doi:10.1002/ijc.28943
Ma S, Cheng Q, Cai Y et al (2014) IL-17A produced by gammadelta T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res 74:1969–1982. doi:10.1158/0008-5472.CAN-13-2534
Schmidt K, Zilio S, Schmollinger JC, Bronte V, Blankenstein T, Willimsky G (2013) Differently immunogenic cancers in mice induce immature myeloid cells that suppress CTL in vitro but not in vivo following transfer. Blood 121:1740–1748. doi:10.1182/blood-2012-06-436568
Talmadge JE, Gabrilovich DI (2013) History of myeloid-derived suppressor cells. Nat Rev Cancer 13:739–752. doi:10.1038/nrc3581
Li H, Han Y, Guo Q, Zhang M, Cao X (2009) Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells through Membrane-Bound TGF-1. J Immunol 182:240–249. doi:10.4049/jimmunol.182.1.240
Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI (2012) Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol 91:167–181. doi:10.1189/jlb.0311177
Fridlender ZG, Sun J, Mishalian I et al (2012) Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS One 7:e31524. doi:10.1371/journal.pone.0031524
Haverkamp JM, Crist SA, Elzey BD, Cimen C, Ratliff TL (2011) In vivo suppressive function of myeloid-derived suppressor cells is limited to the inflammatory site. Eur J Immunol 41:749–759. doi:10.1002/eji.201041069
Bronte V, Zanovello P (2005) Regulation of immune responses by l-arginine metabolism. Nat Rev Immunol 5:641–654. doi:10.1038/nri1668
Medina-Echeverz J, Ma C, Duffy A, Eggert T, Hawk N, Kleiner DE, Korangy F, Greten TF (2015) Systemic agonistic anti-CD40 treatment of tumor bearing mice modulates hepatic myeloid suppressive cells and causes immune-mediated liver damage. Cancer Immunol Res 3(5):557–566. doi:10.1158/2326-6066.CIR-14-0182
Lu T, Gabrilovich DI (2012) Molecular pathways: tumor-infiltrating myeloid cells and reactive oxygen species in regulation of tumor microenvironment. Clin Cancer Res 18:4877–4882. doi:10.1158/1078-0432.CCR-11-2939
Lalor PF, Adams DH (2002) The liver: a model of organ-specific lymphocyte recruitment. Expert Rev Mol Med 4:1–16. doi:10.1017/S1462399402004155
Majumdar A, Curley SA, Wu X et al (2012) Hepatic stem cells and transforming growth factor beta in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 9:530–538. doi:10.1038/nrgastro.2012.114
Thorn M, Point GR, Burga RA, Nguyen CT, Joseph Espat N, Katz SC (2014) Liver metastases induce reversible hepatic B cell dysfunction mediated by Gr-1+ CD11b+ myeloid cells. J Leukoc Biol 96:883–894. doi:10.1189/jlb.3A0114-012RR
Schneider C, Teufel A, Yevsa T et al (2012) Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut 61:1733–1743. doi:10.1136/gutjnl-2011-301116
Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D (2004) Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol 172:464–474
Ostrand-Rosenberg S, Sinha P (2009) Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 182:4499–4506. doi:10.4049/jimmunol.0802740
Sawanobori Y, Ueha S, Kurachi M et al (2008) Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood 111:5457–5466. doi:10.1182/blood-2008-01-136895
Murdoch C, Muthana M, Coffelt SB, Lewis CE (2008) The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 8:618–631. doi:10.1038/nrc2444
Lim SY, Gordon-Weeks AN, Zhao L et al (2013) Recruitment of myeloid cells to the tumor microenvironment supports liver metastasis. Oncoimmunology 2:e23187. doi:10.4161/onci.23187
Wu D, Wu P, Huang Q, Liu Y, Ye J, Huang J (2013) Interleukin-17: a promoter in colorectal cancer progression. Clin Dev Immunol 2013:436307. doi:10.1155/2013/436307
Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, Schwabe RF, Brenner DA (2009) CCR2 promotes hepatic fibrosis in mice. Hepatology 50:185–197. doi:10.1002/hep.22952
Si Y, Tsou CL, Croft K, Charo IF (2010) CCR2 mediates hematopoietic stem and progenitor cell trafficking to sites of inflammation in mice. J Clin Invest 120:1192–1203. doi:10.1172/JCI40310
Zimmermann HW, Trautwein C, Tacke F (2012) Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol 3:56. doi:10.3389/fphys.2012.00056
Lim SY, Gordon-Weeks A, Allen D, Kersemans V, Beech J, Smart S, Muschel RJ (2015) CD11b myeloid cells support hepatic metastasis through downregulation of Angiopoietin-like 7 in cancer cells. Hepatology. doi:10.1002/hep.27838
Greten TF, Manns MP, Korangy F (2011) Myeloid derived suppressor cells in human diseases. Int Immunopharmacol 11:802–807. doi:10.1016/j.intimp.2011.01.003
Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, Greten TF, Korangy F (2008) A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 135:234–243. doi:10.1053/j.gastro.2008.03.020
Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y (2013) Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res 73:2435–2444. doi:10.1158/0008-5472.CAN-12-3381
Hoechst B, Voigtlaender T, Ormandy L et al (2009) Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 50:799–807. doi:10.1002/hep.23054
Hochst B, Schildberg FA, Sauerborn P et al (2013) Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J Hepatol 59:528–535. doi:10.1016/j.jhep.2013.04.033
Resheq YJ, Li KK, Ward ST et al (2015) Contact-dependent depletion of hydrogen peroxide by catalase is a novel mechanism of myeloid-derived suppressor cell induction operating in human hepatic stellate cells. J Immunol 194:2578–2586. doi:10.4049/jimmunol.1401046
Duffy A, Zhao F, Haile L et al (2013) Comparative analysis of monocytic and granulocytic myeloid-derived suppressor cell subsets in patients with gastrointestinal malignancies. Cancer Immunol Immunother 62:299–307. doi:10.1007/s00262-012-1332-3
Yen BL, Yen ML, Hsu PJ, Liu KJ, Wang CJ, Bai CH, Sytwu HK (2013) Multipotent human mesenchymal stromal cells mediate expansion of myeloid-derived suppressor cells via hepatocyte growth factor/c-met and STAT3. Stem Cell Rep 1:139–151. doi:10.1016/j.stemcr.2013.06.006
Oleinika K, Nibbs RJ, Graham GJ, Fraser AR (2013) Suppression, subversion and escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol 171:36–45. doi:10.1111/j.1365-2249.2012.04657.x
Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY (2007) Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 25:2586–2593. doi:10.1200/JCO.2006.09.4565
Fu J, Xu D, Liu Z et al (2007) Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 132:2328–2339. doi:10.1053/j.gastro.2007.03.102
Yang XH, Yamagiwa S, Ichida T et al (2006) Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J Hepatol 45:254–262. doi:10.1016/j.jhep.2006.01.036
Ma C, Kapanadze T, Gamrekelashvili J, Manns MP, Korangy F, Greten TF (2012) Anti-Gr-1 antibody depletion fails to eliminate hepatic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol 92:1199–1206. doi:10.1189/jlb.0212059
Qin H, Lerman B, Sakamaki I et al (2014) Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med 20:676–681. doi:10.1038/nm.3560
Cao M, Xu Y, Youn JI, Cabrera R, Zhang X, Gabrilovich D, Nelson DR, Liu C (2011) Kinase inhibitor Sorafenib modulates immunosuppressive cell populations in a murine liver cancer model. Lab Invest 91:598–608. doi:10.1038/labinvest.2010.205
Chen Y, Ramjiawan RR, Reiberger T et al (2015) CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 61(5):1591–1602. doi:10.1002/hep.27665
Cheng AL, Kang YK, Lin DY et al (2013) Sunitinib versus sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J Clin Oncol 31:4067–4075. doi:10.1200/JCO.2012.45.8372
Ozao-Choy J, Ma G, Kao J et al (2009) The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res 69:2514–2522. doi:10.1158/0008-5472.CAN-08-4709
Eisenstein S, Coakley BA, Briley-Saebo K et al (2013) Myeloid-derived suppressor cells as a vehicle for tumor-specific oncolytic viral therapy. Cancer Res 73:5003–5015. doi:10.1158/0008-5472.CAN-12-1597
Cheng L, Wang J, Li X, Xing Q, Du P, Su L, Wang S (2011) Interleukin-6 induces Gr-1+ CD11b+ myeloid cells to suppress CD8+ T cell-mediated liver injury in mice. PLoS One 6:e17631. doi:10.1371/journal.pone.0017631
Califano JA, Khan Z, Noonan KA et al (2015) Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res 21:30–38. doi:10.1158/1078-0432.CCR-14-1716
Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH (2012) Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21:822–835. doi:10.1016/j.ccr.2012.04.025
Yamada D, Rizvi S, Razumilava N et al (2015) IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology 61(5):1627–1642. doi:10.1002/hep.27687
MacKinnon AC, Farnworth SL, Hodkinson PS et al (2008) Regulation of alternative macrophage activation by galectin-3. J Immunol 180:2650–2658
Hassan MM, Abdel-Wahab R, Kaseb A et al (2015) Obesity Early in Adulthood Increases Risk but Does Not Affect Outcomes of Hepatocellular Carcinoma. Gastroenterology. doi:10.1053/j.gastro.2015.03.044
Deng ZB, Liu Y, Liu C et al (2009) Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology 50:1412–1420. doi:10.1002/hep.23148
Acknowledgments
We especially thank Joselyn Natasha Allen for artwork design and the NIH Fellows Editorial Board for editorial assistance. This work was supported by the Intramural Research Program of the National Cancer Institute, NIH.
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Medina-Echeverz, J., Eggert, T., Han, M. et al. Hepatic myeloid-derived suppressor cells in cancer. Cancer Immunol Immunother 64, 931–940 (2015). https://doi.org/10.1007/s00262-015-1736-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-015-1736-y