
Abstract
During the last decades, obesity and osteoporosis have become important global health problems, and the belief that obesity is protective against osteoporosis has recently come into question. In fact, the latest epidemiologic and clinical studies have shown that a high level of fat mass might be a risk factor for osteoporosis and fragility fractures, and several potential mechanisms have been proposed to explain the complex relationship between the adipose tissue and bone. This chapter considers recent data in the literature to further evaluate the relationship between fat and bone tissue.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
During the last decades, obesity and osteoporosis have become important global health problems with an increasing prevalence worldwide [1,2,3,4], and the belief that obesity is protective against osteoporosis has recently come into question. In fact, the latest epidemiologic and clinical studies have shown that a high level of fat mass might be a risk factor for osteoporosis and fragility fractures [5,6,7,8].
Several potential mechanisms have been proposed to explain the complex relationship between the adipose tissue and bone.
For instance, fat has long been viewed as a passive energy reservoir, but since the discovery of leptin and the identification of other adipose tissue-derived hormones and serum mediators [9,10,11], it has come to be considered as an active endocrine organ involved in the modulation of the energy homeostasis. Adipose tissue, in fact, secretes various inflammatory cytokines, including interleukin (IL)-6, tumor necrosis factor-alpha (TNF-α) resistin, leptin, and adiponectin, which affect human energy and metabolic homeostasis and are involved in bone metabolism [12,13,14,15]. Moreover, fat tissue is one of the major sources of aromatase, an enzyme also expressed in the gonads, which synthesizes estrogens from androgen precursors. As known estrogens are steroid hormones which play a pivotal role in the maintenance of skeletal homeostasis, protecting against osteoporosis by reducing bone resorption and stimulating bone formation, and in obese postmenopausal women, increased estrogen synthesis by adipose tissue has been suggested as one of the potential mechanisms for the protective effect of fat mass on the bone. Thus, the pathophysiological role of adipose tissue in skeletal homeostasis lies in the production of several adipokines and hormones which modulate bone remodeling via their effects on either bone formation or resorption.
On the other hand, since the demonstration that bone cells express several specific hormone receptors, the skeleton is considered an endocrine target organ [13,14,15,16], and since recent observations have shown that bone-derived factors, such as osteocalcin and osteopontin, affect body weight control and glucose homeostasis [17,18,19], the bone has come to be considered an endocrine organ itself [20]. These considerations suggest a possible role of the bone as a player of a potential feedback mechanism between the skeleton and the other endocrine organs [20]. Thus, the cross talk between fat and bone likely constitutes a homeostatic feedback system in which adipokines and bone-derived molecules represent the link of an active bone-adipose axis.
Finally, adipocytes and osteoblasts originate from a common progenitor, a pluripotential mesenchymal stem cell (MSC) [21], which has an equal propensity for differentiation into adipocytes or osteoblasts (or other lines) under the influence of several cell-derived transcription factors. This process is complex, suggesting significant plasticity and multifaceted mechanism(s) of regulation within different cell lineages, among which are adipocytes and osteoblasts [22, 23].
2 Obesity and Osteoporosis: Fat and Bone Metabolism Interplay
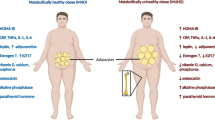
Obesity is recognized as a risk factor for metabolic and cardiovascular diseases [2]. However, it has been considered a protective factor for bone loss and osteoporosis, which is defined as a bone metabolic disease, characterized by a decrease in bone strength leading to an increased risk of developing spontaneous and traumatic fractures. Even though body fat and lean mass are linked with bone mineral density (BMD), with obesity apparently exerting protection against bone loss, especially after menopause, during the last decades numerous evidences have described an opposite event, suggesting an inverse relationship between obesity and osteoporosis. In fact recent studies have shown that an increased abdominal fat tissue might be considered a risk factor for osteoporosis [5, 7, 8].
The mechanisms whereby increased central adiposity leads to metabolic alterations, cardiovascular morbidity, and bone loss have been largely based on the demonstration that adipose tissue secretes a number of cytokines and bioactive compounds, named adipokines.
The adipokines, which include a variety of pro-inflammatory peptides, are involved in many physiological or pathological processes, and their disregulation is a strong determinant of the low-grade inflammatory state of obesity, which promotes a cascade of metabolic alterations leading to cardiovascular complications, insulin resistance or diabetes mellitus, and bone loss [9, 11].
Leptin, the first identified adipose tissue-derived factor, is an anorexigenic hormone secreted by adipocytes in proportion to body fat content. Leptin levels are typically elevated in obesity, which is considered a leptin-resistant state [24]. In obese subjects hyperleptinemia has been widely recognized as an independent cardiovascular risk factor associated with hyperinsulinemia and insulin resistance [25] while its effect on the bone is complex, and both negative and positive actions have been reported on BMD [26, 27]. Leptin-deficient ob/ob mice and leptin receptor-deficient db/db mice are extremely obese, with increased vertebral trabecular bone volume due to increased bone formation [28]. Interestingly, intracerebroventricular infusion of leptin in both ob/ob and wild-type mice was shown to decrease vertebral trabecular bone mass [28]. In vivo studies indicate that the effect of leptin might depend on its site and mode of action [29], and it has been proposed that peripheral administration of leptin could increase bone mass by inhibiting bone resorption and increasing bone formation, while inhibiting bone formation through a central nervous system effect [26]. In vitro studies also found that leptin can act directly on bone marrow-derived mesenchymal stem cells (BMSCs) to enhance their differentiation into osteoblasts and to inhibit their differentiation into adipocytes [30]. Finally, leptin inhibits the expression of neuropeptide Y (NPY), a hypothalamus-derived peptide, essential for the regulation of food consumption, energy homeostasis, and bone remodeling [31]. Specific NPY-knockout mice show a significant decrease in body weight, a significant increase in food intake, and twofold increase in trabecular bone volume compared with wild-type animals [32].
Adiponectin exerts a protective role on cardiovascular system and glucose metabolism, and in contrast with leptin, serum adiponectin levels are reduced in obese and diabetic subjects and increase after weight loss [33]. Low levels of adiponectin are a common feature of obesity and correlate with insulin resistance [34]. Adiponectin levels are inversely related to the circulating levels of C-reactive protein (CRP), TNF-α, and IL-6, which are powerful inhibitors of adiponectin expression and secretion in cultured human adipose cells [35]. Human osteoblasts express adiponectin and its receptors, and in vivo and in vitro studies show that adiponectin increases bone mass by suppressing osteoclastogenesis and activating osteoblastogenesis [36], likely indicating that a rise in adiponectin levels, caused by fat reduction, could have a beneficial effect on BMD.
Resistin is produced by macrophages and visceral adipocytes. It is elevated in obesity and regulates insulin sensitivity in the skeletal muscle and liver, and it is positively associated with insulin resistance and glucose tolerance in both human and animal models [37]. Resistin might also play a role in bone remodeling, increasing osteoblast proliferation, cytokine release, and osteoclast differentiation [38].
TNF-α is a pro-inflammatory cytokine which plays important regulatory effects on lipid metabolism, adipocyte function, insulin signaling, and bone remodeling [39]. Its expression has been shown to correlate with percent body fat and insulin resistance in humans [40], and it was further recognized that inflammatory processes predispose to bone loss, giving rise to speculation that inflammatory cytokines, such as IL-6 and TNF-α, may play critical roles in osteoclast activity [41]. Osteoclasts are the unique cells of the body tasked with resorbing the bone, and in the late 1990s, the identification of three different molecules built the bases of the modern bone biology: an osteoclastogenic cytokine, the receptor activator of NF-kB ligand (RANKL), its receptor (RANK), and its inhibitor osteoprotegerin (OPG) [42]. It is now clear that RANKL is the key osteoclastogenic cytokine effector, inducing osteoclast formation and promoting osteoclast resorptive activity, while OPG functions as a decoy receptor, preventing association of RANKL with RANK receptor, thus moderating osteoclastogenesis and bone resorption [43]. It has also become clear that TNF-α promotes RANKL production by BMSCs and mature osteoblasts, reduces OPG production, and upregulates the receptor RANK on osteoclast precursors, increasing their sensitivity to prevailing RANKL concentrations [44]. Additionally, TNF-α turns out to have another property that is relatively unique among the inflammatory cytokines; it has potent effects on osteoclastogenesis as it not only promotes RANKL production but synergizes with RANKL to amplify osteoclastogenesis and to intensify osteoclastic resorption by directly modulating RANKL-induced signal transduction pathways [45]. These effects are likely a consequence of the fact that RANKL is a TNF-superfamily member and functions through many of the same pathways induced by TNF-α itself.
IL-6 is a cytokine, which has a wide range of actions; it is secreted by several cell types, including fibroblast, endothelial cells, and adipocytes; and its plasma levels are significantly upregulated in human obesity and insulin resistance [46]. As TNF-α also IL-6 is a well-recognized stimulator of osteoclastogenesis and bone resorption. Several data show that IL-6 mRNA is expressed in preosteoblasts and osteoblasts [47] and that it stimulates osteoblast proliferation and differentiation by controlling the production of local factor [48]. In addition, IL-6 may play a role in bone formation in conditions of high bone turnover [49].
Emerging evidence points to a critical role for the skeleton in several homeostatic processes including energy balance and adipose metabolism, and the connection between fuel utilization and skeletal remodeling seems to begin in the bone marrow with lineage allocation of MSCs into adipocytes or osteoblasts.
Mature bone cells secrete factors that modulate insulin sensitivity and glucose metabolism, such as osteocalcin (OCN), by which the skeleton could function as an endocrine organ itself [50]. OCN is an osteoblast-specific protein and a major non-collagenous protein in the extracellular matrix. Karsenty and colleagues recently demonstrated that uncarboxylated OCN, acting as a prohormone, can increase β-cell proliferation, insulin secretion, insulin sensitivity, and adiponectin expression [51]. Thus, osteoblasts may be able to regulate glucose metabolism by modulating the bioactivity of OCN. In addition, more recent studies showed that OCN bioactivity is modulated by enhanced sympathetic tone driven by leptin, which has been shown to suppress insulin secretion by β-cells [52], and three recent studies have demonstrated an inverse correlation between serum OCN and plasma glucose levels, supporting a role for this pathway in humans [53]. Thus, a novel picture has emerged linking glucose metabolism, adipose stores, and skeletal activity.
Since its first description more than 20 years ago, osteopontin (OPN) has emerged as an active player in many physiological and pathological processes, including biomineralization, tissue remodeling, and inflammation. Modulation of immune cell response by OPN has been associated with various inflammatory diseases and may play a pivotal role in the development of adipose tissue inflammation, insulin resistance, and diabetes [54]. OPN expression is significantly upregulated by 40- and 80-fold in adipose tissue from diet-induced and genetically obese mice, respectively [55]. Moreover, it has been demonstrated that OPN expression in adipose tissue and circulating OPN levels were substantially elevated in obese, diabetic, and insulin-resistant patients compared with lean subjects and conversely that dietary weight loss significantly decreased OPN concentrations [56, 57].
3 Fat Bone Marrow and Osteoporosis: Cause or Consequence?
Adipocytes and osteoblasts originate from a common progenitor, a pluripotential MSC [58], which has an equal propensity for differentiation into adipocytes or osteoblasts or other lines, such as chondrocytes, fibroblast, and endothelial cells, under the influence of several cell-derived transcription factors. This process is complex, suggesting significant plasticity and multifaceted mechanism(s) of regulation within different cell lineages, among which are adipocytes and osteoblasts [24, 59].
Transdifferentiation is the irreversible switching of differentiated cells that sometimes occurs during disease [60], and it interests partially differentiated cells (e.g., preosteoblasts) that switches to another lineage (e.g., adipocytes) [61].
Fat bone marrow is indicative of aging and it is frequently observed in the presence of osteoporosis, especially in postmenopausal women [62]. One possible cause of bone marrow fat deposition is the aberrant commitment of BMMSCs into adipocytes due to their inability to differentiate into other cell lineages, such as osteoblasts. There exists an inverse relationship between bone marrow fat production and bone formation during osteoporosis; in fact an inhibited adipogenesis in patients with a high bone mass has been observed [63].
Recently, a correlation between the osteo-adipogenic transdifferentiation of bone marrow cells and numerous bone metabolism diseases has been established. Human BMMSC-derived osteoblasts, adipocytes, and chondrocytes had the potential to transdifferentiate to each lineage, and these findings provided new insights on the pathogenesis of skeletal diseases such as osteoporosis [64].
Estrogens can regulate several molecular signals within bone metabolism and play an important role in the development of bone marrow fat [65,66,67,68]. After menopause an increase in adipogenic switches in bone marrow and a decrease in bone mass have been observed [69, 70]. Several human and animal studies have examined the function of adipocytes in bone marrow. Mesenchymal stem cells isolated from bone marrow in postmenopausal osteoporotic patients express more adipose differentiation markers than those from subjects with normal bone mass [25], and pronounced fatty infiltration in the bone marrow of rats following oophorectomy has been observed, suggesting a pivotal role of estrogen in regulating adipocyte and osteoblast recruitment [26]. More recent studies have shown that estrogens are negative regulators of adipogenesis, and they are essential for osteogenic commitment; in particular, it seems that estrogens simultaneously induce osteogenesis and inhibits adipogenesis both in vivo and in vitro [71,72,73], and it has been demonstrated that estrogens suppress osteo-adipogenic transdifferentiation via canonical Wnt signaling, an important system which regulates bone development, adipogenic differentiation, and gene expression in whole process of bone metabolism [63, 74]. Specifically, canonical Wnt/β-catenin signaling is highly expressed in mesenchymal precursor cells and pluripotent cells, especially toward the osteoblast lineage, while it inhibits adipogenic differentiation [75]. Canonical Wnt signaling stabilizes and promotes cellular and nuclear β-catenin levels, which inhibits adipogenesis [75], and the suppression of Wnt signaling is essential for PPARγ induction and preadipocyte differentiation [76].
PPARγ plays a central role in initiating adipogenesis, and mutations of the PPARγ gene are associated with an altered balance between bone and fat formation in the bone marrow [59]. PPARγ insufficiency led to increased osteoblastogenesis in vitro and higher trabecular bone volume in vivo, confirming the key role of mesenchymal stem cell lineage allocation in the skeleton [58]. Interestingly, aged mice exhibit fat infiltration into bone marrow and enhanced expression of PPARγ, along with reduced mRNA expression of bone differentiation factors [77], and mice with premature aging (the SAM-P/6 model) show nearly identical patterns of adipocyte infiltration, with impaired osteoblastogenesis [78], indicating that aging or events that accelerate aging result in significant bone marrow adiposity and a defect in osteoblastogenesis in mice [79].
Conclusions
Body fat and bone interplay through several adipokines and bone-derived molecules, which modulate bone remodeling, adipogenesis, body weight control, and glucose homeostasis.
Thus, the existence of a cross talk between fat and the skeleton suggests a homeostatic feedback system in which adipokines and bone-derived molecules form part of an active bone-adipose axis, which due also its peculiarity to the common origin of osteoblasts and adipocytes from a pluripotent mesenchymal stem cell.
When specific conditions occur, such as aging, menopause, or diseases as osteoporosis, obesity, or metabolic alterations, it has been observed an osteo-adipogenic transdifferentiation and an aberrant commitment of BMMSCs into adipocytes because of their inability to differentiate into other cell lineages, such as osteoblasts.
However, the mechanism(s) by which all these events occur remains unclear, and this molecular control could be crucial to understand the pathogenesis of both obesity and osteoporosis.
References
Kado DM, Huang MH, Karlamangla AS, Barrett-Connor E, Greendale GA. Hyperkyphotic posture predicts mortality in older community-dwelling men and women: a prospective study. J Am Geriatr Soc. 2004;52:1662–7.
Rossner S. Obesity: the disease of the twenty-first century. Int J Obes Relat Metab Disord. 2002;26(Suppl 4):S2–4.
Hu FB. Overweight and obesity in women: health risks and consequences. J Women Health (Larchmt). 2003;12(2):163–72.
Obesity: preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ Tech Rep Ser. 2000;894:1–253.
Greco EA, Fornari R, Rossi F, Santiemma V, Prossomariti G, Annoscia C, Aversa A, Brama M, Marini M, Donini LM, Spera G, Lenzi A, Lubrano C, Migliaccio S. Is obesity protective for osteoporosis? Evaluation of bone mineral density in individuals with high body mass index. Int J Clin Pract. 2010;64(6):817–20.
Kim KC, Shin DH, Lee SY, Im JA, Lee DC. Relation between obesity and bone mineral density and vertebral fractures in Korean postmenopausal women. Yonsei Med J. 2010;51(6):857–63.
Greco EA, Francomano D, Fornari R, Marocco C, Lubrano C, Papa V, Wannenes F, Di Luigi L, Donini LM, Lenzi A, Aversa A, Migliaccio S. Negative association between trunk fat, insulin resistance and skeleton in obese women. World J Diabetes. 2013;4(2):31–9.
Compston JE, Flahive J, Hosmer DV, Watts NB, Siris ES, Silverman S, Saag KG, Roux C, Rossini M, Pfeilschiffer J, Nieves JW, Netelenbos JC, March L, LaCroix AZ, Hooven FH, Greenspan SL, Gehlbach SH, Diez-Perez A, Cooper C, Chapurlat RD, Boonen S, Anderson FA Jr, Adami S, Adachi JD, GLOW Investigators. Relationship of weight, height, and body mass index with fracture risk at different sites in postmenopausal women: the Global Longitudinal study of Osteoporosis in Women (GLOW). J Bone Miner Res. 2014;29(2):487–93.
Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–51.
Steppan CM, Crawford DT, Chidsey-Frink KL, Ke H, Swick AG. Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Pept. 2000;92:73–8.
Vendrell J, Broch M, Vilarrasa N, Molina A, Gomez JM, Gutierrez C, Simon I, Soler J, Richart C. Resistin, adiponectin, ghrelin, leptin, and proinflammatory cytokines: relationships in obesity. Obes Res. 2004;12:962–71.
Magni P, Dozio E, Galliera E, Ruscica M, Corsi MM. Molecular aspects of adipokine-bone interactions. Curr Mol Med. 2010;10(6):522–32.
Eriksen EF, Colvard DS, Berg NJ, Graham ML, Mann KG, Spelsberg TC, Riggs BL. Evidence of estrogen receptors in normal human osteoblast-like cells. Science. 1988;241(4861):84–6.
Kim HJ. New understanding of glucocorticoid action in bone cells. BMB Rep. 2010;43(8):524–9.
Komm BS, Terpening CM, Benz DJ, Graeme KA, Gallegos A, Korc M, Greene GL, O’Malley BW, Haussler MR. Estrogen binding, receptor mRNA, and biologic response in osteoblast-like osteosarcoma cells. Science. 1988;241(4861):81–4.
Migliaccio S, Davis VL, Gibson MK, Gray TK, Korach KS. Estrogens modulate the responsiveness of osteoblast-like cells (ROS 17/2.8) stably transfected with estrogen receptor. Endocrinology. 1992;130(5):2617–24.
Gomez-Ambrosi J, Rodrıguez A, Catalan V, Fruhbeck G. The bone-adipose axis in obesity and weight loss. Obes Surg. 2008;18:1134–43.
Takeda S. Effect of obesity on bone metabolism. Clin Calcium. 2008;18:632–7.
Gimble JM, Zvonic S, Floyd ZE, Kassem M, Nuttall ME. Playing with bone and fat. J Cell Biochem. 2006;98:251–66.
Fukumoto S, Martrin TJ. Bone as an endocrine organ. Trends Endocrinol Metab. 2009;20(5):230–6.
Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, Terauchi Y, Harada Y, Azuma Y, Nakamura K, Kadowaki T, Kawaguchi H. PPARγ insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–55.
Gimble JM, Robinson CE, Wu X, Kelly KA, Rodriguez BR, Kliewer SA, Lehmann JM, Morris DC. Peroxisome proliferator activated receptor-gamma activation by thiazolidinediones induces adipogenesis in bone marrow stromal cells. Mol Pharmacol. 1996;50:1087–94.
Rodriguez JP, Montecinos L, Rios S, Reyes P, Martinez J. Mesenchymal stem cells from osteoporotic patients produce a type I collagen-deficient extracellular matrix favoring adipogenic differentiation. J Cell Biochem. 2000;79:557–65.
Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, Caro JF. Serum immunoreactive leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;34:292–5.
Martin SS, Qasim A, Reilly MP. Leptin resistance: a possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J Am Coll Cardiol. 2008;52:1201–10.
Kontogianni MD, Dafni UG, Routsias JG, Skopouli FN. Blood leptin and adiponectin as possible mediators of the relation between fat mass and BMD in perimenopausal women. J Bone Miner Res. 2004;19:546–55.
Goulding A, Taylor RW. Plasma leptin values in relation to bone mass and density and to dynamic biochemical markers of bone resorption and formation in postmenopausal women. Calcif Tissue Int. 1998;63:456–8.
Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, Shen J, Vinson C, Rueger JM, Karsenty G. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100:197–207.
Thomas T. The complex effects of leptin on bone metabolism through multiple pathways. Curr Opin Pharmacol. 2004;4:295–300.
Thomas T, Gori F, Khosla S, Jensen MD, Burguera B, Riggs BL. Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology. 1999;140:1630–8.
Baldock PA, Sainsbury A, Couzens M, Enriquez RF, Thomas GP, Gardiner EM, Herzog H. Hypothalamic Y2 receptors regulate bone formation. J Clin Invest. 2002;109:915–21.
Sainsbury A, Schwarzer C, Couzens M, Fetissov S, Furtinger S, Jenkins A, cox HM, Sperk G, Hokfelt T, Herzog H. Important role of hypothalamic Y2 receptors in body weight regulation revealed in conditional knockout mice. Proc Natl Acad Sci U S A. 2002;99:8938–43.
Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, Engel J, Brownlee M, Scherer PE. Structure-function studies of the adipocytes-secreted hormone Acrp30/adiponectin. Implications for metabolic regulation and bioactivity. J Biol Chem. 2003;278:9073–85.
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudio K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with body lipoatrophy and obesity. Nat Med. 2001;7:941–6.
Fasshauer M, Klein J, Neumann S, Eszlinger M, Paschke R. Hormonal regulation of adiponectin gene expression in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2002;290:1084–9.
Jurimae J, Rembel K, Jurimae T, Rehand M. Adiponectin is associated with bone mineral density in perimenopausal women. Horm Metab Res. 2005;37:297–302.
Ukkola O. Resistin–a mediator of obesity-associated insulin resistance or an innocent bystander? Eur J Endocrinol. 2002;147:571–4.
Thommesen L, Stunes AK, Monjo M, Grosvik K, Tamburstuen MV, Kjobli E, Lyngstadaas SP, Reseland JE, Syversen U. Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J Cell Biochem. 2006;99(3):824–34.
Fasshauer M, Klein J, Krahlisch S, Lossner U, Klier M, Bluher M, Paschke R. GH is a positive regulator of tumor necrosis factor-alpha-induced adipose related protein in 3T3-L1 adipocytes. J Endocrinol. 2003;178:523–31.
Hotamisligil G, Arner P, Caro J, Atkinson R, Spiegelman B. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–15.
Pacifici R, Brown C, Puscheck E, Friedrich E, Slatopolsky E, Maggio D, McCracken R, Avioli LV. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc Natl Acad Sci U S A. 1991;88(12):5134–8.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennet L, Boone T, Shimamoto G, DeRose M, Elliot R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–19.
Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390(6656):175–9.
Wei S, Kitaura H, Zhou P, Ross P, Teitelbaum SL. IL-1 mediates TNFα-induce osteoclastogenesis. J Clin Invest. 2005;115(2):282–90.
Cenci S, Weitzmann MN, Roggia C, Namba N, Novack D, Woodring J, Pacifici R. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-α. J Clin Invest. 2000;106(10):1229–37.
Vozarova B, Weyer C, Hanson K, Tataranni PA, Bogardus C, Pratley RE. Circulating IL-6 in relation to adiposity, insulin action and insulin secretion. Obes Res. 2001;9:414–7.
Dodds A, Merry K, Littlewood A, Gowen M. Expression of mRNA for IL1 beta, IL6 and TGF beta 1 in developing human bone and cartilage. J Histochem Cytochem. 1994;42:733–44.
Taguchi Y, Yamamoto M, Yamate T, Lin SC, Mocharla H, DeTogni P, Nakayama N, Boyce BF, Abe E, Manolagas SC. Interleukin-6-type cytokines stimulate mesenchymal progenitor differentiation toward the osteoblastic lineage. Proc Assoc Am Physicians. 1998;110:559–74.
Sims NA, Jenkins BJ, Quinn JM, Nakamura A, Glatt M, Gillespie MT, Ernst M, Martin TJ. Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J Clin Invest. 2004;113:379–89.
Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, Zhang Z, Kim JK, Mauvais-Jarvis F, Ducy P, Karsenty G. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–69.
Ferron M, Hinoi E, Karsenty G, Ducy P. Osteocalcin differentially regulates beta cell and dipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci U S A. 2008;105:5266–70.
Hinoi E, Gao N, Jung DY, Yadav V, Yoshizawa T, Myers MG Jr, Chua SC Jr, Kim JK, Kaestner KH, Karsenty G. The sympathetic tone mediates leptin’s inhibition of insulin secretion by modulating osteocalcin bioactivity. J Cell Biol. 2008;183:1235–42.
Covey SD, Wideman RD, McDonald C, Unniappan S, Huynh F, Asadi A, Speck M, Webber T, Chua SC, Kieffer TJ. The pancreatic beta cell is a key site for mediating the effects of leptin on glucose homeostasis. Cell Metab. 2006;4:291–302.
Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:2302–9.
Kiefer FW, Zeyda M, Todoric J, Huber J, Geyeregger R, Weichhart T, Aszmann O, Ludvik B, Silberhumer GR, Prager G, Stulnig TM. Osteopontin expression in human and murine obesity: extensive local up-regulation in adipose tissue but minimal systemic alterations. Endocrinology. 2008;149:1350–7.
Sarac F, Basoglu OK, Gunduz C, Bayrak H, Biray Avci C, Akcicek F. Association of osteopontin and tumor necrosis factor-alpha levels with insulin resistance in obese patients with obstructive sleep apnea syndrome. J Endocrinol Invest. 2011;34:528–33.
You JS, Ji HI, Chang KJ, Yoo MC, Yang HI, Jeong IK, Kim KS. Serum osteopontin concentration is decreased by exercise-induced fat loss but is not correlated with body fat percentage in obese humans. Mol Med Rep. 2013;8:579–84.
Sekiya I, Larson BL, Vuoristo JT, Cui JG, Prockop DJ. Adipogenic differentiation of human adult stem cells from bone marrow stroma (MSCs). J Bone Miner Res. 2004;19:256–64.
Martin RB, Zissimos SL. Relationships between marrow fat and bone turnover in ovariectomized and intact rats. Bone. 1991;12:123–31.
Burke ZD, Tosh D. Therapeutic potential of transdifferentiated cells. Clin Sci (Lond). 2005;108:309–21.
Schilling T, Kuffner R, Klein-Hitpass L, Zimmer R, Jakob F, Schutze N. Microarray analyses of transdifferentiated mesenchymal stem cells. J Cell Biochem. 2008;103:413–33.
Menagh PJ, Turner RT, Jump DB, Wong CP, Lowry MB, Yakar S, Rosen CJ, Iwaniec UT. Growth hormone regulates the balance between bone formation and bone marrow adiposity. J Bone Miner Res. 2010;25:757–68.
Gao B, Huang Q, Lin Y-S, Wei B-Y, Guo Y-S, Sun Z, Wang L, Fan J, Zhang H-Y, Han Y-H, Li X-J, Shi J, Liu J, Yang L, Luo Z-J. Dose-dependent effect of estrogen suppresses the osteo-adipogenic transdifferentiation of osteoblasts via canonical wnt signaling pathway. PLoS One. 2014;9(6):e-99137.
Song L, Tuan RS. Transdifferentiation potential of human mesenchymal stem cells derived from bone marrow. FASEB J. 2004;18:980–2.
Abdallah BM, Ditzel N, Mahmood A, Isa A, Traustadottir GA, Schilling AF, ruiz-hidalgo MJ, Laborda J, Amling M, Kassem M. DLK1 is a novel regulator of bone mass that mediates estrogen deficiency induced bone loss in mice. J Bone Miner Res. 2011;26:1457–71.
Kamiya Y, Chen J, Xu M, Utreja A, Choi T, Drissi H, Wadhwa S. Increased mandibular condylar growth in mice with estrogen receptor beta deficiency. J Bone Miner Res. 2013;28:1127–34.
Song L, Zhao J, Zhang X, Li H, Zhou Y. Icariin induces osteoblast proliferation, differentiation and mineralization through estrogen receptor mediated ERK and JNK signal activation. Eur J Pharmacol. 2013;714:15–22.
Pierroz DD, Rufo A, Bianchi EN, Glatt V, Capulli M, Rucci N, Cavat F, Rizzoli R, Teti A, Bouxsein ML, Ferrari SL. Beta Arrestin2 regulates RANKL and ephrins gene expression in response to bone remodeling in mice. J Bone Miner Res. 2009;24:775–84.
Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology. 2001;2:165–71.
Gambacciani M, Ciaponi M, Cappagli B, Piaggesi L, De Simone L, Orlandi R, genazzani AR. Body weight, body fat distribution, and hormonal replacement therapy in early postmenopausal women. J Clin Endocrinol Metab. 1997;82:414–7.
Guo YF, Xiong DH, Shen H, Zhao LJ, Xiao P, guo Y, Wang W, Yang TL, Recker RR, Deng HW. Polymorphisms of the low-density lipoprotein receptor-related protein 5 (LRP5) gene are associated with obesity phenotypes in a large family-based association study. J Med Genet. 2006;43:798–803.
Mani A, Radhakrishnan J, Wang H, Mani A, Mani MA, Nelson-William C, Carew KS, Mane S, Najmabadi H, Wu D, Lifton RP. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278–82.
Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–3.
Colaianni G, Brunetti G, Faienza MF, Colucci S, Grano M. Osteoporosis and obesity: role of Wnt pathway in human and murine models. World J Orthop. 2014;5(3):242–6.
Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–9.
Qiu W, Chen L, Kassem M. Activation of non-canonical Wnt/JNK pathway by Wnt3a is associated with differentiation fate determination of human bone marrow stromal (mesenchymal) stem cells. Biochem Biophys Res Commun. 2011;413:98–104.
Moerman EJ, Teng K, Lipschitz DA, Lecka-Czernik B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-γ2-transcription factor and TGF-β/BMP signaling pathways. Aging Cell. 2004;3:379–89.
Kajkenova O, Lecka-Czernik B, Gubrij I, Hauser SP, Takahashi K, Parfitt AM, Jilka RL, Manolagas SC, Lipschitz DA. Increased adipogenesis and myelopoiesis in the bone marrow of SAMP6: a murine model of defective osteoblastogenesis and low turnover osteopenia. J Bone Miner Res. 1997;12:1772–9.
Duque G, Macoritto M, Kremer R. Vitamin D treatment of senescence accelerated mice (SAM-P/6) induces several regulators of stromal cell plasticity. Biogerontology. 2004;5:421–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Greco, E.A., Fornari, R., Lenzi, A., Migliaccio, S. (2018). Obesity and Osteoporosis: Is the Paradigm Changing?. In: Lenzi, A., Migliaccio, S. (eds) Multidisciplinary Approach to Osteoporosis. Springer, Cham. https://doi.org/10.1007/978-3-319-75110-8_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-75110-8_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75108-5
Online ISBN: 978-3-319-75110-8
eBook Packages: MedicineMedicine (R0)