
Abstract
Although ammonia is considered the main factor involved in the pathogenesis of hepatic encephalopathy (HE), it correlates well with the severity of HE in acute liver failure, but not in chronic liver disease. Oxidative stress is another factor believed to play a role in the pathogenesis of this syndrome; it represents an imbalance between the production and neutralization of reactive oxygen species, which leads to cellular dysfunction. In the setting of liver disease, oxidative stress represents a systemic phenomenon induced by several mechanisms: decreased antioxidant synthesis, increased systemic release of oxidant enzymes, generation of reactive oxygen species, and impaired neutrophil function. High ammonia concentrations induce cerebral oxidative stress, thus contributing to severe hepatic encephalopathy, as observed in acute liver failure. In chronic liver disease, significantly lower degrees of hyperammonemia (<500 μM) do not induce cerebral nor systemic oxidative stress. Data from both animal and human studies sustain that there is a synergistic effect between systemic oxidative stress, and ammonia that is implicated in the pathogenesis of hepatic encephalopathy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Oxidative stress
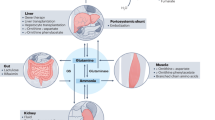
Oxidative stress occurs when the balance between the production of free radicals and their neutralization is tipped, thus favoring the generation of reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), hydroxyl radical (.OH), superoxide ion (O2 .-) and peroxynitrite (ONOO-). These reactive oxygen intermediates, which play important roles in cell signaling (Valko et al. 2007), are highly reactive due to the presence of unpaired valence shell electrons and are constantly produced during oxygen metabolism. However, in excess, ROS are very harmful to the cell, for their reaction with cellular structures and macromolecules leads to cellular dysfunction (Fig. 1).

Schematic representation of the effects of reactive oxygen species leading to cellular dysfunction: lipid oxidation leads to alterations in membrane permeability, protein oxidation affects the function of enzymes, transporters and receptors and nucleic acids oxidation alters protein synthesis, thus affecting cellular metabolism
Proteins, formed of polypeptides comprising various amino acids with several chemical groups, such as amino, carboxyl, hydroxyl or sulfhydryl, are vulnerable to oxidation and thus easily fall victim to ROS. Oxidative post-translational modifications such as carboxylation, nitration or S-nitrosylation lead to inflicting consequences, such as fragmentation of the polypeptide chain and generation of protein-protein cross linkages. As a result, functional alterations of enzymes, transporters and receptors arise (Stadtman and Levine 2000). ROS also target lipids, especially polyunsaturated fatty acids containing methylene (-CH2-) groups that can donate electrons to ROS, which results in lipid peroxidation and aldehyde formation (4-hydroxy-2-noneal and malondialdehyde). These, in turn, can bind several proteins at the time, forming abnormal protein-protein complexes. Furthermore, with lipids being the main component of plasma membranes, lipid peroxidation leads to alterations in membrane permeability (Poon et al. 2004). In addition, nucleic acids including DNA and RNA are also prone to oxidation, with guanine being the most sensitive, since it has a higher oxidation potential than cytosine, thymine and adenine. For this reason, 8-oxo-2′deoxyguanosine is recognized as an important marker of oxidative stress (Cooke et al. 2003; Görg et al. 2008). In any case, the interaction of ROS with nucleic acids consequently leads to impaired gene translation and abnormal protein synthesis.
Found in several organs, upregulation and activation of oxidant enzymes, such as NADPH-, xanthine-, monoamine-, and aldehyde-oxidase or nitric oxide synthetase, are implicated in various diseases, as their activation stresses and overwhelms the intrinsic antioxidant defense system, which results in an accumulation of ROS. Antioxidant enzymes such as superoxide dismutase (SOD) and catalase (CAT), or ROS neutralizing molecules, such as glutathione and albumin, are important oxidant defense mechanisms, and are present in most organs. As such, a decrease in antioxidant defense, for example decreased glutathione synthesis, accompanies several diseases (Valko et al. 2007), resulting in ROS-induced cellular damage.
Sources of oxidative stress in cirrhosis
Because it is a major producer of antioxidants, the liver is an important organ in oxidative stress regulation. During the setting of liver disease, liver protein synthesis is decreased, thus diminishing the antioxidant capacity. Albumin, which constitutes close to 50 % of serum protein, is synthesized in the liver and significantly decreases during chronic liver disease (CLD) (Chen et al. 1997). Albumin, an important transporter for several molecules and a regulator of plasma osmotic pressure, is also a powerful antioxidant, due to its cysteine and methionine residues (Roche et al. 2008). In addition to albumin, glutathione is also synthesized by the liver. Both enzymes in the glutathione synthesis pathway, glutamate-cysteine ligase and glutathione synthetase, have been shown to decrease in later stages in the livers of bile duct-ligated mice, which is accompanied by a decrease in plasmatic glutathione as well (Yang et al. 2009). N-acetylcysteine, a glutathione precursor, shows extraordinary beneficial effects in acute liver failure, especially if administered during early stages when functional hepatocytes are still present (Lee et al. 2009). N-acetylcysteine has little effect in CLD, probably due to impaired hepatic glutathione synthesis.
Hepatocyte necrosis is associated with an increase in ROS and the systemic release of oxidant enzymes (Kmieć 2001). Liver-derived oxidant xanthine oxidase intervenes in the purine metabolism by oxidizing hypoxanthine to xanthine, and xanthine to uric acid, reactions which lead to H2O2 release. Xanthine oxidase activity in plasma has been demonstrated to be increased in cirrhotic patients (Battelli et al. 2001). Therefore, inhibiting this enzyme with the hypoxanthine analog allopurinol significantly attenuates plasmatic oxidative stress (Spahr et al. 2007). Another important oxidant enzyme, aldehyde oxidase, which converts aldehyde into carboxylic acids and H2O2, further contributes to an increase of ROS in cirrhotic patients, especially those afflicted with alcoholic-induced cirrhosis (Shaw and Jayatilleke 1990).
Moreover, the ailing liver can trigger other oxidative mechanisms, such as neutrophil dysfunction. Neutrophils are immune cells that eliminate microbes through phagocytosis. During this procedure, neutrophils produce ROS in response to microbial aggression, a process termed oxidative burst. The neutrophils of patients with cirrhosis presented increased oxidative burst, indicating neutrophil activation, along with reduced phagocytic capacity (Mookerjee et al. 2007).
All the above-mentioned mechanisms contribute to systemic oxidative stress in cirrhosis. As circulating levels of superoxide are significantly increased in decompensated versus compensated cirrhotic patients (Chen et al. 1997), in cirrhosis oxidative stress represents a systemic phenomenon associated with the severity of the liver disease.
Systemic oxidative stress in hepatic encephalopathy
Hepatic encephalopathy (HE) is a complex neuropsychiatric syndrome that defines the spectrum of cerebral complications associated with both acute liver failure and CLD. Ammonia is considered to be an important factor involved in the pathogenesis of HE, and has been shown to correlate well with intracranial hypertension and brain stem herniation in acute liver failure (Clemmesen et al. 1999). However, in the setting of CLD, the correlation between ammonia and severity of HE remains inconclusive (Ong et al. 2003; Kundra et al. 2005), suggesting that other pathogenic factors may be implicated.
Data from the literature suggests that the relationship between ammonia and oxidative stress in the pathogenesis of HE is complex. In rats with acute liver failure (induced by hepatic devascularization), the development of brain edema and severe HE (coma stage) occurs in association with hyperammonemia, as well as high ammonia levels in the brain (approx. 800 μM), together with systemic and central oxidative stress (Jiang et al. 2009). However, in the setting of chronic hyperammonemia, portacaval shunted (PCA) rats demonstrate significantly lower levels of blood and brain ammonia (approx. 200 μM), which are neither associated with systemic and cerebral oxidative stress, or with brain edema (Bosoi et al. 2012). Using cultured astrocytes, it has been shown that ammonia toxicity, at concentrations >500 μM, leads to the generation of ROS. Ammonia concentrations, 100 and 200 μM, did not induce oxidative stress (Görg et al. 2008; Mehrotra and Trigun 2012). Combined, these results suggest that a threshold of ammonia is needed in order to induce oxidative stress in the brain.
Systemically, the relationship between hyperammonemia and oxidative stress differs from that depicted in the brain. Our group has observed the presence of systemic oxidative stress in cirrhotic rats afflicted with hyperammonemia, minimal HE and brain edema. No signs, however, of oxidative stress were found in the brain. In the same cirrhotic rat model, we pursued to demonstrate that systemic oxidative stress and blood ammonia were independent factors, since bile duct-ligated (BDL) rats that had been treated with the oral carbon microspheres, AST-120, showed an attenuation of blood ammonia and brain edema, although systemic oxidative stress remained present (Bosoi et al. 2011). Furthermore, we observed an increase in the activity of xanthine oxidase in plasma which, following treatment with the xanthine oxidase inhibitor allopurinol, lead to an attenuation in systemic oxidative stress and brain edema (Bosoi et al. 2012). These results, together with the non-existence of oxidative stress in hyperammonemic PCA rats, suggest that systemic oxidative stress is not induced by elevated blood ammonia levels but instead arises from the ailing liver in CLD. Taken togeher, the results stated above along with similar degrees of hyperammonemia existing in PCA and BDL rats, suggest that systemic oxidative stress and hyperammonemia, two independent pathogenic factors (as observed in BDL rats), are synergistically implicated in the pathogenesis of brain edema in CLD.
These results are supported through a recent study conducted in Spain, in which Dr. Felipo’s group described, in a cohort of 212 patients (63 controls, 87 cirrhotics without minimal HE, 62 with minimal HE), that systemic oxidative stress is the pathogenic factor dissociating cirrhotic patients with minimal HE from those without minimal HE (Montoliu et al. 2011). In this study, in which the degree of hyperammonemia was similar in cirrhotic patients with or without minimal HE (152 ± 9 vs 139 ± 7 μM respectively), only cirrhotic patients with minimal HE demonstrated an 8.6 fold increase in the oxidative stress marker 3-nitrotyrosine in the plasma. This fact established that serum 3-nitro-tyrosine is a good marker in identifying the presence of minimal HE in patients with CLD. The results of this clinical study confirm the findings in animal models, and sustain the existence of a synergistic effect of systemic oxidative stress and ammonia in the pathogenesis of HE.
Conclusions
Systemic oxidative stress is an entity associated with liver disease. An imbalance between increased production and decreased antioxidant defence is a direct consequence of liver dysfunction. High ammonia levels (≥500 μM) induce cerebral oxidative stress, which is primarily associated with severe HE, while inferior concentrations of brain ammonia, <500 μM, do not induce cerebral oxidative stress. However, the latter in combination with systemic oxidative stress contributes to the progression of HE suggesting that a synergistic effect between these two factors is implicated in the pathogenesis of minimal HE in CLD (Fig. 2).

A synergistic effect between ammonia and systemic oxidative stress is proposed in the pathogenesis of brain edema in hepatic encephalopathy. Ammonia concentrations <500 μM in the brain do not induce cerebral oxidative stress, and lead to minimal hepatic encephalopathy. Ammonia concentrations ≥ 500 μM provoke oxidative stress in the brain, causing severe hepatic encephalopathy
References
Battelli MG, Musiani S, Valgimigli M, Gramantieri L, Tomassoni F, Bolondi L, Stirpe F (2001) Serum xanthine oxidase in human liver disease. Am J Gastroenterol 96:1194–1199
Bosoi CR, Parent-Robitaille C, Anderson K, Tremblay M, Rose CF (2011) AST-120 (spherical carbon adsorbent) lowers ammonia levels and attenuates brain edema in bile duct-ligated rats. Hepatology 53:1995–2002
Bosoi CR, Yang X, Huynh J, Parent-Robitaille C, Jiang W, Tremblay M, Rose CF (2012) Systemic oxidative stress is implicated in the pathogenesis of brain edema in rats with chronic liver failure. Free Radic Biol Med 52:1228–1235
Chen M-F, Mo L-R, Lin R-C, Kuo J-Y, Chang K-K, Liao C, Lu F-J (1997) Increase of resting levels of superoxide anion in the whole blood of patients with decompensated liver cirrhosis. Free Radic Biol Med 23:672–679
Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P (1999) Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 29:648–653
Cooke MS, Evans MD, Dizdaroglu M, Lunec J (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17:1195–1214
Görg B, Qvartskhava N, Keitel V, Bidmon HJ, Selbach O, Schliess F, Häussinger D (2008) Ammonia induces RNA oxidation in cultured astrocytes and brain in vivo. Hepatology 48:567–579
Jiang W, Desjardins P, Butterworth RF (2009) Hypothermia attenuates oxidative/nitrosative stress, encephalopathy and brain edema in acute (ischemic) liver failure. Neurochem Int 55:124–128
Kmieć Z (2001) Cooperation of liver cells in health and disease. Adv Anat Embryol Cell Biol 161, III–XIII, 1–151
Kundra A, Jain A, Banga A, Bajaj G, Kar P (2005) Evaluation of plasma ammonia levels in patients with acute liver failure and chronic liver disease and its correlation with the severity of hepatic encephalopathy and clinical features of raised intracranial tension. Clin Biochem 38:696–699
Lee WM, Hynan LS, Rossaro L, Fontana RJ, Stravitz RT, Larson AM, Davern TJ 2nd, Murray NG, McCashland T, Reisch JS, Robuck PR (2009) Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 137:856–864, 864.e1
Mehrotra A, Trigun SK (2012) Moderate grade hyperammonemia induced concordant activation of antioxidant enzymes is associated with prevention of oxidative stress in the brain slices. Neurochem Res 37:171–181
Montoliu C, Cauli O, Urios A, ElMlili N, Serra MA, Giner-Duran R, González-Lopez O, Del Olmo JA, Wassel A, Rodrigo JM, Felipo V (2011) 3-nitro-tyrosine as a peripheral biomarker of minimal hepatic encephalopathy in patients with liver cirrhosis. Am J Gastroenterol 106:1629–1637
Mookerjee RP, Stadlbauer V, Lidder S, Wright GAK, Hodges SJ, Davies NA, Jalan R (2007) Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology 46:831–840
Ong JP, Aggarwal A, Krieger D, Easley KA, Karafa MT, Van Lente F, Arroliga AC, Mullen KD (2003) Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med 114:188–193
Poon HF, Calabrese V, Scapagnini G, Butterfield DA (2004) Free radicals and brain aging. Clin Geriatr Med 20:329–359
Roche M, Rondeau P, Singh NR, Tarnus E, Bourdon E (2008) The antioxidant properties of serum albumin. FEBS Lett 582:1783–1787
Shaw S, Jayatilleke E (1990) The role of aldehyde oxidase in ethanol-induced hepatic lipid peroxidation in the rat. Biochem J 268:579–583
Spahr L, Bresson-Hadni S, Amann P, Kern I, Golaz O, Frossard J-L, Hadengue A (2007) Allopurinol, oxidative stress and intestinal permeability in patients with cirrhosis: an open-label pilot study. Liver Int 27:54–60
Stadtman ER, Levine RL (2000) Protein oxidation. Ann N Y Acad Sci 899:191–208
Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39:44–84
Yang H, Ramani K, Xia M, Ko KS, Li TWH, Oh P, Li J, Lu SC (2009) Dysregulation of glutathione synthesis during cholestasis in mice: molecular mechanisms and therapeutic implications. Hepatology 49:1982–1991
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bosoi, C.R., Rose, C.F. Oxidative stress: a systemic factor implicated in the pathogenesis of hepatic encephalopathy. Metab Brain Dis 28, 175–178 (2013). https://doi.org/10.1007/s11011-012-9351-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-012-9351-5