
Abstract
Prostate cancer is a leading cause of morbidity and mortality among men in the United States and Western Europe. From 5% to 10% of prostate cancer cases are believed to be primarily caused by high-risk inherited genetic factors or prostate cancer susceptibility genes. Case-control studies, linkage analyses, admixture mapping and genome-wide association studies (GWAS) have identified a number of candidate genes associated with prostate cancer susceptibility; however, the replication of these findings remains inconsistent, with numerous genes likely to be involved. Studies of ethnic and founder populations have identified inherited genetic factors associated with a higher risk of prostate cancer. These include the African-American population and 8q24 locus, the Ashkenazi Jewish and BRCA gene mutations and Northern Europeans and the HOXB13 gene. Furthermore, prostate cancer risk appears to be increased by the loss of effective DNA repair processes which occurs in populations with BRCA gene mutations, Lynch syndrome and Fanconi anaemia. Tumours with a disrupted DNA repair pathway are hypersensitive to PARP inhibitors, and these agents have been shown to be efficacious in prostate cancer. The formation of DNA adducts can lead to DNA replication errors and the potential for carcinogenesis; however, whether ethnicity or an inherited susceptibility is associated with an increased risk of prostate cancer is uncertain. Genes identified through GWAS may have a role in prostate cancer screening and as targets for therapeutic targets.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

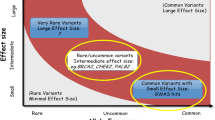

Keywords
Introduction
Prostate cancer is a leading cause of morbidity and mortality among men in the United States and Western Europe [1]. Widespread screening with prostate-specific antigen (PSA) and early treatment of localized prostate cancer have contributed to a decrease in age-adjusted rates of death due to prostate cancer [2]. Advancing age, diet, lifestyle-related factors, family history and ethnicity have long been recognized as contributors to the risk of prostate cancer [3]. Recent discoveries in the genetics of prostate cancer and in the acquired mutations that accumulate in prostate cancer cells have improved our understanding of the development of prostate cancer [1]. Evidence supporting the role of genetic factors comes from studies of relatives of patients with prostate cancer, founder populations, genome-wide association studies (GWAS), case-control studies and linkage analyses and from studies in patients with abnormalities in known cancer syndrome-associated genes such as BRCA1 and BRCA2 [4].
Hereditary Prostate Cancer
The risk of prostate cancer is increased approximately twofold in men with affected first-degree relatives [5]. This risk is increased further if more than one family member is affected or if there is early age of onset in a family member [6, 7]. In a cohort study of 44,000 pairs of Scandinavian twins, concordance for cancer in identical twins was higher for prostate cancer than either breast or colorectal cancer [8]. This study estimated that as much as 42% of the risk of prostate cancer could be explained by heritable factors. Interestingly, in addition to increasing the risk of developing prostate cancer, genetic factors may also influence the prognosis in these men. In a Swedish study in men with prostate cancer whose fathers also had prostate cancer, the survival of sons was similar to that of their fathers [9].
As with other cancers, familial clustering of prostate cancer has been reported. The Massachusetts Male Aging Study of 1149 Boston-area men found a much higher risk for prostate cancer among men with a family history of the disease which appeared to be independent of environmental factors [10]. Further associations between family history and risk of prostate cancer were characterized in a population-based case-control study of 1557 men aged 40–86 years [11]. At baseline, 4.6% of the cohort reported a family history of prostate cancer in a brother or father, and this was positively associated with prostate cancer risk after adjustment for age, alcohol and dietary factors. However, at least some of this familial clustering is due to increased prostate cancer screening in families thought to be at high risk [12].
This recognition that prostate cancer clusters within families has led investigators to collect multiple-case families in order to localize prostate cancer susceptibility genes through linkage analysis. It is now estimated that 5–10% of prostate cancer cases are primarily caused by high-risk inherited genetic factors or prostate cancer susceptibility genes [13,14,15]. Linkage analysis studies have mapped several susceptibility loci, and a number of genes have been cloned at these loci [5]. These include 1q24–25 (HPC1/RNASEL), 1q42.2–43 (PCAP), Xq27-q28 (HPCX), 1p36 (CAPB), 20q13 (HPC20), 8p22–23 (MSR1), 8q24 and 17p11 (HPC2/ELAC2) [16,17,18,19,20,21,22,23,24,25,26] (see Table 5.1). However, the replication of these findings remains inconsistent, with numerous genes likely to be involved in prostate cancer.
Ethnic groups and founder populations are of particular interest for genetic mapping of complex traits due to a lack of genetic heterogeneity. African-American men have the world’s highest incidence of prostate cancer and a twofold higher mortality rate compared to Caucasians [27]. Admixture mapping has identified a number of chromosomal regions associated with prostate cancer in African Americans including the inherited variation at the 8q24 risk locus which appears to contribute to differences in African-American and European-American incidence of the disease [28,29,30]. Ashkenazi Jewish men have a higher incidence of mutations in BRCA1 and BRCA2 genes than the general population, and studies have reported a significant increased risk of prostate cancer in these men [31].
Although linkage studies have provided evidence that prostate cancer has a strong genetic component, identifying specific genes that contribute to the development of the disease has proven more difficult. Using gene sequencing technology, it has been possible to identify rare genes associated with an increased cancer risk [32]. Susceptibility genes with an associated increased risk for prostate cancer that have been identified include ZNF652, PRAC, EMSY, KLF6, AMACR, NBS1, SRD5A2, ER-beta, E-cadherin (CDH1), CHEK2, BRCA1 and BRCA2 [33,34,35,36,37,38,39,40,41,42,43,44,45,46] (see Table 5.2).
Single nucleotide polymorphisms (SNPs) can be used to identify candidate genes by identifying alleles that are associated with an increased susceptibility to prostate cancer [47]. Using GWAS that include SNPs, more than 100 prostate cancer susceptibility loci have been identified, explaining an estimated 30% of the familial risk for this disease [48]. Based on combined risks conferred by known risk loci, the top 1% of the risk distribution has a 4.7-fold higher risk than the average of the population [49]. Among the genes that have been identified in this way are HOXB13, MSMB, LMTK2, KLK3, CPNE3, IL16, CDH13, and HNF1B [49,50,51,52,53].
BRCA1 and BRCA2 Genes
The BRCA1 and BRCA2 genes are tumour suppressor genes inherited in an autosomal dominant pattern with reduced penetrance [54, 55]. The development of cancer in individuals with germline mutations in the BRCA genes requires somatic mutation of the remaining wild-type allele [56]. The BRCA1 and BRCA2 genes encode proteins that maintain genomic stability by promoting repair of DNA double-strand breaks [57]. The main functions of BRCA1 are DNA damage response and repair, transcriptional regulation and chromatin modelling [58, 59]. The role of BRCA2 is more limited to DNA repair by homologous recombination including regulating RAD51 activity, an important component of the DNA repair process [60]. Therefore, functional loss of BRCA1 or BRCA2 leads to a deficiency in repairing DNA double-strand breaks by conservative mechanisms, allowing cells to repair these lesions through other methods which are potentially mutagenic. This genomic instability may explain the increased risk of cancer caused by deleted mutations in the BRCA genes, although it is unclear why these mutations are particularly associated with certain cancers such as breast and ovarian cancer and, less commonly, prostate cancer [61].
BRCA Genes and Cancer Risk
Germline mutations in the BRCA genes have been shown to be associated with an increased risk of breast and ovarian cancer [33, 62]. The extent to which BRCA mutation carriers are at an increased risk of other cancers has been less clear; however, the presence of BRCA1 or BRCA2 mutations in men has been shown to be associated with an increased risk of developing prostate cancer [31, 43, 63,64,65,66,67,68,69,70]. As an example, in a cohort study that involved 3728 men from 173 breast–ovarian cancer families with BRCA2 mutations, the estimated relative risk (RR) of prostate cancer among BRCA2 carriers was 4.7-fold greater than controls [71] (see Table 5.3). The risk of developing prostate cancer in BRCA1 mutation carriers appears to be lower (see Table 5.4). In a multinational cohort study of 11,847 individuals with BRCA1 mutations, the risk of prostate cancer was elevated 1.8-fold in men under the age of 65, but this increase was not observed in older men [73].
Prostate cancer in men with BRCA2 mutations also appears to be associated with more aggressive histology and a substantially worse prognosis [63, 74,75,76]. In a study from Iceland that included 30 men with a mutation in BRCA2, prostate cancer was diagnosed at an earlier age (69 versus 74 years) and was associated with a significantly shorter survival (2.1 versus 12.4 years) [77]. Similarly, in a multinational cohort study of men with prostate cancer that included 183 men from known BRCA2 families and 119 from BRCA1 families, those from BRCA2 families had a significantly shorter survival (4.0 versus 8.0 years) [78]. A Spanish study of 2,000 men with prostate cancer confirmed the worse prognosis in mutated BRCA2 patients with a significant survival advantage if patients were noncarriers (15.7 versus 8.6 years) [76].
The IMPACT trial (Identification of Men with a genetic predisposition to ProstAte Cancer: Targeted screening) is looking at the feasibility and role of PSA screening in men who are carriers for BRCA1 or BRCA2 mutations [79]. Results from the initial screening round in this study showed a detection rate for prostate cancer of 2.4%. There was an evidence of a more aggressive phenotype in these patients with more than two-thirds of the prostate cancer detected in the BRCA2 carriers being classified as intermediate or high risk. Furthermore, the only cancers detected in men younger than 50 years of age were in BRCA1 and BRCA2 carriers. A study by Castro et al. showed that BRCA carriers treated for localized prostate cancer have worse outcomes than noncarriers because they relapse and progress earlier to lethal metastatic disease [75]. This data adds to the increasing evidence that BRCA1 and BRCA2 mutation carriers develop more aggressive disease at a younger age suggesting that screening may be beneficial in this subgroup [79].
BRCA and Tumourigenesis
It has been proposed that the BRCA genes may act as tumour suppressors in prostate cells and that their functional loss predisposes to the development of premalignant prostatic lesions [80, 81]. It has been shown in animal studies that the simultaneous deletion of BRCA2 and the tumour suppressor p53 give rise to focal hyperplasia and high-grade PIN [80]. Furthermore, evidence has shown that functional BRCA1 and BRCA2 proteins may limit the metastatic potential of neoplastic cells [81]. This is achieved by downregulating MMP-9 production through inhibition of PI3-kinase/AKT and activation of MAPK/ERK pathways, which prevents cancer cell migration and invasion [82, 83].
Poly(ADP-ribose) Polymerase 1 (PARP1)
The DNA repair defect associated with mutations in BRCA1 or BRCA2 is being used to develop new targeted therapeutic approaches for prostate cancer [84, 85]. Poly(ADP-ribose) polymerase 1 (PARP1) is a nuclear enzyme which assists in the maintenance of genomic stability by identifying sites of DNA damage and recruiting repair mechanisms [86]. A number of studies indicate that tumour cells with a defect in homologous recombination, such as tumours bearing BRCA1 or BRCA2 mutations, depend on compensatory DNA repair of double-strand breaks, for which the enzyme PARP1 is essential [87, 88]. Consequently, tumours with defects in homologous recombination are hypersensitive to drugs that inhibit PARP [89]. PARP has also been implicated in the transcription regulation of the androgen receptor (AR) and has also shown antitumor activity in preclinical models of TMPRSS2-ERG-rearranged prostate cancer [90]. Additionally, PARP inhibitors suppress AR-target gene expression and tumour proliferation [91]. This had led to several studies examining the role of PARP inhibitors in prostate cancer [92,93,94]. Olaparib is a PARP inhibitor which has shown antitumor activity in both germline and sporadic cases of metastatic, castration-resistant prostate cancer with DNA-repair defects [93].
Susceptibility Genes
Androgen Receptor (AR) Gene
The androgen pathway and its function in the development and progression of prostate cancer has been well established, and overexpression of the AR gene has been associated with poor prognosis [95]. Altered activity of the androgen receptor caused by inherited variants of the AR gene, located on the X chromosome, may increase the risk of prostate cancer [96]. The length of the polymorphic trinucleotide CAG and GGN microsatellite repeats in exon 1 of the AR gene has been associated with an increased risk of prostate cancer; however, data from other studies has been conflicting [97, 98]. Germline mutations in the AR gene associated with an increased risk of prostate cancer have been identified. In a Finnish study, the R726L substitution in the AR gene may confer up to sixfold increased risk of prostate cancer and may contribute to cancer development in up to 2% of prostate cancer patients [99]; however, a subsequent Finnish study did not replicate these results [100]. Therefore, germline AR mutations may only contribute to a small fraction of familial and early-onset cases of prostate cancer.
RNASEL Gene
The RNASEL gene (encodes for RNase L enzyme) has been mapped to the HPC1 (hereditary prostate cancer 1) region at 1q24–25 and regulates cell proliferation and apoptosis through the interferon-regulated 2–5A pathway [101, 102]. Interestingly, much of the evidence for a role for RNASEL in prostate cancer seems to be in cases with a positive family history, supporting the initial discovery in hereditary patients [103]. Multiple variants of the RNASEL gene have been described including Arg462G, 471delAAAG, R462Q, E265X and D541E and may be involved in up to 13% of prostate cancer cases, though the true role of RNASEL genetic variation and its influence on prostate cancer risk have been controversial [104, 105]. The R462Q variant was originally associated with an increasing risk of prostate cancer due to a significant decrease in RNASE L enzymatic activity; however, this finding has not been universally replicable [104, 106, 107]. Furthermore, results of a meta-analysis of ten independent RNASEL genotyping studies for the variants E265X, R462Q and D541E suggested that although there was no overall effect on prostate cancer risk, there was a less than twofold increase in the risk of developing prostate cancer in Caucasians with the D541E variant [108]. Missense mutations in R462Q and D541E have been shown to be associated with an increased risk of advanced-stage disease only in the pre-PSA era with no effect on survival [109].
TMPRSS2-ERG Gene Fusion
Fusions of the androgen-regulated gene TMPRSS2 to the oncogenic ETS transcription factor ERG occur in over 50% of prostate cancers [110]. It has been found to vary according to ethnic groups, to be associated with p53 mutation expression and to have a more aggressive phenotype [111, 112]. Significant association of TMPRSS2-ERG fusion-positive prostate cancer with rare variants in the DNA repair genes POLI (variant F532S) and ESC01 (variant N191S) has also been found [113]. Furthermore, linkage analysis has found the presence of an inherited susceptibility to develop the TMPRSS2-ERG fusion with several loci located on chromosomes #9, #18 and X [114, 115]. Therefore, familial aggregation of TMPRSS2-ERG could be due to an inherited chromosomal instability caused by variations in the DNA repair pathway leading to genomic instability. ERG has been also shown to interact with the PARP1 enzymes in the DNA repair pathway, and interestingly PARP1 inhibitors have been shown to inhibit ERG-positive prostate cancer xenograft growth in a manner similar to that of BRCA1/2 deficiency [90].
HOXB13
The homeobox B13 (HOXB13) gene codes for a transcription factor that is important in prostate development [116,117,118]. Linkage to 17q21–22 was initially reported by the Prostate Cancer Genetics Project at the University of Michigan from pedigrees of families with hereditary prostate cancer [119, 120]. Next-generation sequencing of the 17q21–22 region identified the G84E variant of the HOXB13 gene in families with hereditary prostate cancer [116, 121]. Researchers have demonstrated that the HOXB13 G84E mutation is present in about 5% of prostate cancer families, predominantly of European descent, and have shown it to be associated with an increased prostate cancer risk [122] (see Table 5.5). In Europe, the prevalence of the HOXB13 G84E is highest in the Nordic countries, especially Finland and Sweden with a prevalence among men diagnosed with familial prostate cancer of 8.4% [125]. In the Reduction by Dutasteride of Prostate Cancer Events (REDUCE) study, an international multicentre chemoprevention trial of 3508 subjects, the HOXB13 G84E mutation was only present in Caucasians, with the highest prevalence in Northern Europeans, followed by Western Europeans and North Americans with no carriers identified in Africa, Australia, Latin America and the rest of the European population [124]. A number of studies have confirmed an increased risk of prostate cancer in patients with the HOXB13 G84E variant [116, 122, 124, 126]. In a study of 5083 unrelated subjects with prostate cancer and 1401 controls, there was a 20-fold increase in the frequency of the HOXB13 G84E mutation in men with prostate cancer compared with those without it (1.4 versus 0.1 percent) [116]. Similarly, in the 4-year follow-up of the REDUCE study, the prostate cancer detection rate was 53.8% among mutation carriers and 22.0% among noncarriers, with a relative risk of 2.45 [124]. In a second case-control study of familial prostate cancer, investigators genotyped 928 familial prostate cancer probands and 930 control probands without a personal or family history of prostate cancer and found the point estimate of the odds ratio, adjusted for age, was 7.9 among carriers of the mutation [122]. The estimate was greater among cases with a family history of three or more relatives affected (OR = 11.8), compared to a family history of only two affected (OR = 5.8). In a British case-control study assessing the prevalence of HOXB13 G84E, investigators identified the variant in 0.5% of healthy controls and 1.5% of prostate cancer cases and found the presence of HOXB13 G84E to be associated with a 2.93-fold increased risk of prostate cancer [126]. The risk was even higher among men with family history of prostate cancer supporting the hereditary link.
The penetrance estimates for prostate cancer development in HOXB13 G84E mutation carriers have also been reported. A study from Sweden found HOXB13 G84E to be prevalent in more than 1% of the population and to be associated with a 3.5-fold increased risk of prostate cancer with an estimated 33% lifetime risk of prostate cancer [127]. Furthermore, an Australian study reported age-specific cumulative risk of prostate cancer of up to 60% by the age 80 years [128].
HOXB13 expression has been linked to advanced pT stage, high Gleason grade, positive lymph node status, high preoperative PSA levels, TMPRSS2:ERG fusion, PTEN deletions, AR expression, cell proliferation, reduced PSA expression and early PSA recurrence; however, it has not been found to have an effect on prognostic outcomes and overall or cancer-specific survival [126, 131, 132]. It has also been demonstrated that the prostate cancer risk-associated T allele of rs339331 enhances HOXB13 chromatin binding and drives allele-specific upregulation of the rs339331-associated gene RFX6 which might have a role in prostate cancer cellular transformation [130]. It appears that HOXB13 has an important role in prostate cancer development; however, the mechanism by which it contributes to the pathogenesis of prostate cancer remains unknown.
Mismatch Repair (MMR) Genes and Prostate Cancer
Lynch Syndrome
Lynch syndrome is an autosomal dominant disorder caused by a germline mutation in one of the mismatch repair (MMR) genes , MLH1, MSH2, MSH6 or PMS2 [133]. Chromosomal deletion, point mutation or epigenetic inactivation by hypermethylation in a second allele predisposes to a lack of MMR protein function, leading to an accumulation of mutations [134]. This can lead to malignant transformation of cells and tumour formation with a mutated phenotype, demonstrated by the presence of microsatellite instability (MSI) and lack of one or more of the four MMR proteins on staining by immunohistochemistry (IHC) [135]. There is an increased risk of several cancers in patients with Lynch syndrome including colorectal, endometrial, ovarian, gastric, small intestinal, pancreatic, ureteral, brain and sebaceous gland adenocarcinomas [136]. Screening for colorectal cancer and prophylactic surgery for gynaecological cancers have been shown to improve outcomes in these patients [137, 138]. Prostate cancer is currently not considered part of the Lynch syndrome spectrum, and data for the association has been inconclusive [139,140,141]. However, a number of studies have shown the cumulative lifetime risk of prostate cancer to be increased in individuals with Lynch syndrome, ranging from twofold to fivefold higher than in the general population [142,143,144,145].
Loss of MMR protein expression has been shown in prostate cancer tumours in patients with Lynch syndrome [146, 147]; however, this has been rarely detected in patients with hereditary prostate cancer [148], suggesting that Lynch syndrome is unlikely to be implicated in the majority of cases of familial prostate cancer [139]. Furthermore, patients with Lynch syndrome do not appear to have an earlier onset of prostate cancer or a more aggressive phenotype [143].
MSH2
There is some evidence that prostate cancer is more commonly diagnosed in men with an MSH2 mutation compared to men with a mutation in one of the other MMR genes [142, 143, 149,150,151,152]. A German study identified cases of prostate cancer among men who were positive or obligate carriers of MSH2 mutations; however, they found no increased incidence of prostate cancer [153]. The investigators noted a median age of 59 years at diagnosis, younger than the average age at diagnosis, suggesting a marginal association between MSH2 mutation and risk of prostate cancer. Rosty et al. have shown that MMR gene mutation carriers have at least a twofold or greater increased risk of developing MMR-deficient prostate cancer, with the risk being highest for MSH2 mutation carriers [154]. Except for Rosty et al. most studies have been underpowered to observe any differences in prostate cancer risk by specific MMR gene mutations [145]. Large cohorts will be required to measure separate prostate cancer risks for specific MMR gene mutation carriers.
Fanconi Anaemia
Fanconi anaemia (FA) is a rare disorder of chromosomal instability characterized by bone marrow failure, developmental anomalies and an increased incidence of myelodysplasia, leukaemia and solid tumours [155, 156]. The prevalence of FA is 1–5 cases per 1 million persons, and the heterozygous carrier frequency is about 1 case per 300 persons [157]. Germline mutations, somatic mutations and epigenetic silencing have all been shown to occur in FA genes [158]. FA is caused by biallelic mutation of any 1 of the 16 known genes and can be either autosomal or X-linked recessive, depending on the inherited gene. Of the 16 genes (FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, FANCN/PALB2, FANCJ/BRIP1, FANCO/RAD51C, FANCP/SLX4 and FANCQ/ERCC4), three of them, FANCD1, FANCN and FANCJ, are identical to the DNA repair genes BRCA2, PALB2 and BRIP1 [159,160,161]. The protein products of these genes function cooperatively in the FA-BRCA pathway which plays a central role in DNA repair and the maintenance of genomic integrity [159].
The FA Pathway
After DNA damage, FA proteins form a nuclear complex that mediates the monoubiquitylation of the FA protein FANCD2 [159,160,161]. This monoubiquitylated FANCD2 colocalizes in nuclear foci with proteins involved in DNA repair, including BRCA1, FANCD1/BRCA2, FANCN/PALB2 and RAD51 [161,162,163,164,165,166,167,168]. FANCJ interacts directly with BRCA1 and is a member of the DNA helicase family [169]. FANCN interacts with FANCD1/BRCA2 and is required for its homologous recombination and checkpoint functions [170]. In the absence of an intact FA pathway, cells are sensitive to spontaneous and DNA damage-induced chromosomal breaks leading to tumourigenesis [171]. Clinical trials are now testing the use of PARP inhibitors in patients with FA pathway defects [172].
FA Genes and Prostate Cancer
Initial studies of cancer risk in FA heterozygotes found a higher rate of cancers; however, subsequent studies have not confirmed this risk [173,174,175,176]. Due to the conflicting data from other studies and the relative rarity of FA, it is difficult to confirm these findings. In a British study of FA families, there was no higher incidence of cancer detected; however, 2 prostate cancer cases were observed in 33 obligate carriers, with an overall relative risk of prostate cancer in carriers which was calculated to be 3.089, an incidence which was higher than expected [177]. In a founder population cohort study of Finnish FA patients, the prevalence of 6 FA-causing mutations in over 1800 breast cancer and 565 prostate cancer cases was analysed [178]. All mutations were recurrent, but no significant association with cancer susceptibility was observed for any. Further analysis from the prostate cancer cohort revealed several carriers both among affected and unaffected males, but the frequencies were roughly the same and without any statistical significance. Although clearly deleterious, the tested heterozygous mutations in the FA pathway do not act as high- or moderate-risk alleles for prostate cancer in the general population; however, there could be a modest increased risk in prostate cancer in some FA heterozygotes which merits further investigation in larger cohort studies [179].
DNA Adducts
Polycyclic aromatic hydrocarbons (PAH) and heteroc yclic amines (HCA) are environmental contaminants and known carcinogens (1). PAHs and HCAs are thought to derive their carcinogenic properties through their ability to form DNA adducts. 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) is the major HCA generated from cooking meats at high temperatures, and exposure has been shown to induce prostate cancer in animal studies. PhIP induces cancer by the formation of PhIP-DNA adducts [180]. This formation of DNA adducts can lead to DNA replication errors and increase the potential for carcinogenesis. DNA adducts have been detected in prostate cells, but the exact nature of adducts with respect to prostate cancer risk factors and histology is unclear [181]. African ancestry is strongly associated with PhIP-DNA adduct levels in non-tumour prostate cells [182]. Furthermore, the SULT1A1 genotype and enzyme activity has been suggested to be associated with DNA adduct levels and ethnicity [183]. However, further studies indicate the SULT1A1 genotype does not appear to be associated with increased genetic susceptibility to prostate cancer [182, 184], and overall, elevated levels of PhIP-DNA adducts do not appear to significantly increase prostate cancer risk, independent of ethnicity [185].
Conclusion
Case-control studies, linkage analyses , admixture mapping and GWAS have identified a number of candidate genes associated with prostate cancer susceptibility. Similarly, studies of ethnic and founder populations have identified inherited genetic factors associated with a higher risk of prostate cancer. However, the replication of these findings remains inconsistent, with numerous genes likely to be involved. GWAS have provided evidence supporting the genetic complexity of prostate cancer. It is also likely that there could be significant variation in the contribution of various genes and SNPs to prostate cancer risk in various ethnic groups. Additional studies will be required to determine whether genes or SNPs can be combined with PSA levels and other clinical factors to identify men who are at particularly high risk of being diagnosed with prostate cancer. The finding that the FA–BRCA pathway is intimately involved in the response to DNA damage and repair and may confer potential susceptibility to prostate cancer has spurred further research in this area. Furthermore, tumour cells with a disrupted DNA repair pathway are hypersensitive to PARP inhibitors, and these agents have been shown to be efficacious in prostate cancer. Moving forward, genes identified through GWAS may eventually have a role in prostate cancer screening and as targets for therapeutic targets.
References
Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N Engl J Med. 2003;349:366–81.
Bartsch G, Horninger W, Klocker H, Reissigl A, Oberaigner W, Schonitzer D, Severi G, Robertson C, Boyle P. Prostate cancer mortality after introduction of prostate-specific antigen mass screening in the Federal State of Tyrol, Austria. Urology. 2001;58:417–24.
Gann PH. Risk factors for prostate cancer. Rev Urol. 2002;4:S3–S10.
Eeles R, Goh C, Castro E, Bancroft E, Guy M, Olama AAA, Easton D, Kote-Jarai Z. The genetic epidemiology of prostate cancer and its clinical implications. Nat Rev Urol. 2014;11:18–31.
Watkins Bruner D, Moore D, Parlanti A, Dorgan J, Engstrom P. Relative risk of prostate cancer for men with affected relatives: systematic review and meta-analysis. Int J Cancer. 2003;107:797–803.
Zeegers MP, Jellema A, Ostrer H. Empiric risk of prostate carcinoma for relatives of patients with prostate carcinoma: a meta-analysis. Cancer. 2003;97:1894–903.
Hemminki K, Czene K. Age specific and attributable risks of familial prostate carcinoma from the family-cancer database. Cancer. 2002;95:1346–53.
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85.
Hemminki K, Ji J, Forsti A, Sundquist J, Lenner P. Concordance of survival in family members with prostate cancer. J Clin Oncol. 2008;26:1705–9.
Kalish LA, McDougal WS, McKinlay JB. Family history and the risk of prostate cancer. Urology. 2000;56:803–6.
Cerhan JR, Parker AS, Putnam SD, Chiu BC, Lynch CF, Cohen MB, Torner JC, Cantor KP. Family history and prostate cancer risk in a population-based cohort of Iowa men. Cancer Epidemiol Biomarkers Prev. 1999;8:53–60.
Bratt O, Garmo H, Adolfsson J, Bill-Axelson A, Holmberg L, Lambe M, Stattin P. Effects of prostate-specific antigen testing on familial prostate cancer risk estimates. J Natl Cancer Inst. 2010;102:1336–43.
Steinberg GD, Carter BS, Beaty TH, Childs B, Walsh PC. Family history and the risk of prostate cancer. Prostate. 1990;17:337–47.
Carter BS, Beaty TH, Steinberg GD, Childs B, Walsh PC. Mendelian inheritance of familial prostate cancer. Proc Natl Acad Sci U S A. 1992;89:3367–71.
Ghadirian P, Howe GR, Hislop TG, Maisonneuve P. Family history of prostate cancer: a multi-center case-control study in Canada. Int J Cancer. 1997;70:679–81.
Smith JR, Freije D, Carpten JD, Gronberg H, Xu J, Isaacs SD, Brownstein MJ, Bova GS, Guo H, Bujnovszky P, et al. Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science. 1996;274:1371–4.
Berthon P, Valeri A, Cohen-Akenine A, Drelon E, Paiss T, Wohr G, Latil A, Millasseau P, Mellah I, Cohen N, et al. Predisposing gene for early-onset prostate cancer, localized on chromosome 1q42.2-43. Am J Hum Genet. 1998;62:1416–24.
Xu J, Meyers D, Freije D, Isaacs S, Wiley K, Nusskern D, Ewing C, Wilkens E, Bujnovszky P, Bova GS, et al. Evidence for a prostate cancer susceptibility locus on the X chromosome. Nat Genet. 1998;20:175–9.
Gibbs M, Stanford JL, McIndoe RA, Jarvik GP, Kolb S, Goode EL, Chakrabarti L, Schuster EF, Buckley VA, Miller EL, et al. Evidence for a rare prostate cancer-susceptibility locus at chromosome 1p36. Am J Hum Genet. 1999;64:776–87.
Berry R, Schroeder JJ, French AJ, McDonnell SK, Peterson BJ, Cunningham JM, Thibodeau SN, Schaid DJ. Evidence for a prostate cancer-susceptibility locus on chromosome 20. Am J Hum Genet. 2000;67:82–91.
Tavtigian SV, Simard J, Teng DH, Abtin V, Baumgard M, Beck A, Camp NJ, Carillo AR, Chen Y, Dayananth P, et al. A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet. 2001;27:172–80.
Lu L, Cancel-Tassin G, Valeri A, Cussenot O, Lange EM, Cooney KA, Farnham JM, Camp NJ, Cannon-Albright LA, Tammela TL, et al. Chromosomes 4 and 8 implicated in a genome wide SNP linkage scan of 762 prostate cancer families collected by the ICPCG. Prostate. 2012;72:410–26.
Xu J, Zheng SL, Komiya A, Mychaleckyj JC, Isaacs SD, Hu JJ, Sterling D, Lange EM, Hawkins GA, Turner A, et al. Germline mutations and sequence variants of the macrophage scavenger receptor 1 gene are associated with prostate cancer risk. Nat Genet. 2002;32:321–5.
Chang BL, Liu W, Sun J, Dimitrov L, Li T, Turner AR, Zheng SL, Isaacs WB, Xu J. Integration of somatic deletion analysis of prostate cancers and germline linkage analysis of prostate cancer families reveals two small consensus regions for prostate cancer genes at 8p. Cancer Res. 2007;67:4098–103.
Xu J, Dimitrov L, Chang BL, Adams TS, Turner AR, Meyers DA, Eeles RA, Easton DF, Foulkes WD, Simard J, et al. A combined genomewide linkage scan of 1,233 families for prostate cancer-susceptibility genes conducted by the international consortium for prostate cancer genetics. Am J Hum Genet. 2005;77:219–29.
El Gammal AT, Bruchmann M, Zustin J, Isbarn H, Hellwinkel OJ, Kollermann J, Sauter G, Simon R, Wilczak W, Schwarz J, et al. Chromosome 8p deletions and 8q gains are associated with tumor progression and poor prognosis in prostate cancer. Clin Cancer Res. 2010;16:56–64.
Edwards BK, Brown ML, Wingo PA, Howe HL, Ward E, Ries LA, Schrag D, Jamison PM, Jemal A, XC W, et al. Annual report to the nation on the status of cancer, 1975-2002, featuring population-based trends in cancer treatment. J Natl Cancer Inst. 2005;97:1407–27.
Freedman ML, Haiman CA, Patterson N, McDonald GJ, Tandon A, Waliszewska A, Penney K, Steen RG, Ardlie K, John EM, et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African—American men. Proc Natl Acad Sci U S A. 2006;103:14068–73.
Han Y, Signorello LB, Strom SS, Kittles RA, Rybicki BA, Stanford JL, Goodman PJ, Berndt SI, Carpten J, Casey G, et al. Generalizability of established prostate cancer risk variants in men of African ancestry. Int J Cancer. 2015;136:1210–7.
Bock CH, Schwartz AG, Ruterbusch JJ, Levin AM, Neslund-Dudas C, Land SJ, Wenzlaff AS, Reich D, McKeigue P, Chen W, et al. Results from a prostate cancer admixture mapping study in African-American men. Hum Genet. 2009;126:637–42.
Kirchhoff T, Kauff ND, Mitra N, Nafa K, Huang H, Palmer C, Gulati T, Wadsworth E, Donat S, Robson ME, et al. BRCA mutations and risk of prostate cancer in Ashkenazi Jews. Clin Cancer Res. 2004;10:2918–21.
Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20.
Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast cancer linkage consortium. Lancet. 1994;343:692–5.
Edwards SM, Kote-Jarai Z, Meitz J, Hamoudi R, Hope Q, Osin P, Jackson R, Southgate C, Singh R, Falconer A, et al. Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet. 2003;72:1–12.
Dong X, Wang L, Taniguchi K, Wang X, Cunningham JM, McDonnell SK, Qian C, Marks AF, Slager SL, Peterson BJ, et al. Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet. 2003;72:270–80.
Hale V, Weischer M, Park JY. CHEK2 ( *) 1100delC mutation and risk of prostate cancer. Prostate Cancer. 2014;294575:6.
Abate-Shen C, Shen MM, Gelmann E. Integrating differentiation and cancer: the Nkx3.1 homeobox gene in prostate organogenesis and carcinogenesis. Differentiation. 2008;76:717–27.
Callen DF, Ricciardelli C, Butler M, Stapleton A, Stahl J, Kench JG, Horsfall DJ, Tilley WD, Schulz R, Nesland JM, et al. Co-expression of the androgen receptor and the transcription factor ZNF652 is related to prostate cancer outcome. Oncol Rep. 2010;23:1045–52.
Olsson P, Motegi A, Bera TK, Lee B, Pastan I. PRAC2: a new gene expressed in human prostate and prostate cancer. Prostate. 2003;56:123–30.
Nurminen R, Lehtonen R, Auvinen A, Tammela TL, Wahlfors T, Schleutker J. Fine mapping of 11q13.5 identifies regions associated with prostate cancer and prostate cancer death. Eur J Cancer. 2013;49:3335–43.
Chiam K, Ryan NK, Ricciardelli C, Day TK, Buchanan G, Ochnik AM, Murti K, Selth LA, Butler LM, Tilley WD, Bianco-Miotto T. Characterization of the prostate cancer susceptibility gene KLF6 in human and mouse prostate cancers. Prostate. 2013;73:182–93.
FitzGerald LM, Thomson R, Polanowski A, Patterson B, McKay JD, Stankovich J, Dickinson JL. Sequence variants of alpha-methylacyl-CoA racemase are associated with prostate cancer risk: a replication study in an ethnically homogeneous population. Prostate. 2008;68:1373–9.
Cybulski C, Wokolorczyk D, Kluzniak W, Jakubowska A, Gorski B, Gronwald J, Huzarski T, Kashyap A, Byrski T, Debniak T, et al. An inherited NBN mutation is associated with poor prognosis prostate cancer. Br J Cancer. 2013;108:461–8.
Cicek MS, Conti DV, Curran A, Neville PJ, Paris PL, Casey G, Witte JS. Association of prostate cancer risk and aggressiveness to androgen pathway genes: SRD5A2, CYP17, and the AR. Prostate. 2004;59:69–76.
Holt SK, Kwon EM, Fu R, Kolb S, Feng Z, Ostrander EA, Stanford JL. Association of variants in estrogen-related pathway genes with prostate cancer risk. Prostate. 2013;73:1–10.
Qiu LX, Li RT, Zhang JB, Zhong WZ, Bai JL, Liu BR, Zheng MH, Qian XP. The E-cadherin (CDH1)—160 C/A polymorphism and prostate cancer risk: a meta-analysis. Eur J Hum Genet. 2009;17:244–9.
Hoffmann TJ, Passarelli MN, Graff RE, Emami NC, Sakoda LC, Jorgenson E, Habel LA, Shan J, Ranatunga DK, Quesenberry CP, et al. Genome-wide association study of prostate-specific antigen levels identifies novel loci independent of prostate cancer. Nat Commun. 2017;8:14248.
Berndt SI, Wang Z, Yeager M, Alavanja MC, Albanes D, Amundadottir L, Andriole G, Beane Freeman L, Campa D, Cancel-Tassin G, et al. Two susceptibility loci identified for prostate cancer aggressiveness. Nat Commun. 2015;6:6889.
Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, Mulholland S, Leongamornlert DA, Edwards SM, Morrison J, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–21.
Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, Ghoussaini M, Luccarini C, Dennis J, Jugurnauth-Little S, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet. 2013;45:385–91.
Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–5.
Sun J, Zheng SL, Wiklund F, Isaacs SD, Purcell LD, Gao Z, Hsu FC, Kim ST, Liu W, Zhu Y, et al. Evidence for two independent prostate cancer risk-associated loci in the HNF1B gene at 17q12. Nat Genet. 2008;40:1153–5.
Yeager M, Deng Z, Boland J, Matthews C, Bacior J, Lonsberry V, Hutchinson A, Burdett LA, Qi L, Jacobs KB, et al. Comprehensive resequence analysis of a 97 kb region of chromosome 10q11.2 containing the MSMB gene associated with prostate cancer. Hum Genet. 2009;126:743–50.
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71.
Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265:2088–90.
Dan R, Van Allen EM, Y-M W, Schultz N, Lonigro RJ, Mosquera J-M, Montgomery B, Taplin M-E, Pritchard CC, Attard G, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28.
Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, King MC. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250:1684.
Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864–74.
Boulton SJ. Cellular functions of the BRCA tumour-suppressor proteins. Biochem Soc Trans. 2006;34:633–45.
Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–82.
Turner N, Tutt A, Ashworth A. Hallmarks of 'BRCAness' in sporadic cancers. Nat Rev Cancer. 2004;4:814–9.
Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329–33.
Agalliu I, Gern R, Leanza S, Burk RD. Associations of high-grade prostate cancer with BRCA1 and BRCA2 founder mutations. Clin Cancer Res. 2009;15:1112–20.
Gallagher DJ, Gaudet MM, Pal P, Kirchhoff T, Balistreri L, Vora K, Bhatia J, Stadler Z, Fine SW, Reuter V, et al. Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res. 2010;16:2115–21.
Thompson D, Easton D. Variation in cancer risks, by mutation position, in BRCA2 mutation carriers. Am J Hum Genet. 2001;68:410–9.
Giusti RM, Rutter JL, Duray PH, Freedman LS, Konichezky M, Fisher-Fischbein J, Greene MH, Maslansky B, Fischbein A, Gruber SB, et al. A twofold increase in BRCA mutation related prostate cancer among Ashkenazi Israelis is not associated with distinctive histopathology. J Med Genet. 2003;40:787–92.
Eerola H, Pukkala E, Pyrhönen S, Blomqvist C, Sankila R, Nevanlinna H. Risk of cancer in BRCA1 and BRCA2 mutation-positive and -negative breast cancer families (Finland). Cancer Causes Control. 2001;12:739–46.
Leongamornlert D, Mahmud N, Tymrakiewicz M, Saunders E, Dadaev T, Castro E, Goh C, Govindasami K, Guy M, O'Brien L, et al. Germline BRCA1 mutations increase prostate cancer risk. Br J Cancer. 2012;106:1697–701.
Johannesdottir G, Gudmundsson J, Bergthorsson JT, Arason A, Agnarsson BA, Eiriksdottir G, Johannsson OT, Borg A, Ingvarsson S, Easton DF, et al. High prevalence of the 999del5 mutation in icelandic breast and ovarian cancer patients. Cancer Res. 1996;56:3663–5.
Kote-Jarai Z, Leongamornlert D, Saunders E, Tymrakiewicz M, Castro E, Mahmud N, Guy M, Edwards S, O'Brien L, Sawyer E, et al. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: implications for genetic testing in prostate cancer patients. Br J Cancer. 2011;105:1230–4.
Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91:1310–6.
Agalliu I, Kwon EM, Zadory D, McIntosh L, Thompson J, Stanford JL, Ostrander EA. Germline mutations in the BRCA2 gene and susceptibility to hereditary prostate cancer. Clin Cancer Res. 2007;13:839–43.
Thompson D, Easton DF. Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358–65.
Mitra A, Fisher C, Foster CS, Jameson C, Barbachanno Y, Bartlett J, Bancroft E, Doherty R, Kote-Jarai Z, Peock S, et al. Prostate cancer in male BRCA1 and BRCA2 mutation carriers has a more aggressive phenotype. Br J Cancer. 2008;98:502–7.
Castro E, Goh C, Leongamornlert D, Saunders E, Tymrakiewicz M, Dadaev T, Govindasami K, Guy M, Ellis S, Frost D, et al. Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localised prostate cancer. Eur Urol. 2015;68:186–93.
Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, Mahmud N, Dadaev T, Govindasami K, Guy M, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31:1748–57.
Tryggvadottir L, Vidarsdottir L, Thorgeirsson T, Jonasson JG, Olafsdottir EJ, Olafsdottir GH, Rafnar T, Thorlacius S, Jonsson E, Eyfjord JE, Tulinius H. Prostate cancer progression and survival in BRCA2 mutation carriers. J Natl Cancer Inst. 2007;99:929–35.
Narod SA, Neuhausen S, Vichodez G, Armel S, Lynch HT, Ghadirian P, Cummings S, Olopade O, Stoppa-Lyonnet D, Couch F, et al. Rapid progression of prostate cancer in men with a BRCA2 mutation. Br J Cancer. 2008;99:371–4.
Bancroft EK, Page EC, Castro E, Lilja H, Vickers A, Sjoberg D, Assel M, Foster CS, Mitchell G, Drew K, et al. Targeted prostate cancer screening in BRCA1 and BRCA2 mutation carriers: results from the initial screening round of the IMPACT study. Eur Urol. 2014;66:489–99.
Francis JC, McCarthy A, Thomsen MK, Ashworth A, Swain A. Brca2 and Trp53 deficiency cooperate in the progression of mouse prostate tumourigenesis. PLoS Genet. 2010;6:e1000995.
Bednarz N, Eltze E, Semjonow A, Rink M, Andreas A, Mulder L, Hannemann J, Fisch M, Pantel K, Weier HU, et al. BRCA1 loss preexisting in small subpopulations of prostate cancer is associated with advanced disease and metastatic spread to lymph nodes and peripheral blood. Clin Cancer Res. 2010;16:3340–8.
Moro L, Arbini AA, Yao JL, di Sant'Agnese PA, Marra E, Greco M. Loss of BRCA2 promotes prostate cancer cell invasion through up-regulation of matrix metalloproteinase-9. Cancer Sci. 2008;99:553–63.
Schmidt H, DeAngelis G, Eltze E, Gockel I, Semjonow A, Brandt B. Asynchronous growth of prostate cancer is reflected by circulating tumor cells delivered from distinct, even small foci, harboring loss of heterozygosity of the PTEN gene. Cancer Res. 2006;66:8959–65.
Lee JM, Ledermann JA, Kohn EC. PARP inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25:32–40.
O’Sullivan CC, Moon DH, Kohn EC, Lee J-M. Beyond breast and ovarian cancers: PARP inhibitors for BRCA mutation-associated and BRCA-like solid tumors. Front Oncol. 2014;4:42.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
Howard SM, Yanez DA, Stark JM. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015;11:e1004943.
Beck C, Boehler C, Guirouilh Barbat J, Bonnet M-E, Illuzzi G, Ronde P, Gauthier LR, Magroun N, Rajendran A, Lopez BS, et al. PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res. 2014;42:5616–32.
Lombardi AJ, Hoskins EE, Foglesong GD, Wikenheiser-Brokamp KA, Wiesmuller L, Hanenberg H, Andreassen PR, Jacobs AJ, Olson SB, Keeble WW, et al. Acquisition of relative interstrand crosslinker resistance and PARP inhibitor sensitivity in Fanconi Anemia head and neck cancers. Clin Cancer Res. 2015;21:21.
Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, Patel S, Wang X, Liang H, Yu J, et al. Mechanistic rationale for inhibition of Poly(ADP-Ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–78.
Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, Liu F, Planck JL, Ravindranathan P, Chinnaiyan AM, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–49.
Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14:882–92.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. DNA-repair defects and Olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–708.
Konecny GE, Kristeleit RS. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: current practice and future directions. Br J Cancer. 2016;115:1157–73.
Li R, Wheeler T, Dai H, Frolov A, Thompson T, Ayala G. High level of androgen receptor is associated with aggressive clinicopathologic features and decreased biochemical recurrence-free survival in prostate: cancer patients treated with radical prostatectomy. Am J Surg Pathol. 2004;28:928–34.
Fromont G, Yacoub M, Valeri A, Mangin P, Vallancien G, Cancel-Tassin G, Cussenot O. Differential expression of genes related to androgen and estrogen metabolism in hereditary versus sporadic prostate cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:1505–9.
Giovannucci E, Stampfer MJ, Krithivas K, Brown M, Dahl D, Brufsky A, Talcott J, Hennekens CH, Kantoff PW. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci U S A. 1997;94:3320–3.
Lange EM, Sarma AV, Ray A, Wang Y, Ho LA, Anderson SA, Cunningham JM, Cooney KA. The androgen receptor CAG and GGN repeat polymorphisms and prostate cancer susceptibility in African-American men: results from the Flint Men's Health Study. J Hum Genet. 2008;53:220–6.
Mononen N, Syrjakoski K, Matikainen M, Tammela TL, Schleutker J, Kallioniemi OP, Trapman J, Koivisto PA. Two percent of Finnish prostate cancer patients have a germ-line mutation in the hormone-binding domain of the androgen receptor gene. Cancer Res. 2000;60:6479–81.
Koivisto PA, Hyytinen ER, Matikainen M, Tammela TL, Ikonen T, Schleutker J. Germline mutation analysis of the androgen receptor gene in Finnish patients with prostate cancer. J Urol. 2004;171:431–3.
Squire J, Zhou A, Hassel BA, Nie H, Silverman RH. Localization of the interferon-induced, 2-5A-dependent RNase gene (RNS4) to human chromosome 1q25. Genomics. 1994;19:174–5.
Malathi K, Paranjape JM, Ganapathi R, Silverman RH. HPC1/RNASEL mediates apoptosis of prostate cancer cells treated with 2′,5′-oligoadenylates, topoisomerase I inhibitors, and tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2004;64:9144–51.
Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J, Faruque M, Moses T, Ewing C, Gillanders E, et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet. 2002;30:181–4.
Casey G, Neville PJ, Plummer SJ, Xiang Y, Krumroy LM, Klein EA, Catalona WJ, Nupponen N, Carpten JD, Trent JM, et al. RNASEL Arg462Gln variant is implicated in up to 13% of prostate cancer cases. Nat Genet. 2002;32:581–3.
Rennert H, Bercovich D, Hubert A, Abeliovich D, Rozovsky U, Bar-Shira A, Soloviov S, Schreiber L, Matzkin H, Rennert G, et al. A novel founder mutation in the RNASEL gene, 471delAAAG, is associated with prostate cancer in Ashkenazi Jews. Am J Hum Genet. 2002;71:981–4.
Robbins CM, Hernandez W, Ahaghotu C, Bennett J, Hoke G, Mason T, Pettaway CA, Vijayakumar S, Weinrich S, Furbert-Harris P, et al. Association of HPC2/ELAC2 and RNASEL non-synonymous variants with prostate cancer risk in African American familial and sporadic cases. Prostate. 2008;68:1790–7.
Orr-Urtreger A, Bar-Shira A, Bercovich D, Matarasso N, Rozovsky U, Rosner S, Soloviov S, Rennert G, Kadouri L, Hubert A, et al. RNASEL mutation screening and association study in Ashkenazi and non-Ashkenazi prostate cancer patients. Cancer Epidemiol Biomarkers Prev. 2006;15:474–9.
Li H, Tai BC. RNASEL gene polymorphisms and the risk of prostate cancer: a meta-analysis. Clin Cancer Res. 2006 Oct 1;12(19):5713–9.
Meyer MS, Penney KL, Stark JR, Schumacher FR, Sesso HD, Loda M, Fiorentino M, Finn S, Flavin RJ, Kurth T, et al. Genetic variation in RNASEL associated with prostate cancer risk and progression. Carcinogenesis. 2010;31:1597–603.
Chaux A, Albadine R, Toubaji A, Hicks J, Meeker A, Platz EA, De Marzo AM, Netto GJ. Immunohistochemistry for ERG expression as a surrogate for TMPRSS2-ERG fusion detection in prostatic adenocarcinomas. Am J Surg Pathol. 2011;35:1014–20.
Nishijima J, Hara T, Ikemoto K, Oga A, Kobayashi K, Kawai Y, Matsumoto H, Nagao K, Sasaki K, Gkoleizakis V, et al. Clinical significance of ERG rearrangement subtype and its association with increased p53 expression in Japanese and German prostate cancer. Neoplasma. 2015;62:278–87.
Wang J, Cai Y, Ren C, Ittmann M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006;66:8347–51.
Luedeke M, Linnert CM, Hofer MD, Surowy HM, Rinckleb AE, Hoegel J, Kuefer R, Rubin MA, Vogel W, Maier C. Predisposition for TMPRSS2-ERG fusion in prostate cancer by variants in DNA repair genes. Cancer Epidemiol Biomarkers Prev. 2009;18:3030–5.
Hofer MD, Kuefer R, Maier C, Herkommer K, Perner S, Demichelis F, Paiss T, Vogel W, Rubin MA, Hoegel J. Genome-wide linkage analysis of TMPRSS2-ERG fusion in familial prostate cancer. Cancer Res. 2009;69:640–6.
Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, Varambally S, Palanisamy N, Chinnaiyan AM. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230.
Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, Tembe WD, Wiley KE, Isaacs SD, Johng D, Wang Y, et al. Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med. 2012;366:141–9.
Gudmundsson J, Sulem P, Gudbjartsson DF, Masson G, Agnarsson BA, Benediktsdottir KR, Sigurdsson A, Magnusson OT, Gudjonsson SA, Magnusdottir DN, et al. A study based on whole-genome sequencing yields a rare variant at 8q24 associated with prostate cancer. Nat Genet. 2012;44:1326–9.
Stott-Miller M, Karyadi DM, Smith T, Kwon EM, Kolb S, Stanford JL, Ostrander EA. HOXB13 mutations in a population-based, case-control study of prostate cancer. Prostate. 2013;73:634–41.
Lange EM, Gillanders EM, Davis CC, Brown WM, Campbell JK, Jones M, Gildea D, Riedesel E, Albertus J, Freas-Lutz D, et al. Genome-wide scan for prostate cancer susceptibility genes using families from the University of Michigan prostate cancer genetics project finds evidence for linkage on chromosome 17 near BRCA1. Prostate. 2003;57:326–34.
Zuhlke KA, Madeoy JJ, Beebe-Dimmer J, White KA, Griffin A, Lange EM, Gruber SB, Ostrander EA, Cooney KA. Truncating BRCA1 mutations are uncommon in a cohort of hereditary prostate cancer families with evidence of linkage to 17q markers. Clin Cancer Res. 2004;10:5975–80.
Xu J, Lange EM, Lu L, Zheng SL, Wang Z, Thibodeau SN, Cannon-Albright LA, Teerlink CC, Camp NJ, Johnson AM, et al. HOXB13 is a susceptibility gene for prostate cancer: results from the International Consortium for Prostate Cancer Genetics (ICPCG). Hum Genet. 2013;132:5–14.
Breyer JP, Avritt TG, McReynolds KM, Dupont WD, Smith JR. Confirmation of the HOXB13 G84E Germline mutation in familial prostate cancer. Cancer Epidemiol Biomarkers Prev. 2012;21:1348–53.
Akbari MR, Trachtenberg J, Lee J, Tam S, Bristow R, Loblaw A, Narod SA, Nam RK. Association between germline HOXB13 G84E mutation and risk of prostate cancer. J Natl Cancer Inst. 2012;104:1260–2.
Chen Z, Greenwood C, Isaacs WB, Foulkes WD, Sun J, Zheng SL, Condreay LD, Xu J. The G84E mutation of HOXB13 is associated with increased risk for prostate cancer: results from the REDUCE trial. Carcinogenesis. 2013;34:1260–4.
Laitinen VH, Wahlfors T, Saaristo L, Rantapero T, Pelttari LM, Kilpivaara O, Laasanen S-L, Kallioniemi A, Nevanlinna H, Aaltonen L, et al. HOXB13 G84E mutation in Finland: population-based analysis of prostate, breast, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2013;22:452–60.
Kote-Jarai Z, Mikropoulos C, Leongamornlert DA, Dadaev T, Tymrakiewicz M, Saunders EJ, Jones M, Jugurnauth-Little S, Govindasami K, Guy M, et al. Prevalence of the HOXB13 G84E germline mutation in British men and correlation with prostate cancer risk, tumour characteristics and clinical outcomes. Ann Oncol. 2015;26(4):756–61.
Karlsson R, Aly M, Clements M, Zheng L, Adolfsson J, Xu J, Gronberg H, Wiklund F. A population-based assessment of germline HOXB13 G84E mutation and prostate cancer risk. Eur Urol. 2014;65:169–76.
MacInnis RJ, Severi G, Baglietto L, Dowty JG, Jenkins MA, Southey MC, Hopper JL, Giles GG. Population-based estimate of prostate cancer risk for carriers of the HOXB13 missense mutation G84E. PLoS One. 2013;8:15.
Hoffmann TJ, Sakoda LC, Shen L, Jorgenson E, Habel LA, Liu J, Kvale MN, Asgari MM, Banda Y, Corley D, et al. Imputation of the rare HOXB13 G84E mutation and cancer risk in a large population-based cohort. PLoS Genet. 2015;11:e1004930.
Huang Q, Whitington T, Gao P, Lindberg JF, Yang Y, Sun J, Vaisanen M-R, Szulkin R, Annala M, Yan J, et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat Genet. 2014;46:126–35.
Payton S. Prostate cancer: HOXB13 and a SNP collaborate to increase risk. Nat Rev Urol. 2014;11:64.
Zabalza CV, Adam M, Burdelski C, Wilczak W, Wittmer C, Kraft S, Krech T, Steurer S, Koop C, Hube-Magg C, et al. HOXB13 overexpression is an independent predictor of early PSA recurrence in prostate cancer treated by radical prostatectomy. Oncotarget. 2015;6(14):12822–34.
Papadopoulos N, Lindblom A. Molecular basis of HNPCC: mutations of MMR genes. Hum Mutat. 1997;10:89–99.
The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Pawlik TM, Raut CP, Rodriguez-Bigas MA. Colorectal carcinogenesis: MSI-H versus MSI-L. Dis Markers. 2004;20:199–206.
Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomaki P, Mecklin JP, Jarvinen HJ. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–8.
Jarvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomaki P, De La Chapelle A, Mecklin JP. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–34.
Schmeler KM, Lynch HT, Chen LM, Munsell MF, Soliman PT, Clark MB, Daniels MS, White KG, Boyd-Rogers SG, Conrad PG, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354:261–9.
Ryan S, Jenkins MA, Win AK. Risk of prostate cancer in lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2014;23:437–49.
Pande M, Wei C, Chen J, Amos CI, Lynch PM, KH L, Lucio LA, Boyd-Rogers SG, Bannon SA, Mork ME, Frazier ML. Cancer spectrum in DNA mismatch repair gene mutation carriers: results from a hospital based Lynch syndrome registry. Fam Cancer. 2012;11:441–7.
Scott RJ, McPhillips M, Meldrum CJ, Fitzgerald PE, Adams K, Spigelman AD, du Sart D, Tucker K, Kirk J. Hereditary nonpolyposis colorectal cancer in 95 families: differences and similarities between mutation-positive and mutation-negative kindreds. Am J Hum Genet. 2001;68:118–27.
Raymond VM, Mukherjee B, Wang F, Huang SC, Stoffel EM, Kastrinos F, Syngal S, Cooney KA, Gruber SB. Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol. 2013;31:1713–8.
Haraldsdottir S, Hampel H, Wei L, Wu C, Frankel W, Bekaii-Saab T, de la Chapelle A, Goldberg RM. Prostate cancer incidence in males with Lynch syndrome. Genet Med. 2014;16:553–7.
Dominguez-Valentin M, Joost P, Therkildsen C, Jonsson M, Rambech E, Nilbert M. Frequent mismatch-repair defects link prostate cancer to Lynch syndrome. BMC Urol. 2016;16:15.
da Silva FC, de Oliveira LP, Santos ÉM, Nakagawa WT, Junior SA, Valentin MD, Rossi BM, de Oliveira Ferreira F. Frequency of extracolonic tumors in Brazilian families with Lynch syndrome: analysis of a hereditary colorectal cancer institutional registry. Fam Cancer. 2010;9:563–70.
Grindedal EM, Moller P, Eeles R, Stormorken AT, Bowitz-Lothe IM, Landro SM, Clark N, Kvale R, Shanley S, Maehle L. Germ-line mutations in mismatch repair genes associated with prostate cancer. Cancer Epidemiol Biomarkers Prev. 2009;18:2460–7.
Bauer CM, Ray AM, Halstead-Nussloch BA, Dekker RG, Raymond VM, Gruber SB, Cooney KA. Hereditary prostate cancer as a feature of Lynch syndrome. Fam Cancer. 2011;10:37–42.
Ahman AK, Jonsson BA, Damber JE, Bergh A, Gronberg H. Low frequency of microsatellite instability in hereditary prostate cancer. BJU Int. 2001;87:334–8.
Win AK, Lindor NM, Young JP, Macrae FA, Young GP, Williamson E, Parry S, Goldblatt J, Lipton L, Winship I, et al. Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J Natl Cancer Inst. 2012;104:1363–72.
Engel C, Loeffler M, Steinke V, Rahner N, Holinski-Feder E, Dietmaier W, Schackert HK, Goergens H, von Knebel Doeberitz M, Goecke TO, et al. Risks of less common cancers in proven mutation carriers with lynch syndrome. J Clin Oncol. 2012;30:4409–15.
Maul JS, Warner NR, Kuwada SK, Burt RW, Cannon-Albright LA. Extracolonic cancers associated with hereditary nonpolyposis colorectal cancer in the Utah Population Database. Am J Gastroenterol. 2006;101:1591–6.
Maillard F, Manouvrier S, Biardeau X, Ouzzane A, Villers A. Lynch syndrome and risk of prostate cancer; review of the literature. Prog Urol. 2015;29:00002–0.
Goecke T, Schulmann K, Engel C, Holinski-Feder E, Pagenstecher C, Schackert HK, Kloor M, Kunstmann E, Vogelsang H, Keller G, et al. Genotype-phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with lynch syndrome: a report by the German HNPCC consortium. J Clin Oncol. 2006;24:4285–92.
Rosty C, Walsh MD, Lindor NM, Thibodeau SN, Mundt E, Gallinger S, Aronson M, Pollett A, Baron JA, Pearson S, et al. High prevalence of mismatch repair deficiency in prostate cancers diagnosed in mismatch repair gene mutation carriers from the colon cancer family registry. Fam Cancer. 2014;13:573–82.
D'Andrea AD. Susceptibility pathways in Fanconi's anemia and breast cancer. N Engl J Med. 2010;362:1909–19.
Ceccaldi R, Sarangi P, D'Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17:337–49.
Alan D, D'Andrea MD. The Fanconi anemia and breast cancer susceptibility pathways. N Engl J Med. 2010;362:1909–19.
D'Andrea AD. The Fanconi road to cancer. Genes Dev. 2003;17:1933–6.
Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–48.
Boisvert RA, Rego MA, Azzinaro PA, Mauro M, Howlett NG. Coordinate nuclear targeting of the FANCD2 and FANCI proteins via a FANCD2 nuclear localization signal. PLoS One. 2013;8(11):e81387.
Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–408.
Jacquemont C, Taniguchi T. The Fanconi anemia pathway and ubiquitin. BMC Biochem. 2007;22.
Garcia MJ, Benitez J. The Fanconi anaemia/BRCA pathway and cancer susceptibility. Searching for new therapeutic targets. Clin Transl Oncol. 2008;10:78–84.
Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald Iii ER, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ. Identification of the FANCI protein, a Monoubiquitinated FANCD2 paralog required for DNA Repair. Cell. 2007;129:289–301.
Sy SMH, Huen MSY, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci. 2009;106:7155–60.
Moldovan GL, D'Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49.
Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17:331–9.
Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell. 2007;28:798–809.
Wu W, Togashi Y, Johmura Y, Miyoshi Y, Nobuoka S, Nakanishi M, Ohta T. HP1 regulates the localization of FANCJ at sites of DNA double-strand breaks. Cancer Sci. 2016;107:1406–15.
Michl J, Zimmer J, Tarsounas M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016;35:909–23.
Brosh RM Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat Rev Cancer. 2013;13:542–58.
Gojo I, Beumer JH, Pratz KW, McDevitt MA, Baer MR, Blackford AL, Smith BD, Gore SD, Carraway HE, Showel MM, et al. A Phase 1 Study of the PARP Inhibitor Veliparib in Combination with Temozolomide in Acute Myeloid Leukemia. Clin Cancer Res. 2017;23:697–706.
Swift M. Fanconi's anaemia in the genetics of neoplasia. Nature. 1971;230:370–3.
Swift M, Caldwell RJ, Chase C. Reassessment of cancer predisposition of Fanconi anemia heterozygotes. J Natl Cancer Inst. 1980;65:863–7.
Potter NU, Sarmousakis C, Li FP. Cancer in relatives of patients with aplastic anemia. Cancer Genet Cytogenet. 1983;9:61–5.
Berwick M, Satagopan JM, Ben-Porat L, Carlson A, Mah K, Henry R, Diotti R, Milton K, Pujara K, Landers T, et al. Genetic heterogeneity among Fanconi anemia heterozygotes and risk of cancer. Cancer Res. 2007;67:9591–6.
Tischkowitz M, Easton DF, Ball J, Hodgson SV, Mathew CG. Cancer incidence in relatives of British Fanconi Anaemia patients. BMC Cancer. 2008;8:1471–2407.
Mantere T, Haanpaa M, Hanenberg H, Schleutker J, Kallioniemi A, Kahkonen M, Parto K, Avela K, Aittomaki K, von Koskull H, et al. Finnish Fanconi anemia mutations and hereditary predisposition to breast and prostate cancer. Clin Genet. 2014;2:12447.
Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006;25:5875–84.
Tang D, Liu JJ, Rundle A, Neslund-Dudas C, Savera AT, Bock CH, Nock NL, Yang JJ, Rybicki BA. Grilled meat consumption and PhIP-DNA adducts in prostate carcinogenesis. Cancer Epidemiol Biomarkers Prev. 2007;16:803–8.
Rybicki BA, Rundle A, Savera AT, Sankey SS, Tang D. Polycyclic aromatic hydrocarbon-DNA adducts in prostate cancer. Cancer Res. 2004;64:8854–9.
Rybicki BA, Neslund-Dudas C, Bock CH, Nock NL, Rundle A, Jankowski M, Levin AM, Beebe-Dimmer J, Savera AT, Takahashi S, et al. Red Wine Consumption is inversely associated with 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-DNA Adduct Levels in Prostate. Cancer Prev Res (Philadelphia, PA). 2011;4:1636–44.
Nowell S, Ratnasinghe DL, Ambrosone CB, Williams S, Teague-Ross T, Trimble L, Runnels G, Carrol A, Green B, Stone A, et al. Association of SULT1A1 phenotype and genotype with prostate cancer risk in African-Americans and Caucasians. Cancer Epidemiol Biomarkers Prev. 2004;13:270–6.
Koike H, Nakazato H, Ohtake N, Matsui H, Okugi H, Shibata Y, Nakata S, Yamanaka H, Suzuki K. Further evidence for null association of phenol sulfotransferase SULT1A1 polymorphism with prostate cancer risk: a case-control study of familial prostate cancer in a Japanese population. Int Urol Nephrol. 2008;40:947–51.
Tang D, Kryvenko ON, Wang Y, Trudeau S, Rundle A, Takahashi S, Shirai T, Rybicki BA. 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-DNA adducts in benign prostate and subsequent risk for prostate cancer. Int J Cancer. 2013;133:961–71.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Greene, J.P., Finn, S.P. (2018). Genetic Susceptibility. In: Robinson, B., Mosquera, J., Ro, J., Divatia, M. (eds) Precision Molecular Pathology of Prostate Cancer. Molecular Pathology Library. Springer, Cham. https://doi.org/10.1007/978-3-319-64096-9_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-64096-9_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64094-5
Online ISBN: 978-3-319-64096-9
eBook Packages: MedicineMedicine (R0)