
Abstract
The obesity epidemic has highlighted the need to reassess and fundamentally rethink the pathophysiology of obesity-related hypertension and the interplay between adipocytes, the vasculature and blood glucose homeostasis. In reality, the components of the metabolic syndrome do not exist in isolation, rather in an intricate balance which is ultimately disturbed in disease states. In this chapter, we review the role of adipocytes and the perivascular adipose tissue in health and disease in relation to obesity, hypertension and diabetes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Adipocytes
- Adiponectin
- Hypertension
- Inflammation
- Obesity
- Diabetes
- Oxidative stress
- Perivascular adipose tissue
- Sympathetic nervous system
1 The Clinical Problem
Obesity has more than doubled since 1980, with nearly 2 billion overweight (39%) and 600 million obese (13%) adults worldwide [1]. In England nearly a quarter and in the USA close to a third of all adults are obese [2, 3].
Our contemporary understanding of obesity focuses on our ancestors and the fact that fat was the energy store developed in times of plenty which could then be utilised during times of famine. This theory suggests that genes predisposing to obesity would confer survival benefits and such individuals would live long enough to reproduce which is often seen as nature’s ultimate aim. Whilst adaptive variations in weight in the animal kingdom continue to serve their pro-survival purpose, we are observing a worrying trend in weight gain often associated with diseases that shorten the lifespan of the affected individual or at the very least don’t confer any survival benefits. Susceptible individuals are no longer exposed to periods of famine and are instead able to access easily high energy foods without expending much energy [4, 5]. Hunting and gathering food has been replaced by as little effort as a simple click of a button on the computer or a short drive to the nearest supermarket. In the hibernating mammal, short-term obesity and insulin resistance help direct glucose to the brain to keep the animal alive; however obesity has inflicted the modern man with chronic illnesses with significant associated cardiovascular morbidity and mortality, often requiring treatments such as polypharmacy or surgery in the extreme cases [6].
A clue to the complexity of obesity-related disorders is that whilst the presence of obesity is thought to be detrimental to health, simple and intuitive measurements such as BMI, waist circumference and waist-to-hip ratio do not improve cardiovascular disease risk prediction substantially when information such as blood pressure, lipid profile and a history of diabetes are available [7]. This highlights the fact that there is more to obesity than the shape or size of the individual and there may well be obese individuals whose cardiovascular risk profile is not particularly adverse. This cohort of individuals has been labelled as the ‘metabolically healthy obese’. A better understanding of the physiology of these individuals will no doubt help guide future therapies to potentially transform those with MetS into ‘metabolically healthy’ individuals without resorting to drastic measure to achieve significant weight loss. Nonetheless, obesity and metabolic syndrome remain strong predictors of future cardiovascular events. In those with disproportionately high WC for a given BMI, assessment of further cardiometabolic risk factors is encouraged [8], thus highlighting the specific properties of individual fat depots and providing clues that certain depots convey a worse risk profile.
Hypertension affects nearly one third of the US population [9], and in England it was reported in over 45% of those in the obese group, compared with around 30% of the overweight and 15% of those in the normal weight category [2]. Globally over a billion people have raised blood pressure, that is, 24% of the adult male and 20% of adult female population in 2015 [10]. The Framingham Heart Study identifies obesity as a contributory factor in 60–70% of essential hypertension [11], and obese individuals have a 3.5-fold increase in the likelihood of developing hypertension [12].
The co-occurrence of obesity and hypertension has prompted the scientific community to further investigate the pathophysiology of obesity-related hypertension as well as its links to diabetes. Contemporary hypotheses have described prediabetes as a disease of the microvasculature under the influence of its surrounding perivascular adipose tissue (PVAT).
There are many mechanisms via which obesity can lead to hypertension. In the acute phase, both bolus oral ingestion and the intravenous infusion of fat by normotensive obese individuals result in a significant rise in systolic blood pressure, attenuated endothelial function, increased oxidative stress markers and activation of the sympathetic nervous system [13].
Genetic causes of obesity are rare, and the majority of cases are a consequence of indulgence in readily available and calorie-rich foods which provide significant proportion of the recommended daily allowance of salt and fat. High-salt diets accelerate the development of hypertension in diet-induced obese rats without raising the ceiling of the systolic blood pressure beyond that observed in diet-induced obese rats fed a low-salt diet [14]. This effect may be a consequence of the increase in oxidative stress levels in the vasculature as evidenced by significantly higher superoxide levels within aortic rings of high-fat and high-salt diet fed animals.
There are numerous facilitators of obesity-related hypertension. These include the renin-angiotensin-aldosterone system (RAAS), the overactive sympathetic nervous system, metabolic dysregulation including hyperinsulinaemia, adipokine imbalance, and PVAT damage. There is currently no direct evidence to suggest that a loss of PVAT vasorelaxant function leads to systemic hypertension in man, but we have reported a correlation between the loss of PVAT vasorelaxant effect and a rise in BP in a murine model of obesity [15] and believe that such a correlation might also exist in man.
2 Fat Depots
This chapter reviews the role of perivascular adipose tissue in MetS, but it is vital to point out that the distribution of fat around the body determines not only the obese phenotype but also its consequences. Intra-abdominal and visceral fat depots have been linked with an adverse cardiometabolic profile and mortality associated with obesity [16, 17]. The total amount of internal fat rises with increasing subcutaneous adiposity, but even individuals classed as thin may have more visceral fat than some obese individuals. Fat accumulation in some fat depots seems to be more favourable. Increased gluteofemoral fat mass negatively correlates with levels of inflammatory cytokines and is positively linked to raised concentrations of adipokines resulting in decreased metabolic and cardiovascular risk [18]. In human experiments, this is the fat depot that is most easily accessible and studied in ex vivo protocols.
Adipose tissue depots have unique inflammatory profiles. Perivascular adipose tissue from murine aortic arch expresses lower levels of adipocyte-associated genes compared with subcutaneous and visceral fat [19]. Visceral adipose tissue exhibits a more inflammatory profile with a higher macrophage content than subcutaneous fat [20]. This may somewhat explain the stronger link between central obesity and hypertension than between BMI and raised blood pressure [21].
Epicardial adipose tissue thickness correlates well with waist circumference, visceral adipose tissue mass, fasting insulin and diastolic blood pressure [22, 23] and has been shown to be significantly greater in patients with MetS than those without [24].
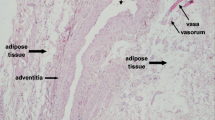
PVAT surrounding human coronary vessels is made up of smaller, more irregularly shaped adipocytes as compared with visceral and subcutaneous fat depots. Coronary PVAT secretes lower levels of adiponectin and higher levels of cytokines such as IL-8 and IL-6 as compared with subcutaneous and visceral adipocytes [19]. Exposure to IL-6 has been linked with a reduction in adiponectin production by human adipocytes [25]. Interestingly, there is a high level of macrophage infiltration and lower adiponectin mRNA levels in the epicardial fat tissue of patients with coronary artery disease [26]. Coronary PVAT contains higher levels of monocyte chemotactic protein-1 (MCP-1) as compared with visceral and subcutaneous tissue [19, 27,28,29]. Lower levels of adiponectin in epicardial tissue have also been associated with hypertension [30, 31] and increased risk of myocardial infarction [32].
In this chapter we will focus mainly on PVAT as the fat depot of interest.
3 Perivascular Adipose Tissue
Adipocytes surround almost every blood vessel in the body. They are biological active cells that produce and secrete a number of molecules called adipokines with metabolic and vasoactive properties. In the adult man, these are predominantly white adipocytes and form the perivascular adipose tissue or PVAT. In 2005, Yudkin et al. proposed that PVAT might be the link between obesity and the development of diabetes and MetS as a consequence of an adverse effect on the microvasculature [33]. In health, PVAT could produce adipokines that influence metabolism and the control of local microvessel tone. They suggested that the loss of these adipokines would result in a change in vessel function and development of insulin resistance. The effect of circulating insulin on NO-mediated vasodilatation is of paramount importance in modulating the postprandial increase in nutritive flow, and the authors postulated that this could be challenged by the paracrine action of adipokines released from local fat stores in obesity. They further highlighted the role inflammation may play and that higher concentrations of TNF-α in obesity could disrupt the crosstalk between fat and blood vessels. Only recently we have been able to provide the evidence in support of the Yudkin hypothesis, but we are far from identifying definitive therapeutic strategies to treat prediabetes and pre-hypertension before the onset of MetS.
The effect of PVAT on its surrounding vasculature is not only dependent on the properties of the adipocytes but also the presence of a variety of cells coexisting within PVAT. In chronic inflammatory conditions such as obesity, there is an increase in numbers of inflammatory cells including macrophages and eosinophils within PVAT which distorts the properties of ‘healthy PVAT’ and leads to a less favourable profile.
At first glance, a simple review of the vast literature on the properties of PVAT can appear somewhat confusing or contradictory. There are reports of both vasorelaxant and pro-contractile properties which can bemuse the casual reader. We know now that PVAT can behave differently, depending on the specific species and vascular beds being studied. The distinctive properties of PVAT also depend on the agents used to the tissue. In health, PVAT from both human and murine mesenteric vessel beds exerts a vasorelaxant effect. That is to say that in a simple organ bath or myography experiment, in the presence of PVAT, the adjacent small vessel constricts significantly less than a skeletonised vessel when stimulated with a vasoconstrictor agent. In obesity this effect is not observed.
There is evidence to suggest that the vasorelaxant property is due to a number of molecules being secreted from PVAT as well as a degree of contribution from the ‘sponging effect’ of PVAT forming a physical barrier and obstructing the flow of the provocative agents from reaching the vessel. We have shown previously that damage to the PVAT vasorelaxant property directly correlates with BP elevation in a murine model of obesity [15]. This is a significant finding as it is the first evidence of a correlation between PVAT function and blood pressure. Quantifying PVAT vasorelaxant effect is a novel endeavour. In brief, animals were fed a high-fat diet to establish an environmental model of obesity. Vessel segments from their mesenteric beds were assessed, and the degree of contraction of their adjacent skeletonised vessel was quantified as a ratio of the degree of contraction of the PVAT-intact segment of the same vessel to KPSS (potassium-rich physiological saline solution). We reported a correlation between this derived figure and systemic BP. This means that as the animals gained weight and lost their PVAT vasorelaxant effect, there was an attendant elevation in blood pressure. This remains the most convincing evidence of a link between weight gain and PVAT function correlating with a rise in BP.
4 PVAT as a Vasorelaxant Organ
We have already discussed that healthy PVAT exerts a vasorelaxant effect on adjacent microvasculature when subject to vasoconstrictors [34]. This is true in the majority of vascular beds. A number of mechanisms have been proposed to describe this phenomenon with just as many outstanding questions requiring further investigation.
Experimental protocols have identified both endothelium-dependent and endothelium-independent [35] mechanisms, and a number of molecules have been implicated which will be discussed briefly in this chapter.
White and brown adipocytes have similar yet distinguishably different secretion profiles [36], but the vasorelaxant property of PVAT has been documented in both white and brown tissues [37, 38].
Adiponectin is the most abundant adipokine with a significant vasorelaxant effect on small arteries and is able to reverse endothelial dysfunction in diet-induced obese rats via the AMPK-eNOS pathway [39]. Adiponectin levels are low in hypertension and improve with antihypertensive treatment [40]. Adiponectin secreted from murine PVAT modulates the tone of the adjoining microvessel segment by functioning as an adipose-derived relaxant factor or ADRF [41]. Further data from our group demonstrated that adiponectin receptor type 1 blockade abolishes PVAT vasorelaxant effect on adjacent small arteries obtained from healthy biopsies [42], thus clearly demonstrating that adiponectin is an ADRF in man. Recently we have reviewed in detail the properties of this adipokine and its role as an ADRF [43].
Adiponectin directly stimulates the production of nitric oxide in endothelial cells using the phosphatidylinositol 3-kinase-dependent pathways involving phosphorylation of eNOS at Ser1179 by AMPK. This vasodilator action of adiponectin may in part explain the effects of adiponectin in augmenting the metabolic actions of insulin in vivo [44].
Serum levels of the adipocyte-derived proteins adiponectin and leptin correlate with insulin resistance (HOMA-IR) and BMI. Levels of expression of MCP-1 and TNF-alpha in visceral adipose tissue are also higher in those with BMI ≥25. Inflammation plays a major role in diabetes, and a growing body of evidence is pointing to obesity-induced PVAT damage as a precursor to the development of diabetes in obesity. In obesity, adipocytes outgrow their blood supply and exist in a state of chronic low-grade hypoxia. We have shown previously that there is increased staining for TNF-alpha receptor in obese compared with lean PVAT [42] and that following bariatric surgery, there is a significant reduction in staining for the TNF-alpha cytokine in the PVAT which correlates with a reduction in adipocyte size following weight loss [45]. It has been shown that TNF-alpha in visceral adipose tissue correlates with HOMA-IR [46] and those with type 2 diabetes have higher circulating levels of TNF-alpha [47].
Treatment with TNF-alpha leads to a reduction in adiponectin mRNA levels in 3T3-L1 adipocytes, and this can be partially recovered by treatment with a c-Jun N-terminal kinase (JNK) inhibitor or the PPAR-gamma agonist rosiglitazone [48].
Decreased total and HMW adiponectin and increased IL-6 and TNF-alpha levels are characteristic of patients with metabolic syndrome and type 2 diabetes [47, 49]. Adiponectin itself can reduce inflammation. The mRNA expression of TNF-alpha, IL-6 and ICAM-1 is elevated in db/db mice, and adiponectin treatment decreases these expressions in the aorta. Adiponectin may contribute to an increase in nitric oxide bioavailability by decreasing superoxide production as well as by inhibiting inflammation and adhesion molecules in the aorta in type 2 diabetic mice [50].
Nacci et al. have used streptozotocin (STZ)-induced diabetic mice and investigated whether treatment with the TNF-alpha blocking antibody infliximab can normalise the expression of adiponectin and adiponectin receptors in different fat depots and if this effect correlates with improved endothelial activity and vasodilator function. The STZ mice were studied at 1 and 2 weeks after diabetes onset and compared to age-matched infliximab-treated diabetic (I-STZ) and control animals (CTRL). In STZ mice, activation of pro-inflammatory JNK signalling was faster in PVAT than in visceral (VAT), epididymal (EAT) and subcutaneous (SAT) adipose depots and associated with reduced adiponectin synthesis and dysregulated AdipoR1/R2 levels. Compared with controls, activation of JNK in aortic endothelial cells and mesenteric arteries was associated with reduced expression/phosphorylation of eNOS and impaired ACh-mediated vasodilation. Infliximab treatment abrogated JNK activation, ameliorated adiponectin protein expression and normalised expression of both AdipoR1 and AdipoR2 in PVAT, concomitantly improving eNOS expression and vessel relaxation in mesenteric arteries. These data highlight the early susceptibility of PVAT to activation of pro-inflammatory JNK signalling and its potential importance in early vascular changes of T1DM [51].
PVAT secretes a number of other adipokines with vasorelaxant properties in addition to adiponectin; these include angiotensin 1–7 (Ang 1–7), nitric oxide (NO), leptin, hydrogen sulphide and palmitic acid methyl ester (PAME).
Angiotensin 1–7 stimulates the release of endothelial NO, activating Ca-dependent potassium channels in arteries [37] and voltage-dependent potassium channels in veins [52]. In keeping with this, Angiotensin 1–7 receptor antagonists attenuate PVAT vasorelaxant function [53]. Ang 1–7 is also able to function via AT2 and Mas receptors to reduce the nerve-stimulated overflow of noradrenaline [54]. This may prove to be of paramount importance as sympathetic nerve over-activity contributes to pathophysiology of obesity-related hypertension, and we shall discuss this further in this chapter. An oral preparation of Ang 1–7 has been produced [55], and assessment of its in vivo effect on vessel tone may provide another therapeutic opportunity in treating obesity-related hypertension.
Healthy white adipose tissue [56] and PVAT [57] produce nitric oxide (NO). Insulin [58] and leptin [59] stimulate NO production in adipocytes, and it follows that the increased levels of insulin and leptin in obesity should enhance NO concentrations in PVAT. In early diet-induced obesity, there is enhanced NO bioavailability in mesenteric PVAT of rats [57], but factors such as elevated superoxide levels in chronic obesity lead to a diminution of NO bioavailability in obese PVAT [15].
Leptin is another molecule secreted from white adipocytes, and its plasma levels are elevated in obesity. Its central actions include its effects on the hypothalamus resulting in appetite suppression as well as an increase in the activity of the sympathetic nervous system [60]. Leptin also has a direct endothelial NO-dependent vasorelaxant effect in health. Leptin-deficient ob/ob mice are severely obese, but remain normotensive [61]. Leptin stimulates endothelial NO release in the vasculature; thus an acute rise in leptin concentrations does not significantly affect blood pressure despite elevated SNS activity. In obesity, leptin levels are chronically elevated and confounded by endothelial dysfunction and a reduction in NO bioavailability [62]; therefore its vasopressor effects become more prominent.
Hydrogen sulphide functions via KCNQ [63], whilst palmitic acid methyl ester (PAME) functions via Kv channels, independent of nitric oxide and endothelium [64], and its release is Ca-dependent. These two molecules have been more recent additions to the list of ADRFs. There is a reduction in the release of PAME from PVAT of 20-week-old SHR as compared with pre-hypertensive SHR and normotensive Wistar-Kyoto rats. Exogenously applied PAME has a reduced vasorelaxant effect on de-endothelialised aortic rings of SHR as compared with its significant vasorelaxant effect on pre-constricted vessels from pre-hypertensive SHR and normotensive rats [64]. Clearly PAME plays a role in pre-hypertension and is worthy of further clinical investigation in this context.
It has become apparent that there is more than one PVAT-derived molecule that satisfies the criteria for ADRF. We have shown that adiponectin is the ADRF from human subcutaneous PVAT [42], but other ADRFs may well play a significant role in human PVAT.
5 PVAT as a Pro-contractile Tissue
In obesity, the vasorelaxant function of PVAT is attenuated or lost completely.
A number of explanations and theories exist as to the cause of this loss of function. Amongst the most likely are the effects of oxidative stress and inflammation, as well as adipokine dysregulation and increased sympathetic nervous system action.
We have shown previously that incubation of healthy PVAT with the inflammatory cytokines TNF-alpha and IL-6 leads to a significant attenuation of PVAT vasorelaxant properties similar to that observed in obesity [42]. In keeping with the complexity of PVAT studies, we have reported that there is no homogenous effect from the presence of different white blood cells within the PVAT. In particular, we have studied both macrophages and eosinophils within PVAT. Macrophages secrete a number of inflammatory cytokines including TNF-α and free radicals such as the superoxide anion. We used experimental hypoxia in tissue baths to approximate obesity-induced PVAT damage and observed that macrophage recruitment and activation in adipose tissue is an essential step resulting in the loss of PVAT vasorelaxant function [65]. On the contrary, mice deficient of eosinophils lack the PVAT vasorelaxant effect, and eosinophil reconstitution did lead to enhanced adiponectin and PVAT-derived NO bioavailability leading to the restoration of PVAT vasorelaxant function [66]. As previously mentioned, fat depots seem to have specific characteristics, and the PVAT surrounding rat thoracic aorta expresses brown adipose tissue genes and appears to resist inflammation and macrophage infiltration in diet-induced obesity [67], so it is possible that in obesity, not all fat depots are affected equally.
The paramount role of macrophages in PVAT damage cannot be overstated; thus macrophage recruitment into PVAT tissue has been studied in some detail. Monocyte chemotactic protein-1 (MCP-1) levels are increased in adipose tissue and in plasma of genetically obese and diet-induced obese mice [68], as well in obese humans [69]. Moreover, insulin increases the secretion of MCP-1 from insulin-resistant 3T3-L1 adipocytes and in ob/ob mice [70]; in this way the hyperinsulinaemic state in obesity leads to PVAT macrophage recruitment and subsequent release of cytokines which attenuate its vasorelaxant function. Fractalkine or CX3CL1 is a protein secreted from adipocytes that promotes monocyte adhesion to human adipocytes [71]. Fractalkine levels are increased in diabetes as well as in obesity [72]. There is also direct evidence for involvement of fractalkine in hypertension. The expression of CX3CL1 receptor gene in blood leukocytes from patients with arterial hypertension has been shown to be significantly increased [73]. This protein may well play a significant facilitator role in the process of initiation of the macrophage-induced loss of PVAT vasorelaxant function and is worthy of further consideration from a therapeutic target viewpoint.
Chemerin is another adipokine that plays a potentially significant role in the loss of PVAT vasorelaxant function by behaving as a possible link between obesity, BMI [74], PVAT, diabetes [75] and hypertension both in adults and children [76, 77].
Adipose tissue explants from obese patients exhibit significantly higher chemerin secretion compared with lean controls, and higher chemerin release is associated with insulin resistance and insulin-induced antilipolysis. Chemerin stimulates vascular smooth muscle cell proliferation and migration via a ROS-dependent signalling pathway, and at least in theory, this vascular remodelling can contribute to raising blood pressure [78]. Moreover, chemerin evokes direct vasoconstriction, as well as enhancing agonist-induced contractions in human and rat vessels through Gi proteins, resulting in the activation of L-type Ca2+ channels, as well as Src, and Rho kinase [79]. Levels of chemerin correlate well with clinical parameters too. Its plasma concentration is raised in obesity, insulin resistance and inflammatory conditions, and levels positively correlate with increases in BMI and abdominal visceral fat accumulation [80]. It has been linked with increasing BP in mice, and importantly, its levels fall with loss of adipocyte mass following exercise or bariatric surgery [81].
We have discussed chemerin’s role in the vasculature, but it also plays a role in disrupting glucose homeostasis. Chemerin induces insulin resistance in human skeletal muscle cells at the level of insulin receptor substrate 1, Akt and glycogen synthase kinase 3 phosphorylation and glucose uptake, and ERK inhibition prevents chemerin-induced insulin resistance [74]. Following weight loss, the significant decrease in chemerin levels in 3 months after bariatric surgery is associated with a decrease in HOMA-IR and blood glucose [82].
From an inflammatory perspective, chemerin induces ICAM-1 and E-selectin expression in endothelial cells [83]. Given that it plays a role in monocytes recruitment, insulin resistance and vasoconstriction and its levels correlate with weight gain and drop following weight loss, chemerin is one of the major adipokines that could be targeted in therapeutic strategies to treat MetS.
No doubt further PVAT-derived pro-contractile entities will be described much the same way as there is now a list of candidates for PVAT-derived vasorelaxant factors.
6 The RAAS Within PVAT
In obesity, there are raised circulating levels of the components of the renin-angiotensin-aldosterone system (RAAS). Adipocytes have an intrinsic RAAS system including ACE and angiotensin type 1 and type 2 receptors, and they secrete angiotensinogen, the levels of which are raised in obesity [84]. The source of the adipocyte renin activity remains controversial and unclear [85].The raised circulating aldosterone levels in obesity correlate with the degree of visceral adiposity and waist-to-hip ratio [86,87,88]. In the context of obesity-related hypertension, the raised aldosterone concentrations have a twofold effect: they contribute to increased blood volume by increasing sodium reabsorption and lead to the generation of reactive oxygen species (ROS). Aldosterone activates NADPH oxidase, thus increasing ROS levels leading to oxidative posttranslational changes to guanylyl cyclase rendering it NO-insensitive [89]. ROS can also reduce NO bioavailability by forming molecules such as peroxynitrite, thus contributing to endothelial dysfunction. ROS can also stimulate the mineralocorticoid receptor (MR) [90], thereby theoretically contributing to further elevations in ROS levels, forming a vicious circle. At the endothelial level, aldosterone decreases glucose-6-phosphate dehydrogenase (G6PD) activity. G6PD is a cytosolic enzyme and the main source of intracellular NADPH which functions to limit ROS activity [91]. There are two aldosterone receptor antagonists in clinical use: spironolactone is a nonselective aldosterone receptor antagonist, whereas eplerenone is a selective aldosterone receptor antagonist which has a lower degree of cross-reactivity with sex-steroid hormones and a longer half-life than spironolactone [92]. Spironolactone increases the expression of G6PD and its activity, as well as raising NADPH levels leading to a reduction in ROS generation in aortas of aldosterone-treated mice [91]. Aldosterone increases the expression of TNF-α from macrophages within PVAT, and we have reviewed the role of macrophages and TNF-alpha in PVAT damage. Eplerenone leads to a reduction of ROS generation and increased levels of adiponectin in obese and diabetic mice [93].
It is not clear to what extent the blood pressure reduction is a result of blood volume and cardiac output reduction secondary to reduced sodium reabsorption, or due to a reduction in sympathetic activity through the direct CNS effect of aldosterone [94, 95]. Certainly, a reduction in ROS generation within PVAT would partly restore the favourable vasorelaxant profile lost, in part, following hypoxia-induced inflammatory damage in obesity.
The ROS-induced PVAT damage in obesity would suggest that antioxidants and free radical scavengers could be therapeutic agents to reverse this damage and possibly lower blood pressure in obesity. We’ve shown in ex vivo experiments that SOD and catalase can restore the PVAT vasorelaxant property in both human and murine models of obesity [15, 45]. A 3-week administration of desmethyltirilazad (lazaroid), a potent antioxidant, significantly ameliorates blood pressure in SHR rats [96].
We have shown also that MR blockade using eplerenone is able to reduce macrophage activation and rescue aldosterone-induced and hypoxia-induced PVAT damage [65]. Intuitively, it has been proposed that prevention of ROS generation using NADPH oxidase inhibitors may be a better way of tackling oxidative stress than scavenging the free radicals once they have been generated, although clinical studies need to assess the feasibility of this theory [97].
Vessel stiffness is another important contributing factor in the pathophysiology of obesity-related hypertension. The association of vessel stiffness is strongest for waist circumference and visceral adiposity, rather than global obesity as measured by BMI [98]. Obesity is a complex multifaceted disorder, and dysregulation of any number of factors can affect vascular stiffness. The adipokine leptin has been linked with impairment of arterial distensibility, and its raised levels in obesity may well be a contributing factor in arterial stiffness [99].
Inflammation, oxidative stress and monocyte recruitment all play their part in initiating endothelial dysfunction in obesity. There is also disruption to the fine balance between the vasoconstrictor action of endothelin-1 and the vasodilator effect of NO in endothelial cells. In health, insulin activates phosphoinositide 3-kinase leading to increased NO production secondary to eNOS phosphorylation [100]. Postprandial physiological surge in insulin concentrations leads to dilatation of precapillary arterioles, thus improving blood flow and delivery of nutrients to tissues, a process known as nutritive flow [33]. In obesity, NO-mediated vasorelaxation is impaired, leading to vasoconstriction via unopposed endothelin-1 action [33, 100]. Reduced endothelial nitric oxide bioavailability in obesity is a significant consequence of the reactions between free radicals and NO. Reactive oxygen species such as the superoxide anion react with nitric oxide to produce peroxynitrite and deplete endothelial NO levels. The role of nitric oxide in vessel tone modulation and its fate in inflammatory diseases have been extensively reviewed by Jin and Loscalzo [101]. We now know that PVAT is a source of NO which contributes to the PVAT vasorelaxant function, but in obesity, the generation of ROS depletes this vital source of NO; the microvessel is faced with reduced NO bioavailability from both outside the vessel, the PVAT, and inside, the endothelium.
There is a close correlation between obesity, obstructive sleep apnoea (OSA) and hypertension. There is a dose-response relationship between sleep-disordered breathing and hypertension, independent of confounding factors [102]. Almost half of all hypertensive patients suffer from sleep apnoea, and half of all sleep apnoea patients are hypertensive [86]. Whilst fat deposition around the upper airway in obesity is thought to be the most significant contributor to the development of OSA in obesity, there are a number of potential mechanisms linking OSA with hypertension, including endothelial dysfunction, CNS stimulation, oxidative stress and inflammation [103]. The most significant factor is thought to be the elevated oxidative stress levels initiated by intermittent hypoxia, coupled with hyperleptinaemia [104] with its direct stimulatory effects on the sympathetic nervous system. The elevated levels of aldosterone in OSA also correlate with severity of OSA. Once again, this highlights the significance of ROS and aldosterone generation, both of which are generated by hypoxic and inflamed adipocytes.
7 Sympathetic Nerves Within PVAT
PVAT is innervated by nerves from the sympathetic nervous system. Obesity has a differential effect on local SNS activity. Hypertensive obese individuals show an increased sympathetic activity in both cardiac and renal nerves [105].There is also evidence of central stimulation of the SNS by reactive oxygen species, the levels of which are raised in obesity. Animal studies suggest that NADPH oxidase-dependent oxidative stress in the brain may be a cause of increased sympathetic tone leading to hypertension in high-fat fed animals [106].
The heightened sympathetic state in obesity and presence of nerve endings within PVAT have led to further evaluation of the effects of SNS on PVAT using electric field stimulation (EFS) protocols. Work by Gao et al. has shown that superoxide generated by NADPH oxidase in response to electric field stimulation enhances the contractile response of adjacent small arteries [107]. It has been shown that candesartan (angiotensin II type 1 receptor antagonist) reduces this PVAT-mediated potentiation of EFS-induced contractile response, thus providing another potential explanation for the increased vascular resistance in obesity where there are both increased sympathetic nerve activity and increased angiotensin II levels [108].
Circulating adiponectin levels also increase by nearly 70% post gastric bypass and by around 36% post gastric banding procedures. The greatest increase is after the loss of 35% of the original body weight, with a strong correlation between percentage increase in adiponectin levels and percentage decrease in BMI [109]. This is not true of weight loss by other surgical means. After liposuction, despite a 10% weight reduction, no improvements in adiponectin or insulin resistance have been noted [110]. This is likely due to the differing qualities of adipose tissue depots with visceral fat exhibiting a more inflammatory profile as compared with subcutaneous adipose tissue [111].
Weight loss or bariatric surgery remains the most reliable means to achieve and maintain significant weight loss. Bariatric surgery has also been shown to improve the inflammatory profile of obese individuals [112]. In subcutaneous adipose tissue, the expression of IL-6 and TNF-α mRNA decreases significantly, and expression of adiponectin and its receptors increases after dramatic weight loss post surgery [113]. The significant degree of weight loss, together with improvements in adipokine and inflammatory cytokine profile, as well as resolution or improvement in diabetes status [114] makes this an invaluable procedure in those suffering from morbid obesity and its sequelae. We have reported that 6 months following significant weight loss post bariatric surgery, PVAT regains its vasorelaxant function, despite the individuals still weighing in the obese category. This shows that there is more to weight loss than purely loss of mass, and other factors such as the reduced adipocyte size and a reduction in PVAT inflammation as a consequence of a more balanced oxygen supply may be the fundamental trigger leading to our observation [45].
Vitamin D is a perfect example to highlight the need for tissue-specific or tissue-targeted therapies, given that it can suppress renin transcription, and transgenic animals devoid of vitamin D receptor develop hypertension, but they remain lean with smaller adipocytes on a high-fat diet. Overexpression of the receptors on adipocytes leads to a suppression of lipolysis, thermogenesis and resultant obesity [115, 116]. This is a perfect example of the challenges facing researchers trying to identify a molecule that can treat one condition without causing detrimental off-target effects. The (re)search continues.
Conclusion
Perivascular adipose tissue plays a crucial role in modulating vessel tone and blood glucose homeostasis. In obesity, PVAT is damaged and dysfunctional. Rescuing the damaged PVAT and restoring the healthy PVAT phenotype before the onset of hypertension and diabetes should be the focus of future studies.
Abbreviations
- ACE:
-
Angiotensin converting enzyme
- ADRF:
-
Adipose-derived relaxing factor
- AMPK:
-
5′ Adenosine monophosphate-activated protein kinase
- BMI:
-
Body mass index
- BP:
-
Blood pressure
- CNS:
-
Central nervous system
- eNOS:
-
Endothelial nitric oxide synthase
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- MCP-1:
-
Monocyte chemotactic protein-1
- MetS:
-
Metabolic syndrome
- MR:
-
Mineralocorticoid receptor
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- OSA:
-
Obstructive sleep apnoea
- PAME:
-
Palmitic acid methyl ester
- PVAT:
-
Perivascular adipose tissue
- RAAS:
-
Renin-angiotensin-aldosterone system
- ROS:
-
Reactive oxygen species
- SHR:
-
Spontaneously hypertensive rat
- SNS:
-
Sympathetic nervous system
- WC:
-
Waist circumference
References
WHO. Obesity and overweight (Factsheet 311). 2016 [updated 2016; cited 04 July 2017]. http://www.who.int/mediacentre/factsheets/fs311/en/index.html.
The NHS Information Centre LS. Statistics on Obesity, Physical Activity and Diet: England, 2011. 24 Feb 2011.
CDC. U.S. Obesity Trends. 2011 [updated 2011; cited 2011 18 Aug 2011]. http://www.cdc.gov/obesity/data/trends.html#State.
Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet. 1962;14:353–62.
Diamond J. The double puzzle of diabetes. Nature. 2003;423(6940):599–602.
Scott EM, Grant PJ. Neel revisited: the adipocyte, seasonality and type 2 diabetes. Diabetologia. 2006;49(7):1462–6.
Emerging Risk Factors Collaboration, Wormser D, Kaptoge S, Di Angelantonio E, Wood AM, Pennells L, et al. Separate and combined associations of body-mass index and abdominal adiposity with cardiovascular disease: collaborative analysis of 58 prospective studies. Lancet. 2011;377(9771):1085–95.
Cornier MA, Despres JP, Davis N, Grossniklaus DA, Klein S, Lamarche B, et al. Assessing adiposity: a scientific statement from the American Heart Association. Circulation. 2011;124:1996–2019.
CDC. Hypertension. 2011 [updated 2011; cited 2011 18 Aug 2011]. http://www.cdc.gov/nchs/fastats/hyprtens.htm.
WHO. Blood pressure. 2017 [updated 2017; cited 2017 04 July 2017]. http://apps.who.int/gho/data/view.main.2464GLOBALSTANDARD?lang=en.
Henry SL, Barzel B, Wood-Bradley RJ, Burke SL, Head GA, Armitage JA. The developmental origins of obesity-related hypertension. Clin Exp Pharmacol Physiol. 2012;39:799–806.
Kotchen TA. Obesity-related hypertension: epidemiology, pathophysiology, and clinical management. Am J Hypertens. 2010;23(11):1170–8.
Gosmanov AR, Smiley DD, Robalino G, Siquiera J, Khan B, Le NA, et al. Effects of oral and intravenous fat load on blood pressure, endothelial function, sympathetic activity, and oxidative stress in obese healthy subjects. Am J Physiol Endocrinol Metab. 2010;299(6):E953–8.
Dobrian AD, Schriver SD, Lynch T, Prewitt RL. Effect of salt on hypertension and oxidative stress in a rat model of diet-induced obesity. Am J Physiol Ren Physiol. 2003;285(4):F619–28.
Aghamohammadzadeh R, Unwin RD, Greenstein AS, Heagerty AM. Effects of obesity on perivascular adipose tissue vasorelaxant function: nitric oxide, inflammation and elevated systemic blood pressure. J Vasc Res. 2015;52(5):299–305.
Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116(1):39–48.
Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131(2):242–56.
Manolopoulos KN, Karpe F, Frayn KN. Gluteofemoral body fat as a determinant of metabolic health. Int J Obes. 2010;34(6):949–59.
Chatterjee TK, Stoll LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G, et al. Proinflammatory phenotype of perivascular adipocytes: influence of high-fat feeding. Circ Res. 2009;104(4):541–9.
Dorresteijn JA, Visseren FL, Spiering W. Mechanisms linking obesity to hypertension. Obes Rev. 2011;13:17–26.
Lee CM, Huxley RR, Wildman RP, Woodward M. Indices of abdominal obesity are better discriminators of cardiovascular risk factors than BMI: a meta-analysis. J Clin Epidemiol. 2008;61(7):646–53.
Iacobellis G, Assael F, Ribaudo MC, Zappaterreno A, Alessi G, Di Mario U, et al. Epicardial fat from echocardiography: a new method for visceral adipose tissue prediction. Obes Res. 2003;11(2):304–10.
Iacobellis G, Ribaudo MC, Assael F, Vecci E, Tiberti C, Zappaterreno A, et al. Echocardiographic epicardial adipose tissue is related to anthropometric and clinical parameters of metabolic syndrome: a new indicator of cardiovascular risk. J Clin Endocrinol Metab. 2003;88(11):5163–8.
Iacobellis G, Willens HJ, Barbaro G, Sharma AM. Threshold values of high-risk echocardiographic epicardial fat thickness. Obesity (Silver Spring). 2008;16(4):887–92.
Simons PJ, van den Pangaart PS, Aerts JM, Boon L. Pro-inflammatory delipidizing cytokines reduce adiponectin secretion from human adipocytes without affecting adiponectin oligomerization. J Endocrinol. 2007;192(2):289–99.
Baker AR, Silva NF, Quinn DW, Harte AL, Pagano D, Bonser RS, et al. Human epicardial adipose tissue expresses a pathogenic profile of adipocytokines in patients with cardiovascular disease. Cardiovasc Diabetol. 2006;5:1.
Eiras S, Teijeira-Fernandez E, Shamagian LG, Fernandez AL, Vazquez-Boquete A, Gonzalez-Juanatey JR. Extension of coronary artery disease is associated with increased IL-6 and decreased adiponectin gene expression in epicardial adipose tissue. Cytokine. 2008;43(2):174–80.
Iacobellis G, Pistilli D, Gucciardo M, Leonetti F, Miraldi F, Brancaccio G, et al. Adiponectin expression in human epicardial adipose tissue in vivo is lower in patients with coronary artery disease. Cytokine. 2005;29(6):251–5.
Iacobellis G, di Gioia CR, Cotesta D, Petramala L, Travaglini C, De Santis V, et al. Epicardial adipose tissue adiponectin expression is related to intracoronary adiponectin levels. Horm Metab Res. 2009;41(3):227–31.
Teijeira-Fernandez E, Eiras S, Grigorian-Shamagian L, Fernandez A, Adrio B, Gonzalez-Juanatey JR. Epicardial adipose tissue expression of adiponectin is lower in patients with hypertension. J Hum Hypertens. 2008;22(12):856–63.
Ohashi K, Kihara S, Ouchi N, Kumada M, Fujita K, Hiuge A, et al. Adiponectin replenishment ameliorates obesity-related hypertension. Hypertension. 2006;47(6):1108–16.
Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004;291(14):1730–7.
Yudkin JS, Eringa E, Stehouwer CD. “Vasocrine” signalling from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet. 2005;365(9473):1817–20.
Soltis EE, Cassis LA. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A. 1991;13(2):277–96.
Gao YJ, Lu C, Su LY, Sharma AM, Lee RM. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol. 2007;151(3):323–31.
Galvez-Prieto B, Bolbrinker J, Stucchi P, de Las Heras AI, Merino B, Arribas S, et al. Comparative expression analysis of the renin-angiotensin system components between white and brown perivascular adipose tissue. J Endocrinol. 2008;197(1):55–64.
Lu C, Su LY, Lee RM, Gao YJ. Alterations in perivascular adipose tissue structure and function in hypertension. Eur J Pharmacol. 2011;656(1-3):68–73.
Dubrovska G, Verlohren S, Luft FC, Gollasch M. Mechanisms of ADRF release from rat aortic adventitial adipose tissue. Am J Physiol Heart Circ Physiol. 2004;286(3):H1107–13.
Deng G, Long Y, Yu YR, Li MR. Adiponectin directly improves endothelial dysfunction in obese rats through the AMPK-eNOS Pathway. Int J Obes. 2010;34(1):165–71.
Yilmaz MI, Sonmez A, Caglar K, Celik T, Yenicesu M, Eyileten T, et al. Effect of antihypertensive agents on plasma adiponectin levels in hypertensive patients with metabolic syndrome. Nephrology (Carlton). 2007;12(2):147–53.
Fesus G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC, et al. Adiponectin is a novel humoral vasodilator. Cardiovasc Res. 2007;75(4):719–27.
Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119(12):1661–70.
Aghamohammadzadeh R, Withers SB, Lynch FM, Greenstein AS, Malik R, Heagerty AM. Perivascular adipose tissue from human systemic and coronary vessels: the emergence of a new pharmacotherapeutic target. Br J Pharmacol. 2012;165:670–82.
Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem. 2003;278(45):45021–6.
Aghamohammadzadeh R, Greenstein AS, Yadav R, Jeziorska M, Hama S, Soltani F, et al. The effects of bariatric surgery on human small artery function: evidence for reduction in perivascular adipocyte inflammation, and the restoration of normal anticontractile activity despite persistent obesity. J Am Coll Cardiol. 2013;62:128–35.
Kang YE, Kim JM, Joung KH, Lee JH, You BR, Choi MJ, et al. The roles of adipokines, proinflammatory cytokines, and adipose tissue macrophages in obesity-associated insulin resistance in modest obesity and early metabolic dysfunction. PLoS One. 2016;11(4):e0154003.
Qiao YC, Shen J, He L, Hong XZ, Tian F, Pan YH, et al. Changes of regulatory T cells and of proinflammatory and immunosuppressive cytokines in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. J Diabetes Res. 2016;2016:3694957.
Kim KY, Kim JK, Jeon JH, Yoon SR, Choi I, Yang Y. c-Jun N-terminal kinase is involved in the suppression of adiponectin expression by TNF-alpha in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2005;327(2):460–7.
Lee JM, Kim SR, Yoo SJ, Hong OK, Son HS, Chang SA. The relationship between adipokines, metabolic parameters and insulin resistance in patients with metabolic syndrome and type 2 diabetes. J Int Med Res. 2009;37(6):1803–12.
Lee S, Zhang H, Chen J, Dellsperger KC, Hill MA, Zhang C. Adiponectin abates diabetes-induced endothelial dysfunction by suppressing oxidative stress, adhesion molecules, and inflammation in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2012;303(1):H106–15.
Nacci C, Leo V, De Benedictis L, Potenza MA, Sgarra L, De Salvia MA, et al. Infliximab therapy restores adiponectin expression in perivascular adipose tissue and improves endothelial nitric oxide-mediated vasodilation in mice with type 1 diabetes. Vasc Pharmacol. 2016;87:83–91.
Lu C, Zhao AX, Gao YJ, Lee RM. Modulation of vein function by perivascular adipose tissue. Eur J Pharmacol. 2011;657(1-3):111–6.
Lee RM, Bader M, Alenina N, Santos RA, Gao YJ, Lu C. Mas receptors in modulating relaxation induced by perivascular adipose tissue. Life Sci. 2011;89:467–72.
Byku M, Macarthur H, Westfall TC. Inhibitory effects of angiotensin (1-7) on the nerve stimulation-induced release of norepinephrine and neuropeptide y from the mesenteric arterial bed. Am J Physiol Heart Circ Physiol. 2009;289:H457–65.
Marques FD, Ferreira AJ, Sinisterra RD, Jacoby BA, Sousa FB, Caliari MV, et al. An oral formulation of angiotensin-(1-7) produces cardioprotective effects in infarcted and isoproterenol-treated rats. Hypertension. 2011;57(3):477–83.
Ribiere C, Jaubert AM, Gaudiot N, Sabourault D, Marcus ML, Boucher JL, et al. White adipose tissue nitric oxide synthase: a potential source for NO production. Biochem Biophys Res Commun. 1996;222(3):706–12.
Gil-Ortega M, Stucchi P, Guzman-Ruiz R, Cano V, Arribas S, Gonzalez MC, et al. Adaptative nitric oxide overproduction in perivascular adipose tissue during early diet-induced obesity. Endocrinology. 2010;151:3299–306.
Ribiere C, Jaubert AM, Sabourault D, Lacasa D, Giudicelli Y. Insulin stimulates nitric oxide production in rat adipocytes. Biochem Biophys Res Commun. 2002;291(2):394–9.
Mehebik N, Jaubert AM, Sabourault D, Giudicelli Y, Ribiere C. Leptin-induced nitric oxide production in white adipocytes is mediated through PKA and MAP kinase activation. Am J Phys Cell Physiol. 2005;289(2):C379–87.
Rahmouni K, Correia ML, Haynes WG, Mark AL. Obesity-associated hypertension: new insights into mechanisms. Hypertension. 2005;45(1):9–14.
Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens. 1999;17(12 Pt 2):1949–53.
da Silva AA, do Carmo J, Dubinion J, Hall JE. The role of the sympathetic nervous system in obesity-related hypertension. Curr Hypertens Rep. 2009;11(3):206–11.
Schleifenbaum J, Kohn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T, et al. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens. 2010;28(9):1875–82.
Lee YC, Chang HH, Chiang CL, Liu CH, Yeh JI, Chen MF, et al. Role of perivascular adipose tissue-derived methyl palmitate in vascular tone regulation and pathogenesis of hypertension. Circulation. 2011;124:1160–71.
Withers BS, Agabiti-Rosei C, Linvingstone DM, Little MC, Aslam R, Malik RA, et al. Macrophage activation is responsible for loss of anticontractile function in inflamed perivascular fat. Arterioscler Thromb Vasc Biol. 2011;31:908–13.
Withers SB, Forman R, Meza-Perez S, Sorobetea D, Sitnik K, Hopwood T, et al. Eosinophils are key regulators of perivascular adipose tissue and vascular functionality. Sci Rep. 2017 Mar 17;7:44571.
Fitzgibbons TP, Kogan S, Aouadi M, Hendricks GM, Straubhaar J, Czech MP. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am J Physiol Heart Circ Physiol. 2011;301(4):H1425–37.
Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116(6):1494–505.
Kim CS, Park HS, Kawada T, Kim JH, Lim D, Hubbard NE, et al. Circulating levels of MCP-1 and IL-8 are elevated in human obese subjects and associated with obesity-related parameters. Int J Obes. 2006;30(9):1347–55.
Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci U S A. 2003;100(12):7265–70.
Shah R, Hinkle CC, Ferguson JF, Mehta NN, Li M, Qu L, et al. Fractalkine is a novel human adipochemokine associated with type 2 diabetes. Diabetes. 2011;60(5):1512–8.
Sirois-Gagnon D, Chamberland A, Perron S, Brisson D, Gaudet D, Laprise C. Association of common polymorphisms in the fractalkine receptor (CX3CR1) with obesity. Obesity (Silver Spring). 2010;19(1):222–7.
Timofeeva AV, Goryunova LE, Khaspekov GL, Kovalevskii DA, Scamrov AV, Bulkina OS, et al. Altered gene expression pattern in peripheral blood leukocytes from patients with arterial hypertension. Ann N Y Acad Sci. 2006;1091:319–35.
Sell H, Laurencikiene J, Taube A, Eckardt K, Cramer A, Horrighs A, et al. Chemerin is a novel adipocyte-derived factor inducing insulin resistance in primary human skeletal muscle cells. Diabetes. 2009;58(12):2731–40.
Ouwens DM, Bekaert M, Lapauw B, Van Nieuwenhove Y, Lehr S, Hartwig S, et al. Chemerin as biomarker for insulin sensitivity in males without typical characteristics of metabolic syndrome. Arch Physiol Biochem. 2012;118(3):135–8.
Schipper HS, Nuboer R, Prop S, van den Ham HJ, de Boer FK, Kesmir C, et al. Systemic inflammation in childhood obesity: circulating inflammatory mediators and activated CD14++ monocytes. Diabetologia. 2012;55(10):2800–10.
Verrijn Stuart AA, Schipper HS, Tasdelen I, Egan DA, Prakken BJ, Kalkhoven E, et al. Altered plasma adipokine levels and in vitro adipocyte differentiation in pediatric type 1 diabetes. J Clin Endocrinol Metab. 2011;97(2):463–72.
Kunimoto H, Kazama K, Takai M, Oda M, Okada M, Yamawaki H. Chemerin promotes the proliferation and migration of vascular smooth muscle and increases mouse blood pressure. Am J Physiol Heart Circ Physiol. 2015;309(5):H1017–28.
Ferland DJ, Darios ES, Neubig RR, Sjogren B, Truong N, Torres R, et al. Chemerin-induced arterial contraction is Gi- and calcium-dependent. Vasc Pharmacol. 2017;88:30–41.
Shin H-Y, Lee DC, Chu SH, Jeon JY, Lee MK, Im JA, et al. Chemerin levels are positively correlated with abdominal visceral fat accumulation. Clin Endocrinol. 2012;77(1):47–50.
Chakaroun R, Raschpichler M, Kloting N, Oberbach A, Flehmig G, Kern M, et al. Effects of weight loss and exercise on chemerin serum concentrations and adipose tissue expression in human obesity. Metabolism. 2011;61(5):706–14.
Sell H, Divoux A, Poitou C, Basdevant A, Bouillot JL, Bedossa P, et al. Chemerin correlates with markers for fatty liver in morbidly obese patients and strongly decreases after weight loss induced by bariatric surgery. J Clin Endocrinol Metab. 2010;95(6):2892–6.
Landgraf K, Friebe D, Ullrich T, Kratzsch J, Dittrich K, Herberth G, et al. Chemerin as a mediator between obesity and vascular inflammation in children. J Clin Endocrinol Metab. 2012;97(4):E556–64.
Van Harmelen V, Ariapart P, Hoffstedt J, Lundkvist I, Bringman S, Arner P. Increased adipose angiotensinogen gene expression in human obesity. Obesity. 2000;8(4):337–41.
Engeli S, Negrel R, Sharma AM. Physiology and pathophysiology of the adipose tissue renin-angiotensin system. Hypertension. 2000;35(6):1270–7.
Goodfriend TL, Calhoun DA. Resistant hypertension, obesity, sleep apnea, and aldosterone: theory and therapy. Hypertension. 2004;43(3):518–24.
Goodfriend TL, Egan BM, Kelley DE. Aldosterone in obesity. Endocr Res. 1998;24(3-4):789–96.
Goodfriend TL, Kelley DE, Goodpaster BH, Winters SJ. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res. 1999;7(4):355–62.
Maron BA, Zhang YY, Handy DE, Beuve A, Tang SS, Loscalzo J, et al. Aldosterone increases oxidant stress to impair guanylyl cyclase activity by cysteinyl thiol oxidation in vascular smooth muscle cells. J Biol Chem. 2009;284(12):7665–72.
Wang H, Shimosawa T, Matsui H, Kaneko T, Ogura S, Uetake Y, et al. Paradoxical mineralocorticoid receptor activation and left ventricular diastolic dysfunction under high oxidative stress conditions. J Hypertens. 2008;26(7):1453–62.
Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13(2):189–97.
Maron BA, Leopold JA. Aldosterone receptor antagonists: effective but often forgotten. Circulation. 2010;121(7):934–9.
Guo C, Ricchiuti V, Lian BQ, Yao TM, Coutinho P, Romero JR, et al. Mineralocorticoid receptor blockade reverses obesity-related changes in expression of adiponectin, peroxisome proliferator-activated receptor-gamma, and proinflammatory adipokines. Circulation. 2008;117(17):2253–61.
de Paula RB, da Silva AA, Hall JE. Aldosterone antagonism attenuates obesity-induced hypertension and glomerular hyperfiltration. Hypertension. 2004;43(1):41–7.
Rahmouni K, Barthelmebs M, Grima M, Imbs JL, De Jong W. Involvement of brain mineralocorticoid receptor in salt-enhanced hypertension in spontaneously hypertensive rats. Hypertension. 2001;38(4):902–6.
Vaziri ND, Ni Z, Oveisi F, Trnavsky-Hobbs DL. Effect of antioxidant therapy on blood pressure and NO synthase expression in hypertensive rats. Hypertension. 2000;36(6):957–64.
Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10(6):453–71.
Safar ME, Czernichow S, Blacher J. Obesity, arterial stiffness, and cardiovascular risk. J Am Soc Nephrol. 2006;17(4 Suppl 2):S109–11.
Singhal A, Farooqi IS, Cole TJ, O’Rahilly S, Fewtrell M, Kattenhorn M, et al. Influence of leptin on arterial distensibility: a novel link between obesity and cardiovascular disease? Circulation. 2002;106(15):1919–24.
Kotsis V, Stabouli S, Papakatsika S, Rizos Z, Parati G. Mechanisms of obesity-induced hypertension. Hypertens Res. 2010;33(5):386–93.
Jin RC, Loscalzo J. Vascular nitric oxide: formation and function. J Blood Med. 2010;2010(1):147–62.
Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342(19):1378–84.
Wolk R, Shamsuzzaman AS, Somers VK. Obesity, sleep apnea, and hypertension. Hypertension. 2003;42(6):1067–74.
Yang R, Sikka G, Larson J, Watts VL, Niu X, Ellis CL, et al. Restoring leptin signaling reduces hyperlipidemia and improves vascular stiffness induced by chronic intermittent hypoxia. Am J Physiol Heart Circ Physiol. 2011;300(4):H1467–76.
Rumantir MS, Vaz M, Jennings GL, Collier G, Kaye DM, Seals DR, et al. Neural mechanisms in human obesity-related hypertension. J Hypertens. 1999;17(8):1125–33.
Nagae A, Fujita M, Kawarazaki H, Matsui H, Ando K, Fujita T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in obesity-induced hypertension. Circulation. 2009;119(7):978–86.
Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM, et al. Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovasc Res. 2006;71(2):363–73.
Lu C, Su LY, Lee RM, Gao YJ. Mechanisms for perivascular adipose tissue-mediated potentiation of vascular contraction to perivascular neuronal stimulation: the role of adipocyte-derived angiotensin II. Eur J Pharmacol. 2011;634(1-3):107–12.
Butner KL, Nickols-Richardson SM, Clark SF, Ramp WK, Herbert WG. A review of weight loss following Roux-en-Y gastric bypass vs restrictive bariatric surgery: impact on adiponectin and insulin. Obes Surg. 2010;20(5):559–68.
Compher C, Badellino KO. Obesity and inflammation: lessons from bariatric surgery. JPEN J Parenter Enteral Nutr. 2008;32(6):645–7.
Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev. 2009;11(1):11–8.
Forsythe LK, Wallace JM, Livingstone MB. Obesity and inflammation: the effects of weight loss. Nutr Res Rev. 2008;21(2):117–33.
Moschen AR, Molnar C, Geiger S, Graziadei I, Ebenbichler CF, Weiss H, et al. Anti-inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor {alpha} expression. Gut. 2010;59:1259–64.
Blackburn GL, Wollner SB, Jones DB. Bariatric surgery as treatment for type 2 diabetes. Curr Diab Rep. 2010;10(4):261–3.
Wong KE, Szeto FL, Zhang W, Ye H, Kong J, Zhang Z, et al. Involvement of the vitamin D receptor in energy metabolism: regulation of uncoupling proteins. Am J Physiol Endocrinol Metab. 2009;296(4):E820–8.
Wong KE, Kong J, Zhang W, Szeto FL, Ye H, Deb DK, et al. Targeted expression of human vitamin D receptor in adipocytes decreases energy expenditure and induces obesity in mice. J Biol Chem. 2011;286:33804–10.
Acknowledgments
Sources of Funding: Dr. Aghamohammadzadeh is an NIHR Academic Clinical Lecturer in Cardiology at the University of Manchester.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Aghamohammadzadeh, R., Heagerty, A.M. (2019). The Role of Perivascular Fat in Raising Blood Pressure in Obesity and Diabetes. In: Zimlichman, R., Julius, S., Mancia, G. (eds) Prehypertension and Cardiometabolic Syndrome. Updates in Hypertension and Cardiovascular Protection. Springer, Cham. https://doi.org/10.1007/978-3-319-75310-2_20
Download citation
DOI: https://doi.org/10.1007/978-3-319-75310-2_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75309-6
Online ISBN: 978-3-319-75310-2
eBook Packages: MedicineMedicine (R0)