
Abstract
In liver disease, a number of systemic pathogenic factors are responsible for the onset and progression of hepatic encephalopathy. A reduced capacity of the liver to clear gut-derived ammonia leads to an increase in blood ammonia and neurotoxicity. Portal-systemic shunting as well as a reduction in muscle quality and/or muscle mass loss further increase the risk of developing hyperammonemia. Systemic oxidative stress and inflammation together with hyperammonemia can trigger cognitive decline. These factors can cross the blood-brain barrier and affect the brain directly or alter the permeability and signaling across the blood-brain barrier. Long considered a reversible disorder, there is ample amount of evidence demonstrating that repeated episodes of overt HE can prompt neuronal cell injury/death leading to irreversibility. This chapter describes a patient who embarks on the continuum of HE, describing precipitating factors involved in the pathogenesis of cognitive decline.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
Clinical Scenario
Part 1: A 58-year-old male with nonalcoholic steatohepatitis (NASH) cirrhosis presents for routine follow-up. He has no history of decompensation of his liver disease and is currently taking furosemide to control lower limb edema. When questioned, he has no specific complaints. However, his wife reports that for the past 3 months, she has noticed changes in his behavior. She states that he is moody, often wakes up during the night (interrupting her sleep), is forgetful, and has missed a few days at work. In addition, he was recently involved in a minor car accident. She also states that her husband has lost 12 pounds over the last 6 months. Physical examination reveals an alert and oriented patient with normal vital signs, no ascites, or flapping tremor. Body mass index is at 31 kg/m 2 . The physician notices a clear loss of muscle mass since the last visit, including temporal muscle wasting. He has no signs of gastrointestinal bleeding or infection. Blood work reveals moderate thrombocytopenia, normal hemoglobin, white blood cell count and C-reactive protein levels, preserved liver function (INR 1.2 and total bilirubin 1.9 mg/dL), and preserved kidney function. Blood level of ammonia is at 87 μM. The ultrasound reports no focal liver lesion, minimal perihepatic ascites, and a patent portal vein with a large spontaneous splenorenal shunt.
Question 1: How is the concentration of blood ammonia regulated? What are the contributing factors to hyperammonemia in chronic liver disease?
Causes of Hyperammonemia
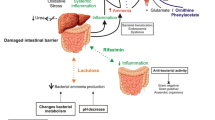
A large amount of ammonia is generated as a by-product in the gut through the degradation of urea, protein digestion, and amino acid deamination. Subsequently, high concentrations of gut-derived ammonia are absorbed into portal venous system and filtered through the liver via the portal vein. The liver plays a vital role in regulating the circulating concentrations of ammonia (extracting 80% of ammonia) with the urea cycle (found within periportal hepatocytes) removing two molecules of ammonia for every molecule of urea produced (a high-capacity, low-affinity system). In addition, perivenous hepatocytes contain glutamine synthetase (GS), an enzyme which removes ammonia via the amination of glutamate to glutamine (a low-capacity, high-affinity system). Therefore, inevitably, a reduction in liver function leads to an increase in blood ammonia which consequently causes alterations in whole-body ammonia metabolism [1]. Alternatively, portal-systemic shunting (portal vein blood bypassing the liver) can also lead to hyperammonemia. In patients with liver disease, portosystemic collateral pathways can become significant following dilatation of preexisting anastomoses between the portal and systemic venous systems. This facilitates shunting of blood away from the liver into the systemic venous system as a means for reducing portal venous pressure (portal hypertension). In addition, hyperammonemia also frequently develops in patients with congenital portal-systemic shunting (even without any signs of liver disease) [2]. If spontaneous portosystemic shunts do not develop and portal hypertension and ascites persist, cirrhotic patients may undergo a transjugular intrahepatic portosystemic shunt (TIPS) which often leads to hyperammonemia [3]. Aside the liver, skeletal muscle, since it expresses GS [4], also has the capacity to eliminate ammonia which becomes important in regulating the circulating concentrations of ammonia during liver failure [5]. However, muscle mass loss as well as increased intramuscular adipose tissue (myosteatosis) leading to poor muscle quality weaken the capacity of muscle to clear ammonia. As a result cirrhotic patients who present with myosteatosis (due to overweight or obesity) and/or malnutrition are at a higher risk of developing hyperammonemia [6, 7].
Following the wife’s observations of her husband changes in behavior, the patient undergoes the EncephalApp Stroop test with a result of 232 s (90 s with Stroop effect “off” and 142 s with Stroop effect “on”), expected result below 162 s for age, gender, and education level (based on American norms). A diagnosis of covert HE is made.
Question 2: How can ammonia explain altered mental status in this patient?
Neurotoxicity of Ammonia
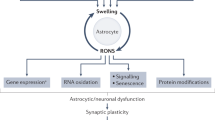
Ammonia is considered an important factor in the pathogenesis of HE as hyperammonemia consequently leads to increased brain ammonia. Ammonia, at physiological pH (7.4), is primarily found (98%) in ionic form (NH4 +), with the remaining 2% as a gas (NH3). Ammonia, as a gas and ion, acts as a weak base and a weak acid, respectively, and therefore ammonia can accept and donate protons which denotes that changes in ammonia cause fluctuations in pH. Ammonia is quite distinct from other weak bases and acids, since NH4 + is very comparable in size and contains similar properties to K+ [8]. This entails that ammonia can cross all cell membranes both as NH3 via diffusion and as NH4 + via K+ channels and transporters [9]. Therefore, due to the latter, increased concentrations of ammonia can cause shifts in cellular resting membrane [10]. Overall, throughout the body, in various organs and cell types, ammonia is produced and consumed in a number of biochemical reactions. Hence, elevated concentrations of ammonia, in addition to influencing pH and membrane potential, can also impact on cellular metabolism [11].
It is well documented that elevated levels of ammonia are toxic to the brain [12]. However, the initial effects of ammonia are not specific to the brain and it has recently been described that elevated ammonia levels can impact the function of other organs [13] (including the muscle) [14]. Nevertheless, the brain is the most sensitive organ to elevated concentrations of ammonia due to the high metabolic rate of this complex organ. A plethora of pathophysiological mechanisms and pathways have been described to underlie the neurotoxic effects of ammonia [9]. Elevated brain ammonia has been shown to disrupt neuron-neuron signaling consequently causing variations in extracellular concentration of neurotransmitters and affecting neurotransmission (glutamatergic, GABAergic, dopaminergic, serotonergic, etc.) [15]. Several lines of evidence demonstrate support for increased GABAergic tone in HE, particularly due to increased synthesis of neurosteroids in the brain which have potent positive allosteric modulatory action on the GABA-A receptor complex [16]. Neurotransmitter transporter and receptor systems have been shown to be affected by elevated ammonia, including changes in membrane expression and initiation of posttranslational protein modifications contributing to disturbed signal transduction pathways [17]. An increase in brain water has been demonstrated to be associated with HE [18] with the plausible cause due to astrocyte swelling [19]. Since the astrocyte is the sole cell within the brain that is capable of removing ammonia since it expresses GS, the accumulation of ammonia-derived glutamine within the astrocyte leading to hypertonicity is believed to be the underlying cause of astrocyte swelling. However, recent evidence suggests that lactate may also play an important role in the onset of astrocyte swelling and brain edema [20, 21]. Independent of the cause, a swollen astrocyte per se can alter astrocyte function precipitating impairment in neuron-astrocyte signaling [22]. It remains controversial on whether breakdown of the blood-brain barrier (BBB) is responsible for the development of brain edema [23]. However, an increase in BBB permeability (independent of breakdown) has been recognized as a contributing factor to the onset of HE.
The patient is prescribed lactulose at 30 mL three times daily, regularly, and is referred to a dietitian for assessment of nutritional status.
Part 2: Two months later, the patient presents to the emergency department with fever, chills, and altered mental status with gross disorientation and slurred speech. Physical examination reveals a temperature of 38.3°C, abundant ascites, and asterixis with no lateralization of brain function on neurological examination. The wife reports that the patient has not been taking lactulose because of abdominal bloating and diarrhea. Blood work shows neutrophilia, no important deterioration of liver function (INR 1.2 and total bilirubin 2.1 mg/dL), and preserved kidney function with hyponatremia (127 mmol/L). The abdominal tap draws turbid liquid with 1800 white blood cells per mm 3 (90% polymorphonuclear cells) and Gram-negative rods. Ammonia is measured at 92 μM.
A diagnosis of spontaneous bacterial peritonitis is made, precipitating overt HE. The patient is admitted to the medical ward. He is treated with intravenous antibiotics, isotonic hydration, albumin and lactulose is cautiously reintroduced.
Question 3: How does systemic inflammation/oxidative stress lead to neurologic impairment in patients with chronic liver disease?
Systemic Inflammation and Oxidative Stress
Systemic inflammation and oxidative stress have been identified as important factors in the onset of HE [24, 25]. Blood inflammatory mediators as well as markers of oxidative stress are often detected in patients with cirrhosis primarily arising from the ailing liver. Liver disease also hastens immune impairment, consequently rendering patients with cirrhosis susceptible to infection. An increase in reactive oxygen species (ROS) production from hepatocyte necrosis together with a reduction in the liver’s capacity to synthesize antioxidants magnifies the induction of systemic oxidative stress. Circulating pro-inflammatory cytokines and ROS can influence the neurological function by impacting the BBB [26, 27]. The BBB, the interface between the blood and brain, is affected by ROS and inflammatory mediators as they can impact the expression of transporters and receptors along the luminal side endothelial cells lining the BBB. In turn, signaling pathways across the BBB are disturbed. Increased levels or accelerated generation of ROS, such as superoxide anion and hydroxyl radical, have been reported in the plasma of patients with liver disease [28]. ROS are highly reactive and can bind to proteins, DNA, RNA, and lipids affecting their function. Pro-inflammatory cytokines can directly pass into the brain via various transport systems and furthermore can bind with their respective receptors expressed along the cerebral endothelium causing alterations in BBB permeability [29, 30].
It has been described that “moderate” elevations of ammonia, systemic inflammation, and oxidative stress independently do not lead to HE. However, the “moderate” levels of these pathogenic factors synergistically can precipitate neurological decline. It has been demonstrated that systemic inflammation exacerbates the neuropsychological effects of hyperammonemia [31] and contributes to advanced HE [32, 33]. Similarly, in both animal and human studies, a synergistic effect of systemic oxidative stress and hyperammonemia has been shown to induce HE [25]. Plasma levels of 3-nitrotyrosine (a marker of oxidative stress) differentiated cirrhotic patients with and without minimal hepatic encephalopathy with similar degrees of hyperammonemia [34]. Bosoi et al. elegantly demonstrated that systemic oxidative stress and hyperammonemia are independent factors and that lowering either systemic oxidative stress or hyperammonemia in BDL rats leads to an attenuation in brain edema [35, 36]. In addition, inducing systemic oxidative stress in hyperammonemic portacaval shunted rats leads to the onset of brain edema [37]. A two-hit hypothesis has been proposed to be involved in the onset of HE and it has been shown that ammonia sensitizes the brain to the effects of systemic inflammation [38]; however whether the order of the pathogenic events and/or the preconditioning of a pathogenic factor has any important significance remains to be determined.
Markers of neuroinflammation and cerebral oxidative stress have also been demonstrated to be associated with HE and it is speculated that these factors may be related to HE severity. In vitro studies (cultured astrocytes) have demonstrated that high concentrations of ammonia (5 mM) can induce oxidative stress [39]. However, in hyperammonemic cirrhotic rats with MHE, no signs of oxidative stress in the brain [36] were observed, suggesting that lower blood ammonia levels (between 100 and 250 μM) do not stimulate cerebral oxidative stress. Furthermore, oxidative modifications of cerebral proteins and RNA as well as microglial activation were found in postmortem brain tissue from cirrhotic patients who died with overt HE [40, 41]. Nevertheless, cirrhotic rats with MHE show evidence of neuroinflammation [42] and therefore it is speculated that nitrosative and oxidative modifications (proteins, RNA, DNA, and lipids) may be the triggering factor in the development of overt HE.
Six days later, the infection is resolved and the patient is back to his normal functioning. He is discharged home with lactulose and instructed to take the dose that will produce two to three bowel movements in a day, and ciprofloxacin as secondary prophylaxis of spontaneous bacterial peritonitis.
Part 3: Three months later, the patient consults again at the emergency department for diarrhea that has been present for 4 days, along with fever and altered mental status. The patient has been observant to his lactulose prescription, except for the previous 24 h because of the new-onset diarrhea. Physical examination discloses hyperthermia (38.2°C), slurred speech, no gross disorientation, and flapping tremor. Abdominal tenderness is present, but with no obvious ascites.
Blood work reveals neutrophilia, deterioration of liver function (total bilirubin at 4.7 mg/dL and INR at 1.6), and kidney function. C-reactive protein is elevated at 42 mg/L and blood ammonia at 72 μM. A stool sample is positive for the presence of Clostridium difficile antigen.
A diagnosis of Clostridium difficile colitis is made. The patient is admitted to the medical ward and treated with oral vancomycin and intravenous hydration. Ciprofloxacin prophylaxis is stopped.” Eight days later, after improvement in his neurological status and clinical resolution of the infection, he is discharged home with long-term treatment with lactulose and rifaximin. Outpatient evaluation for liver transplantation is planned because of the decline in liver function.
Question 4: Is hepatic encephalopathy fully reversible or do repeated episodes of overt HE lead to permanent sequelae?
Impact of Recurrent HE
HE is defined as a metabolic disorder and therefore the brain is expected to fully recover following liver transplantation (LT). However, even following the implantation of a new liver, persisting neurological complications remain a problem affecting 8–47% of liver-transplant recipients [500,501,502,503,504,48]. Retrospective studies documented that cirrhotic patients with a history of existing bouts of overt HE were associated with impaired neurological resolution, with an increased risk of mortality and morbidity following LT [49]. In addition, pretransplant severe HE results in prolonged hospital stays post-LT which renders patients more vulnerable to nosocomial infections [49]. Persistent cognitive impairment related to learning, tested using the psychometric hepatic encephalopathy score (PHES), inhibitory control test (ICT), and EncephalApp Stroop test, has been demonstrated to occur in cirrhotic patients with preexisting bouts of OHE [46,47,52]. As a result, these patients are difficult to treat as they respond poorly to adequate medical therapy [53]. There is increasing evidence that recurrent HE leads to permanent cell damage. Cerebral cortical and white matter lesions have been demonstrated in patients with chronic HE [54]. Furthermore, cirrhotic patients with preexisting HE are associated with reduction in brain volume post-LT indicating signs of neuronal loss [44, 55]. Recently, an animal model of recurrent HE was characterized where episodes of overt HE were induced in portacaval shunted rats. Modulation of neurodegeneration-related genes as well as existence of neuronal cell loss in cerebellum [56] were observed to occur following multiple episodes of overt HE. Interestingly, increased mRNA expression of senescence-associated genes was found in postmortem brain samples from cirrhotic patients who died with OHE, an indication of cell cycle arrest and cell loss [57]. In addition, persistent altered mental status post-LT may lead to a reduced dose of immunosuppression (calcineurin inhibitors) and therefore may increase the risk of rejection [58]. However, in addition to preexisting episodes of OHE possibly leading to permanent cell damage or injury, it has also been demonstrated that the strongest predictor of postoperative neurological morbidity is the presence of HE at the time of LT [43]. It is understood that the invasiveness of LT can lead to perioperative conditions that include various degrees and prolongation of anesthesia, blood loss, hypotension, and ischemia. Therefore, a compromised “frail” HE brain becomes predisposed to what would normally be an innocuous perioperative insult resulting in cell injury and death.
Conclusion
This clinical case described the importance of the interplay between hyperammonemia, systemic oxidative stress, and inflammation and their role in the development and advancement of HE. These pathogenic factors can impinge in the BBB as well as cause alterations in metabolism, cell-cell communication, and neurotransmission and lead to cognitive dysfunction. The risk of hyperammonemia rises depending on the degree of liver impairment, portal-systemic shunting, kidney function, and muscle mass quantity/quality. Liver disease increases the susceptibility of infection which can aggravate the neurotoxic effects of ammonia. It is recognized that repeated episodes of overt HE can lead to a poor prognosis, survival, as well as poor neurological outcome following LT. The latter implies that a history of HE precipitates irreversible cell injury and death. In addition, the impact of HE at the time of LT should not be neglected as a “frail” HE brain may be susceptible to minor perioperative insults occurring during surgery. While the effects of ammonia, oxidative stress, and inflammation on the brain still remain unresolved, the pathophysiological pathways underlying the continuum of HE (covert to overt to recurrent to irreversible) merit to be thoroughly investigated.
References
Olde Damink SWM, Deutz NEP, Dejong CHC, et al. Interorgan ammonia metabolism in liver failure. Neurochem Int. 2002;41:177–88.
Sokollik C, Bandsma RHJ, Gana JC, et al. Congenital portosystemic shunt: characterization of a multisystem disease. J Pediatr Gastroenterol Nutr. 2013;56:675–81. https://doi.org/10.1097/MPG.0b013e31828b3750.
Pereira K, Carrion AF, Martin P, et al. Current diagnosis and management of post-transjugular intrahepatic portosystemic shunt refractory hepatic encephalopathy. Liver Int. 2015;35:2487–94. https://doi.org/10.1111/liv.12956.
He Y, Hakvoort TBM, Vermeulen JLM, et al. Glutamine synthetase deficiency in murine astrocytes results in neonatal death. Glia. 2010;58:741–54. https://doi.org/10.1002/glia.20960.
Chatauret N, Desjardins P, Zwingmann C, et al. Direct molecular and spectroscopic evidence for increased ammonia removal capacity of skeletal muscle in acute liver failure. J Hepatol. 2006;44:1083–8. https://doi.org/10.1016/j.jhep.2005.11.048.
Merli M, Giusto M, Lucidi C, et al. Muscle depletion increases the risk of overt and minimal hepatic encephalopathy: results of a prospective study. Metab Brain Dis. 2013;28:281–4. https://doi.org/10.1007/s11011-012-9365-z.
Montano-Loza AJ, Angulo P, Meza-Junco J, et al. Sarcopenic obesity and myosteatosis are associated with higher mortality in patients with cirrhosis. J Cachexia Sarcopenia Muscle. 2016;7:126–35. https://doi.org/10.1002/jcsm.12039.
Nagaraja TN, Brookes N. Intracellular acidification induced by passive and active transport of ammonium ions in astrocytes. Am J Phys. 1998;274:C883–91.
Bosoi CR, Rose CF. Identifying the direct effects of ammonia on the brain. Metab Brain Dis. 2009;24:95–102. https://doi.org/10.1007/s11011-008-9112-7.
Allert N, Köller H, Siebler M. Ammonia-induced depolarization of cultured rat cortical astrocytes. Brain Res. 1998;782:261–70.
Cooper AJ, Plum F. Biochemistry and physiology of brain ammonia. Physiol Rev. 1987;67:440–519.
Felipo V, Butterworth RF. Neurobiology of ammonia. Prog Neurobiol. 2002;67:259–79. https://doi.org/10.1016/S0301-0082(02)00019-9.
Sawhney R, Holland-Fischer P, Rosselli M, et al. Role of ammonia, inflammation, and cerebral oxygenation in brain dysfunction of acute-on-chronic liver failure patients. Liver Transpl. 2016;22:732–42. https://doi.org/10.1002/lt.24443.
Davuluri G, Allawy A, Thapaliya S, et al. Hyperammonaemia-induced skeletal muscle mitochondrial dysfunction results in cataplerosis and oxidative stress. J Physiol. 2016;594:7341–60. https://doi.org/10.1113/JP272796.
Felipo V. Hepatic encephalopathy: effects of liver failure on brain function. Nat Rev Neurosci. 2013;14:851–8. https://doi.org/10.1038/nrn3587.
Butterworth RF. Neurosteroids in hepatic encephalopathy: novel insights and new therapeutic opportunities. J Steroid Biochem Mol Biol. 2016;160:94–7. https://doi.org/10.1016/j.jsbmb.2015.11.006.
Görg B, Morwinsky A, Keitel V, et al. Ammonia triggers exocytotic release of L-glutamate from cultured rat astrocytes. Glia. 2010a;58:691–705. https://doi.org/10.1002/glia.20955.
Shah NJ, Neeb H, Kircheis G, et al. Quantitative cerebral water content mapping in hepatic encephalopathy. NeuroImage. 2008;41:706–17. https://doi.org/10.1016/j.neuroimage.2008.02.057.
Wright G, Soper R, Brooks HF, et al. Role of aquaporin-4 in the development of brain oedema in liver failure. J Hepatol. 2010;53:91–7. https://doi.org/10.1016/j.jhep.2010.02.020.
Bosoi CR, Zwingmann C, Marin H, et al. Increased brain lactate is central to the development of brain edema in rats with chronic liver disease. J Hepatol. 2014b;60:554–60. https://doi.org/10.1016/j.jhep.2013.10.011.
Hadjihambi A, De Chiara F, Hosford PS, et al. Ammonia mediates cortical hemichannel dysfunction in rodent models of chronic liver disease. Hepatology. 2017;65:1306–18. https://doi.org/10.1002/hep.29031.
Sofroniew MV. Astrocyte failure as a cause of CNS dysfunction. Mol Psychiatry. 2000;5:230–2.
Bosoi CR, Rose CF. Brain edema in acute liver failure and chronic liver disease: similarities and differences. Neurochem Int. 2013a;62:446–57. https://doi.org/10.1016/j.neuint.2013.01.015.
Aldridge DR, Tranah EJ, Shawcross DL. Pathogenesis of hepatic encephalopathy: role of ammonia and systemic inflammation. J Clin Exp Hepatol. 2015;5:S7–S20. https://doi.org/10.1016/j.jceh.2014.06.004.
Gimenez-Garzó C, Urios A, Agustí A, et al. Is cognitive impairment in cirrhotic patients due to increased peroxynitrite and oxidative stress? Antioxid Redox Signal. 2015;22:871–7. https://doi.org/10.1089/ars.2014.6240.
Bosoi CR, Rose CF. Oxidative stress: a systemic factor implicated in the pathogenesis of hepatic encephalopathy. Metab Brain Dis. 2013b;28:175–8. https://doi.org/10.1007/s11011-012-9351-5.
Coltart I, Tranah TH, Shawcross DL. Inflammation and hepatic encephalopathy. Arch Biochem Biophys. 2013;536:189–96. https://doi.org/10.1016/j.abb.2013.03.016.
Chen MF, Mo LR, Lin RC, et al. Increase of resting levels of superoxide anion in the whole blood of patients with decompensated liver cirrhosis. Free Radic Biol Med. 1997;23:672–9. https://doi.org/10.1016/S0891-5849(97)00057-9.
de Vries HE, Blom-Roosemalen MC, van Oosten M, et al. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol. 1996;64:37–43.
Didier N, Romero IA, Créminon C, et al. Secretion of interleukin-1beta by astrocytes mediates endothelin-1 and tumour necrosis factor-alpha effects on human brain microvascular endothelial cell permeability. J Neurochem. 2003;86:246–54.
Shawcross DL, Davies NA, Williams R, Jalan R. Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J Hepatol. 2004;40:247–54. https://doi.org/10.1016/j.jhep.2003.10.016.
Odeh M, Sabo E, Srugo I, Oliven A. Relationship between tumor necrosis factor-alpha and ammonia in patients with hepatic encephalopathy due to chronic liver failure. Ann Med. 2005;37:603–12. https://doi.org/10.1080/07853890500317414.
Shawcross DL, Sharifi Y, Canavan JB, et al. Infection and systemic inflammation, not ammonia, are associated with Grade 3/4 hepatic encephalopathy, but not mortality in cirrhosis. J Hepatol. 2011;54:640–9.
Montoliu C, Cauli O, Urios A, et al. 3-Nitro-tyrosine as a peripheral biomarker of minimal hepatic encephalopathy in patients with liver cirrhosis. Am J Gastroenterol. 2011;106:1629–37. https://doi.org/10.1038/ajg.2011.123.
Bosoi CR, Parent-Robitaille C, Anderson K, et al. AST-120 (spherical carbon adsorbent) lowers ammonia levels and attenuates brain edema in bile-duct ligated rats. Hepatology. 2011;53:1995–2002. https://doi.org/10.1002/hep.24273.
Bosoi CR, Yang X, Huynh J, et al. Systemic oxidative stress is implicated in the pathogenesis of brain edema in rats with chronic liver failure. Free Radic Biol Med. 2012;52:1228–35. https://doi.org/10.1016/j.freeradbiomed.2012.01.006.
Bosoi CR, Tremblay M, Rose CF. Induction of systemic oxidative stress leads to brain oedema in portacaval shunted rats. Liver Int. 2014a;34:1322–9. https://doi.org/10.1111/liv.12414.
Marini JC, Broussard SR. Hyperammonemia increases sensitivity to LPS. Mol Genet Metab. 2006;88:131–7. https://doi.org/10.1016/j.ymgme.2005.12.013.
Rama Rao KV, Jayakumar AR, Norenberg MD. Role of oxidative stress in the ammonia-induced mitochondrial permeability transition in cultured astrocytes. Neurochem Int. 2005;47:31–8. https://doi.org/10.1016/j.neuint.2005.04.004.
Görg B, Qvartskhava N, Bidmon H-J, et al. Oxidative stress markers in the brain of patients with cirrhosis and hepatic encephalopathy. Hepatology. 2010b;52:256–65. https://doi.org/10.1002/hep.23656.
Zemtsova I, Görg B, Keitel V, et al. Microglia activation in hepatic encephalopathy in rats and humans. Hepatology. 2011;54:204–15. https://doi.org/10.1002/hep.24326.
Rodrigo R, Cauli O, Gomez-Pinedo U, et al. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology. 2010;139:675–84. https://doi.org/10.1053/j.gastro.2010.03.040.
Planas R, Ballesté B, Alvarez MA, et al. Natural history of decompensated hepatitis C virus-related cirrhosis. A study of 200 patients. J Hepatol. 2004;40:823–30. https://doi.org/10.1016/j.jhep.2004.01.005.
Dhar R, Young GB, Marotta P. Perioperative neurological complications after liver transplantation are best predicted by pre-transplant hepatic encephalopathy. Neurocrit Care. 2008;8:253–8. https://doi.org/10.1007/s12028-007-9020-4.
Sotil EU, Gottstein J, Ayala E, et al. Impact of preoperative overt hepatic encephalopathy on neurocognitive function after liver transplantation. Liver Transplant. 2009;15:184–92. https://doi.org/10.1002/lt.21593.
Campagna F, Biancardi A, Cillo U, et al. Neurocognitive-neurological complications of liver transplantation: a review. Metab Brain Dis. 2010;25:115–24. https://doi.org/10.1007/s11011-010-9183-0.
Garcia-Martinez R, Rovira A, Alonso J, et al. Hepatic encephalopathy is associated with posttransplant cognitive function and brain volume. Liver Transpl. 2011;17:38–46. https://doi.org/10.1002/lt.22197.
Tryc AB, Pflugrad H, Goldbecker A, et al. New-onset cognitive dysfunction impairs the quality of life in patients after liver transplantation. Liver Transpl. 2014;20:807–14. https://doi.org/10.1002/lt.23887.
Brandman D, Biggins SW, Hameed B, et al. Pretransplant severe hepatic encephalopathy, peritransplant sodium and post-liver transplantation morbidity and mortality. Liver Int. 2012;32:158–64. https://doi.org/10.1111/j.1478-3231.2011.02618.x.
Acharya C, Wade JB, Fagan A, et al. Overt hepatic encephalopathy impairs learning on the EncephalApp stroop which is reversible after liver transplantation. Liver Transpl. 2017;23:1396–403. https://doi.org/10.1002/lt.24864.
Bajaj JS, Schubert CM, Heuman DM, et al. Persistence of cognitive impairment after resolution of overt hepatic encephalopathy. Gastroenterology. 2010;138:2332–40. https://doi.org/10.1053/j.gastro.2010.02.015.
Riggio O, Ridola L, Pasquale C, et al. Evidence of persistent cognitive impairment after resolution of overt hepatic encephalopathy. Clin Gastroenterol Hepatol. 2011;9:181–3. https://doi.org/10.1016/j.cgh.2010.10.002.
Nardelli S, Allampati S, Riggio O, et al. Hepatic encephalopathy is associated with persistent learning impairments despite adequate medical treatment: a multicenter, international study. Dig Dis Sci. 2017;62:794–800. https://doi.org/10.1007/s10620-016-4425-6.
Matsusue E, Kinoshita T, Ohama E, Ogawa T. Cerebral cortical and white matter lesions in chronic hepatic encephalopathy: MR-pathologic correlations. Am J Neuroradiol. 2005;26:347–51.
Zeneroli ML, Cioni G, Vezzelli C, et al. Prevalence of brain atrophy in liver cirrhosis patients with chronic persistent encephalopathy: evaluation by computed tomography. J Hepatol. 1987;4:283–92.
García-Lezana T, Oria M, Romero-Giménez J, et al. Cerebellar neurodegeneration in a new rat model of episodic hepatic encephalopathy. J Cereb Blood Flow Metab. 2017;37:927–37. https://doi.org/10.1177/0271678X16649196.
Görg B, Karababa A, Shafigullina A, et al. Ammonia-induced senescence in cultured rat astrocytes and in human cerebral cortex in hepatic encephalopathy. Glia. 2015;63:37–50. https://doi.org/10.1002/glia.22731.
Senzolo M, Marco S, Ferronato C, et al. Neurologic complications after solid organ transplantation. Transpl Int. 2009;22:269–78. https://doi.org/10.1111/j.1432-2277.2008.00780.x.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Bissonnette, J., Rose, C.F. (2018). Hepatic Encephalopathy: Pathophysiology—Brain. In: Bajaj, J. (eds) Diagnosis and Management of Hepatic Encephalopathy. Springer, Cham. https://doi.org/10.1007/978-3-319-76798-7_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-76798-7_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-76797-0
Online ISBN: 978-3-319-76798-7
eBook Packages: MedicineMedicine (R0)